

Abstract
Purpose of review:
To summarize recent evidence on the pathogenic effects of neutrophils and neutrophil extracellular traps (NETs) in autoimmune diseases, including systemic lupus erythematosus and rheumatoid arthritis.
Recent findings:
NETs can orchestrate innate and adaptive immune dysregulation through diverse mechanisms. NETs induce potent inflammatory responses and represent sources of many autoantigens, creating a feed-forward loop that may perpetuate disease and lead to organ damage. NETs are also increasingly relevant in atherosclerosis and could contribute to the increased risk of premature cardiovascular disease in patients with autoimmunity.
Summary:
NET formation is increased in a variety of autoimmune and autoinflammatory diseases and can have remarkable effects on cell and tissue-specific pathology. Novel therapeutics that target NET formation or clearance are a promising strategy for clinical management of autoimmune diseases and may prevent chronic complications associated with these conditions.
Keywords: neutrophils, NETs, autoimmunity, lupus, rheumatoid arthritis
Introduction:
Neutrophils are the most abundant leukocytes in human blood and are important effector cells in innate immunity. Neutrophils can kill microorganisms through various strategies, including phagocytosis or degranulation with release of cytotoxic enzymes and proteases. Neutrophils can also engage in a unique form of cell death, resulting in the formation of neutrophil extracellular traps (NETs). During this process, strands of DNA in complex with nuclear, cytoplasmic, and granule proteins are released into the extracellular space where they can neutralize microbes [1]. In addition to their antimicrobial function, NETs have recently been implicated as an important mechanism in the pathogenesis of autoimmune diseases, including systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA). In this review, we discuss new evidence linking NETs to dysregulation of innate and adaptive immunity. Additionally, we discuss potential therapeutic targets for clinical intervention in these diseases.
NET formation:
Several pathways of NET formation, including reactive oxygen species (ROS)-dependent and ROS-independent pathways, have been described and are reviewed in detail elsewhere [2]. Notably, NET formation can involve citrullination, a post-translational modification catalyzed by the protein arginine deiminase (PAD) family enzymes, whereby native arginine residues are converted into citrulline. Citrullination of histones disrupts chromatin structure and promotes decondensation of nuclear material, contributing to the formation of NET structures [3]. Citrullinated proteins, including those contained within NETs, have also been described as prominent autoantigens in RA, and will be discussed in the following sections. Recent studies have identified gasdermin D, a pore-forming protein activated during pyroptosis, as a novel pathway towards NET formation [*4,*5]. Gasdermin D can be cleaved by caspases or neutrophil proteases, leading to nuclear expansion and release of NETs. Further studies, including conditional knockouts in neutrophils and treatment with chemical inhibitors, may be warranted to determine the role of gasdermin D-mediated NET formation in autoimmunity. Interestingly, it appears that different types of stimulation activate different pathways of NET formation. The role of these specific pathways in neutrophil dysregulation and autoimmunity has not been systematically determined.
NETs in systemic lupus erythematosus:
SLE is a complex autoimmune disease that can affect multiple organ systems, including the skin, kidneys, and vasculature. A defining immunological feature of SLE is the production of autoantibodies against nuclear antigens, including nucleic acids (e.g. double-stranded DNA) and associated proteins. Patients with SLE also develop exaggerated type I interferon responses that disrupt immune homeostasis. Early studies identified that a subset of SLE patients are unable to degrade NETs efficiently due to impaired DNAse1 function [6]. Since then, multiple reports have linked NETs to the pathogenesis of SLE. Genetic variants associated with SLE, including single nucleotide polymorphisms in TNFAIP3 (A20) have also been linked to citrullination and increased NET formation [7].
Lupus-associated autoantibodies and immune complexes can induce NET formation that subsequently activates type I interferon production by plasmacytoid dendritic cells (pDCs) [8–10]. Moreover, a subset of neutrophils termed low density granulocytes (LDGs) are elevated in SLE and display an increased capacity for NET formation. Lupus LDGs produce NETs that are enriched in oxidized mitochondrial DNA and enhance type I interferon production by human peripheral blood cells through a STING-dependent pathway [10]. Inhibition of mitochondrial ROS (leading to decreased nucleic acid oxidation) significantly improves clinical features and immune dysregulation in murine lupus. In addition to type I interferons, NETs can also induce other pro-inflammatory cytokines in macrophages. The cathelicidin LL-37 contained in NETs can activate the NLRP3 inflammasome via P2X7 receptor, leading to increased IL-1b and IL-18 production [11]. IL-18 further stimulates NET formation by neutrophils in a positive feedback loop that amplifies inflammation. NETs can also be internalized by SLE macrophages, leading to increased TNF-a and IL-6 production [*12]. Taken together, these findings demonstrate that NETs coordinate multiple aspects of the innate immune response in SLE (figure 1).
Figure 1. NETs orchestrate innate and adaptive autoimmune responses in SLE.
