

Abstract
Although Duchenne muscular dystrophy is primarily categorised as a skeletal muscle disease, deficiency in the membrane cytoskeletal protein dystrophin also affects the heart. The central transsarcolemmal linker between the actin membrane cytoskeleton and the extracellular matrix is represented by the dystrophin-associated dystroglycans. Chemical cross-linking analysis revealed no significant differences in the dimeric status of the α-/β-dystroglycan subcomplex in the dystrophic mdx heart as compared to normal cardiac tissue. In analogy to skeletal muscle fibres, heart muscle also exhibited a greatly reduced abundance of both dystroglycans in dystrophin-deficient cells. Immunoblotting demonstrated that the degree of reduction in α-dystroglycan is more pronounced in matured mdx skeletal muscle as contrasted to the mdx heart. The fact that the deficiency in dystrophin triggers a similar pathobiochemical response in both types of muscle suggests that the cardiomyopathic complications observed in x-linked muscular dystrophy might be initiated by the loss of the dystrophin-associated surface glycoprotein complex.
INTRODUCTION
Dystrophinopathies are caused by mutations in the dystrophin gene on chromosome Xp21.1 [1] and comprise severe Duchenne muscular dystrophy, benign Becker's muscular dystrophy, and x-linked dilated cardiomyopathy [2, 3, 4]. Deficiency in the Dp427 isoform of the membrane cytoskeletal protein dystrophin affects skeletal muscle, the heart, and the central nervous system to varying degrees [5, 6, 7]. Cardiac involvement in dystrophinopathies is represented by the pathoanatomical replacement of cardiac fibres by connective and fatty tissue [8]. During the clinical progression of Duchenne muscular dystrophy, approximately 90% of patients develop a serious impairment of cardiac function and cardiomyopathy is the cause of death in about 20% of cases [9]. With respect to enhancing our knowledge of the molecular pathogenesis of the most common gender-specific genetic disease in humans, the significant proportion of Duchenne patients suffering from cardiac involvement warrants a detailed investigation of the dystrophic heart muscle.
In normal muscle fibres, dystrophin is tightly associated with several surface glycoproteins [10]. The dystrophin-glycoprotein complex is involved in the stabilisation of the fibre periphery by providing a transsarcolemmal linkage between the actin membrane cytoskeleton and laminin [11]. The central element of this supramolecular assembly is represented by the dystroglycan subcomplex [12]. The extracellular 156 kd glycoprotein α-dystroglycan and the integral membrane protein β-dystroglycan of apparent 43 kd are encoded by a single mRNA [13]. Since the actin-binding protein dystrophin is directly linked to β-dystroglycan, which in turn interacts with the laminin-binding element α-dystroglycan, this complex provides a linkage between the sarcolemma and the extracellular matrix. In dystrophin-deficient skeletal muscle fibres, the dystrophin-associated glycoproteins are greatly reduced [14] triggering a loss of a proper linkage between the plasma membrane and the extracellular matrix component laminin. This pathobiochemical defect is thought to render muscle fibres more susceptible to surface microrupturing which triggers a Ca2+-dependent membrane resealing process at the sites of sarcolemmal disintegration [15]. The newly introduced plasmalemmal patches contain Ca2+ leak channels causing an increased flux of Ca2+ ions into the cytoplasm of dystrophic fibres. Increased cytosolic Ca2+ levels then activate Ca2+-dependent proteases resulting in the net degradation of muscle proteins in muscular dystrophy [16].
Here we used the mdx animal model of x-linked muscular dystrophy [17], which lacks the Dp427 isoform of dystrophin due to a point mutation in exon 23 [18], to analyse the fate of the dystroglycan complex in the dystrophic heart. Although cardiac mdx fibres do not represent a perfect replica of the human cardiomyopathic pathology, several studies have shown that their phenotype is of clinical relevancy. The mdx mice exhibit an abnormal electrocardiogram [19] and their hearts show necrotic changes and inflammation to a varying degree [20]. The mdx heart displays significant tachycardia and decreased heart rate variability [21], and has markedly altered contractile properties [22]. A progression of the dystrophic phenotype is observed in mdx hearts during aging [23]. Interestingly, physical exercise appears to accelerate the dystrophic process in cardiac tissues. Exercised mdx hearts show extensive infiltration of inflammatory cells, as well as an increase in adipose tissue and interstitial fibrosis [24]. This makes it a suitable model system to study the molecular and cellular effects of deficiency in dystrophin on cardiac tissues. Our chemical cross-linking analysis and immunoblotting survey suggests that the lack of cardiac dystrophin clearly affects the abundance of the α-/β-dystroglycan complex but not its dimeric status. Hence, in analogy to skeletal muscle fibres, dystrophic heart muscle also exhibits a greatly reduced abundance of dystroglycans suggesting a similar pathobiochemical pathway in both types of muscle.
MATERIALS AND METHODS
Materials
Primary antibodies were obtained from Novocastra Laboratories Ltd, Newcastle upon Tyne, UK (mAb NCL-43 against β-dystroglycan and mAb DYS-2 to the carboxy-terminus of the dystrophin isoform Dp427), Affinity Bioreagents, Golden, Colo (mAb 20A4 to the α2-subunit of the dihydropyridine receptor, mAb IID8 to the slow/cardiac SERCA2 isoform of the sarcoplasmic reticulum Ca2+-ATPase, and mAb C3-33 to the cardiac RyR2 isoform of the ryanodine receptor Ca2+ release channel), Sigma Chemical Company, Poole, Dorset, UK (pAb to laminin), and Upstate Biotechnology, Lake Placid, NY (mAb C464.6 to the α1-subunit of the Na+/K+-ATPase and mAb VIA41 to α-dystroglycan). Peroxidase-conjugated secondary antibodies were from Chemicon International (Temecula, Calif). Ultrapure Protogel acrylamide stock solutions were obtained from National Diagnostics (Atlanta, Ga). Chemiluminescence substrates and chemical cross-linkers were purchased from Perbio Science UK (Tattenhall, Cheshire). Protran nitrocellulose membranes were from Schleicher and Schuell (Dassel, Germany). Protease inhibitors were obtained from Roche Diagnostics GmbH (Mannheim, Germany). All other chemicals used were of analytical grade and purchased from Sigma Chemical Company.
