

Abstract
Importance.
MRI demonstrates disease dissemination in space and time and may exclude multiple sclerosis (MS) mimics when applied in diagnosis of MS. Less effort has been expended in the application of MRI to progressive MS (PMS), including diagnosis of primary progressive (PP) MS and identifying patients with relapsing-remitting (RR) MS at risk to develop secondary progressive (SP) MS. We address clinical application of MRI in PMS for both diagnosis and prognosis. We also consider novel MRI indicators that reflect PMS pathophysiology.
Observations.
Although nonspecific, some spinal cord imaging features (diffuse abnormalities and lesions involving the gray matter -GM- and ≥2 white matter columns) are typical of PPMS. Both in PPMS and relapse-onset MS patients, the location of lesions in critical CNS regions (spinal cord, infratentorial regions, GM) and MRI-detected high inflammatory activity in the first years after the diagnosis predict long-term disability and future progressive disease course. These measures are currently evaluable in clinical practice.
In patients with established MS, GM involvement and neurodegeneration is associated with accelerated clinical worsening. Subpial demyelination and slowly expanding lesions are novel and promising indicators of progressive MS.
Conclusions and Relevance.
Diagnosis of PPMS is more challenging than of RRMS. No qualitative clinical, immunological, histopathological and neuroimaging features differentiate PPMS and SPMS; both are characterized by imaging findings reflecting neurodegeneration and are also influenced by aging and comorbidities.
Identification of MRI markers capable of distinguishing PPMS from RRMS and predicting evolution of RRMS to SPMS remain unmet needs. Integration of multiple parameters will likely be essential to achieve these aims.
Keywords: Progressive Multiple Sclerosis, Neurodegeneration, Magnetic Resonance Imaging, Diagnosis, Prognosis
INTRODUCTION
Magnetic resonance imaging (MRI) is central in the diagnostic work up of patients with suspected MS given its high sensitivity to demonstrate disease dissemination in space (DIS) and time and its substantial, albeit imperfect, ability to exclude other mimics of MS. From 2001 until 2017,1 successive iterations of the McDonald criteria expended great effort to define imaging features typical for MS in patients presenting with a clinically isolated syndrome (CIS). Diagnosis of primary progressive (PP) MS remains challenging, however, and is only possible retrospectively, based on clinical assessment.
Identification of imaging features associated with PPMS, as well as features that predict evolution from relapsing-remitting (RR) to secondary progressive (SP) MS are important unmet needs. Given the advent of effective therapies for RRMS that may reduce development of SPMS and limit disability worsening in progressive MS (PMS),2 the need for such imaging indicators is all the greater. Diagnosis of PMS is limited by difficulties distinguishing accumulating disability due to inflammatory disease activity from that attributable to degenerative processes associated with SPMS. Moreover, there are no accepted clinical criteria for SPMS.3,4
This has promoted extensive research in the imaging field, facilitated by novel MRI sequences, to image pathophysiological mechanisms relevant to the pathobiology of PMS.
METHODS
A workshop was held in November 2019 in Milan, which involved neurologists and neuroradiologists. Five main clinically relevant questions regarding the role of MRI in PMS diagnosis and prognosis were identified (Box 1).
Box 1. Summary of the key questions addressed in the review and of the proposed MRI features for the identification of PMS.
| Key questions and summary conclusions | |
|---|---|
| (1) Is there any MRI feature that can support PPMS and SPMS diagnosis? | |
| PPMS | Diffuse signal abnormalities in the spinal cord Spinal cord lesions involving the GM and ≥2 WM columns (axial plane) Atrophy of the lower portion of the cervical cord |
| SPMS | Spinal cord lesions involving the GM and ≥2 WM columns (axial plane) Atrophy of the lower portion of the cervical cord Cord GM atrophy |
| (2) Are there specific MRI features at disease onset able to predict disability and a progressive course? | |
| PPMS | Gd-enhancing lesions at baseline Spinal cord lesions at baseline |
| SPMS | Number and volume of baseline brain T2-hyperintense lesions Increase of brain T2-hyperintense lesion volume during the first 5 years ≥2 Gd-enhancing lesions at baseline ≥1 spinal cord lesion at baseline ≥1 infratentorial lesion at baseline ≥1 cortical lesion at baseline ≥1 spinal cord lesion within 1 or 3 years ≥1 infratentorial lesion within 1 or 3 years ≥1 deep WM lesion within 1 year |
| (3) Are there MRI markers able to identify disability progression? In relapse-onset MS, are there MRI markers able to identify evolution to SPMS? | |
| PPMS | New T1-hypointense lesions Cortical lesion number and volume Baseline GM damage Rate of brain atrophy |
| SPMS | T2 lesion volume Increase of T1-hypointense lesion volume Cortical lesion number and volume Conversion of dirty-appearing WM into focal WM lesions GM volume Rate of brain GM and deep GM atrophy |
| (4) Are there distinguishing MRI features between PPMS and SPMS? | |
| None | |
| (5) Are there candidate MRI biomarkers promising to identify MS progression? | |
| ≥4 paramagnetic rim lesions Subpial demyelination | |
Abbreviations: Gd=gadolinium; GM=gray matter; MRI=magnetic resonance imaging; MS=multiple sclerosis; P=progressive; PP=primary progressive; SP=secondary progressive; WM=white matter.
Experts provided a summary related to each topic (see eTable 1 for search strategy and selection criteria). A group consensus was reached during the workshop and summarized in a first draft, which was circulated among the meeting participants and additional experts in the field for critical discussion and revision. Here, we present the final conclusions from this workshop.
We also briefly discuss other promising biomarkers, including neurofilament light chain (NfL) levels, specific radiotracers used with positron emission tomography (PET), and optical coherence tomography (OCT), which have been investigated for similar purposes.
OBSERVATIONS
Diagnostic criteria for PMS
PPMS.
About 10–15% of MS patients exhibit a gradual progression of disability from disease onset.5 According to the 2017 revision of the McDonald criteria,1 PPMS can be diagnosed in patients with ≥12 months of disability progression (retrospectively or prospectively determined), independent of clinical relapses, who additionally satisfy two of the three following criteria: (a) one or more T2-hyperintense lesions characteristic of MS in one or more of periventricular, cortical/juxtacortical, or infratentorial brain regions; (b) ≥2 T2-hyperintense lesions in the spinal cord; (c) CSF-specific oligoclonal bands. Distinction between symptomatic and asymptomatic lesions, an element of the 2010 iteration of the McDonald criteria, was eliminated in 2017.
SPMS.
Diagnosis is based on retrospective determination of progressive disability worsening unrelated to clinical relapses over ≥6–12 months.3,4
Clinical criteria for disability progression
Gradual worsening of disability independently from relapses over at least 6 or 12 months in relapse-onset MS and 12 months in PPMS is the gold standard to identify disability progression.4,6
The Expanded Disability Status Scale (EDSS) score with its functional system sub-scores is the most widely used scale to quantify MS-related disability.7 A gradual increase of EDSS score allows to demonstrate disability progression, which should be sustained for at least 3 or 6 months (i.e., confirmed disability progression [CDP]) to exclude possible confounding effects due to recent relapses or assessment errors.4,6
However, EDSS score has limited accuracy in identifying MS progression. Inter- and intra-rater reliability are low; using self-reported walking distance adds to problems with reliability; gait is the major indicator for worsening when scores exceed 3.5 and gait can be influenced by many non-MS issues; EDSS has poor sensitivity when scores exceed 6.0–6.5.
In a study of 16636 MS patients, the progression criteria based on 3- or 6-month CDP were confirmed only in 70% (95% CI=68,71 %) and 74% (95% CI=72,75 %) at 5 years, suggesting up to 30% overestimation of disability progression.3
A recent evaluation of 17356 patients proposed a set of combined criteria to define disability progression that had an accuracy of 87% to identify patients evolving to SPMS over 5 years. The criteria included: (a) evidence of disability progression by 1 step in patients with EDSS ≤5.5 or 0.5 steps in patients with EDSS ≥6.0, in the absence of a relapse; (b) a minimum EDSS score of 4.0 and pyramidal functional system score of 2; (c) CDP for at least 3 months, including confirmation within the leading functional system.4
Another study of 273 MS patients with EDSS score between 4.0 and 7.5 showed that self-reported walking distance was misclassified when compared to the actual walking distance in 145 patients (53%). Such errors were more frequent in patients using walking aids (64% vs 44%, p<.05) and in patients with PPMS (69%, p<.05).8
Quantitative functional composite scores have been proposed to more sensitively and objectively investigate MS disability progression. The Multiple Sclerosis Functional Composite (MSFC) score comprises quantitative measures to assess leg function/ambulation (Timed 25-Foot Walk [T25FW]), arm/hand function (9-hole Peg test [9-HPT]) and cognition (Paced Auditory Serial Addition Test).9
Other composite outcomes combining overall (EDSS), upper (≥20% worsening of 9-HPT) and lower (≥20% worsening of T25FW) extremity disability have been proposed to capture MS progression.10,11 The composite scores, defined as progression on ≥1 of 3 components (EDSS, T25FW, and/or 9HPT) with a ≥20% minimum threshold change for T25FW and 9HPT, detected 6-month CDP in 59.5% of 215 SPMS patients compared to 24.7% detected with EDSS alone.11
Clinical identification of MS progression is typically retrospective, challenging and delayed by months to years after onset. Moreover, clinical examination does not allow an early identification of the neurodegenerative phenomena that start from the earliest phases of the disease, several years before the occurrence of overt disability progression.
MRI features for PMS diagnosis
PPMS.
Signal characteristics on conventional brain and spinal cord MRI do not differ qualitatively between early PPMS and relapse-onset patients, although PPMS patients may have fewer brain lesions. Focal cortical lesions (CLs) have also the same MR signal features.12,13 As a single set of criteria to demonstrate DIS in relapse-onset MS and PPMS was feasible,14 MRI criteria for diagnosis have been harmonized for both MS subgroups,15 with the exception of requiring two rather than one spinal cord lesions to define spinal cord involvement for PPMS. By reducing the number of spinal cord lesions from two to one in PPMS, the sensitivity would improve to 84% (74/88) from 77% (68/88) using the 2010 McDonald criteria.16 However, the specificity of this modification would need further investigation before implementation.
SPMS.
No reliable MRI indicators of SPMS are available. A 10-year longitudinal study of 480 relapse-onset MS patients identified a subgroup of RRMS patients with disability progression without disease activity (i.e., relapses and/or new T2 lesions) (34/480, 7.1%), who experienced higher rates of whole brain, white matter (WM), and gray matter (GM) atrophy compared to those observed in stable patients.17 The investigators referred to this subgroup as experiencing ‘silent progression’ or disease-activity-free neurodegeneration independently from focal inflammation in patients with RRMS. Atrophy evaluation may identify SPMS patients.
Other MRI features for PMS identification.