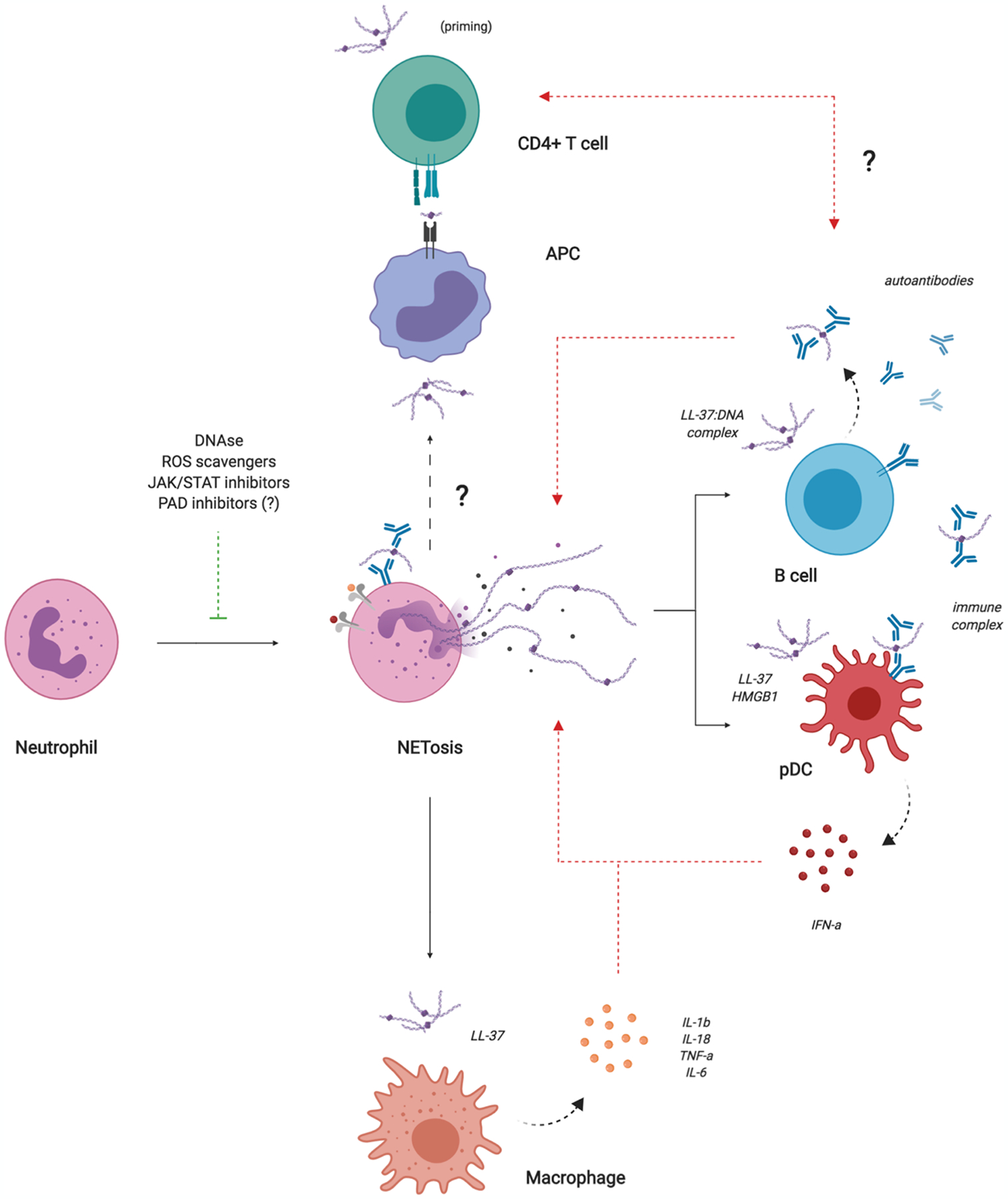
NETs can induce type I interferon production by pDCs and other cell types and pro-inflammatory cytokine production by macrophages. NETs can also activate memory B cells, leading to autoantibody production that activates pDCs and further induces NET formation. NETs may be important in T cell priming; however, the interaction between NETs and antigen presenting cells (APCs) in SLE requires further investigation.