Cardiac and skeletal muscle preparations
Hearts and hind limb skeletal muscles from 6-, 8-, 15-, and 24-week-old mice of the Dmdmdx strain (Jackson Laboratory, Bar Harbor, Me) and age-matched controls were obtained through the Biomedical Facility of the National University of Ireland, Dublin. Heart muscle specimens from the mdx-3cv animal model were a generous gift from Dr. Harald Jockusch (University of Bielefeld, Germany). For two-dimensional immunoblotting, total protein extracts from dystrophic and age-matched normal hearts were prepared by the method of Dowling et al [25]. For chemical cross-linking and one-dimensional immunoblotting, the crude microsomal membrane fraction was isolated from normal and dystrophic heart and skeletal muscle specimens by an established protocol [26]. All buffers contained a mixture of protease inhibitors (0.2 mM Pefabloc, 1.4 μM pepstatin A, 0.3 μM E-64, 1 μM leupeptin, 1 mM EDTA, 0.5 μM soybean trypsin inhibitor) [25] and all preparative steps were performed at 0–4°C. Membranes were resuspended at a protein concentration of 10 mg/mL and used immediately for biochemical analyses.
Chemical cross-linking analysis
Chemical cross-linking of cardiac membrane proteins was carried out with the 0.15 nm heterobifunctional probe N-succinimidyl-iodoacetate (SIA) and the 0.3 nm homobifunctional probe 1,5-difluoro-2,4-dinitrobenzene (DFDNB) using protocols optimised for muscle membrane proteins [27]. Membranes were incubated for 30 minutes at room temperature in 50 mM Hepes, pH 8.0 with 50 μg cross-linker per mg cardiac protein. Subsequently, the chemical cross-linking reaction was terminated by the addition of 50 μL of 1 M ammonium acetate per mL of reaction mixture [26] and an equal volume of sodium dodecyl sulfate-containing sample buffer [28] was added to the suspension. The mixture was warmed for 10 minutes at 37°C and then proteins were electrophoretically separated [25]. Cross-linker-stabilised complexes, as indicated by DFDNB-induced shifts to a lower electrophoretic mobility, were determined by immuno-decoration of protein bands.
Immunoblotting
Using a Mini-MP3 electrophoresis system from Bio-Rad Laboratories (Hemel Hempstead, Hertfordshire, UK), the gel electrophoretic separation of microsomal heart and skeletal muscle proteins was carried out under reducing conditions in the presence of sodium dodecyl sulfate [28]. Polyacrylamide gels (7% (w/v)) were run for 280 Vh with 25 μg protein per lane. Two-dimensional gel electrophoresis of total tissue extracts, using isoelectric focusing in the first dimension and sodium dodecyl sulfate gel electrophoresis in the second dimension, was performed as previously described in detail [25]. The method of Towbin et al [29] was employed to electrophoretically transfer proteins to nitrocellulose paper using a Bio-Rad Mini-MP3 blotting cell system (Bio-Rad Laboratories). Incubation with 1 : 1 000 diluted primary antibodies, washing steps, incubation with 1 : 1 000 diluted secondary antibodies, and the visualization of immuno-decorated protein bands by enhanced chemiluminescence (ECL) were performed as previously described in detail [30]. Densitometric scanning of ECL blots was performed on a Molecular Dynamics 300S computing densitometer (Sunyvale, Calif) using ImageQuant V3.0 software.
RESULTS AND DISCUSSION
A well-established pathobiochemical feature of dystrophic skeletal muscle fibres is a drastic reduction in the expression levels of dystrophin-associated glycoproteins [5, 11, 14, 31]. In order to determine whether deficiency in the full-length cardiac Dp427 isoform of dystrophin has a similar effect on the dystrophic heart, we have biochemically investigated the relative abundance and the oligomeric status of the backbone of the cardiac dystrophin complex, the dystroglycans, using immunoblotting and chemical cross-linking analysis.
REDUCED EXPRESSION OF THE DYSTROGLYCAN COMPLEX IN DYSTROPHIC HEART MUSCLE
Prior to an in-depth immunoblotting and chemical cross-linking analysis comparing the abundance and oligomeric status of the dystroglycan subcomplex in normal versus dystrophic heart membranes, the mutant status of the mdx specimens was confirmed. As illustrated in Figure 1(a), lane 2, mdx cardiac microsomes completely lack the Dp427 isoform of dystrophin. Figures 1(b) and 1(c), lane 2, show that mdx hearts exhibit a greatly reduced expression of both α- and β-dystroglycan. In contrast, the relative density of the extracellular matrix component laminin, the surface membrane marker Na+/K+-ATPase, and the α2-subunit of the transverse-tubular dihydropyridine receptor is not markedly changed in the dystrophic heart (Figures 1(d) to 1(f), lane 2). This shows that the loss of dystrophin-associated glycoproteins is most likely due to the specific lack of dystrophin and is not a consequence of general cardiac muscle fibre degradation. These findings agree with the analysis of another animal model, the mdx-3cv mouse, that has a mutation in exon 65 that affects the splicing of both the 4.8 kb and 14 kb dystrophin mRNAs causing a loss of all dystrophin isoforms [32]. Deficiency of full-length cardiac dystrophin in mdx-3cv membranes causes a similar reduction in the α-/β-dystroglycan subcomplex as compared to mdx specimens, while the expression of laminin, the Na+/K+-ATPase, and the α2-dihydropyridine receptor is not drastically affected (Figures 1(a) to 1(f), lane 3). Since abnormal Ca2+ handling has been proposed to occur in dystrophic fibres [15, 16], we have performed an additional immunoblot analysis of two major ion-regulatory elements in the heart, the sarcoplasmic reticulum Ca2+-ATPase and the Ca2+ release channel. Both cardiac isoforms, the SERCA2 Ca2+-ATPase and the RyR2 Ca2+ release channel were found to be decreased in their expression (Figures 1(g) and 1(h), lane 2).
