


Abstract
Synthesis of new ribosomes is an energy costly and thus highly regulated process. Ribosomal protein synthesis is controlled by regulating translation of the corresponding ribosomal protein (rp)mRNAs. In mammalian cells a 5′-terminal oligopyrimidine tract (TOP) is a conserved feature of these mRNAs that has been demonstrated to be essential for their translational regulation. Translation of TOP mRNAs has been proposed to be regulated by phosphorylation of ribosomal protein S6, which is a common effect of mitogenic stimulation of cells. However, as demonstrated here, S6 phosphorylation is not detectable in murine erythroleukemia (MEL) or other hematopoietic cells. The absence of S6 phosphorylation appears to be due to the action of a phosphatase that acts downstream of S6 kinase, presumably on S6 itself. Despite the absence of changes in S6 phosphorylation, translation of TOP mRNAs is repressed during differentiation of MEL cells. These data demonstrate the existence of a mechanism for regulating S6 phosphorylation that is distinct from kinase activation, as well as the existence of mechanisms for regulating translation of TOP mRNAs that are independent of S6 phosphorylation.
INTRODUCTION
During growth and proliferation a substantial proportion of total cellular energy is devoted to the synthesis of new ribosomes (1,2). Thus, synthesis of both the RNA and protein components of the ribosome is highly regulated. Ribosomal protein synthesis is primarily regulated by translational regulation of ribosomal protein (rp)mRNAs. In mammalian cells a short sequence of 5–15 pyrimidine nucleotides immediately following the m7GTP cap (the 5′-terminal oligopyrimidine tract or TOP) is an invariant feature of rpmRNAs. This sequence has been demonstrated to be essential for translational regulation of these mRNAs in response to different growth stimuli, including mitogens, amino acid starvation and differentiation, and mutation of this sequence abrogates this regulation (for a review see 3). Thus, coordinate regulation of ribosomal protein synthesis appears to be due to the presence of a TOP sequence on these mRNAs.
Phosphorylation of ribosomal protein S6 is a common effect of mitogens and has been proposed to regulate translation of TOP mRNAs (4–6). S6 phosphorylation requires activation of the S6 kinase, S6K (S6K1 or its isoform S6K2) (7,8), which is dependent upon activation of both phosphatidylinositol 3-kinase (PI3K) and the mammalian target of rapamycin (mTOR) (9–11). This latter kinase has also been demonstrated to phosphorylate S6K1 in vitro at a physiologically relevant site (12), suggesting that it directly regulates S6K activity. Exposure of cells to the mTOR inhibitor, rapamycin, inactivates S6K (13) and results in translational repression of TOP mRNAs (4,5). Embryonic stem cells that lack S6K are viable but grow at a reduced rate (14). S6 is not detectably phosphorylated in these cells and translational regulation of TOP mRNAs in these cells in response to rapamycin and serum deprivation is nearly absent. In addition, translation of these mRNAs under optimal growth conditions also appears to be reduced (14). These data suggest that S6K is required for translational regulation of TOP mRNAs.
While mitogens increase translation rate (15), translation is generally decreased during cell differentiation (16–21). In differentiating murine erythroleukemic (MEL) cells a 2-fold decrease in translation rate occurs (16–18). While little is known regarding the regulation of translation in differentiating MEL cells, the acuteness of this change (18) suggests that inducers may affect normal cell signaling pathways, such as those used by mitogenic receptors to stimulate translation (for a review see 22). In MEL cells the erythropoietin receptor (EpoR) exhibits ligand-independent activation (23), raising the possibility that inducers might affect signaling initiated by this receptor. However, in previous studies we determined that inducers had no effect upon initiation factor phosphorylation, including eIF2α, eIF2β, eIF4E and eIF4B (J.O.Hensold, R.Duncan, J.Hershey and D.Barth-Baus, unpublished observations). This suggests that inducers do not affect EpoR signaling to the pathways that regulate phosphorylation of these initiation factors.