NETs can also target adaptive immune cells in SLE. A recent study reported that NETs can activate human memory B cells to proliferate and secrete polyclonal IgG through a TLR-9 dependent mechanism [**13]. NETs were also able to induce memory B cell responses against neutrophil-associated antigens through the B-cell receptor (BCR) in lupus patients, but not in healthy controls. Consistent with these data, NETs positively correlate with anti-LL-37 autoantibodies in lupus patients. These findings suggest that NETs can re-activate memory B cells and potentiate autoantibody responses in lupus (figure 1); however, this study did not investigate the ability of NETs to directly activate naïve B cells and initiate autoantibody production or clonal expansion of autoreactive cells. Another unexplored possibility is that NETs can be internalized by B cells via BCR-engagement or other endocytic mechanisms, including toll-like receptors (TLRs), and presented on MHC-II molecules to autoreactive CD4+ T cells that provide important cytokines and co-stimulatory signals. There is also some evidence that NETs can prime CD4+ T cells directly via phosphorylation of Zap70 and reduce their activation threshold to stimuli they would not otherwise respond to under resting conditions [14]. In the context of autoimmunity, priming by NETs could be an important factor in the loss of immunological tolerance toward self-antigens (figure 1). Detailed mechanistic studies are necessary to further investigate the complex relationship between NETs and the adaptive immune system.
Moreover, NETs can have deleterious effects on cells in the tissue microenvironment, leading to organ damage. A recent study demonstrated that SLE neutrophils release NETs containing tissue factor (TF) and IL-17a [**15]. Confirming previous reports, NETs were also abundant in skin and kidney sections from patients with active disease. Importantly, human skin fibroblasts treated with SLE NETs acquire a pro-fibrotic phenotype with increased collagen production and migration rate. Treatment with anti-TF or anti-IL-17a antibodies blocked fibrosis activity, indicating that these molecules may be involved in NET-mediated tissue injury. Although this study was limited to skin fibroblasts, it is likely that NETs also have important effects on other structural cells. A separate study demonstrated that NETs can activate keratinocytes to produce cytokines and chemokines that promote skin inflammation in psoriasis [16]. These exciting new data suggest that NETs have diverse functions in autoimmunity beyond their obvious effects on immune cells.
NETs in rheumatoid arthritis:
RA is a chronic, systemic autoimmune disease characterized by synovial joint inflammation, cartilage damage, and bone erosion. Like SLE, NETs have been strongly implicated in the pathogenesis of RA. Neutrophils are highly abundant in the synovium of RA patients and neutrophils from both the synovium and peripheral blood of RA patients demonstrate increased NET formation [17]. Major risk factors for RA, including smoking, periodontal disease, and genetic variants, have also been associated with NET formation [18–20]. Notably, a significant proportion of patients with RA develop autoantibodies against citrullinated antigens (ACPAs). Histones and other NET-associated proteins can be citrullinated by PAD enzymes during the process of NET formation. Indeed, citrullinated NET proteins have been described as important autoantigens in RA [17,21]. NETs also correlate with ACPA in the sputum of at-risk individuals and first-degree relatives of RA patients, suggesting that they may be involved in the initiation or early stages of disease [22,*23].
Previous work has shown that NETs can activate fibroblast-like synoviocytes (FLS) to produce pro-inflammatory cytokines, including IL-6 and IL-8, that further promote NET formation and joint inflammation [17]. Recently, Carmona-Rivera et al. demonstrated that IL-17b contained in NETs endows FLS with antigen presenting capabilities by inducing expression of MHC-II [24]. Moreover, NET-associated proteins can be internalized by FLS into endosomal compartments through the receptor for advanced glycation end products (RAGE)/TLR-9 pathway and loaded onto MHC-II molecules, where they are capable of activating antigen-specific CD4+ T cells. Injection of NET-loaded FLS into humanized mice also induces T cell responses and ACPA production in vivo, highlighting the importance of the NET-FLS-T cell axis in RA immunobiology. Downstream, ACPA can form immune complexes that activate synovial macrophages to produce inflammatory cytokines, providing an additional feed-forward mechanism that can amplify inflammation in the joint and further induce NET formation (figure 2) [25].
Figure 2. NET promote pathogenic immunity and amplify inflammation in rheumatoid arthritis.