Figure 1.
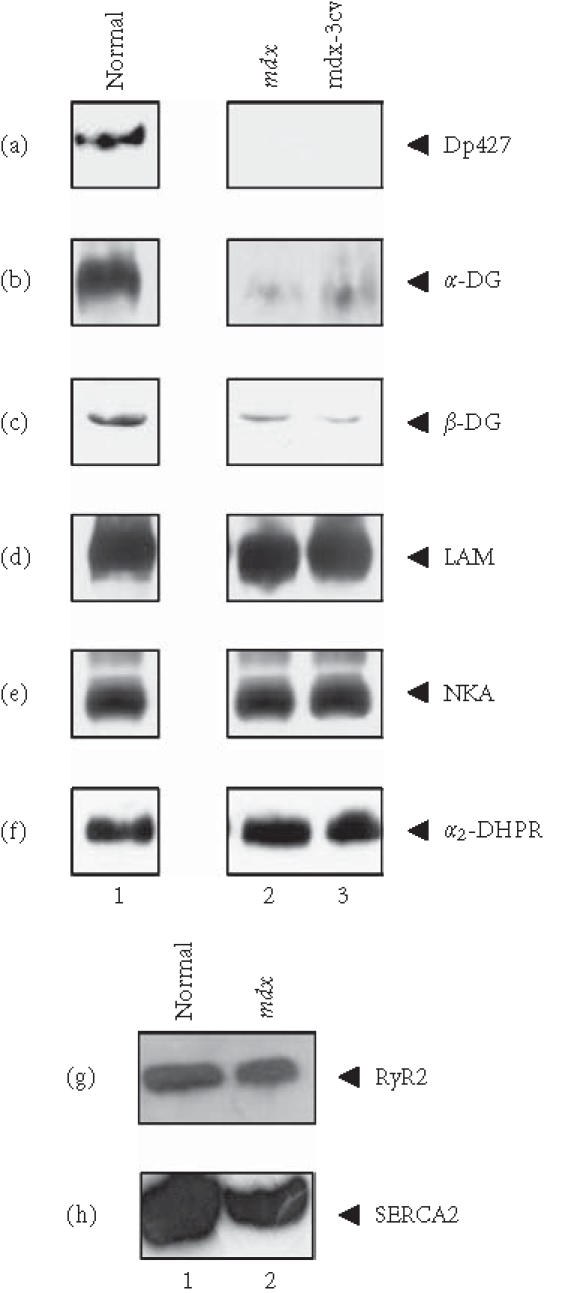
Reduced expression of the dystroglycan complex in dystrophic heart membranes. Shown are immunoblots of normal (lane 1), 15-week-old mdx (lane 2), and 15-week-old mdx-3cv (lane 3) cardiac muscle membranes, labelled with antibodies to the Dp427 isoform of dystrophin (a), α-dystroglycan (α-DG) (b), β-dystroglycan (β-DG) (c), laminin (LAM) (d), the α-subunit of the Na+/K+-ATPase (NKA) (e), the α2-subunit of the dihydropyridine receptor (α2-DHPR) (f), the RyR2 isoform of the ryanodine receptor Ca2+ release channel (g), and the slow/cardiac SERCA2 isoform of the sarcoplasmic reticulum Ca2+-ATPase (h). Dystrophin-deficient cardiac microsomes clearly exhibit a drastic reduction in both dystroglycans.
Since subcellular fractionation protocols may introduce artefacts, the immunoblotting analysis of dystroglycans was also performed with total cardiac extracts. As illustrated in Figures 2a and 2b, the two-dimensional silver-staining pattern of the total protein complement from normal mouse heart versus dystrophic mdx heart did not differ significantly. With the exception of a few low-molecular-mass spots, overall protein expression is relatively comparable. However, this technique only visualizes protein species of relatively high abundance and lacks sensitivity to properly identify proteins that exist at a low density in cardiac membranes. We therefore used immunoblotting to evaluate the status of the dystroglycans in total cardiac fibre extracts. In agreement with our findings from one-dimensional immunoblotting of microsomes (Figure 1(f)), the expression of the α2-subunit of the dihydropyridine receptor was found not to be affected in dystrophin-deficient mdx heart as judged by two-dimensional immunoblotting of total tissue extracts (Figures 2(c), 2(d)). In contrast, the immuno-decoration of α-dystroglycan (Figures 2(e), 2(f)) and β-dystroglycan (Figures 2(g), 2(h)) revealed a clear reduction of both proteins in the dystrophic heart. This agrees with the above-described analysis of cardiac membrane preparations from normal heart and mdx heart. The loss in membrane-associated dystroglycans is therefore not limited to a dissociation from the fibre periphery, but probably also includes a rapid degradation of unbound dystroglycans in the cytosol. If dystrophin is missing as a molecular anchor in the sarcolemma and transverse tubules of cardiac fibres, dystroglycan units appear to dissociate and subsequently disintegrate.
Figure 2.
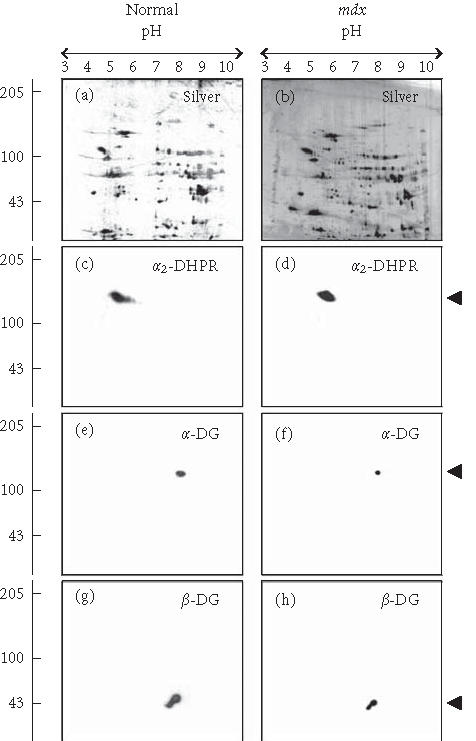
Reduced expression of the dystroglycan complex in dystrophic heart muscle. Shown are silver-stained gels ((a), (b)) and identical immunoblots ((c), (d), (e), (f), (g), and (h)) of 24-week-old normal ((a), (c), (e), (g)) and age-matched mdx ((b), (d), (f), (h)) total heart extracts. Immunoblots were labelled with antibodies to the α2-subunit of the dihydropyridine receptor (α2-DHPR) ((c), (d)), α-dystroglycan (α-DG) ((e), (f)), and β-dystroglycan (β-DG) ((g), (h)). The position of immuno-decorated spots is marked by arrow heads. The pH values of the first-dimensional gel system and molecular mass standards (in kd) of the second dimension are indicated on the top and on the left of the panels, respectively.