PMS patients, particularly PPMS, on average, have fewer and smaller gadolinium (Gd)-enhancing lesions compared with RRMS.18,19 However, the number of Gd-enhancing lesions do not discriminate RRMS from PMS at an individual level; 19/45 (42%) with early PPMS had at least one Gd-enhancing lesion.20
Compared with RRMS and PPMS patients, those with SPMS have a higher proportion of brain T1-hypointense lesions (i.e., ‘black holes’) (Box 2).21,22 However, the diagnostic value of black holes in PMS is still not adequately evaluated.21,22
Box 2. Glossary.
| Black holes |
| Black holes are defined as areas with unequivocal hypointensity compared with normal-appearing WM and GM matter on T1-weighted images obtained with spin-echo sequences at 1.5 T and have a corresponding T2-hyperintense WM lesion. Black holes are classified as acute or chronic (persistent or permanent). Whereas acute black holes reflect transient edema and inflammation, chronic black hole are those T1-hypointesities persisting ≥6 months after their first appearance on MRI and are characterized, pathologically, by severe demyelination and axonal loss.126,127 |
| Diffuse spinal cord abnormalities |
| Abnormal areas of subtle increases of signal intensity on PD-weighted or STIR images, between that of focal lesions and normal-appearing spinal cord, lacking well-demarcated borders from adjacent normal-appearing cord.128 |
| Slowly expanding lesions |
| Pathologically, slowly expanding (also known as chronic active or smoldering) lesions represent up to 57% of chronic lesions (although credible estimates are in the range of 25%). They are often characterized by a ‘rim’ of iron-laden activated microglia/macrophages, although not in all lesions,98 and signs of peripheral slow but ongoing demyelination and axonal loss around an inactive core without substantial BBB damage, thus reflecting a compartmentalized pathological process.79,81,94 Slowly expanding lesions have been investigated in vivo in lesions that progressively increase in size and show a paramagnetic rim on susceptibility-based MRI,85,94–96 corresponding, pathologically, to peripheral iron-laden microglia, or by evaluating the gradual expansion on conventional T1- and T2-weighted sequences of lesions showing a progressive decline of T1-hypointensity.97,100 |
| Dirty-appearing white matter |
| DAWM (or diffusely abnormal WM) is defined as area with ill-defined borders and with signal intensity on T2- and/or PD-weighted MRI between that of focal WM lesions and the surrounding normal-appearing WM and isointense to the signal of the nearby cortical GM.62–64 Typically, DAWM is noted in the periventricular regions especially parieto-occipital or in the centrum semiovale. Pathologically, DAWM is characterized by inflammatory infiltrates, BBB disruption, demyelination, gliosis and axonal loss, which are less severe compared to focal WM lesions.129 |
Abbreviations: BBB=blood-brain barrier; DAWM=dirty-appearing white matter; GM=gray matter; MRI=magnetic resonance imaging; PD=proton density; STIR=short tau inversion recovery.
Spinal cord diffuse T2 signal abnormality21 (Box 2, Figure 1) is commoner in PPMS (19/31, 61%) and SPMS (10/32, 31%) than in RRMS (6/28, 21%) (p<.01).21 It is associated with spinal cord atrophy, sensorimotor and bowel/bladder manifestations and disability.21 Diffuse abnormality has not been incorporated into MRI diagnostic criteria1,15,23 because clinical scans are insufficiently reliable or specific to identify it, and it is dependent on the type of T2-weighted sequence used. This abnormality has not been described in CIS patients.24,25 However, it was the only spinal cord alteration in 3/10 (30%) PPMS patients and in 8/147 (5%) RRMS patients without focal lesions.25
Figure 1. Summary of the MRI features supporting PPMS and SPMS diagnosis.
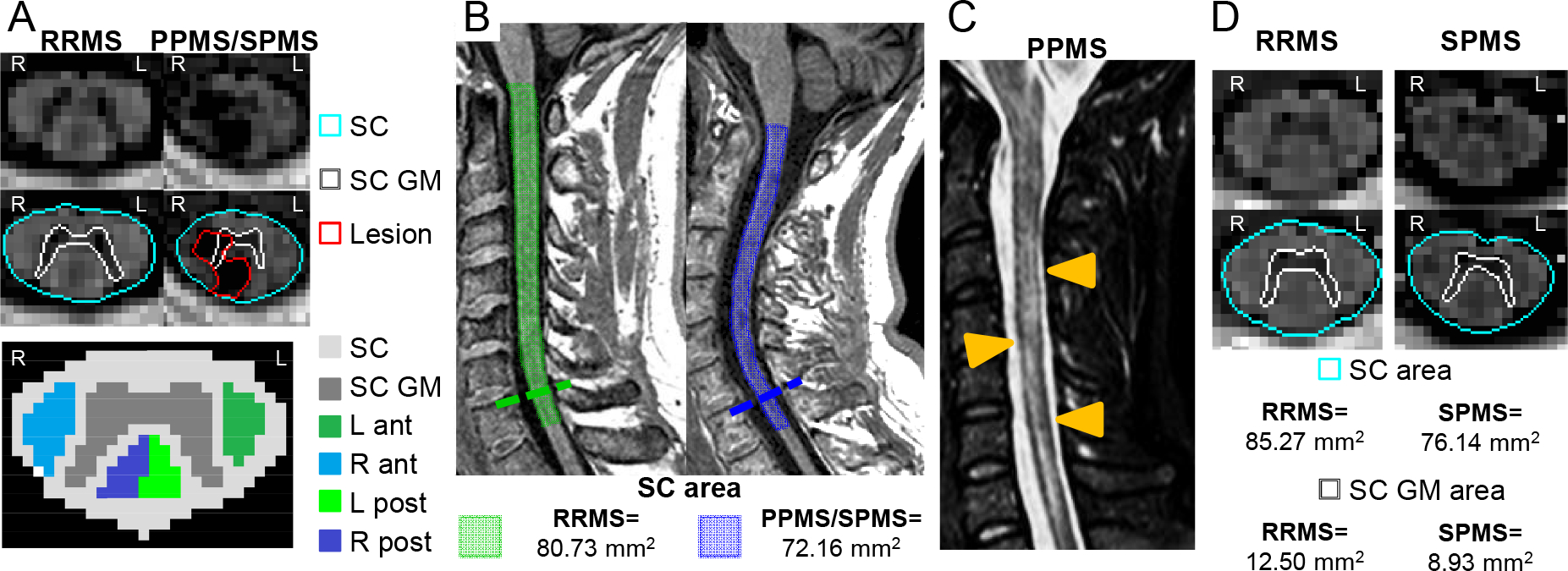
Features supporting diagnosis for both PPMS and SPMS include: A) PSIR axial sequence of the spinal cord (SC) (top and middle rows), lesions involving the GM and ≥2 WM columns on axial plane. No lesions detected in a RRMS female patient with 4 years of disease duration (left), whereas a lesion affecting the right anterior and posterior column, the left posterior column and the central GM is visible at C3-C4 level in a SPMS female patient with a disease duration of 22 years (right). A schematic representation of spinal cord subdivisions of GM and WM columns is shown in the bottom row. B) atrophy of the lower portion of the cervical SC. On the right, a sagittal 3D T1-weighted sequence reveals atrophy of the lower portion of the cervical SC (C7) (cord surface in blue) in a SPMS male patient with disease duration of 26 years. On the left, a sagittal 3D T1-weighted sequence (cord surface in green) of a RRMS male patient with disease duration of 6 years is shown. SC atrophy was quantified using the cord finder toolbox (Jim 7.0). C) Diffuse signal abnormalities in the SC (orange arrowheads) on a T2-weighted sequence in a patient with PPMS. D) GM atrophy of the SC in SPMS.. Compared to a RRMS female patient with a disease duration of 7 years (left), greater SC GM atrophy at C3/C4 level is visible on PSIR sequence in a SPMS patient with a disease duration of 19 years (right) using a local thresholding segmentation technique (Jim 7.0).
Abbreviations: ant=anterior column; GM=gray matter; L=left; mm=millimeter; MS=multiple sclerosis; post=posterior column; PP=primary progressive; PSIR=phase-sensitive inversion recovery; R=right; RR=relapsing-remitting; SC=spinal cord; SP=secondary progressive; WM=white matter.
Confluent cord lesions may resemble longitudinally extensive myelitis and diffuse cord changes seen in other inflammatory disorders or vascular disorders. Axial T2 sequences typically show that diffuse lesions in MS represent confluence of discrete lesions, many peripherally located, rather than a single, homogeneous longitudinally extensive lesion. Diffuse cord changes may occur in inflammatory disorders such as HTLV-1 or HIV-associated myelitis and systemic vasculitis, or in vascular disorders such as dural arteriovenous fistula, in which abnormal flow voids indicate dilated veins and suggest the diagnosis.
A study that separately visualized spinal cord GM and WM reported that more patients with focal lesions extending to GM and involving at least two WM columns had RRMS vs. CIS (odds ratio [OR]=8.3, 95% confidence interval [CI]=2.5–27.6; p=.001) and SPMS vs. RRMS (OR=10.1, 95% CI=1.2–85.3; p=.03) (Figure 1);24 there was no difference between SPMS and PPMS (OR=3.1, 95% CI=0.3–31.8; p=.34).24 In a multicenter study, the central cord, which is primarily GM, was more often affected in PPMS than RRMS patients (peak t value=4.5 at C3; p<.05).26
Spinal cord atrophy may be related to PMS. In the cervical cord, C7 area was smaller in patients with SPMS within 1 year of transition from RRMS (n=25, 56.0 ± 8.8 mm2) than other groups (RIS=63.7 ± 9.8 mm, n=34; RRMS=64.8 ± 10 mm2 n=31; p≤.003) and was associated with SPMS (β=7.6, p=.004).27 The lower cervical cord is selectively more vulnerable in SPMS for uncertain reasons; nonetheless, a craniocaudal pattern of spinal cord atrophy may herald SPMS (Figure 1). A recent multicenter study found that atrophy between C1/C2 and C5 was found in RRMS, whereas extension to C6-C7 was detected in PMS; C5/C6 involvement was more typical of SPMS than PPMS.28 Measurement of cervical and thoracic cord GM area (Figure 1) together with brain GM volume may be superior to measurement of whole brain or cord atrophy in distinguishing PMS from RRMS (area under the curve=0.90).29,30
Early predictors of progression
In relapse-onset MS, the number and volume of brain T2-hyperintense WM lesions at disease onset predicts long-term disability (Figure 2).31–33 T2-hyperintense lesion numbers and volumes (LVs) on initial MRI (median [range] T2 LV=2.5 [0.0,55.0] cm3 vs. 0.7 [0.0,13.7] cm3) and increase in brain T2-hyperintense LV within the first five years after disease onset were greater in CIS patients who developed SPMS after 20 years compared to those who remained RRMS (T2 LV growth rate per year [bootstrap 95% CI]=2.89 [1.78,4.01] cm3 vs. 0.80 [0.63,0.99] cm3; p<.001) (Figure 2).31
Figure 2. Summary of MRI predictors of subsequent disability progression and evolution to SPMS at disease onset.