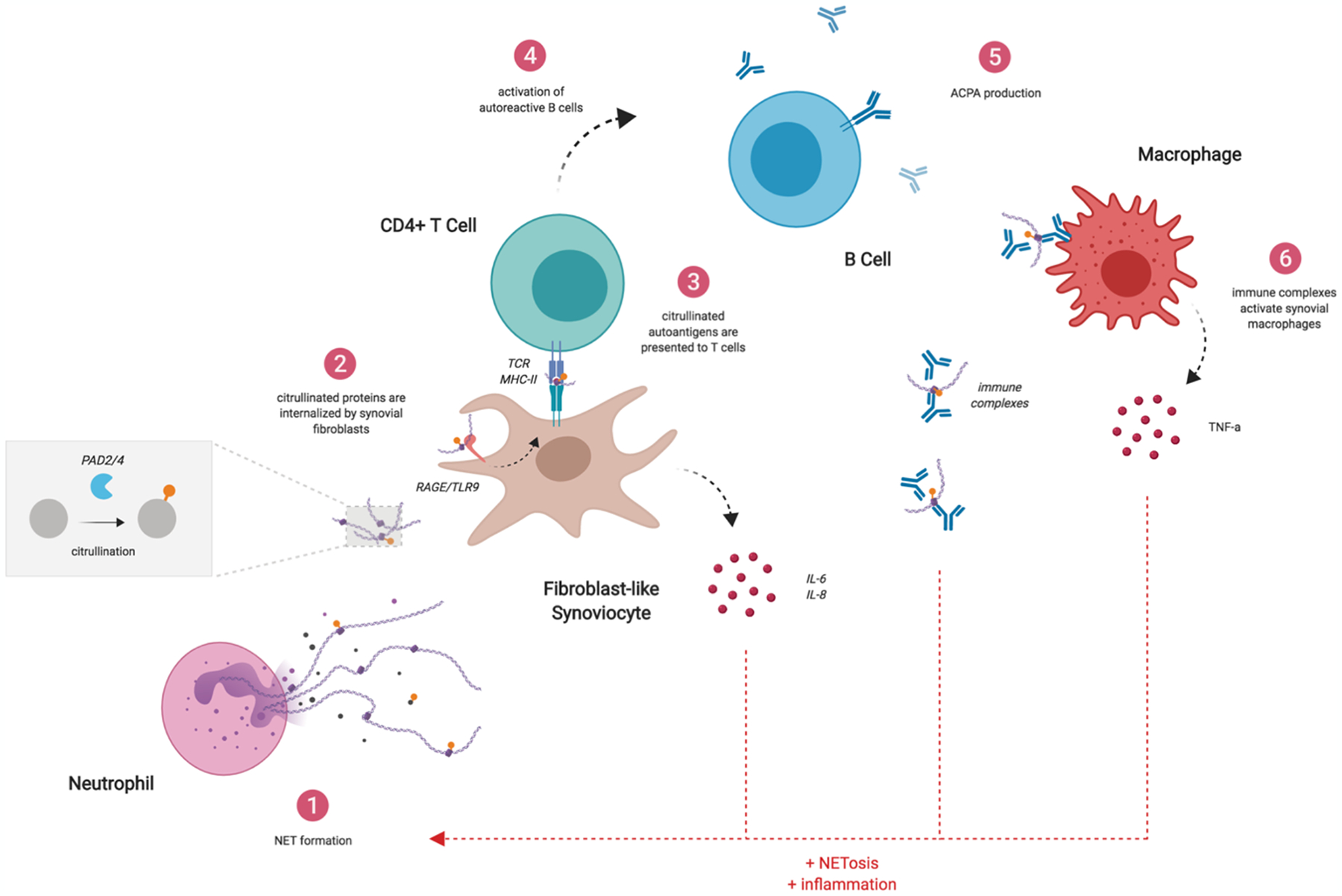
NETs can induce pro-inflammatory cytokines in FLS. Citrullinated NET proteins can also be internalized by FLS and presented to Ag-specific CD4+ T cells, promoting ACPA production by autoreactive B cells. ACPAs can form immune complexes that stimulate synovial macrophages to produce pro-inflammatory cytokines. These cytokines and immune complexes further induce NET formation, creating a feed-forward loop that promotes autoimmunity.
In addition to citrullinated NET proteins, autoantibodies and T cells recognizing citrullinated cartilage proteoglycans, including aggrecan, have also been described in patients with RA [26]. How these antigens get citrullinated and presented to initiate the autoimmune response remains an open question in the field. Active PAD enzymes are released during NET formation (both diffuse and embedded within the NET scaffold) [27]. Therefore, it is possible that neutrophil-derived PADs are capable of citrullinating other proteins in the synovium, providing a rich source of citrullinated autoantigens in addition to those that are directly generated and contained within NETs.
NETs in other immune-mediated diseases:
In addition to SLE and RA, NETs have been implicated in a myriad of other immune-mediated diseases. Most notably, NETs have been identified as key effectors in atherosclerosis. Cholesterol crystals can induce NET formation, which in turn activate macrophages to release pro-inflammatory cytokines (IL-1b, IL-6) that promote immune cell recruitment and responses in atherosclerotic plaques [28]. Treatment with pan-PAD inhibitors or myeloid-specific deletion of PAD4 reduces NET formation, plaques, and inflammatory responses in mouse models of atherosclerosis, providing further evidence that NETs are important drivers in atherogenesis [29,*30]. More recently, a study by Silvestre-Roig et al demonstrated that NETs can damage vascular smooth muscle cells directly through a histone 4-dependent mechanism, resulting in the formation of membrane pores that induce lytic cell death [**31]. Of note, patients with autoimmune diseases, including SLE and RA, are at increased risk for atherosclerosis. NETs can damage endothelial cells in SLE [32]. Low density granulocytes (LDGs), a distinct subset of proinflammatory neutrophils that are increased in SLE and spontaneously undergo NET formation are also significantly associated with vascular inflammation and non-calcified coronary plaque burden in SLE [**33]. These data suggest that NETs, which are significantly elevated in autoimmune diseases, may have secondary effects on cardiovascular health. As such, therapeutic strategies targeting NETs could have added benefit in preventing vasculopathy and premature atherosclerosis in patients with autoimmunity.