During previous analyses of the translational effect of inducers of MEL cell differentiation it was demonstrated that mRNAs possessing a TOP were primarily affected (18,24,25). Since this suggests that inducers affect S6 phosphorylation this possibility was investigated. The present study demonstrates that S6 is not phosphorylated in MEL cells under either normal growth conditions or following inducer exposure. This is not a unique property of MEL cells but also occurs in other hematopoietic cell lines, as well as in primary granulocytes. Thus, S6 phosphorylation is regulated in a lineage-specific manner in hematopoietic cells. In MEL cells S6 phosphorylation appears to be repressed by a phosphatase that acts downstream of S6K since: (i) PHAS I (4EBP1), which shares upstream activation pathways with S6K, is constitutively phosphorylated; (ii) S6K is constitutively phosphorylated and active; (iii) S6 phosphorylation is easily detectable if the cells are exposed to a phosphatase inhibitor during metabolic labeling. Further, despite the absence of changes in S6 phosphorylation, TOP mRNAs are translationally repressed in MEL cells in response to differentiation signals. These data demonstrate that S6 phosphorylation is not an absolute requirement for translational regulation of TOP mRNAs and that alternative mechanisms of regulation exist.
MATERIALS AND METHODS
Materials
Tissue culture supplies were from Life Sciences (Grand Island, NY), except fetal bovine serum, which was obtained from Intergen (Purchase, NY). Biochemical reagents were from Sigma (St Louis, MO), except acrylamide, which was from Life Sciences. Membranes for immunoblotting were from Millipore (Bedford, MA) and immunoblot detection reagents from Amersham/Pharmacia Biotech (Piscataway, NJ). [32P]orthophosphate and [γ-32P]ATP were obtained from ICN (Costa Mesa, CA). The inhibitors of signal transduction pathways, PD98059, LY294002 and bis-indolylmaleimide, were obtained from Alexis (San Diego, CA). Rapamycin was from Calbiochem (San Diego, CA) and calyculin A from Sigma. Recombinant erythropoietin, stem cell factor (SCF) and G-CSF were obtained from Amgen (Thousand Oaks, CA) and GM-CSF from Immunex (Seattle, WA). Antibodies for cell surface analysis of expanded CD34+ cells were obtained from Becton Dickinson Immunocytometry Systems (San Jose, CA).
Cell culture
Da3 cells that expressed a wild-type EpoR cDNA (Da3/EpoR) (26) were obtained from Taolin Yi (Cleveland Clinic Research Foundation) and maintained in RPMI 1640 medium containing 10% fetal bovine serum and 10% WEHI-3 conditioned medium. To assess the effects of erythropoietin on S6 and PHAS I phosphorylation cells were grown overnight (16 h) in medium containing 0.5% serum in the absence of other mitogens. The following morning the cells were transferred to serum-free medium and incubated for an additional 30 min. Erythropoietin was then added at a concentration of 5 U/ml and incubations continued for the indicated times. Where indicated, kinase inhibitors were added at the following concentrations: rapamycin, 20 ng/ml; LY294002, 25 µM; PD98059, 20 µM; bis-indolylmaleimide, 2 µM. MEL cells were maintained in DMEM supplemented with 12% fetal bovine serum. Conditions for assessing S6 and PHAS I phosphorylation were similar to those described above, excepting the addition of erythropoietin. Inducers of differentiation, DMSO (1.6%) or A23187 (0.75 µg/ml), were added as indicated (27). BaF3 cells that expressed a wild-type EpoR cDNA (BaF3/EpoR) were obtained from Alan D’Andrea (Dana Farber Cancer Institute) and maintained in RPMI 1640 medium containing 10% fetal bovine serum and 0.5 U/ml erythropoietin. To assess the affects of erythropoietin, cells were preincubated in medium containing 0.5% serum without erythropoietin for 6 h, followed by 30 min incubation in serum-free medium. Erythropoietin was then added at 2 U/ml for 30 min.
Normal human CD34 cells, obtained from the University/Ireland Cancer Center Stem Cell Facility, were purified by immunoaffinity using a VarioMACS separation column (Miltenyi Biotec, Auburn, CA), as described by the manufacturer. The cells were split into two equal aliquots and grown in Iscove’s modified Dulbecco’s medium, supplemented with 20% fetal bovine serum and either SCF (10 ng/ml) and erythropoietin (5 U/ml) or GM-CSF (10 ng/ml) and G-CSF (5 U/ml). Fresh growth factors were added every 3 days. After 8 days the cells were washed and resuspended in medium containing serum and either erythropoietin (5 U/ml) or G-CSF (5 U/ml) and incubated for an additional 24 h. Cell phenotype was determined by surface marker expression on ∼1 × 105 cells and the remainder used to assess S6 phosphorylation. For phenotyping the cells were incubated for 20 min in RPMI at room temperature with the indicated monoclonal antibody (5 µg/ml), then washed twice in phosphate-buffered saline, resuspended in 400 µl of the same buffer and analyzed on a FACScalibur flow cytometer using Cellquest software (Becton Dickinson). The monoclonal antibodies used in these studies were directed against CD13, CD14, CD33 and CD71. Background staining was determined using isotype-matched control antibodies.