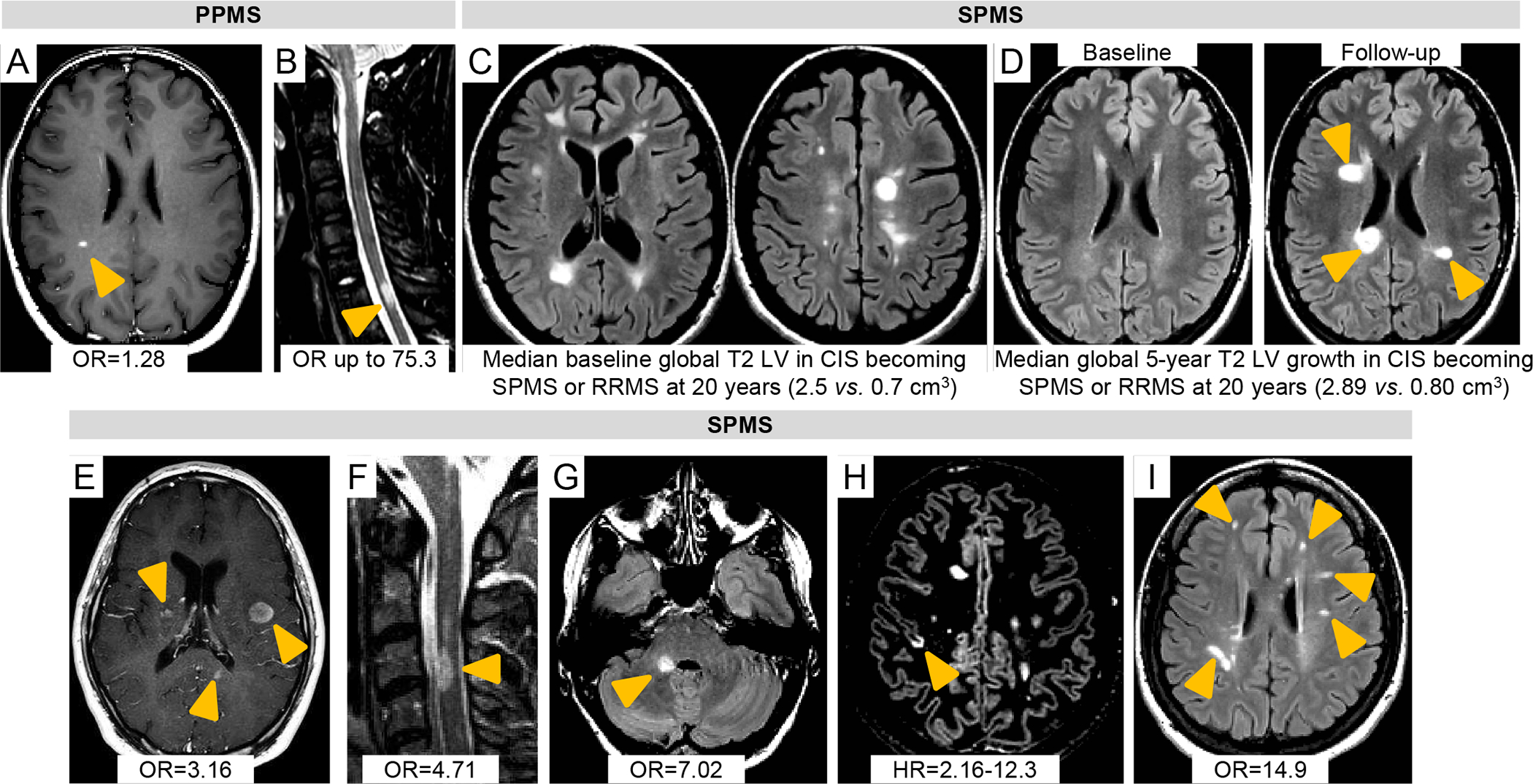
In PPMS early predictors are A) gadolinium (Gd)-enhancing (orange arrowheads) on post-contrast T1-weighted sequences35 and B) spinal cord (SC) lesions at baseline (orange arrowheads),40 STIR sequence. In SPMS early predictors are C) the number and volume of baseline brain lesions (T2-FLAIR sequences);31 D) increase of brain lesion volume (LV) during the first 5 years (orange arrowheads) (T2-FLAIR sequences);31 E) ≥2 Gd-enhancing lesions at baseline (orange arrowheads) (post-contrast T1-weighted sequence);34 F) ≥1 SC lesion at baseline or ≥1 SC lesion within 1–3 years (orange arrowheads) (STIR sequence);34 G)≥1 infratentorial lesion at baseline or ≥1 infratentorial lesion within 1–3 years (orange arrowheads) (T2-FLAIR sequence);34 H) ≥1 cortical lesion at baseline (orange arrowheads) (DIR sequence);44 I) ≥1 new deep WM lesion within 1 year (orange arrowheads) (T2-FLAIR sequence).34 Relevant OR, HRs or global T2 LV that are associated with disability progression and evolution to SPMS at disease onset and that were derived from the literature and discussed in the text are also reported.
Abbreviations: cm=centimeter; DIR=double inversion recovery; FLAIR=fluid-attenuated inversion recovery; Gd=gadolinium; HR=hazard ratio; LV=lesion volume; MRI=magnetic resonance imaging; MS=multiple sclerosis; OR=odds ratio; PP=primary progressive; SC=spinal cord; SP=secondary progressive; STIR=short tau inversion recovery; WM=white matter.
Having at least two Gd-enhancing lesions (OR [95% CI]=3.16 [1.08,9.23]; p=.035) and one spinal cord lesion (OR [95% CI]=4.71 [1.72,12.92], p=.003) at CIS onset, and the occurrence of one spinal cord lesion (at 1 year, OR [95% CI]=5.72 [1.67,19.56], p=.005) or one infratentorial lesion within one or 3 years (at 1 year, OR [95% CI]=7.02 [2.06,23.94], p=.002) after disease onset predicted conversion to SPMS after 15 years (C-statistic=0.86 and accuracy=91% at 1 year; 0.89 and 88% at 3 years) (Figure 2).34 The number of Gd-enhancing lesions on baseline scans was associated with disability progression over 5 years in patients with PPMS (Figure 2) (OR [95% CI]=1.28 [1.04,1.58]; p=.02).35
Lesion topography at disease onset, particularly location in brainstem33,36,37 and spinal cord34,38,39 (Figure 2), is associated with greater disability worsening. A 30-year longitudinal study confirmed the association of baseline infratentorial lesions (≥1 vs. 0) (OR [95% CI]=20.3 [5.4,75.6]; accuracy=78%; p<.001), new infratentorial (≥1 vs. 0) (OR [95% CI]=19.3 [5.7,65.6]; accuracy=65%; p<.001) and new deep WM lesions (≥1 vs. 0) (OR [95% CI]=14.9 [3.3,68.1]; accuracy=65%; p<.001) one year after MS onset with long-term disability progression and SPMS evolution (Figure 2).33
Spinal cord lesions predict worse disease evolution when present early in the disease course, independent of classification as RIS40,41 or CIS34,39 (Figure 2). Twenty-five of 71 (35%) subjects with RIS had asymptomatic spinal cord lesions.40 Asymptomatic spinal cord lesions increased the risk of clinical conversion up to 75.3 fold (95% CI=16.1,350.0; p<.001); 21 out of 25 (84%) with RIS and asymptomatic cord lesions progressed to CIS (n=19) or PPMS (n=2) over 1.6 years.40 In another study, spinal cord lesions were more common in RIS cases who developed PPMS (15/15, 100%) compared to those who developed relapse-onset MS (72/113, 64%) (p=.005) or remained without clinical evidence of MS (i.e., RIS) (74/324, 23%) (p<.001) over 5 years.41 CIS patients with spinal cord lesions had a 5.6-fold higher risk (95% CI=0.9–35.4, p=.065) of reaching an Expanded Disability Status scale (EDSS) score ≥3.0; this risk was greatest in those whose initial attack was non-spinal (hazard ratio [HR] [95% CI]=29.8 [1.1,786.5]; p=.042).39 Baseline spinal cord lesion number (β [95% CI]=0.40 [0.26,0.54]; p<.001), increase in spinal cord lesion number (β [95% CI]=0.19 [0.11,0.28]; p<.001) and higher rate of spinal cord atrophy (β [95% CI]=−0.15 [−0.21,−0.09]; p<.001) are associated independently with higher EDSS (R2=0.53) 5 years after disease onset in non-spinal CIS patients.42 In RRMS within 2 years from disease onset, spinal cord lesions were an independent predictor of EDSS score ≥4.0 after 7 years (β [95% CI]=4.4 [2.1,9.0; p<.001]).43
In 219 relapse-onset MS patients, the number of CLs at disease onset predicted conversion to SPMS after 7 years (HR=2.16, 4.79, and 12.3 for 2, 5, and 7 CLs, respectively; p<.001), and time to progression (up to 4 years earlier on average) (Figure 2).44 No patient without CLs at baseline entered the SP phase and few (1.8%) reached an EDSS score ≥4.0 at last follow-up.44
Markers of disease worsening
WM lesions.
WM LV of untreated MS patients increases by 5–10% per year,45 higher in those with PMS than with RRMS.18 In patients with longstanding, severe MS, multifocal T2 lesions expand, and new ones appear, ultimately leading to confluent T2 lesions in the WM. WM LV and confluent lesions have been proposed as predictors of SPMS onset and disease progression (Figure 3). However, the correlations between T2 LV and disability are only moderate at best. Progressive accumulation of WM lesions in patients with severe MS has been reported in some studies46,47 but not in others.48 Differences in the degree of concomitant accumulation of new lesions, enlargement of pre-existing lesions and shrinkage of others in the cohorts analyzed could explain the discrepant conclusions of these studies.
Figure 3. Summary of the MRI markers to identify disability progression and SPMS evolution during the disease course.
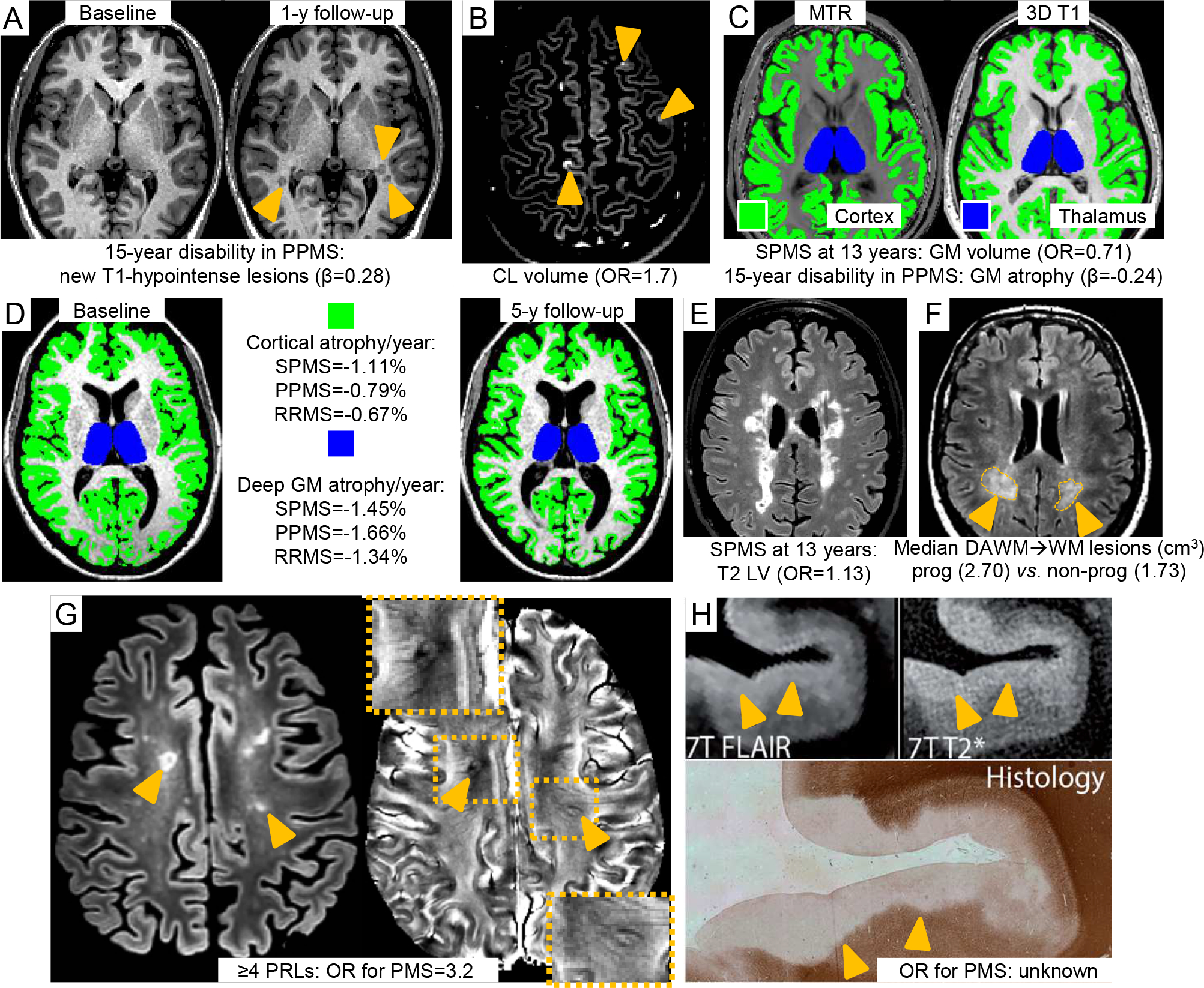
In both PPMS and SPMS MRI markers are A) increase of number or volume of brain T1-hypointense lesions.54 Baseline and 1-year follow-up T1-weighted sequences of a PPMS patient. At follow-up, three new T1-hypointense lesions are visible (orange arrowheads); B) cortical lesion number and volume.55 A DIR sequence showing three cortical lesions (orange arrowheads) from a PPMS patient is shown; C) baseline thalamic (blue) and cortical (green) damage and atrophy.51 Quantification on MT imaging (left) and 3D T1-weighted sequences (right); D) rate of whole brain, brain GM (green) and deep GM (blue) atrophy.69 Quantification on a 3D T1-weighted sequence using SIENA and FIRST software. In SPMS also E) brain T2-hyperintense lesion volume (LV) (T2-FLAIR sequence)51 and F) conversion of DAWM (orange arrowheads and dotted orange areas) into focal WM lesions (T2-FLAIR sequence).65 Further promising MRI markers are G) the presence of ≥4 rim positive lesions on susceptibility-based MRI (orange arrowheads)85 and H) presence of subpial demyelination (orange arrowheads) on T2* sequence at ultra-high field (i.e., 7 Tesla).101 Figure 3H is adapted from Kilsdonk et al.101 with permission. Relevant OR, HRs or global T2 LV that were suggested to identify disability progression and SPMS evolution during the disease course and that were derived from the literature and discussed in the text are also reported.