NETs have also been implicated in other diseases that affect the vasculature. NETs were recently identified in patients with deficiency of adenosine deaminase 2 (DADA2), a severe form of vasculitis caused by loss of function mutations in the ADA2 enzyme. Accumulation of adenosine in this disease induces NET formation via adenosine A1 and A3 receptors, leading to enhanced TNF-a production by macrophages [**34]. This is relevant because anti-TNF agents control clinical features of this condition. NETs can also activate the clotting cascade and lead to thrombosis [35]. This mechanism appears to be especially relevant in anti-phospholipid syndrome (APS), where antiphospholipid antibodies induce formation of NETs that subsequently promote generation of thrombin [36]. Furthermore, treatment with an adenosine A2A receptor agonist (which has opposite functions of A1 and A3 receptors) suppresses NET formation and reduces venous thrombosis in a mouse model of APS, indicating that nucleic acid metabolites are important regulators of neutrophil responses in inflammatory diseases [*37].
Increased NET formation has also been reported in pyogenic arthritis, pyoderma gangrenosum and acne (PAPA) syndrome, a rare genetic disease that results in inflammation of the joints and skin. Neutrophils from PAPA patients form NETs more readily compared to healthy controls [*38]. This effect is enhanced by IL-1b, a pathogenic cytokine involved in PAPA syndrome. NETs are also detected in skin biopsies from PAPA patients in association with increased levels of IL-1b and IL-18. Another recent study characterized the role of NETs in Wiskott-Aldrich syndrome (WAS), a rare immunodeficiency complicated by high rates of autoimmunity. Similar to SLE neutrophils, WAS neutrophils have increased spontaneous NET formation, induce type I interferon production by pDCs, and activate autoreactive B cells [*39]. Together, these findings further highlight the unique contribution of NETs to immune dysregulation in multiple cells and tissues. As the literature continues to expand, we expect NETs to be defined in a variety of other immune-mediated diseases.
Targeting NETs for therapeutic benefit:
Given the importance of NETs in immunopathology, there has been significant interest in developing new therapeutics that target pathways involved in NET formation or clearance. The DNAse enzyme can degrade both NETs and apoptotic cell microparticles and has been found to suppresses autoreactivity [40]. Antimalarials are common treatments for SLE/RA and have been shown to inhibit NET formation in vitro, as well as decrease uptake of NETs by FLS [24,41]. Additionally, the JAK/STAT inhibitor (jakinib) tofacitinib is effective at reducing NET formation and other disease manifestations in lupus-prone mice [42]. Recent clinical trial data also indicate that baricitinib (another jakinib) can improve arthritis and rash in patients with active SLE [*43]. Other strategies, including mitochondrial ROS scavengers and drugs that disrupt neutrophil immunometabolism are exciting possibilities for clinical intervention and merit further investigation.
PAD inhibitors have also been studied in autoimmune disease. Because citrullination of histones has been implicated in chromatin decondensation during NET formation, PAD inhibitors are thought to block NET formation. Indeed, PAD inhibitors have been effective at reducing NET formation and disease severity in several mouse models of SLE and RA [44–46]. Of note, there has been some controversy regarding the importance of NETs and PAD enzymes in lupus autoimmunity. Genetic deletion of PAD4 protects mice from type I interferon responses, autoantibody production, immune complex deposition, vasculopathy, and other clinical manifestations in a model of TLR-7 induced lupus [**47]. In contrast, a separate study reported that NOX2 and PAD4-deficient mice had impaired NET formation, but more severe disease in a model of pristane-induced lupus [48]. Another study reported that PAD4 deficiency had no effect on lupus autoimmunity in the MRL/lpr model [49]. These discrepancies may be explained by the experimental model in which they were observed. At high neutrophil densities, NETs can form aggregates that have paradoxical anti-inflammatory effects by degrading cytokines and chemokines [50]. Although injection of pristane into the peritoneal cavity reproduces many of the clinical features of lupus, it is possible that this model induces aggregation of NETs in the peritoneum that limit inflammation. Similarly, the MRL/lpr model is primarily driven by unregulated expansion of autoreactive T cells due to a mutation in Fas and the relative contribution of NETs to pathology in this model is somewhat ambiguous. Regardless, further studies – especially those testing the efficacy of PAD inhibitors in human SLE – are necessary to determine the clinical benefit of such drugs.
Conclusions:
NETs are complex structures with important effects in both immune defense and autoimmune disease. Recent studies have provided valuable insights into the interactions between NETs and other immune or structural cells. Most notably, NETs can potentiate inflammation and are sources of modified autoantigens. NETs also contribute to atherosclerosis and may explain the increased burden of cardiovascular disease in patients with autoimmunity. Drugs that target NET formation have shown promise in pre-clinical models and are an exciting possibility for the treatment of autoimmune diseases.