Determination of PHAS I phosphorylation
PHAS I phosphorylation was detected by gel shift (28). Cell extracts were prepared by boiling in Laemmli sample buffer and sonicated to reduce viscosity. Proteins were separated by electrophoresis in 12.5% polyacrylamide Laemmli gels and transferred to PVDF membranes in 10 mM CAPS buffer at 400 mA for 20 min. Membranes were blocked by incubation in phosphate-buffered saline, 5% (w/v) milk, 0.2% Tween-20 and 0.05% Antifoam A, antisera to PHAS I added at a dilution of 1/1000 and incubated for 3 h at room temperature. Bound antibody was detected with horseradish peroxidase-conjugated goat anti-rabbit antisera and visualized by chemiluminesence. Antisera to PHAS I was a gift of J. Lawrence (University of Virginia).
Determination of ribosomal protein S6 phosphorylation
Cells were incubated for 2 h in phosphate-free medium lacking serum or growth factors then loaded with [32P]orthophosphate (500 µCi/ml) for 2 h. The [32P]phosphate-loaded cells were then split into equal aliquots and either erythropoietin (5 U/ml) (for Da3/EpoR or BaF3/EpoR cells) or inducers (MEL cells) added to one of the aliquots and incubations continued for an additional 30 min. Where indicated, anisomycin (500 nM) or calyculin A (100 nM) were added for the final 30 min incubation. Cells were pelleted in a pre-cooled centrifuge, washed three times in ice-cold phosphate-buffered saline and lysed in TMK buffer (100 mM KCl, 5 mM MgCl2, 10 mM Tris pH 7.5, 2 mM dithiothreitol) containing 1% Triton X-100 and 0.5% deoxycholate in the presence of protease, phosphatase and RNase inhibitors (0.25 µM phenylmethylsulfonyl fluoride, 2 µg/ml aprotinin, 20 mM p-nitrophenyl phosphate, 1 U/µl RNasin). The nuclei were pelleted by a 5 min centrifugation in a refrigerated microcentrifuge. The supernatants were removed and layered onto 22% sucrose cushions in an Sw50.1 rotor and centrifuged at 49 000 r.p.m. for 7.5 h. Gradient composition was as previously described (18), but with inclusion of 20 mM p-nitrophenyl phosphate. As indicated in the figure legends, in later experiments the gradient KCl concentration was increased to 500 mM to decrease recovery of phosphoproteins that were loosely associated with the ribosomes. Ribosomal pellets were resuspended in ice-cold TMK buffer and proteins extracted with 2 vol glacial acetic acid. Soluble ribosomal proteins were precipitated with 5 vol acetone, resuspended in Laemmli sample buffer and analyzed by electrophoresis in 12% polyacrylamide Laemmli gels. Phosphoproteins were identified by autoradiography.
To assess S6 phosphorylation in unfractionated cytosolic lysates, phosphoproteins were metabolically labeled with [32P]orthophosphate as described above. Cytosolic lysates were prepared from equal numbers of control and calyculin A-treated cells in TMK lysis buffer as described above. Glacial acetic acid (2 vol) was added directly to these preparations and acid-insoluble material removed by two consecutive 20 min centrifugations in a refrigerated microcentrifuge. The acid-soluble proteins were precipitated with acetone and separated by electrophoresis in 12–15% polyacrylamide Laemmli gels. Gel loads were standardized for input cell number. Phosphoproteins were identified by autoradiography.