CHEMICAL CROSS-LINKING ANALYSIS OF THE CARDIAC DYSTROGLYCAN COMPLEX
Chemical cross-linking is an established biochemical technique for the analysis of multimolecular aggregates in biological membranes [33], widely employed for the elucidation of the quaternary structure of oligomeric proteins and their native organisation in membrane systems [34]. Cross-linkers of various length and solubility such as N-succinimidyl-iodoacetate (SIA) and 1,5-difluoro-2,4-dinitrobenzene (DFDNB) have been established as effective tools and immunoblotting has proven to be highly suitable for the analysis of cross-linked products. While DFNB reacts with primary amines, SIA contains an amine-reactive functional group at one end and a sulfhydryl-active group at the other end. In order to keep artefacts of random cross-linking and hydrolysis of cross-linkers to a minimum, it is essential to use controlled conditions with respect to concentration ratios between membrane proteins and cross-linkers, buffer composition, temperature, pH, and length of incubation time [35]. These parameters have been previously optimised by our laboratory for muscle membrane proteins in order to achieve highly reproducible and optimal results with relatively small amounts of muscle tissue [25, 36, 37, 38].
In order to determine whether the reduction in dystrophin-associated glycoproteins has a modulatory effect on protein-protein interactions within the remaining surface assembly, chemical cross-linking was performed with cardiac membranes. The 0.3 nm cross-linker probe DFDNB clearly induced a shift of the α-dystroglycan protein band to a slower electrophoretic mobility (Figure 3(a)) indicating stabilisation of the native membrane complex. In analogy, the monomeric 43 kd β-dystroglycan species was also cross-linked to a high-molecular-mass complex (Figure 3(b)), whereby the bands representing the two different dystroglycans clearly overlapped following treatment with DFDNB (Figures 3(a), 3(b)). Hence, the cross-linker-stabilised band of approximately 200 kd probably consists of the α-/β-dystroglycan dimer. The cross-linker-induced band of apparent 600 kd possibly represents a complex between dystrophin of 427 kd and the 200 kd dystroglycan dimer. A weak band of extremely high molecular mass might consist of the entire dystrophin-glycoprotein complex. An important control experiment was performed with the shorter probe SIA that exhibits a spacer arm length of 0.15 nm. This cross-linker can capture functional groups only in very close proximity, probably too short for the proper stabilisation of the dystroglycan membrane complex (Figures 3(a), 3(b)). The second β-dystroglycan-positive band of higher molecular mass probably represents a dimerisation of the 43 kd dystrophin-associated glycoprotein (Figures 3(b), 3(d); lane 2). Therefore, the DFDNB-induced oligomerisation of α-/β-dystroglycan is probably not due to nonspecific aggregation, but a result of controlled stabilisation via a cross-linker probe with the optimum reactive functional groups and spacer arm length. On the other hand, the absence of accessible binding groups in dystroglycans might also be a reason for the lack of the formation of very-high-molecular-mass bands by SIA.
Figure 3.
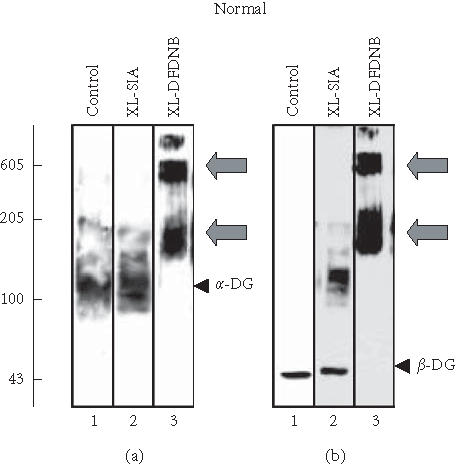
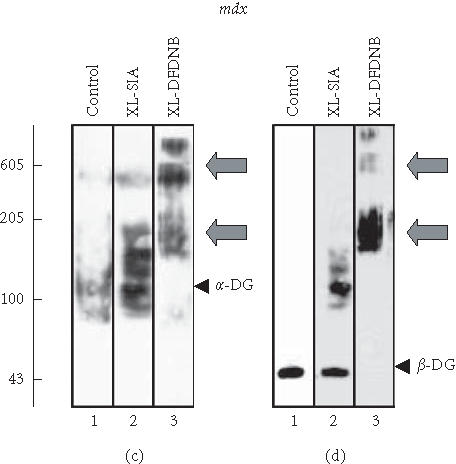
Chemical cross-linking analysis of cardiac dystroglycan complex. Shown are immunoblots of normal ((a), (b)) and mdx ((c), (d)) membranes from 15-week-old mice labelled with antibodies to α-dystroglycan (α-DG) ((a), (c)) and β-dystroglycan (β-DG) ((b), (d)). Lanes from 1 to 3 represent cardiac membranes incubated with no cross-linker probe (control), with 50 μg cross-linker N-succinimidyl-iodoacetate (XL-SIA) and 50 μg cross-linker 1,5-difluoro-2,4-dinitrobenzene (XL-DFDNB) per mg membrane protein, respectively. Monomeric immuno-decorated protein bands are indicated by arrow heads and cross-linker-stabilised oligomeric forms are marked by arrows. For the direct comparison of cross-linker-induced changes in the electrophoretic mobility of dystroglycans, equal amounts of dystroglycan molecules were analysed. Hence, a higher concentration of mdx membranes was used to perform chemical cross-linking as compared to normal membranes. Molecular mass standards (× 10−3) are indicated on the left.