Key points:
NETs are increased in autoimmune diseases, including SLE and RA
NETs promote inflammation and are a potential source of autoantigens
NETs are pathogenic in atherosclerosis and may explain premature cardiovascular disease in patients with autoimmunity
Drugs that target NET formation or their clearance may be effective for patients with autoimmune diseases
Acknowledgements:
Supported by the Intramural Research Program at NIAMS/NIH (ZIAAR041199)
Footnotes
Conflicts of interest:
There are no conflicts of interest.
References
- 1.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A: Neutrophil extracellular traps kill bacteria. Science 2004, 303:1532–1535. [DOI] [PubMed] [Google Scholar]
- 2.Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, Brinkmann V, Bernuth HV, Zychlinsky A: Diverse stimuli engage different neutrophil extracellular trap pathways. Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, et al. : Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 2009, 184:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *4.Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, von Pein JB, Broz P, Sweet MJ, Schroder K: Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol 2018, 3. [DOI] [PubMed] [Google Scholar]; This study demonstrates that neutrophils use an inflammasome- and GSDMD-dependent mechanism to activate NETosis as a defense response against cytosolic bacteria
- *5.Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes, Menninger S, Eickhoff J, Nussbaumer P, Klebl B, et al. : Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol 2018, 3. [DOI] [PubMed] [Google Scholar]; This study demonstrates that gasdermin D can be cleaved by neutrophil proteases, leading to NET formation.
- 6.Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A: Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A 2010, 107:9813–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Odqvist L, Jevnikar Z, Riise R, Oberg L, Rhedin M, Leonard D, Yrlid L, Jackson S, Mattsson J, Nanda S, et al. : Genetic variations in A20 DUB domain provide a genetic link to citrullination and neutrophil extracellular traps in systemic lupus erythematosus. Ann Rheum Dis 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, et al. : Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 2011, 3:73ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, et al. : Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med 2011, 3:73ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ: Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 2016, 22:146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ: Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol 2013, 190:1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *12.Barrera-Vargas A, Gomez-Martin D, Carmona-Rivera C, Merayo-Chalico J, Torres-Ruiz J, Manna Z, Hasni S, Alcocer-Varela J, Kaplan MJ: Differential ubiquitination in NETs regulates macrophage responses in systemic lupus erythematosus. Ann Rheum Dis 2018, 77:944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that NETs can be internalized by macrophages and induce production of pro-inflammatory cytokines. Differences in ubiquitination of NET proteins may also be an important mechanism in macrophage activation.
- **13.Gestermann N, Di Domizio J, Lande R, Demaria O, Frasca L, Feldmeyer L, Di Lucca J, Gilliet M: Netting Neutrophils Activate Autoreactive B Cells in Lupus. J Immunol 2018, 200:3364–3371. [DOI] [PubMed] [Google Scholar]; This study demonstrates that NETs can activate memory B cells from human SLE patients and enhance autoantibody production via TLR9 and BCR signaling.
- 14.Tillack K, Breiden P, Martin R, Sospedra M: T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J Immunol 2012, 188:3150–3159. [DOI] [PubMed] [Google Scholar]
- **15.Frangou E, Chrysanthopoulou A, Mitsios A, Kambas K, Arelaki S, Angelidou I, Arampatzioglou A, Gakiopoulou H, Bertsias GK, Verginis P, et al. : REDD1/autophagy pathway promotes thromboinflammation and fibrosis in human systemic lupus erythematosus (SLE) through NETs decorated with tissue factor (TF) and interleukin-17A (IL-17A). Ann Rheum Dis 2019, 78:238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that NETs from SLE patients contain TF/IL-17a and are present in skin and kidney biopsies. Moreover, SLE NETs induce fibrosis activity in skin fibroblasts in a TF and IL-17a-dependent manner.