Determination of S6K activity
Quiescent and erythropoietin-stimulated Da3/EpoR and serum-starved MEL cells were washed three times in ice-cold phosphate-buffered saline and the cell pellets lysed by incubation for 25 min on ice in MLB (250 mM NaCl, 5 mM EDTA, 1 mM dithiothreitol, 50 mM MOPS, 0.1% IGEPAL CA-630, pH 7.0) containing both protease (2.5 µg/ml leupeptin, 2.5 µg/ml aprotinin and 50 µg/ml phenylmethylsulfonyl fluoride) and phosphatase (10 mM NaF, 5 mM Na4P2O7, 1 mM Na3VO4 and 10 mM β-glycerol phosphate) inhibitors. Lysates were clarified by centrifugation for 10 min at 13 000 r.p.m. in a refrigerated microcentrifuge. S6K was immunoprecipitated from the cleared lysates by incubation for 4 h at 4°C with 20 µl of an 80% slurry of protein A–Sepharose coupled to a rabbit polyclonal anti-S6K IgG (CWR23) recognizing a synthetic C-terminal peptide. The immunoprecipitates were washed with MLB containing protease and phosphatase inhibitors and 2 M lithium chloride and then washed twice in 1 mM dithiothreitol, 50 mM Tris, pH 7.4. The wash in lithium chloride removes a contaminating kinase activity commonly found in the immunoprecipitates.
In vitro kinase assays were performed on the immunoprecipitates by adding 20 µl of kinase buffer (20 mM MgCl2, 1 mM dithiothreitol, 20 µM ATP, 10 µCi [γ-32P]ATP, 40 mM Tris, pH 7.4) to 2 µg rabbit 40S ribosomal subunits (a gift of William Merrick, case Western Reserve University School of Medicine, Cleveland, OH) and incubating at room temperature for 30 min. Reactions were terminated by adding an equal volume of 2× Laemmli sample buffer and the proteins separated by electrophoresis in 12.5% polyacrylamide gels. The proteins were electroblotted to membranes and the radiolabeled protein detected with a Packard Instant Imager. To assess phosphorylation of the immunoprecipitated S6K the blotted proteins were incubated with the antibody described above and bound antibody detected by an alkaline phosphatase-conjugated anti-rabbit IgG. S6K phospyhorylation was detected by gel shift, as the phosphorylated isoforms migrate more slowly during electrophoresis in SDS gels.
Assessment of translation of TOP mRNAs by gradient sedimentation
To analyze the effects of wild-type and mutant TOP sequences on translational regulation in MEL cells, previously described reporter constructs were used (29,30). In both constructs a human growth hormone (HGH) cDNA was cloned 3′ to the rpS16 promoter and 5′-UTR. For the mutant TOP construct a single C→A mutation at the cap-proximal nucleotide (the first transcribed nucleotide) had been introduced. Plasmid DNAs containing the wild-type or mutant TOP constructs were stably introduced into MEL cells by electroporation. The effect of A23187 on translation of the expressed HGH mRNAs was determined by assessing distribution in translating polysomes and non-translating subpolysomal fractions. Extracts were sedimented for 4 h in 10–50% sucrose gradients containing 500 mM KCl, as previously described (18,31). Fractions were collected with continuous monitoring of UV absorbance at 254 nm. RNA was extracted from the fractions with phenol and chloroform, precipitated with ethanol and analyzed by northern blotting. Results were quantified with a phosphorimager.
RESULTS
S6 phosphorylation is repressed in MEL cells at a step distal to S6K activation
A 50–70% decrease in translation rate is a common effect of inducers of MEL cell differentiation (16–18), although the mechanisms regulating this decrease are unknown. We have previously demonstrated that rpmRNAs are translationally repressed following inducer exposure (18,24,25). Since phosphorylation of ribosomal protein S6 has been proposed to regulate the translation of these mRNAs, we determined if inducers affected S6 phosphorylation. Cellular phosphoproteins were metabolically labeled with [32P]orthophosphate. The ribosomes were washed and pelleted by sedimentation through sucrose, the associated proteins extracted and S6 phosphorylation assessed by gel electrophoresis and autoradiography. Unexpectedly, S6 phosphorylation was not detectable in logarithmically growing MEL cells or in cells exposed to inducers, either A23187 (Fig. 1) or DMSO (data not shown). This was not due to constitutive S6 phosphorylation with the resultant inability to incorporate additional phosphate experimentally, since phosphorylation could not be detected even with [32P]orthophosphate incubation times of up to 23 h (approximately two generation times) (data not shown). In addition, phosphorylation was detected in the acidic ribosomal proteins P1 and P2, demonstrating that the inability to detect S6 phosphorylation was not due to excessive phosphatase activity in the extracts. To determine if EpoR activation was capable of stimulating S6 phosphorylation, a growth factor-dependent Da3 cell line that expresses a wild-type EpoR was utilized. In these cells a 30 min exposure to erythropoietin resulted in a >12-fold increase in S6 phosphorylation (Fig. 1). Thus, S6 phosphorylation is repressed in MEL cells, despite constitutive activation of the EpoR.