Interestingly, a slightly different pattern of band shifting was observed with cardiac mdx membranes following incubation with the 0.3 nm probe DFDNB (Figure 3(d)). The high-molecular-mass band of β-dystroglycan was weaker in dystrophin-deficient membranes as compared to normal vesicles (Figures 3(c), 3(d); lane 3). If the cross-linker-induced band of 600 kd contains dystrophin and the two dystroglycans, this would indicate a potential difference in the interaction of β-dystroglycan and the Dp427 isoform in muscular dystrophy. However, chemical cross-linking is a relatively crude biochemical technique for determining subtle changes in protein-protein interactions and more refined biochemical analyses would be necessary to clarify this point. Overall, the immunoblotting and cross-linking analysis indicates that the deficiency in cardiac dystrophin causes a severe reduction in the levels of membrane-associated α- and β-dystroglycan, but has no major effect on the oligomeric status of the dimeric dystroglycan complex.
COMPARATIVE ANALYSIS OF DYSTROGLYCAN EXPRESSION IN DYSTROPHIC HEART AND SKELETAL MUSCLE
Based on the initial immunoblot analysis of dystroglycans in dystrophin-deficient heart (Figure 1), we performed a more in-depth study of dystroglycan expression profiles in differing age groups of the mdx model. To determine potential differences in the degree of reduced expression of dystroglycans between maturing mdx cardiac muscle and mdx skeletal muscle, membranes were isolated from 6-week-, 8-week-, and 24-week-old animals. Membranes isolated from younger animals did not result in the isolation of sufficient material for proper immuno-decoration above background levels (not shown). We therefore focused our investigation on 6- to 24-week-old muscle fibres. The graphical presentation of the densitometric analysis of immuno-decorated protein bands, visualised by enhanced chemiluminescence, shows relatively comparable levels of laminin expression in normal versus dystrophic specimens from both the heart and skeletal muscles (Figure 4(a) and (b)). In stark contrast, both dystroglycans are drastically reduced in dystrophic mdx tissues. Both α- and β-dystroglycan exhibited a gradual decrease in their expression during tissue maturation (Figure 4(c)–(f)). The degree of reduction in α-dystroglycan is more pronounced in matured 24-week-old mdx skeletal muscle as contrasted to the age-matched mdx heart (Figure 4(c) and (d)).
Figure 4.
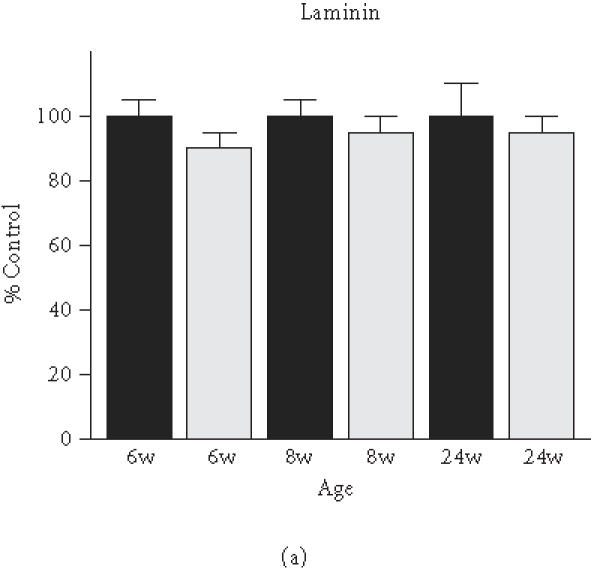
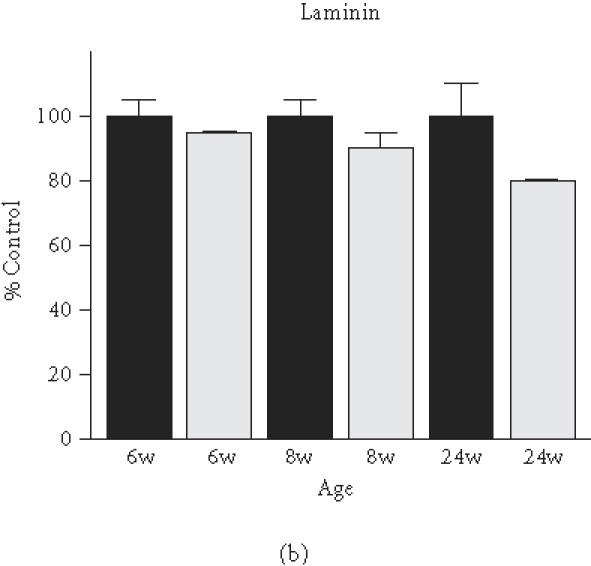
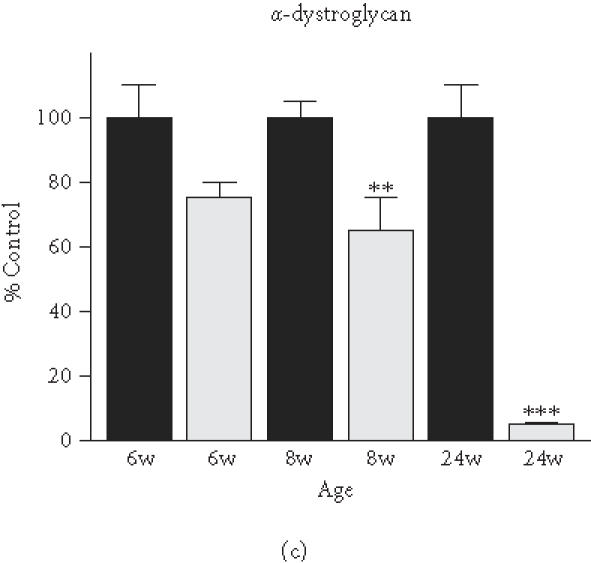
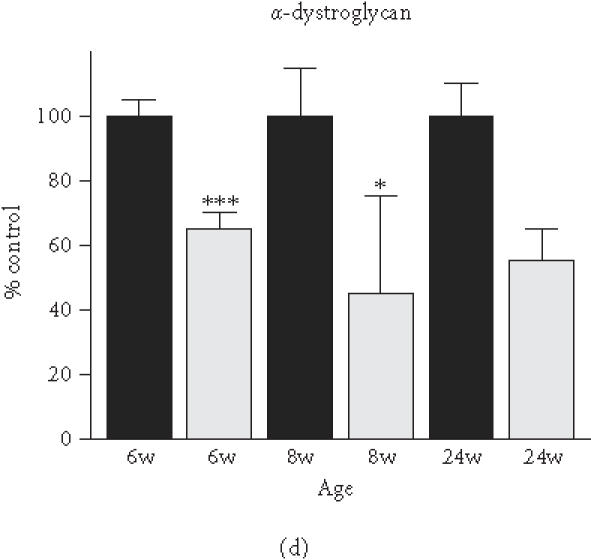
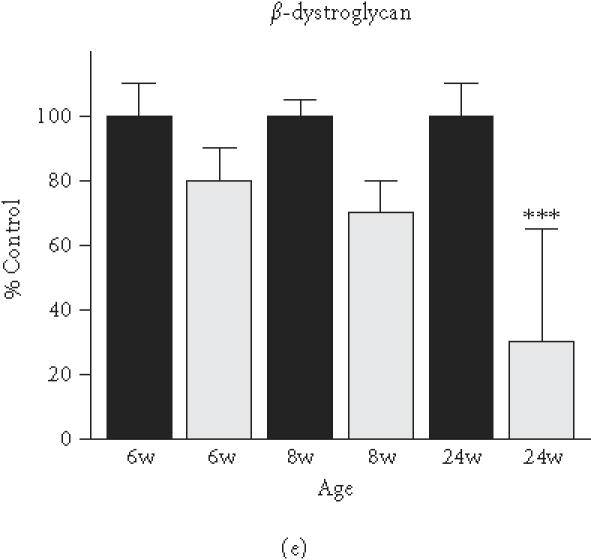
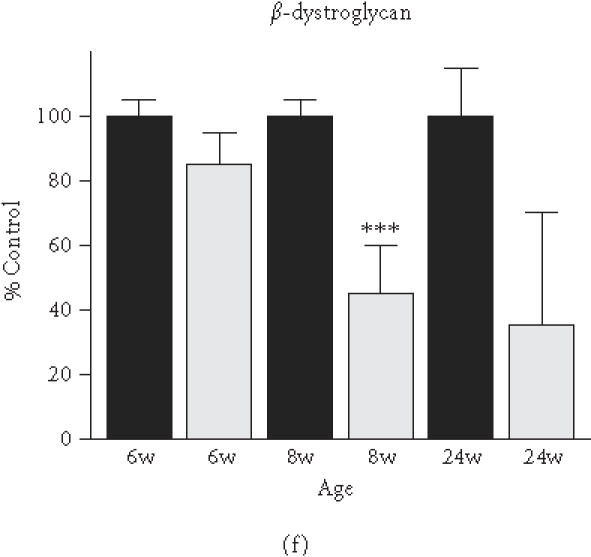
Comparative immunoblot analysis of dystroglycan expression in dystrophic heart and skeletal muscle. Shown is the graphical presentation of the immuno-decoration of laminin ((a),(b)), α-dystroglycan ((c),(d)), and β-dystroglycan ((e),(f)) in normal (black bars) and dystrophic mdx (grey bars) membrane preparations from both skeletal ((a),(c),(e)) and cardiac ((b),(d),(f)) muscles (n = 5; SEM; *P < .05; **P < .01; ***P < .001). Specimens were taken from 6-, 8-, and 24-week-(w-)old mice.