- 16.Shao S, Fang H, Dang E, Xue K, Zhang J, Li B, Qiao H, Cao T, Zhuang Y, Shen S, et al. : Neutrophil Extracellular Traps Promote Inflammatory Responses in Psoriasis via Activating Epidermal TLR4/IL-36R Crosstalk. Front Immunol 2019, 10:746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, Friday S, Li S, Patel RM, Subramanian V, et al. : NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med 2013, 5:178ra140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang HH, Dwivedi N, Nicholas AP, Ho IC: The W620 Polymorphism in PTPN22 Disrupts Its Interaction With Peptidylarginine Deiminase Type 4 and Enhances Citrullination and NETosis. Arthritis Rheumatol 2015, 67:2323–2334. [DOI] [PubMed] [Google Scholar]
- 19.Qiu SL, Zhang H, Tang QY, Bai J, He ZY, Zhang JQ, Li MH, Deng JM, Liu GN, Zhong XN: Neutrophil extracellular traps induced by cigarette smoke activate plasmacytoid dendritic cells. Thorax 2017, 72:1084–1093. [DOI] [PubMed] [Google Scholar]
- 20.Magan-Fernandez A, O’Valle F, Abadia-Molina F, Munoz R, Puga-Guil P, Mesa F: Characterization and comparison of neutrophil extracellular traps in gingival samples of periodontitis and gingivitis: A pilot study. J Periodontal Res 2019, 54:218–224. [DOI] [PubMed] [Google Scholar]
- 21.Corsiero E, Bombardieri M, Carlotti E, Pratesi F, Robinson W, Migliorini P, Pitzalis C: Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann Rheum Dis 2016, 75:1866–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Demoruelle MK, Harrall KK, Ho L, Purmalek MM, Seto NL, Rothfuss HM, Weisman MH, Solomon JJ, Fischer A, Okamoto Y, et al. : Anti-Citrullinated Protein Antibodies Are Associated With Neutrophil Extracellular Traps in the Sputum in Relatives of Rheumatoid Arthritis Patients. Arthritis Rheumatol 2017, 69:1165–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *23.Demoruelle MK, Bowers E, Lahey LJ, Sokolove J, Purmalek M, Seto NL, Weisman MH, Norris JM, Kaplan MJ, Holers VM, et al. : Antibody Responses to Citrullinated and Noncitrullinated Antigens in the Sputum of Subjects With Rheumatoid Arthritis and Subjects at Risk for Development of Rheumatoid Arthritis. Arthritis Rheumatol 2018, 70:516–527. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that individuals at risk for RA develop ACPA in correlation with NETs in sputum.
- 24.Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, Liu Y, Bicker KL, Wahamaa H, Hoffmann V, et al. : Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol 2017, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sokolove J, Zhao X, Chandra PE, Robinson WH: Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcgamma receptor. Arthritis Rheum 2011, 63:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rims C, Uchtenhagen H, Kaplan MJ, Carmona-Rivera C, Carlucci P, Mikecz K, Markovics A, Carlin J, Buckner JH, James EA: Citrullinated Aggrecan Epitopes as Targets of Autoreactive CD4+ T Cells in Patients With Rheumatoid Arthritis. Arthritis Rheumatol 2019, 71:518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spengler J, Lugonja B, Ytterberg AJ, Zubarev RA, Creese AJ, Pearson MJ, Grant MM, Milward M, Lundberg K, Buckley CD, et al. : Release of Active Peptidyl Arginine Deiminases by Neutrophils Can Explain Production of Extracellular Citrullinated Autoantigens in Rheumatoid Arthritis Synovial Fluid. Arthritis Rheumatol 2015, 67:3135–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V: Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349:316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knight JS, Luo W, O’Dell AA, Yalavarthi S, Zhao W, Subramanian V, Guo C, Grenn RC, Thompson PR, Eitzman DT, et al. : Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res 2014, 114:947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *30.Liu Y, Carmona-Rivera C, Moore E, Seto NL, Knight JS, Pryor M, Yang ZH, Hemmers S, Remaley AT, Mowen KA, et al. : Myeloid-Specific Deletion of Peptidylarginine Deiminase 4 Mitigates Atherosclerosis. Front Immunol 2018, 9:1680. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that genetic deletion of PAD4 in myeloid lineage cells reduces NET formation, plaques, and inflammatory responses in the aorta of ApoE−/− mice.
- **31.Silvestre-Roig C, Braster Q, Wichapong K, Lee EY, Teulon JM, Berrebeh N, Winter J, Adrover JM, Santos GS, Froese A, et al. : Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that NETs can damage the vasculature directly by killing vascular smooth muscle cells. Activated SMCs produce chemokines that attract neutrophils and induce NET formation. Moreover, histone 4 contained within these NET structures can form membrane pores in SMCs that induce lytic cell death.
- 32.Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ: Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis 2015, 74:1417–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **33.Carlucci PM, Purmalek MM, Dey AK, Temesgen-Oyelakin Y, Sakhardande S, Joshi AA, Lerman JB, Fike A, Davis M, Chung JH, et al. : Neutrophil subsets and their gene signature associate with vascular inflammation and coronary atherosclerosis in lupus. JCI Insight 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that LDGs are signficantly associated with arterial wall inflammation and coronary atherosclerosis in SLE.
- **34.Carmona-Rivera C, Khaznadar SS, Shwin KW, Irizarry-Caro JA, O’Neil LJ, Liu Y, Jacobson KA, Ombrello AK, Stone DL, Tsai WL, et al. : Deficiency of adenosine deaminase 2 triggers adenosine-mediated NETosis and TNF production in patients with DADA2. Blood 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies a novel mechanism by which excess adenosine in DADA2 patients induces enhanced NET formation leading to increased TNF-alpha production.