Abbreviations: cm=centimeter; DAWM=dirty-appearing white matter; DIR=double inversion recovery; FLAIR=fluid-attenuated inversion recovery; GM=gray matter; LV=lesion volume; MRI=magnetic resonance imaging; MS=multiple sclerosis; MT=magnetization transfer; MTR=magnetization transfer ratio; OR=odds ratio; PP=primary progressive; SP=secondary progressive; y=year; WM=white matter.
Black holes.
The relationship between black holes and disability has been assessed with conflicting results.21,22,45,49–54 In cross-sectional studies, black hole LV showed mild-to-moderate correlation with EDSS, especially when only the most hypointense voxels were evaluated.21,22,45,49,50
Longitudinally, in relapse-onset MS, EDSS worsening over 10 years was most strongly associated with the combination of higher baseline black hole numbers (r=0.42; p<.001) and increasing black hole LV (r=0.53; p<.001).52 The change in black hole LV over 1-year was the only predictor of MS severity score (MSSS) after 12 years, explaining 20% of the variance in MSSS; baseline and 1-year changes of T2 and Gd-enhancing LV, and of brain and ventricular volume fractions were not retained in the model.53 Conversely, baseline black hole volume did not predict worsening of disability assessed using MSSS or EDSS over up to 13 years.51 In PPMS, new T1-hypointense lesions after 15 months were associated with EDSS worsening after 15 years (β=0.28; p=.003) (Figure 3).54
GM lesions.
CLs have been detected in all MS clinical phenotypes, but their burden and accumulation over time are higher in PMS and are associated with clinical worsening (Figure 3).12,13,55,56 In a 5-year longitudinal study, CL volume (OR [95% CI]=1.7 [1.4,2.3]; p<.001), age (OR [95% CI]=1.2 [1.1,1.3]; p=.001) and cerebellar cortical volume (OR [95% CI]=0.2 [0.1,0.4]; p<.001) predicted evolution from RRMS to SPMS.55 Focal lesions frequently affect the thalamus.57,58 PMS patients have a higher thalamic lesion burden, especially lesions with a discrete/ovoid shape,57 and are preferentially located in regions close to the CSF.58 In PPMS and SPMS, thalamic lesion burden correlates with clinical disability.57,58
Spinal cord lesions.
Greater spinal cord lesion burden is consistently associated with more severe disability, a progressive phenotype and faster disability progression.26,28,59
The relevance of spinal cord lesions for clinical disability is supported by the recent recognition of PPMS phenotype in the setting of an isolated CNS demyelinating lesion, termed progressive solitary sclerosis.60 Patients typically have a single critical lesion in the CNS, located in the spinal cord typically at the cervico-medullary junction, oligoclonal bands and experience progressive motor impairment.60
Dirty-appearing WM.
Some investigators have reported that dirty-appearing (DA) WM (Box 2, Figure 3) is associated with higher EDSS score61,62 while others have not.63–65
A recent study found DAWM on proton density (PD)- and T2-weighted images in 88/348 (25.3%) RRMS patients.64 After 8 years, DAWM burden was unchanged in the majority of RRMS patients (61/88, 69.3%), decreased in 25/88 (28.4%) and increased in 2/88 (2.3%). DAWM was associated with more severe brain atrophy (−4.8% vs. −4.2%; p=.038), but not with baseline and longitudinal changes of WM lesion burden and EDSS score.64 The lack of association with disability could be explained by a gradual evolution of DAWM into WM lesions. A recent study of 589 SPMS patients showed greater transformation of DAWM into focal WM lesions after 3 years in those with disability progression than those without (2.70 cm3 vs. 1.76 cm3; p<.001).65
DAWM can be found in up to 6% of healthy controls (HC)64 and also occurs in other conditions such as cerebrovascular disease,66 and therefore may not be MS-specific66 and could be influenced by altered cerebrospinal fluid pulsatility linked to hypertension.67 Furthermore, the prevalence of DAWM is associated with MR field strength.64
Atrophy.
Brain17,68–71 and spinal cord27,28,72–74 atrophy is associated with clinical disability and are useful prognostic markers (Figure 3). This is particularly relevant to PMS, a disease phenotype characterized by prominent brain and spinal cord atrophy.
Cerebral GM atrophy is an important indicator of clinical disability (Figure 3).68–71 Compared with HC (mean [standard deviation, SD] annualized GM fraction loss=−0.028 [0.24] %), the 4-year GM fraction change was 8.1 times greater in RRMS (−0.23 [0.34] %), 12.4 times greater in RRMS converting to SPMS (−0.35 [0.37] %), and 14 times greater in established SPMS (−0.39 [0.50] %).70 A large multicenter 2.4-year longitudinal study showed the annualized rate of deep GM and cortical atrophy was faster in SPMS (−1.45% and −1.11%, respectively) and PPMS (−1.66% and −0.79%, respectively) compared to HC (−0.94% and −0.34%, respectively) and RRMS (−1.34% and −0.67%, respectively). Deep GM atrophy occurred most rapidly and was associated with disability progression (β [95% CI]=−0.04 [−0.02,−0.06]; p=.006).69,71 The pattern of GM atrophy progression was similar in relapse-onset MS and PPMS, first manifest in deep GM and subsequently spreading to involve an increasing number of cortical regions in PMS.68 The cerebellum and the striatum atrophied earlier in relapse-onset MS than in PPMS.68
GM damage also predicts long-term prognosis. In relapse-onset MS, a combined model including baseline GM volume (OR [95% CI] =0.71 [0.51,1.00]; p=.04) and T2 LV (OR [95% CI]=1.13 [1.04,1.24]; p=.005) predicted evolution to SPMS after 13 years (C-index=0.84).51 In PPMS, a model including GM mean diffusivity (a measure of microstructural abnormalities) (β=3.86; p=.03), GM atrophy (β=−0.24; p=.05) and new T1-hypointense lesions during the first 15 months of follow-up (β=0.28; p=.003) predicted 15-year disability (Figure 3) (R2=0.61).54
Spinal cord atrophy progresses at a faster rate in PMS patients and in those experiencing disability worsening than those who do not.27,28,72,74
PPMS and SPMS: similar or not?
Despite their historical separation, whether PPMS and SPMS are different in terms of pathophysiology is uncertain. The ratio of women to men in relapse-onset MS is greater than in PPMS; an excess of women does not occur in PPMS.75–77 PPMS patients are older and more likely to have comorbid conditions and age may contribute to disability and to MRI changes in PPMS.75–78 By contrast, genetic, epidemiological, immunological, histopathological and MRI findings are similar arguing against a fundamental distinction between PPMS and SPMS.75,76,79–82 Members of multiplex pedigrees may present with different clinical phenotypes.80 RIS cases can evolve to either RRMS or PPMS41 and 28% of PPMS patients experience relapses.83
Disability progression is a function of age and not pre-progressive disease course.75,76,82,83 The rate of disease progression is similar in PPMS and SPMS patients after reaching EDSS≥4.0.75,76
The burden of T2-hyperintense, T1-hypointense and Gd-enhancing lesions has been reported to be lower in PPMS vs. SPMS18,19but this is now disputed.35,84 Similar to what occurs in relapse-onset MS, although with a lower prevalence, Gd-enhancing lesions are more frequent in the earliest phases and decline over time.35
The prevalence of paramagnetic rim lesions (PRLs) on susceptibility-based MRI85 and of CLs12,13 do not differ between PPMS and SPMS. Microstructural tissue abnormalities and irreversible tissue loss consistently occur in PPMS and SPMS and predate clinical progression.86,87
Ageing can contribute similarly to disability progression in PPMS and SPMS. Trophic CNS support, compensatory mechanisms, and remyelinating capacity decline with age, limiting functional recovery and brain plasticity, and contributing to neurodegenerative processes.88,89 Mitochondrial abnormalities and higher oxidative stress have greater impact on PMS than RRMS.90
Comorbidities unrelated to MS also have detrimental effects, especially in older PMS patients, possibly accelerating disease progression.78,91,92 Cerebrovascular diseases accelerate brain ageing, promote hypoperfusion with consequent hypoxic WM lesion accumulation, and contribute to neurodegeneration.93 Cerebrovascular risk factors, including higher body mass index and dyslipidemia, are independently associated with higher EDSS and disability progression.78,91,92
Collectively, these observations suggest that disease progression is similar in PPMS and SPMS, and that age and comorbid diseases may confound any reported differences. A recent revision of MS phenotypic categories combined PPMS and SPMS and classified PMS by activity (presence or absence of relapses or MRI activity),5 reflecting the shift in current thought on this issue.
New MRI markers of progression
Slowly expanding lesions.
Slowly expanding lesions (i.e., with a slow but consistent expansion over time)94 have been proposed as a pathological substrate of disability progression in MS (Box 2).79,81,94 These lesions can show a paramagnetic rim on susceptibility-based sequences (Figure 3),85,94–96 limited repair, lower T1 signal intensity compared to quiescent lesions, and a progressive decline of T1-hypointensity indicating ongoing tissue destruction.97 Patients with ≥4 PRLs have more severe brain atrophy, earlier motor disability and cognitive impairment and a 3.2-fold higher prevalence of PMS than those with ≤3 PRLs (8/29 [28%] vs. 4/45 [9%]; p<.05).85
However, not all chronic active lesions show iron-laden rims in pathological studies.98 Additionally, they are not specific to PMS; rim lesions occur in all MS phenotypes, at widely different reported rates: CIS/RRMS from 6% to 53%, PMS from 7% to 62%. Some studies reported a higher frequency in PMS compared to RRMS99 while others did not.84 Linear expansion over time on T1- and T2-weighted sequences occurs in a similar proportion of patients with RRMS and PPMS (68% vs. 72%, respectively).100
Subpial demyelination.