Figure 1.
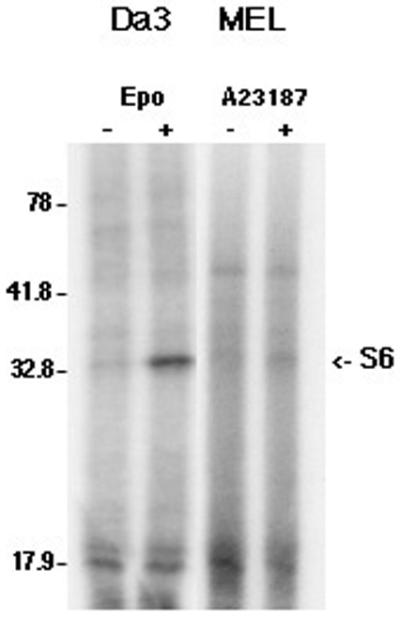
Ribosomal protein S6 is not phosphorylated in MEL cells. Da3/EpoR and MEL cells were incubated overnight in medium containing 0.5% serum, then washed and resuspended in phosphate-free medium for 4 h with [32P]orthophosphate added for the final 2 h of incubation. The cultures were then split into two equal aliquots and erythropoietin added to one of the Da3/EpoR aliquots and A23187 to one of the MEL aliquots. Following an additional 30 min incubation extracts were prepared from the cells. Ribosomes were isolated by sedimentation through sucrose and the recovered ribosomal proteins separated by electrophoresis in 15% polyacrylamide Laemmli gels and visualized by autoradiography. The labels above the lanes indicate the cell type and treatment relevant for the isolated proteins. The positions of migration of the molecular size markers are shown at the left of the autoradiograph. Ribosomal protein S6 has previously been demonstrated to migrate at 32 kDa. The doublet near the 17.9 kDa molecular size marker is the acidic ribosomal phosphoproteins, P1 and P2.
The pathways leading to activation of S6K are similar to those that result in phosphorylation of PHAS I (9,10). Therefore, we determined if PHAS I phosphorylation was also repressed in MEL cells. Phosphorylated isoforms of PHAS I have distinct mobilities during electrophoresis in SDS-containing gels (28) and this mobility shift was utilized to examine PHAS I phosphorylation in both Da3/EpoR and MEL cells. Western blotting detected a shift from the most rapidly migrating, unphosphorylated α-isoform to the more slowly migrating β- and γ-isoforms as early as 5 min following erythropoietin exposure of Da3/EpoR cells (Fig 2A). As determined by metabolic labeling, this reflected a 3-fold increase at 30 min, compared with the unstimulated Da3/EpoR cells (data not shown). In MEL cells PHAS I was constitutively phosphorylated and exposure to inducers of differentiation (DMSO or A23187) did not alter this phosphorylation (Fig. 2B). Exposure to kinase inhibitors for ERK (PD98059), PI3K (LY294002), mTOR (rapamycin) and protein kinase C (bis-indolylmaleimide) had similar effects on PHAS I phosphorylation in these two cell types (Fig. 2C and D), suggesting that this phosphorylation was regulated by similar mechanisms in the two cells.
Figure 2.