MOLECULAR PATHOGENESIS OF MUSCULAR DYSTROPHY-ASSOCIATED CARDIOMYOPATHY
The results of the immunoblot analysis presented in this report indicate that the loss of the Dp427 isoform of dystrophin triggers a similar pathobiochemical response in skeletal muscle and the heart. Our biochemical findings agree with the recent analysis of dystroglycan expression in dystrophic heart muscle fibres by immunofluorescence microscopy [39, 40]. Thus, cardiomyopathic complications associated with x-linked muscular dystrophy might be initiated by a reduced presence of β-dystroglycan in the cellular periphery. We could recently show that the degree of β-dystroglycan reduction is a relative good indicator of the severity of fibre degeneration in skeletal muscles [25]. Despite a proper dimerisation of the dystroglycans, an overall loss of dystroglycan units would trigger a diminished linkage between the extracellular matrix and the actin membrane cytoskeleton. This probably renders cardiac muscle fibres more susceptible to microrupturing of the surface membrane during excitation-contraction-relaxation cycles eventually leading to cellular necrosis. However, since the subcellular distribution of the dystrophin-glycoprotein is not identical between the heart and skeletal muscles [41], the exact histopathological mechanism might vary slightly. In the heart, dystrophin-associated complexes are not restricted to the sarcolemma, as in the case of skeletal muscle, but also localise to the transverse-tubular region of the surface membrane [41, 42]. Thus, contraction-induced membrane rupturing might affect both the plasmalemma and transverse tubules of the dystrophin-deficient cardiac fibre periphery. In analogy to the calcium hypothesis of skeletal muscular dystrophy [15, 16], possibly re-sealing of disrupted membrane patches might also introduce Ca2+ leak channels into the dystrophic heart surface membrane. This could lead to raised cytosolic Ca2+ levels and the activation of Ca2+-dependent proteases causing fibre destruction. In this respect, our immunoblotting result showing a decreased expression of the cardiac RyR2 isoform of the ryanodine receptor Ca2+ release channel and the slow/cardiac SERCA2 isoform of the sarcoplasmic reticulum Ca2+-ATPase agrees with a recent report by Rohman et al [43] that demonstrated a decreased gene expression of both Ca2+ regulatory elements. This suggests that abnormal Ca2+ handling in the dystrophic heart possibly leads to impaired excitation-contraction coupling and cardiac relaxation cycles. It will be of considerable interest to further establish any potential abnormalities in the Ca2+ regulatory apparatus of the dystrophic heart. One potential way forward is the use of proteomics research tools such as the global comparative survey of normal versus dystrophic cardiac tissues employing high-resolution two-dimensional gel electrophoresis in combination with mass spectroscopy. This might lead to the identification of novel candidate proteins involved in the downstream events of x-linked muscular dystrophy and thereby identify new therapeutic targets.