Pathology studies suggest that demyelination extending inward from the brain pial surface may be MS-specific and more widespread in PMS than in RRMS. However, subpial demyelination is very poorly detected at standard field strengths.101 Conventional and quantitative MRI techniques at ultrahigh field strengths could improve the detection (Figure 3). SPMS patients have greater decrease of magnetization transfer ratio values in the outer cortical surface compared to RRMS,102,103 but increased T2*, probably reflecting reduced myelin and iron content, is more widespread in PMS than RRMS.104
Leptomeningeal enhancement.
Focal/linear leptomeningeal enhancement on delayed post-contrast T2-fluid-attenuated inversion recovery (T2-FLAIR) images, presumably reflecting meningeal inflammatory aggregates, has been described in 25% of MS patients using 3T MRI105 and in 90% using 7T MRI.106 Clinical and pathological data suggest an association of these MR findings with cortical atrophy, prior or current inflammation and cortical (subpial) demyelination.105–109 However, focal leptomeningeal enhancement is not MS-specific, and is detected in other inflammatory CNS diseases110 and with ageing.111 It is detectable more frequently in PMS (33%) compared to RRMS (19%)105 but its discriminative power is low; its prognostic value is unknown.
Other candidate biomarkers of progression
Serum neurofilament light chain.
Neurofilaments are structural scaffolding proteins exclusively expressed in neurons and represent promising biomarkers for neuro-axonal injury. They are released into serum upon CNS damage and can be reliably quantified with SIMOA technology.
In a study of 246 MS patients,112 sNfL levels were positively associated with EDSS score (β=1.141 [95% CI=1.106,1.178]; p<.001) and recent EDSS worsening (β=1.294 [95% CI=1.090,1.536]; p=.003). PMS had higher sNfL concentrations compared to CIS/RRMS (41.4 [IQR=32.1,57.2] pg/ml vs. 27.2 [IQR=19.2,57.2] pg/ml; β=1.205 [95% CI=1.106,1.418]; p=.029). This was confirmed in a longitudinal study of 259 MS patients with a median follow-up of 6.5 years (PMS=41.9 [31.9,55.7]) pg/ml vs. RRMS=29.7 [21.2,42.2] pg/ml; β [95% CI]=1.154 [1.059,1.258]; p=.001).113
Higher baseline sNfL levels have been also associated with a greater risk of EDSS worsening over 1 year,112,113 EDSS score after 5 years (r=0.26; q=.012),114 3-month CDP (>60 pg/mL vs. <30 pg/mL: hazard ratio [95% CI]=1.94 [0.97,3.87]; p=.06),115 and SPMS conversion over 5 years (converters vs. non-converters: median sNfL=31.7 vs. 16.9 pg/mL; q=.001).114
Longitudinal sNfL level changes also correlated with EDSS changes. In a study of 42 RRMS patients, EDSS score increased by 0.53 (95% CI=0.14,0.91, p=.009) points with a 10-fold increase in NfL over 2 years.116 In a 12-year longitudinal study of 607 patients, EDSS worsening and change in sNFL levels was correlated (β [95% CI]=1.015 [1.007,1.023]; p< .001).117
sNfL is still limited as a biomarker that identifies progression. Age, disease activity and treatments significantly influence sNfL. Moreover, sNfL changes are not specific for any neurological disease. Short-term variations and the influence of comorbidities still limit its application.
Optical coherence tomography.
OCT yields high-resolution images of the retina and has been proposed to identify PMS and predict disability progression.
A study of 541 MS patients demonstrated that the retinal nerve finer layer (RNFL) was thinner in patients with SPMS (mean [SD]=85.5 [14.3] μm) and PPMS (mean [SD]=80.5 [15.4] μm) compared to CIS (mean [SD]=98.2 [8.4] μm) and RRMS (mean [SD]=92.9 [13.0] μm).118
Similarly, in another cross-sectional study of 113 MS patients and 38 healthy controls, the RNFL the was thinner in those with PMS compared to RRMS (mean [SD]=38.63 [6.55] vs. 42.05 [7.26]; p=.023), as was the ganglion-cell/inner plexiform layer (GCIPL) (mean [SD]=60.01 [8.55] vs. 65.31 [8.87]; p=.006) and outer plexiform layer (OPL) (mean [SD]=17.96 [1.70] vs. 18.51 [1.30]; p=.033).119
Retinal atrophy is accelerated in patients with PMS. A recent study showed that PMS was associated with faster peripapillary RNFL (pRNFL) (β [95% CI]=−0.34% [−0.52,−0.16%] annualized percent rate of change; p<.001), GCIPL (β [95% CI]=−0.27% [−0.41,−0.12%] annualized percent rate of change; p<.001), inner nuclear layer (INL) (β [95% CI]=−0.10% [−0.18,−0.02%] annualized percent rate of change; p=.01),and outer nuclear layer (ONL) (β [95% CI]=−0.13% [−0.24,−0.03%] annualized percent rate of change; p=.01) atrophy, compared to RRMS.120
In a large multicenter trial of 879 MS patients, those with the lowest pRNFL thickness category at baseline (≤87 μm) had increased risk of disability worsening over 3 years (HR [95% CI]=1.75 [1.19,2.59]; p=.005) compared with those in the highest tertile (>98 μm).121
Events of optic neuritis and focal WM lesions in the retro-geniculate portion of the visual pathway can influence OCT measures. Moreover, standardized data acquisitions and rigorous quality control are necessary and further rigorous and prospective studies are required before accepting a role of OCT to identify PMS.
Positron emission tomography. 11C-PK11195 is a first generation tracer for translocator protein (TSPO). Greater 11C-PK11195 uptake, believed to mirror accumulation of activated microglia, was found in SPMS compared to RRMS in the NAWM (mean [SD] 1.38 [0.09] vs. 1.26 [0.05]; p=.018) and in the rim of chronic active lesions (mean [SD] 1.33 [0.08] vs. 1.20 [0.12]; p=.043) but not in GM structures.122 Another study showed a more widespread increased cortical 11C-PK11195 uptake in SPMS compared to RRMS, and an association of this finding with EDSS score (r=0.52; p=.026), especially in SPMS (r=0.87; p=.009).123
Uptake of a second generation tracer for TSPO (11C-PBR28) was greater in SPMS compared to RRMS patients in WM lesions, NAWM, deep GM and cortex but not in cortical lesions, although no formal statistical comparisons were performed.124 Similarly, 18F-PBR06 uptake in deep GM was higher in SPMS compared to RRMS (+10.8%; p=.002).125
PET tracers have been proposed also to investigate demyelination and remyelination processes and neuronal damage; however no study has evaluated specific features of PMS. PET is an encouraging technique for monitoring pathophysiological substrates contributing to MS progression; however, several aspects limit its application to identify PMS. PET has a lower spatial resolution than MRI and uses ionising radiation, thus raising safety concerns regarding serial radionuclide administration. Moreover, specificity of the radiotracers is limited, since some expression has been detected in astrocytes and endothelial cells; PET abnormalities are often not disease-specific. Standardized protocols for acquisition and analysis have not yet been defined. Preliminary results should be confirmed by larger prospective studies with stratification by MS phenotypes.
CONCLUSIONS
No definitive qualitative clinical, immunological, histopathological and neuroimaging features differentiate PPMS and SPMS, whereas both are characterized by neurodegenerative phenomena and a gradual and irreversible accumulation of clinical disability, which is also influenced by ageing and comorbidities.
A confident diagnosis of PPMS is more difficult than RRMS. PPMS is partly a diagnosis of exclusion, since it can be mimicked by other conditions clinically and radiologically. Diffuse signal abnormalities and lesions involving the GM and at least 2 WM columns on spinal cord MRI may be relatively specific to this stage of the disease, but confirmation is required.
Both in PPMS and relapse-onset MS patients, MRI features at disease onset predict long-term disability and a progressive disease course, including lesion location in critical CNS regions (i.e., spinal cord, infratentorial regions, and GM) and a high inflammatory activity in the first years after disease onset (Box 1).
Clinical worsening is associated with imaging measures of GM involvement and neurodegeneration; however, detection validation and standardization need to be implemented at the individual-patient level.
Discovery of MR markers capable of detecting evolution from RRMS to SPMS remains an unmet need. Multiparametric MRI studies are needed, as it is unlikely that a single MR method will be an optimal discriminator.
Novel candidate imaging biomarkers such as subpial demyelination and slowly expanding lesions/PRLs may identify PMS, but should be further investigated.
The contribution of these promising MRI measures, combined with other biomarkers, such as NfL level quantification117 or OCT assessment,120 to improve PMS identification should be explored.
Supplementary Material
Acknowledgments
Funding/Support
This paper reports the conclusions of the “Diagnosis of progressive MS: the imaging perspective” Workshop, which was held in Milan, Italy, from the 25th-26th November 2019, and chaired by Prof. Massimo Filippi. The workshop was supported by an unrestricted education grant from Merck-Serono. FB is supported by the NIHR biomedical research centre at UCLH. ATT is supported by an MRC grant (MR/S026088/1). DSR is supported by the Intramural Research Program of NINDS, NIH.
Role of the Funder/Sponsor
The Sponsor had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Conflict of interest disclosures
Massimo Filippi is Editor-in-Chief of the Journal of Neurology; received compensation for consulting services and/or speaking activities from Bayer, Biogen Idec, Merck-Serono, Novartis, Roche, Sanofi Genzyme, Takeda, and Teva Pharmaceutical Industries; and receives research support from Biogen Idec, Merck-Serono, Novartis, Roche, Teva Pharmaceutical Industries, Italian Ministry of Health, Fondazione Italiana Sclerosi Multipla, and ARiSLA (Fondazione Italiana di Ricerca per la SLA).
Paolo Preziosa received speakers honoraria from Biogen Idec, Novartis, Merck Serono and ExceMED.
Frederik Barkhof acts as a consultant to Biogen-Idec, Janssen, Bayer, Merck, Roche, Novartis, Genzyme, and Apitope Ltd; he has received sponsorship from EU-H2020, Nederlands Wetenschappelijk Onderzoek, SMSR, EU-FP7, Teva, Novartis, and Biogen.
Declan Chard within the last 3 years has received honoraria from Excemed for faculty-led education work. He is a consultant for Biogen and Hoffmann-La Roche. He has received research funding from the International Progressive MS Alliance, the MS Society, and the National Institute for Health Research (NIHR) University College London Hospitals (UCLH) Biomedical Research Centre.
Nicola De Stefano has served as consultant for Immunic Therapeutics, Merck Serono SA, Novartis Pharma AG, Sanofi-Genzyme, Roche and Teva, and has received support for congress participation or speaker honoraria from Biogen Idec, Merck Serono SA, Novartis Pharma AG, Sanofi-Genzyme, Roche and Teva.
Robert J. Fox declares personal consulting fees from Actelion, Biogen, Celgene, EMD Serono, Genentech, Immunic, Novartis, Sanofi, Teva, and TG Therapeutics; have served on clinical trial advisory committees for Actelion, Biogen, Immunic, and Novartis; and have received clinical trial contract and research grant funding from Biogen and Novartis.
Claudio Gasperini has received fees as invited speaker or travel expenses for attending meeting from Biogen, Merck-Serono, Teva, Sanofi, Novartis, Genzyme.
Ludwig Kappos’s institution (University Hospital Basel) has received the following support used exclusively for research support at the department: steering committee, advisory board and consultancy fees from Actelion, Alkermes, Almirall, Bayer, Biogen, Celgene/Receptos, df-mp, Excemed, GeNeuro SA, Genzyme, Japan Tobacco, Merck, Minoryx, Mitsubishi Pharma, Novartis, Roche, Sanofi, Santhera and Teva, as well as license fees for Neurostatus-UHB products; the Research of the MS Centre in Basel has been supported by grants from Bayer, Biogen, Novartis, the European Union, the Roche Research Foundations, the Swiss MS Society, Innoswiss and the Swiss National Research Foundation.