- 35.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., Wrobleski, Wakefield TW, Hartwig JH, Wagner DD: Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 2010, 107:15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Nunez-Alvarez C, Hernandez-Ramirez D, Bockenstedt PL, Liaw PC, Cabral AR, et al. : Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol 2015, 67:2990–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ali RA, Gandhi AA, Meng H, Yalavarthi S, Vreede AP, Estes SK, Palmer OR, Bockenstedt PL, Pinsky DJ, Greve JM, et al. : Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat Commun 2019, 10:1916. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that adenosine receptor agonist (A2A) prevents NET formation and thrombosis in antiphospholipid syndrome.
- *38.Mistry P, Carmona-Rivera C, Ombrello AK, Hoffmann P, Seto NL, Jones A, Stone DL, Naz F, Carlucci P, Dell’Orso S, et al. : Dysregulated neutrophil responses and neutrophil extracellular trap formation and degradation in PAPA syndrome. Ann Rheum Dis 2018, 77:1825–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that NET formation is implicated in immune dysregulation and tissue damage in PAPA syndrome.
- *39.Cervantes-Luevano KE, Caronni N, Castiello MC, Fontana E, Piperno GM, Naseem A, Uva P, Bosticardo M, Marcovecchio GE, Notarangelo LD, et al. : Neutrophils drive type I interferon production and autoantibodies in patients with Wiskott-Aldrich syndrome. J Allergy Clin Immunol 2018, 142:1605–1617 e1604. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports that neutrophils (and NETs) are involved in inante and adaptive immune activation in Wiskott-Aldrich syndrome.
- 40.Sisirak V, Sally B, D’Agati V, Martinez-Ortiz W, Ozcakar ZB, David J, Rashidfarrokhi A, Yeste A, Panea C, Chida AS, et al. : Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell 2016, 166:88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith CK, Vivekanandan-Giri A, Tang C, Knight JS, Mathew A, Padilla RL, Gillespie BW, Carmona-Rivera C, Liu X, Subramanian V, et al. : Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: an additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol 2014, 66:2532–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Furumoto Y, Smith CK, Blanco L, Zhao W, Brooks SR, Thacker SG, Abdalrahman Z, Sciume G, Tsai WL, Trier AM, et al. : Tofacitinib Ameliorates Murine Lupus and Its Associated Vascular Dysfunction. Arthritis Rheumatol 2017, 69:148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *43.Wallace DJ, Furie RA, Tanaka Y, Kalunian KC, Mosca M, Petri MA, Dorner T, Cardiel MH, Bruce IN, Gomez E, et al. : Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392:222–231. [DOI] [PubMed] [Google Scholar]; This study demonstrates that oral JAK/STAT inhibitors may be an effective treatment for human SLE.
- 44.Knight JS, Zhao W, Luo W, Subramanian V, O’Dell AA, Yalavarthi S, Hodgin JB, Eitzman DT, Thompson PR, Kaplan MJ: Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest 2013, 123:2981–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knight JS, Subramanian V, O’Dell AA, Yalavarthi S, Zhao W, Smith CK, Hodgin JB, Thompson PR, Kaplan MJ: Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis 2015, 74:2199–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Willis VC, Banda NK, Cordova KN, Chandra PE, Robinson WH, Cooper DC, Lugo D, Mehta G, Taylor S, Tak PP, et al. : Protein arginine deiminase 4 inhibition is sufficient for the amelioration of collagen-induced arthritis. Clin Exp Immunol 2017, 188:263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **47.Liu Y, Lightfoot YL, Seto N, Carmona-Rivera C, Moore E, Goel R, O’Neil L, Mistry P, Hoffmann V, Mondal S, et al. : Peptidylarginine deiminases 2 and 4 modulate innate and adaptive immune responses in TLR-7-dependent lupus. JCI Insight 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that genetic deletion of PAD2/4 reduces type I interferon signature, autoantibody production, immune complex deposition and other features in murine lupus. Interestingly, neutrophils from PAD4 knockout mice were still able to form NET structures; however these NETs did not contain citrullinated histone 3 and did not induce vascular damage in vitro or in vivo.
- 48.Kienhofer D, Hahn J, Stoof J, Csepregi JZ, Reinwald C, Urbonaviciute V, Johnsson C, Maueroder C, Podolska MJ, Biermann MH, et al. : Experimental lupus is aggravated in mouse strains with impaired induction of neutrophil extracellular traps. JCI Insight 2017, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gordon RA, Herter JM, Rosetti F, Campbell AM, Nishi H, Kashgarian M, Bastacky SI, Marinov A, Nickerson KM, Mayadas TN, et al. : Lupus and proliferative nephritis are PAD4 independent in murine models. JCI Insight 2017, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schauer C, Janko C, Munoz LE, Zhao Y, Kienhofer D, Frey B, Lell M, Manger B, Rech J, Naschberger E, et al. : Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med 2014, 20:511–517. [DOI] [PubMed] [Google Scholar]