ACKNOWLEDGMENT
Research was supported by a project grant from the Irish Health Research Board (HRB/RP01/2001). The authors would like to thank Dr H. Jockusch (University of Bielefeld, Germany) for providing their laboratory with dystrophic animal models.
References
- 1.Ahn A.H, Kunkel L.M. The structural and functional diversity of dystrophin. Nat Genet. 1993;3(4):283–291. doi: 10.1038/ng0493-283. [DOI] [PubMed] [Google Scholar]
- 2.Emery A.E. The muscular dystrophies. Lancet. 2002;359(9307):687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 3.Biggar W.D, Klamut H.J, Demacio P.C, Stevens D.J, Ray P.N. Duchenne muscular dystrophy: current knowledge, treatment, and future prospects. Clin Orthop. 2002;401:88–106. doi: 10.1097/00003086-200208000-00012. [DOI] [PubMed] [Google Scholar]
- 4.Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2(12):731–740. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- 5.Culligan K.G, Mackey A.J, Finn D.M, Maguire P.B, Ohlendieck K. Role of dystrophin isoforms and associated proteins in muscular dystrophy. Int J Mol Med. 1998;2(6):639–648. doi: 10.3892/ijmm.2.6.639. [DOI] [PubMed] [Google Scholar]
- 6.Cox G.F, Kunkel L.M. Dystrophies and heart disease. Curr Opin Cardiol. 1997;12(3):329–343. [PubMed] [Google Scholar]
- 7.Anderson J.L, Head S.I, Rae C, Morley J.W. Brain function in Duchenne muscular dystrophy. Brain. 2002;125(pt 1):4–13. doi: 10.1093/brain/awf012. [DOI] [PubMed] [Google Scholar]
- 8.Muntoni F. Cardiomyopathy in muscular dystrophies. Curr Opin Neurol. 2003;16(5):577–583. doi: 10.1097/01.wco.0000093100.34793.81. [DOI] [PubMed] [Google Scholar]
- 9.Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology. 2003;99(1):1–19. doi: 10.1159/000068446. [DOI] [PubMed] [Google Scholar]
- 10.Sunada Y, Campbell K.P. Dystrophin-glycoprotein complex: molecular organization and critical roles in skeletal muscle. Curr Opin Neurol. 1995;8(5):379–384. [PubMed] [Google Scholar]
- 11.Ohlendieck K. Towards an understanding of the dystrophin-glycoprotein complex: linkage between the extracellular matrix and the membrane cytoskeleton in muscle fibers. Eur J Cell Biol. 1996;69(1):1–10. [PubMed] [Google Scholar]
- 12.Henry M.D, Campbell K.P. Dystroglycan inside and out. Curr Opin Cell Biol. 1999;11(5):602–607. doi: 10.1016/s0955-0674(99)00024-1. [DOI] [PubMed] [Google Scholar]
- 13.Ibraghimov-Beskrovnaya O, Ervasti J.M, Leveille C.J, Slaughter C.A, Sernett S.W, Campbell K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355(6362):696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 14.Ohlendieck K, Matsumura K, Ionasescu V.V, et al. Duchenne muscular dystrophy: deficiency of dystrophin-associated proteins in the sarcolemma. Neurology. 1993;43(4):795–800. doi: 10.1212/wnl.43.4.795. [DOI] [PubMed] [Google Scholar]
- 15.Alderton J.M, Steinhardt R.A. How calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. Trends Cardiovasc Med. 2000;10(6):268–272. doi: 10.1016/s1050-1738(00)00075-x. [DOI] [PubMed] [Google Scholar]
- 16.Alderton J.M, Steinhardt R.A. Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J Biol Chem. 2000;275(13):9452–9460. doi: 10.1074/jbc.275.13.9452. [DOI] [PubMed] [Google Scholar]
- 17.Durbeej M, Campbell K.P. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev. 2002;12(3):349–361. doi: 10.1016/s0959-437x(02)00309-x. [DOI] [PubMed] [Google Scholar]
- 18.Sicinski P, Geng Y, Ryder-Cook A.S, Barnard E.A, Darlison M.G, Barnard P.J. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244(4912):1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 19.Bia B.L, Cassidy P.J, Young M.E, et al. Decreased myocardial nNOS, increased iNOS and abnormal ECGs in mouse models of Duchenne muscular dystrophy. J Mol Cell Cardiol. 1999;31(10):1857–1862. doi: 10.1006/jmcc.1999.1018. [DOI] [PubMed] [Google Scholar]
- 20.Bridges L.R. The association of cardiac muscle necrosis and inflammation with the degenerative and persistent myopathy of MDX mice. J Neurol Sci. 1986;72(2-3):147–157. doi: 10.1016/0022-510x(86)90003-1. [DOI] [PubMed] [Google Scholar]
- 21.Chu V, Otero J.M, Lopez O, et al. Electrocardiographic findings in mdx mice: a cardiac phenotype of Duchenne muscular dystrophy. Muscle Nerve. 2002;26(4):513–519. doi: 10.1002/mus.10223. [DOI] [PubMed] [Google Scholar]
- 22.Sapp J.L, Bobet J, Howlett S.E. Contractile properties of myocardium are altered in dystrophin-deficient mdx mice. J Neurol Sci. 1996;142(1-2):17–24. doi: 10.1016/0022-510x(96)00167-0. [DOI] [PubMed] [Google Scholar]