Xavier Montalban has received a speaker honorarium and travel expenses for participation in scientific meetings or advisory boards in past years from Actelion, Alexion, Bayer, Biogen, Celgene, EMD Serono, Genzyme, Medday, Merck, Nervgen, Novartis, Roche, Sanofi-Genzyme, Teva Pharmaceuticals, TG Therapeutics, Excemed, MSIF, and NMSS.
Bastiaan Moraal has nothing to disclose.
Daniel S. Reich has received unrelated research funding from Vertex Pharmaceuticals. He is supported by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, USA.
Àlex Rovira has received a speaker honorarium from Bayer, Sanofi-Genzyme, Bracco, Merck-Serono, Teva Pharmaceutical Industries Ltd., Novartis, Roche, and Biogen. He serves on scientific advisory boards for Novartis, Sanofi-Genzyme, SyntheticMR, Bayer, Roche, Biogen, Neurodiem, and OLEA Medical.
Ahmed T. Toosy has received speaker honoraria from Biomedia, Sereno Symposia International Foundation, Bayer and meeting expenses from Biogen Idec and is the UK PI for two clinical trials sponsored by MEDDAY pharmaceutical company (MD1003 in optic neuropathy [MS-ON] and progressive MS [MS-SPI2]).
Anthony Traboulsee has research funding from Chugai, Roche, Novartis, Genzyme, Biogen as well as consultancy honoraria from Genzyme, Roche, Teva, Biogen, Serono.
Brian G. Weinshenker reports personal fees from Novartis, MedImmune, Alexion, Chugai, Roche and Mitsubishi-Tanabe has a patent of NMO-IgG for diagnosis of neuromyelitis optica with royalties paid to RSR Ltd, Oxford University, Hospices Civil de Lyon, and MVZ Labor PD Dr Volkmann und Kollegen GbR.
Burcu Zeydan has nothing to disclose. She reports funding from the National Institutes of Health.
Brenda Banwell serves as a centralized MRI reviewer for Novartis, and serves as an unpaid advisor regarding pediatric MS clinical trial design for Novartis, Biogen Idec, and Teva Neuroscience.
Maria A. Rocca received speakers honoraria from Biogen Idec, Novartis, Genzyme, Teva, Merck Serono, Roche, Celgene and Bayer and receives research support from the Italian Ministry of Health, MS Society of Canada and Fondazione Italiana Sclerosi Multipla.
Attendees of the workshop: “Diagnosis of progressive MS: the imaging perspective” (Milan, November 25–26, 2019)
Chair - Massimo Filippi (Neuroimaging Research Unit, Institute of Experimental Neurology, Division of Neuroscience, Neurology Unit, Neurophysiology Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University, Milan, Italy)
Speakers - F. Barkhof (Department of Radiology and Nuclear Medicine, Amsterdam UMC, Location VUmc, MS Center Amsterdam, Amsterdam, Netherlands and Institutes of Neurology and Healthcare Engineering, University College London, London, UK); D. Chard (Nuclear Magnetic Resonance Research Unit, Queen Square Multiple Sclerosis Centre, University College London Institute of Neurology, and National Institute for Health Research, University College London Hospitals, Biomedical Research Centre, London, UK); N. De Stefano (Department of Medicine, Surgery and Neuroscience, University of Siena, Siena, Italy); C. Gasperini (Department of Neurology, San Camillo-Forlanini Hospital, Roma, Italy); M. Filippi (Neuroimaging Research Unit, Institute of Experimental Neurology, Division of Neuroscience, Neurology Unit, Neurophysiology Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy; Vita-Salute San Raffaele University, Milan, Italy); X. Montalban (Department of Neurology, Cemcat, Hospital Vall d’Hebron, Autonomous University of Barcelona, Barcelona, Spain, and Division of Neurology, St Michael’s Hospital, University of Toronto, Toronto, Ontario, Canada); B. Moraal (Department of Radiology and Nuclear Medicine, Amsterdam UMC, Location VUmc, MS Center Amsterdam, Amsterdam, Netherlands); P. Preziosa (Neuroimaging Research Unit, Institute of Experimental Neurology, Division of Neuroscience, and Neurology Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy); D.S. Reich (Translational Neuroradiology Section, Division of Neuroimmunology and Neurovirology, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD, USA); À. Rovira (Neuroradiology Section, Department of Radiology (IDI), Vall d’Hebron University Hospital and Research Institute (VHIR), Autonomous University Barcelona, Spain); A.T. Toosy (Nuclear Magnetic Resonance Research Unit, Queen Square Multiple Sclerosis Centre, University College London Institute of Neurology); A. Traboulsee (MS/MRI Research Group, Djavad Mowafaghian Centre for Brain Health, and Faculty of Medicine, Division of Neurology, University of British Columbia, Vancouver, British Columbia, Canada); B. Zeydan (Department of Neurology and Department of Radiology, Mayo Clinic, Rochester, MN, USA); M.A. Rocca (Neuroimaging Research Unit, Institute of Experimental Neurology, Division of Neuroscience, and Neurology Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy).
Discussants – B. Banwell (Division of Child Neurology, The Children’s Hospital of Philadelphia, Departments of Neurology and Pediatrics, Perelman School of Medicine, University of Pennsylvania); R.J. Fox (Mellen Center for Multiple Sclerosis, Cleveland Clinic, Cleveland, OH, USA); L. Kappos (Neurologic Clinic and Policlinic, Departments of Medicine, Clinical Research, Biomedicine and Biomedical Engineering, University Hospital and University of Basel, Basel, Switzerland); B.G. Weinshenker (Department of Neurology, Mayo Clinic, Rochester, MN, USA);
Access to data and data analysis
Prof. Massimo Filippi had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162–173. [DOI] [PubMed] [Google Scholar]
- 2.Faissner S, Plemel JR, Gold R, Yong VW. Progressive multiple sclerosis: from pathophysiology to therapeutic strategies. Nat Rev Drug Discov. 2019. [DOI] [PubMed] [Google Scholar]
- 3.Kalincik T, Cutter G, Spelman T, et al. Defining reliable disability outcomes in multiple sclerosis. Brain. 2015;138(Pt 11):3287–3298. [DOI] [PubMed] [Google Scholar]
- 4.Lorscheider J, Buzzard K, Jokubaitis V, et al. Defining secondary progressive multiple sclerosis. Brain. 2016;139(Pt 9):2395–2405. [DOI] [PubMed] [Google Scholar]
- 5.Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lublin F, Miller DH, Freedman MS, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10023):1075–1084. [DOI] [PubMed] [Google Scholar]
- 7.Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(11):1444–1452. [DOI] [PubMed] [Google Scholar]
- 8.Skjerbaek AG, Boesen F, Petersen T, et al. Can we trust self-reported walking distance when determining EDSS scores in patients with multiple sclerosis? The Danish MS hospitals rehabilitation study. Mult Scler. 2019;25(12):1653–1660. [DOI] [PubMed] [Google Scholar]
- 9.Fischer JS, Rudick RA, Cutter GR, Reingold SC. The Multiple Sclerosis Functional Composite Measure (MSFC): an integrated approach to MS clinical outcome assessment. National MS Society Clinical Outcomes Assessment Task Force. Mult Scler. 1999;5(4):244–250. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Waubant E, Cutter G, Wolinsky J, Leppert D. Composite end points to assess delay of disability progression by MS treatments. Mult Scler. 2014;20(11):1494–1501. [DOI] [PubMed] [Google Scholar]
- 11.Cadavid D, Cohen JA, Freedman MS, et al. The EDSS-Plus, an improved endpoint for disability progression in secondary progressive multiple sclerosis. Mult Scler. 2017;23(1):94–105. [DOI] [PubMed] [Google Scholar]
- 12.Calabrese M, Rocca MA, Atzori M, et al. Cortical lesions in primary progressive multiple sclerosis: a 2-year longitudinal MR study. Neurology. 2009;72(15):1330–1336. [DOI] [PubMed] [Google Scholar]
- 13.Calabrese M, Rocca MA, Atzori M, et al. A 3-year magnetic resonance imaging study of cortical lesions in relapse-onset multiple sclerosis. Ann Neurol. 2010;67(3):376–383. [DOI] [PubMed] [Google Scholar]
- 14.Montalban X, Sastre-Garriga J, Filippi M, et al. Primary progressive multiple sclerosis diagnostic criteria: a reappraisal. Mult Scler. 2009;15(12):1459–1465. [DOI] [PubMed] [Google Scholar]
- 15.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelly SB, Kinsella K, Duggan M, Tubridy N, McGuigan C, Hutchinson M. A proposed modification to the McDonald 2010 criteria for the diagnosis of primary progressive multiple sclerosis. Mult Scler. 2013;19(8):1095–1100. [DOI] [PubMed] [Google Scholar]
- 17.University of California SFMSET, Cree BAC, Hollenbach JA, et al. Silent progression in disease activity-free relapsing multiple sclerosis. Ann Neurol. 2019;85(5):653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson AJ, Kermode AG, MacManus DG, et al. Patterns of disease activity in multiple sclerosis: clinical and magnetic resonance imaging study. BMJ. 1990;300(6725):631–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson AJ, Kermode AG, Wicks D, et al. Major differences in the dynamics of primary and secondary progressive multiple sclerosis. Ann Neurol. 1991;29(1):53–62. [DOI] [PubMed] [Google Scholar]
- 20.Ingle GT, Sastre-Garriga J, Miller DH, Thompson AJ. Is inflammation important in early PPMS? a longitudinal MRI study. J Neurol Neurosurg Psychiatry. 2005;76(9):1255–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nijeholt GJ, van Walderveen MA, Castelijns JA, et al. Brain and spinal cord abnormalities in multiple sclerosis. Correlation between MRI parameters, clinical subtypes and symptoms. Brain. 1998;121 ( Pt 4):687–697. [DOI] [PubMed] [Google Scholar]
- 22.van Walderveen MA, Lycklama ANGJ, Ader HJ, et al. Hypointense lesions on T1-weighted spin-echo magnetic resonance imaging: relation to clinical characteristics in subgroups of patients with multiple sclerosis. Arch Neurol. 2001;58(1):76–81. [DOI] [PubMed] [Google Scholar]
- 23.Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria”. Ann Neurol. 2005;58(6):840–846. [DOI] [PubMed] [Google Scholar]
- 24.Kearney H, Miszkiel KA, Yiannakas MC, Altmann DR, Ciccarelli O, Miller DH. Grey matter involvement by focal cervical spinal cord lesions is associated with progressive multiple sclerosis. Mult Scler. 2016;22(7):910–920. [DOI] [PubMed] [Google Scholar]
- 25.Weier K, Mazraeh J, Naegelin Y, et al. Biplanar MRI for the assessment of the spinal cord in multiple sclerosis. Mult Scler. 2012;18(11):1560–1569. [DOI] [PubMed] [Google Scholar]
- 26.Eden D, Gros C, Badji A, et al. Spatial distribution of multiple sclerosis lesions in the cervical spinal cord. Brain. 2019;142(3):633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeydan B, Gu X, Atkinson EJ, et al. Cervical spinal cord atrophy: An early marker of progressive MS onset. Neurol Neuroimmunol Neuroinflamm. 2018;5(2):e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rocca MA, Valsasina P, Meani A, et al. Clinically relevant cranio-caudal patterns of cervical cord atrophy evolution in MS. Neurology. 2019;93(20):e1852–e1866. [DOI] [PubMed] [Google Scholar]
- 29.Schlaeger R, Papinutto N, Panara V, et al. Spinal cord gray matter atrophy correlates with multiple sclerosis disability. Ann Neurol. 2014;76(4):568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schlaeger R, Papinutto N, Zhu AH, et al. Association Between Thoracic Spinal Cord Gray Matter Atrophy and Disability in Multiple Sclerosis. JAMA Neurol. 2015;72(8):897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fisniku LK, Brex PA, Altmann DR, et al. Disability and T2 MRI lesions: a 20-year follow-up of patients with relapse onset of multiple sclerosis. Brain. 2008;131(Pt 3):808–817. [DOI] [PubMed] [Google Scholar]
- 32.Tintore M, Rovira A, Rio J, et al. Defining high, medium and low impact prognostic factors for developing multiple sclerosis. Brain. 2015;138(Pt 7):1863–1874. [DOI] [PubMed] [Google Scholar]
- 33.Chung KK, Altmann D, Barkhof F, et al. A 30-Year Clinical and Magnetic Resonance Imaging Observational Study of Multiple Sclerosis and Clinically Isolated Syndromes. Ann Neurol. 2020;87(1):63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brownlee WJ, Altmann DR, Prados F, et al. Early imaging predictors of long-term outcomes in relapse-onset multiple sclerosis. Brain. 2019;142(8):2276–2287. [DOI] [PubMed] [Google Scholar]
- 35.Khaleeli Z, Ciccarelli O, Mizskiel K, Altmann D, Miller DH, Thompson AJ. Lesion enhancement diminishes with time in primary progressive multiple sclerosis. Mult Scler. 2010;16(3):317–324. [DOI] [PubMed] [Google Scholar]
- 36.Minneboo A, Barkhof F, Polman CH, Uitdehaag BM, Knol DL, Castelijns JA. Infratentorial lesions predict long-term disability in patients with initial findings suggestive of multiple sclerosis. Arch Neurol. 2004;61(2):217–221. [DOI] [PubMed] [Google Scholar]
- 37.Tintore M, Rovira A, Arrambide G, et al. Brainstem lesions in clinically isolated syndromes. Neurology. 2010;75(21):1933–1938. [DOI] [PubMed] [Google Scholar]
- 38.Sombekke MH, Wattjes MP, Balk LJ, et al. Spinal cord lesions in patients with clinically isolated syndrome: a powerful tool in diagnosis and prognosis. Neurology. 2013;80(1):69–75. [DOI] [PubMed] [Google Scholar]
- 39.Arrambide G, Rovira A, Sastre-Garriga J, et al. Spinal cord lesions: A modest contributor to diagnosis in clinically isolated syndromes but a relevant prognostic factor. Mult Scler. 2018;24(3):301–312. [DOI] [PubMed] [Google Scholar]
- 40.Okuda DT, Mowry EM, Cree BA, et al. Asymptomatic spinal cord lesions predict disease progression in radiologically isolated syndrome. Neurology. 2011;76(8):686–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kantarci OH, Lebrun C, Siva A, et al. Primary Progressive Multiple Sclerosis Evolving From Radiologically Isolated Syndrome. Ann Neurol. 2016;79(2):288–294. [DOI] [PubMed] [Google Scholar]
- 42.Brownlee WJ, Altmann DR, Alves Da Mota P, et al. Association of asymptomatic spinal cord lesions and atrophy with disability 5 years after a clinically isolated syndrome. Mult Scler. 2017;23(5):665–674. [DOI] [PubMed] [Google Scholar]
- 43.D’Amico E, Patti F, Leone C, Lo Fermo S, Zappia M. Negative prognostic impact of MRI spinal lesions in the early stages of relapsing-remitting multiple sclerosis. Mult Scler J Exp Transl Clin. 2016;2:2055217316631565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scalfari A, Romualdi C, Nicholas RS, et al. The cortical damage, early relapses, and onset of the progressive phase in multiple sclerosis. Neurology. 2018;90(24):e2197–e2118. [DOI] [PubMed] [Google Scholar]
- 45.Caramanos Z, Francis SJ, Narayanan S, Lapierre Y, Arnold DL. Large, nonplateauing relationship between clinical disability and cerebral white matter lesion load in patients with multiple sclerosis. Arch Neurol. 2012;69(1):89–95. [DOI] [PubMed] [Google Scholar]
- 46.Kapoor R, Ho PR, Campbell N, et al. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol. 2018;17(5):405–415. [DOI] [PubMed] [Google Scholar]
- 47.Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018;391(10127):1263–1273. [DOI] [PubMed] [Google Scholar]
- 48.Li DK, Held U, Petkau J, et al. MRI T2 lesion burden in multiple sclerosis: a plateauing relationship with clinical disability. Neurology. 2006;66(9):1384–1389. [DOI] [PubMed] [Google Scholar]
- 49.Thaler C, Faizy T, Sedlacik J, et al. T1- Thresholds in Black Holes Increase Clinical-Radiological Correlation in Multiple Sclerosis Patients. PLoS One. 2015;10(12):e0144693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tam RC, Traboulsee A, Riddehough A, Sheikhzadeh F, Li DK. The impact of intensity variations in T1-hypointense lesions on clinical correlations in multiple sclerosis. Mult Scler. 2011;17(8):949–957. [DOI] [PubMed] [Google Scholar]
- 51.Filippi M, Preziosa P, Copetti M, et al. Gray matter damage predicts the accumulation of disability 13 years later in MS. Neurology. 2013;81(20):1759–1767. [DOI] [PubMed] [Google Scholar]
- 52.Giorgio A, Stromillo ML, Bartolozzi ML, et al. Relevance of hypointense brain MRI lesions for long-term worsening of clinical disability in relapsing multiple sclerosis. Mult Scler. 2014;20(2):214–219. [DOI] [PubMed] [Google Scholar]
- 53.Minneboo A, Uitdehaag BM, Jongen P, et al. Association between MRI parameters and the MS severity scale: a 12 year follow-up study. Mult Scler. 2009;15(5):632–637. [DOI] [PubMed] [Google Scholar]
- 54.Rocca MA, Sormani MP, Rovaris M, et al. Long-term disability progression in primary progressive multiple sclerosis: a 15-year study. Brain. 2017;140(11):2814–2819. [DOI] [PubMed] [Google Scholar]
- 55.Calabrese M, Romualdi C, Poretto V, et al. The changing clinical course of multiple sclerosis: a matter of gray matter. Ann Neurol. 2013;74(1):76–83. [DOI] [PubMed] [Google Scholar]
- 56.Treaba CA, Granberg TE, Sormani MP, et al. Longitudinal Characterization of Cortical Lesion Development and Evolution in Multiple Sclerosis with 7.0-T MRI. Radiology. 2019;291(3):740–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harrison DM, Oh J, Roy S, et al. Thalamic lesions in multiple sclerosis by 7T MRI: Clinical implications and relationship to cortical pathology. Mult Scler. 2015;21(9):1139–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Louapre C, Govindarajan ST, Gianni C, et al. Heterogeneous pathological processes account for thalamic degeneration in multiple sclerosis: Insights from 7 T imaging. Mult Scler. 2018;24(11):1433–1444. [DOI] [PubMed] [Google Scholar]
- 59.Kearney H, Altmann DR, Samson RS, et al. Cervical cord lesion load is associated with disability independently from atrophy in MS. Neurology. 2015;84(4):367–373. [DOI] [PubMed] [Google Scholar]
- 60.Keegan BM, Kaufmann TJ, Weinshenker BG, et al. Progressive solitary sclerosis: Gradual motor impairment from a single CNS demyelinating lesion. Neurology. 2016;87(16):1713–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laule C, Vavasour IM, Leung E, et al. Pathological basis of diffusely abnormal white matter: insights from magnetic resonance imaging and histology. Mult Scler. 2011;17(2):144–150. [DOI] [PubMed] [Google Scholar]
- 62.West J, Aalto A, Tisell A, et al. Normal appearing and diffusely abnormal white matter in patients with multiple sclerosis assessed with quantitative MR. PLoS One. 2014;9(4):e95161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ge Y, Grossman RI, Babb JS, He J, Mannon LJ. Dirty-appearing white matter in multiple sclerosis: volumetric MR imaging and magnetization transfer ratio histogram analysis. AJNR Am J Neuroradiol. 2003;24(10):1935–1940. [PMC free article] [PubMed] [Google Scholar]
- 64.Vertinsky AT, Li DKB, Vavasour IM, et al. Diffusely Abnormal White Matter, T2 Burden of Disease, and Brain Volume in Relapsing-Remitting Multiple Sclerosis. J Neuroimaging. 2019;29(1):151–159. [DOI] [PubMed] [Google Scholar]
- 65.Dadar M, Narayanan S, Arnold DL, Collins DL, Maranzano J. Conversion of diffusely abnormal white matter to focal lesions is linked to progression in secondary progressive multiple sclerosis. Mult Scler. 2020:1352458520912172. [DOI] [PubMed] [Google Scholar]
- 66.Liauw L, van der Grond J, Slooff V, et al. Differentiation between peritrigonal terminal zones and hypoxic-ischemic white matter injury on MRI. Eur J Radiol. 2008;65(3):395–401. [DOI] [PubMed] [Google Scholar]
- 67.Beggs CB, Magnano C, Shepherd SJ, et al. Dirty-Appearing White Matter in the Brain is Associated with Altered Cerebrospinal Fluid Pulsatility and Hypertension in Individuals without Neurologic Disease. J Neuroimaging. 2016;26(1):136–143. [DOI] [PubMed] [Google Scholar]
- 68.Eshaghi A, Marinescu RV, Young AL, et al. Progression of regional grey matter atrophy in multiple sclerosis. Brain. 2018;141(6):1665–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eshaghi A, Prados F, Brownlee WJ, et al. Deep gray matter volume loss drives disability worsening in multiple sclerosis. Ann Neurol. 2018;83(2):210–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fisher E, Lee JC, Nakamura K, Rudick RA. Gray matter atrophy in multiple sclerosis: a longitudinal study. Ann Neurol. 2008;64(3):255–265. [DOI] [PubMed] [Google Scholar]
- 71.Magon S, Tsagkas C, Gaetano L, et al. Volume loss in the deep gray matter and thalamic subnuclei: a longitudinal study on disability progression in multiple sclerosis. J Neurol. 2020;267(5):1536–1546. [DOI] [PubMed] [Google Scholar]
- 72.Casserly C, Seyman EE, Alcaide-Leon P, et al. Spinal Cord Atrophy in Multiple Sclerosis: A Systematic Review and Meta-Analysis. J Neuroimaging. 2018;28(6):556–586. [DOI] [PubMed] [Google Scholar]
- 73.Tsagkas C, Magon S, Gaetano L, et al. Spinal cord volume loss: A marker of disease progression in multiple sclerosis. Neurology. 2018;91(4):e349–e358. [DOI] [PubMed] [Google Scholar]
- 74.Tsagkas C, Magon S, Gaetano L, et al. Preferential spinal cord volume loss in primary progressive multiple sclerosis. Mult Scler. 2019;25(7):947–957. [DOI] [PubMed] [Google Scholar]
- 75.Confavreux C, Vukusic S. Natural history of multiple sclerosis: a unifying concept. Brain. 2006;129(Pt 3):606–616. [DOI] [PubMed] [Google Scholar]
- 76.Kremenchutzky M, Rice GP, Baskerville J, Wingerchuk DM, Ebers GC. The natural history of multiple sclerosis: a geographically based study 9: observations on the progressive phase of the disease. Brain. 2006;129(Pt 3):584–594. [DOI] [PubMed] [Google Scholar]
- 77.Cottrell DA, Kremenchutzky M, Rice GP, et al. The natural history of multiple sclerosis: a geographically based study. 5. The clinical features and natural history of primary progressive multiple sclerosis. Brain. 1999;122 ( Pt 4):625–639. [DOI] [PubMed] [Google Scholar]
- 78.Marrie RA, Rudick R, Horwitz R, et al. Vascular comorbidity is associated with more rapid disability progression in multiple sclerosis. Neurology. 2010;74(13):1041–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frischer JM, Weigand SD, Guo Y, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol. 2015;78(5):710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hensiek AE, Seaman SR, Barcellos LF, et al. Familial effects on the clinical course of multiple sclerosis. Neurology. 2007;68(5):376–383. [DOI] [PubMed] [Google Scholar]