- 23.Lefaucheur J.P, Pastoret C, Sebille A. Phenotype of dystrophinopathy in old mdx mice. Anat Rec. 1995;242(1):70–76. doi: 10.1002/ar.1092420109. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura A, Yoshida K, Takeda S, Dohi N, Ikeda S. Progression of dystrophic features and activation of mitogen-activated protein kinases and calcineurin by physical exercise, in hearts of mdx mice. FEBS Lett. 2002;520(1–3):18–24. doi: 10.1016/s0014-5793(02)02739-4. [DOI] [PubMed] [Google Scholar]
- 25.Dowling P, Lohan J, Ohlendieck K. Comparative analysis of Dp427-deficient mdx tissues shows that the milder dystrophic phenotype of extraocular and toe muscle fibres is associated with a persistent expression of beta-dystroglycan. Eur J Cell Biol. 2003;82(5):222–230. doi: 10.1078/0171-9335-00315. [DOI] [PubMed] [Google Scholar]
- 26.Murray B.E, Ohlendieck K. Cross-linking analysis of the ryanodine receptor and alpha1-dihydropyridine receptor in rabbit skeletal muscle triads. Biochem J. 1997;324(pt 2):689–696. doi: 10.1042/bj3240689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lennon N.J, Harmon S, Mackey A, Ohlendieck K. Oligomerization of the sarcoplasmic reticulum Ca2+-ATPase SERCA2 in cardiac muscle. Mol Cell Biol Res Commun. 1999;1(3):182–187. doi: 10.1006/mcbr.1999.0129. [DOI] [PubMed] [Google Scholar]
- 28.Dunn M.J, Bradd S.J. Separation and analysis of membrane proteins by SDS-polyacrylamide gel electrophoresis. Methods Mol Biol. 1993;19:203–210. doi: 10.1385/0-89603-236-1:203. [DOI] [PubMed] [Google Scholar]
- 29.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76(9):4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glover L, Quinn S, Ryan M, Pette D, Ohlendieck K. Supramolecular calsequestrin complex. Eur J Biochem. 2002;269(18):4607–4616. doi: 10.1046/j.1432-1033.2002.03160.x. [DOI] [PubMed] [Google Scholar]
- 31.Campbell K.P. Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80(5):675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- 32.Cox G.A, Phelps S.F, Chapman V.M, Chamberlain J.S. New mdx mutation disrupts expression of muscle and nonmuscle isoforms of dystrophin. Nat Genet. 1993;4(1):87–93. doi: 10.1038/ng0593-87. [DOI] [PubMed] [Google Scholar]
- 33.Han K.K, Richard C, Delacourte A. Chemical crosslinks of proteins by using bifunctional reagents. Int J Biochem. 1984;16:129–145. [Google Scholar]
- 34.Gaffney B.J. Chemical and biochemical crosslinking of membrane components. Biochim Biophys Acta. 1985;822(3-4):289–317. doi: 10.1016/0304-4157(85)90012-7. [DOI] [PubMed] [Google Scholar]
- 35.Mattson G, Conklin E, Desai S, Nielander G, Savage M.D, Morgensen S. A practical approach to crosslinking. Mol Biol Rep. 1993;17(3):167–183. doi: 10.1007/BF00986726. [DOI] [PubMed] [Google Scholar]
- 36.Froemming G.R, Ohlendieck K. Oligomerisation of Ca2+-regulatory membrane components involved in the excitation-contraction-relaxation cycle during postnatal development of rabbit skeletal muscle. Biochim Biophys Acta. 1998;1387(1-2):226–238. doi: 10.1016/s0167-4838(98)00126-5. [DOI] [PubMed] [Google Scholar]
- 37.Froemming G.R, Murray B.E, Ohlendieck K. Self-aggregation of triadin in the sarcoplasmic reticulum of rabbit skeletal muscle. Biochim Biophys Acta. 1999;1418(1):197–205. doi: 10.1016/s0005-2736(99)00024-3. [DOI] [PubMed] [Google Scholar]
- 38.Froemming G.R, Ohlendieck K. Native skeletal muscle dihydropyridine receptor exists as a supramolecular triad complex. Cell Mol Life Sci. 2001;58(2):312–320. doi: 10.1007/PL00013228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hainsey T.A, Senapati S, Kuhn D.E, Rafael J.A. Cardiomyopathic features associated with muscular dystrophy are independent of dystrophin absence in cardiovasculature. Neuromuscul Disord. 2003;13(4):294–302. doi: 10.1016/s0960-8966(02)00286-9. [DOI] [PubMed] [Google Scholar]
- 40.Yue Y, Li Z, Harper S.Q, Davisson R.L, Chamberlain J.S, Duan D. Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation. 2003;108(13):1626–1632. doi: 10.1161/01.CIR.0000089371.11664.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klietsch R, Ervasti J.M, Arnold W, Campbell K.P, Jorgensen A.O. Dystrophin-glycoprotein complex and laminin colocalize to the sarcolemma and transverse tubules of cardiac muscle. Circ Res. 1993;72(2):349–360. doi: 10.1161/01.res.72.2.349. [DOI] [PubMed] [Google Scholar]
- 42.Peri V, Ajdukovic B, Holland P, Tuana B.S. Dystrophin predominantly localizes to the transverse tubule/Z-line regions of single ventricular myocytes and exhibits distinct associations with the membrane. Mol Cell Biochem. 1994;130(1):57–65. doi: 10.1007/BF01084268. [DOI] [PubMed] [Google Scholar]
- 43.Rohman M.S, Emoto N, Takeshima Y, Yokoyama M, Matsuo M. Decreased mAKAP, ryanodine receptor, and SERCA2a gene expression in mdx hearts. Biochem Biophys Res Commun. 2003;310(1):228–235. doi: 10.1016/j.bbrc.2003.09.005. [DOI] [PubMed] [Google Scholar]