- 81.Luchetti S, Fransen NL, van Eden CG, Ramaglia V, Mason M, Huitinga I. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: a retrospective autopsy cohort analysis. Acta Neuropathol. 2018;135(4):511–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tutuncu M, Tang J, Zeid NA, et al. Onset of progressive phase is an age-dependent clinical milestone in multiple sclerosis. Mult Scler. 2013;19(2):188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kremenchutzky M, Cottrell D, Rice G, et al. The natural history of multiple sclerosis: a geographically based study. 7. Progressive-relapsing and relapsing-progressive multiple sclerosis: a re-evaluation. Brain. 1999;122 ( Pt 10):1941–1950. [DOI] [PubMed] [Google Scholar]
- 84.Kuchling J, Ramien C, Bozin I, et al. Identical lesion morphology in primary progressive and relapsing-remitting MS--an ultrahigh field MRI study. Mult Scler. 2014;20(14):1866–1871. [DOI] [PubMed] [Google Scholar]
- 85.Absinta M, Sati P, Masuzzo F, et al. Association of Chronic Active Multiple Sclerosis Lesions With Disability In Vivo. JAMA Neurol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Preziosa P, Rocca MA, Mesaros S, et al. Intrinsic damage to the major white matter tracts in patients with different clinical phenotypes of multiple sclerosis: a voxelwise diffusion-tensor MR study. Radiology. 2011;260(2):541–550. [DOI] [PubMed] [Google Scholar]
- 87.Ceccarelli A, Rocca MA, Pagani E, et al. A voxel-based morphometry study of grey matter loss in MS patients with different clinical phenotypes. Neuroimage. 2008;42(1):315–322. [DOI] [PubMed] [Google Scholar]
- 88.Frischer JM, Bramow S, Dal-Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132(Pt 5):1175–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Goldschmidt T, Antel J, Konig FB, Bruck W, Kuhlmann T. Remyelination capacity of the MS brain decreases with disease chronicity. Neurology. 2009;72(22):1914–1921. [DOI] [PubMed] [Google Scholar]
- 90.Dutta R, McDonough J, Yin X, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol. 2006;59(3):478–489. [DOI] [PubMed] [Google Scholar]
- 91.Tettey P, Simpson S Jr., Taylor B, et al. An adverse lipid profile is associated with disability and progression in disability, in people with MS. Mult Scler. 2014;20(13):1737–1744. [DOI] [PubMed] [Google Scholar]
- 92.Oliveira SR, Simao AN, Kallaur AP, et al. Disability in patients with multiple sclerosis: influence of insulin resistance, adiposity, and oxidative stress. Nutrition. 2014;30(3):268–273. [DOI] [PubMed] [Google Scholar]
- 93.Geraldes R, Esiri MM, DeLuca GC, Palace J. Age-related small vessel disease: a potential contributor to neurodegeneration in multiple sclerosis. Brain Pathol. 2017;27(6):707–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dal-Bianco A, Grabner G, Kronnerwetter C, et al. Slow expansion of multiple sclerosis iron rim lesions: pathology and 7 T magnetic resonance imaging. Acta Neuropathol. 2017;133(1):25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Absinta M, Sati P, Fechner A, Schindler MK, Nair G, Reich DS. Identification of Chronic Active Multiple Sclerosis Lesions on 3T MRI. AJNR Am J Neuroradiol. 2018;39(7):1233–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Absinta M, Sati P, Schindler M, et al. Persistent 7-tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J Clin Invest. 2016;126(7):2597–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Elliott C, Belachew S, Wolinsky JS, et al. Chronic white matter lesion activity predicts clinical progression in primary progressive multiple sclerosis. Brain. 2019;142(9):2787–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Popescu BF, Frischer JM, Webb SM, et al. Pathogenic implications of distinct patterns of iron and zinc in chronic MS lesions. Acta Neuropathol. 2017;134(1):45–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harrison DM, Li X, Liu H, et al. Lesion Heterogeneity on High-Field Susceptibility MRI Is Associated with Multiple Sclerosis Severity. AJNR Am J Neuroradiol. 2016;37(8):1447–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Elliott C, Wolinsky JS, Hauser SL, et al. Slowly expanding/evolving lesions as a magnetic resonance imaging marker of chronic active multiple sclerosis lesions. Mult Scler. 2019;25(14):1915–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kilsdonk ID, Jonkman LE, Klaver R, et al. Increased cortical grey matter lesion detection in multiple sclerosis with 7 T MRI: a post-mortem verification study. Brain. 2016;139(5):1472–1481. [DOI] [PubMed] [Google Scholar]
- 102.Derakhshan M, Caramanos Z, Narayanan S, Arnold DL, Louis Collins D. Surface-based analysis reveals regions of reduced cortical magnetization transfer ratio in patients with multiple sclerosis: a proposed method for imaging subpial demyelination. Human brain mapping. 2014;35(7):3402–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Samson RS, Cardoso MJ, Muhlert N, et al. Investigation of outer cortical magnetisation transfer ratio abnormalities in multiple sclerosis clinical subgroups. Mult Scler. 2014;20(10):1322–1330. [DOI] [PubMed] [Google Scholar]
- 104.Mainero C, Louapre C, Govindarajan ST, et al. A gradient in cortical pathology in multiple sclerosis by in vivo quantitative 7 T imaging. Brain. 2015;138(Pt 4):932–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Absinta M, Vuolo L, Rao A, et al. Gadolinium-based MRI characterization of leptomeningeal inflammation in multiple sclerosis. Neurology. 2015;85(1):18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Harrison DM, Wang KY, Fiol J, et al. Leptomeningeal Enhancement at 7T in Multiple Sclerosis: Frequency, Morphology, and Relationship to Cortical Volume. J Neuroimaging. 2017;27(5):461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zivadinov R, Ramasamy DP, Vaneckova M, et al. Leptomeningeal contrast enhancement is associated with progression of cortical atrophy in MS: A retrospective, pilot, observational longitudinal study. Mult Scler. 2017;23(10):1336–1345. [DOI] [PubMed] [Google Scholar]
- 108.Makshakov G, Magonov E, Totolyan N, et al. Leptomeningeal Contrast Enhancement Is Associated with Disability Progression and Grey Matter Atrophy in Multiple Sclerosis. Neurol Res Int. 2017;2017:8652463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zurawski J, Tauhid S, Chu R, et al. 7T MRI cerebral leptomeningeal enhancement is common in relapsing-remitting multiple sclerosis and is associated with cortical and thalamic lesions. Mult Scler. 2019:1352458519885106. [DOI] [PubMed] [Google Scholar]
- 110.Absinta M, Cortese IC, Vuolo L, et al. Leptomeningeal gadolinium enhancement across the spectrum of chronic neuroinflammatory diseases. Neurology. 2017;88(15):1439–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Freeze WM, Schnerr RS, Palm WM, et al. Pericortical Enhancement on Delayed Postgadolinium Fluid-Attenuated Inversion Recovery Images in Normal Aging, Mild Cognitive Impairment, and Alzheimer Disease. AJNR Am J Neuroradiol. 2017;38(9):1742–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Disanto G, Barro C, Benkert P, et al. Serum Neurofilament light: A biomarker of neuronal damage in multiple sclerosis. Ann Neurol. 2017;81(6):857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Barro C, Benkert P, Disanto G, et al. Serum neurofilament as a predictor of disease worsening and brain and spinal cord atrophy in multiple sclerosis. Brain. 2018;141(8):2382–2391. [DOI] [PubMed] [Google Scholar]
- 114.Jakimovski D, Zivadinov R, Ramanthan M, et al. Serum neurofilament light chain level associations with clinical and cognitive performance in multiple sclerosis: A longitudinal retrospective 5-year study. Mult Scler. 2019:1352458519881428. [DOI] [PubMed] [Google Scholar]
- 115.Kuhle J, Kropshofer H, Haering DA, et al. Blood neurofilament light chain as a biomarker of MS disease activity and treatment response. Neurology. 2019;92(10):e1007–e1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kuhle J, Nourbakhsh B, Grant D, et al. Serum neurofilament is associated with progression of brain atrophy and disability in early MS. Neurology. 2017;88(9):826–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Canto E, Barro C, Zhao C, et al. Association Between Serum Neurofilament Light Chain Levels and Long-term Disease Course Among Patients With Multiple Sclerosis Followed up for 12 Years. JAMA Neurol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gelfand JM, Goodin DS, Boscardin WJ, Nolan R, Cuneo A, Green AJ. Retinal axonal loss begins early in the course of multiple sclerosis and is similar between progressive phenotypes. PLoS One. 2012;7(5):e36847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Behbehani R, Abu Al-Hassan A, Al-Salahat A, Sriraman D, Oakley JD, Alroughani R. Optical coherence tomography segmentation analysis in relapsing remitting versus progressive multiple sclerosis. PLoS One. 2017;12(2):e0172120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sotirchos ES, Gonzalez Caldito N, Filippatou A, et al. Progressive Multiple Sclerosis Is Associated with Faster and Specific Retinal Layer Atrophy. Ann Neurol. 2020;87(6):885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Martinez-Lapiscina EH, Arnow S, Wilson JA, et al. Retinal thickness measured with optical coherence tomography and risk of disability worsening in multiple sclerosis: a cohort study. Lancet Neurol. 2016;15(6):574–584. [DOI] [PubMed] [Google Scholar]
- 122.Rissanen E, Tuisku J, Vahlberg T, et al. Microglial activation, white matter tract damage, and disability in MS. Neurol Neuroimmunol Neuroinflamm. 2018;5(3):e443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Politis M, Giannetti P, Su P, et al. Increased PK11195 PET binding in the cortex of patients with MS correlates with disability. Neurology. 2012;79(6):523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Herranz E, Gianni C, Louapre C, et al. Neuroinflammatory component of gray matter pathology in multiple sclerosis. Ann Neurol. 2016;80(5):776–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Singhal T, O’Connor K, Dubey S, et al. Gray matter microglial activation in relapsing vs progressive MS: A [F-18]PBR06-PET study. Neurol Neuroimmunol Neuroinflamm. 2019;6(5):e587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.van Walderveen MA, Kamphorst W, Scheltens P, et al. Histopathologic correlate of hypointense lesions on T1-weighted spin-echo MRI in multiple sclerosis. Neurology. 1998;50(5):1282–1288. [DOI] [PubMed] [Google Scholar]
- 127.Bitsch A, Kuhlmann T, Stadelmann C, Lassmann H, Lucchinetti C, Bruck W. A longitudinal MRI study of histopathologically defined hypointense multiple sclerosis lesions. Ann Neurol. 2001;49(6):793–796. [DOI] [PubMed] [Google Scholar]
- 128.Lycklama G, Thompson A, Filippi M, et al. Spinal-cord MRI in multiple sclerosis. Lancet Neurol. 2003;2(9):555–562. [DOI] [PubMed] [Google Scholar]
- 129.Seewann A, Vrenken H, van der Valk P, et al. Diffusely abnormal white matter in chronic multiple sclerosis: imaging and histopathologic analysis. Arch Neurol. 2009;66(5):601–609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.