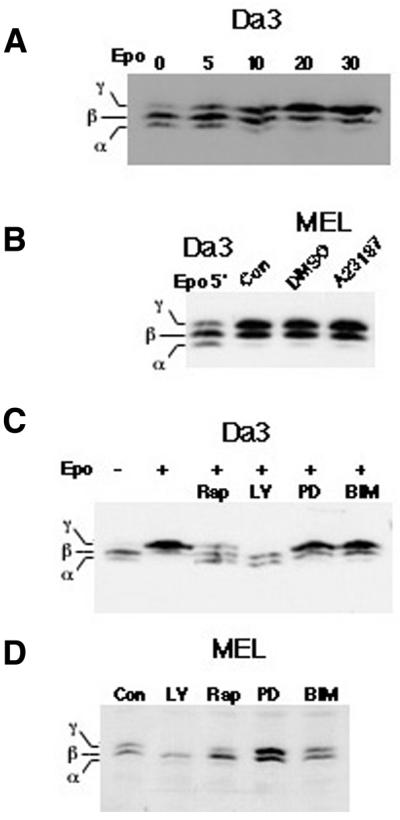
PHAS I is constitutively phosphorylated in MEL cells. (A) Quiescent Da3/EpoR cells were exposed to erythropoietin for the times indicated. Cell extracts were prepared and the proteins separated by electrophoresis in 12.5% polyacrylamide gels under denaturing conditions. The separated proteins were transferred to PVDF membranes and PHAS I proteins detected by immunoblotting. Immunoreactive proteins were visualized by chemiluminscence. The most rapidly migrating α-isoform is unphosphorylated while the β- and γ-isoforms represent singly and doubly phosphorylated forms of PHAS I. (B) MEL cells were maintained overnight in 0.5% serum, then switched to serum-free medium and incubated for an additional 60 min. For the final 30 min incubation inducers of differentiation, either DMSO or A231287, were added as indicated. For reference, an extract of Da3/EpoR cells that had been exposed to erythropoietin for 5 min was included in the left-most lane of the gel that included the MEL cell extracts. (C) Quiescent Da3/EpoR cells were exposed to the indicated inhibitors for 30 min prior to the addition of erythropoietin (Epo). Following an additional 30 min incubation in the presence of both erythropoietin and the indicated inhibitor, cell extracts were prepared and analyzed by western blotting, as previously described. The inhibitors are indicated above the relevant lanes: LY, LY294002; Rap, rapamycin; PD, PD98059; BIM, bis-indolylmaleimide. (D) MEL cells were deprived of growth factors, as described previously, and exposed to signal transduction inhibitors. Extracts were prepared and PHAS I phosphorylation assessed as described above.
To determine where the regulation of S6 and PHAS I phosphorylation diverged in MEL cells, S6K activation was assessed by determination of its phosphorylation state and by in vitro kinase assay. Immunoprecipitated S6K was almost completely inactive in unstimulated Da3/EpoR cells, as assessed by the ability to phosphorylate S6 in purified 40S ribosomal subunits. Exposure to erythropoietin resulted in kinase activation and an increase in S6K phosphorylation, as demonstrated by the presence of the more slowly migrating isoforms (Fig. 3A, right). As expected, kinase activation was inhibited by LY294002 and rapamycin (Fig. 3B). In MEL cells maintained under conditions identical to the unstimulated Da3/EpoR cells S6K was constitutively active, as assessed by in vitro kinase assay, and its phosphorylation state was similar to that in erythropoietin-stimulated Da3/EpoR cells. S6K activation in MEL cells was also inhibited by LY294002 and rapamycin. Thus, similar to the results obtained for PHAS I phosphorylation, activation of S6K in MEL cells is constitutive and dependent upon the activity of the signaling pathways that normally regulate this kinase.
Figure 3.

S6K is constitutively active in MEL cells. Quiescent Da3/EpoR cells were stimulated with erythropoietin for 30 min. MEL cells were maintained under similar conditions, without added erythropoietin. (A) The 70 kDa ribosomal protein S6 kinase (p70 S6k) was immunoprecipitated from cell extracts as described in the text. Kinase activity was assessed using purified 40S ribosomal subunits as substrate (left). Phosphorylation of S6K was assessed by immunoblotting the precipitated protein and is shown on the right. The phosphorylated isoforms of the kinase are detected by their slower mobility during electrophoresis. (B) Cells were grown in the presence of the indicated inhibitor for 60 min prior to extract preparation and p70s6k activity assessed by in vitro kinase assay. The extent of S6 phosphorylation is indicated graphically, relative to the corresponding extract prepared from cells that had not been exposed to inhibitors. Kinase activity in MEL cells was standardized to that in the erythropoietin-stimulated Da3/EpoR cells in the absence of inhibitors, which was arbitrarily set at 100.
In addition to mitogens, exposure to translational inhibitors, such as cycloheximide and anisomycin, can also induce S6 phosphorylation (32). To determine if this alternative means of activation could induce S6 phosphorylation in MEL cells, phosphoproteins were metabolically labeled as described above, with anisomycin added for the final 30 min incubation. Ribosomal proteins were recovered and analyzed by gel electrophoresis. As demonstrated in Figure 4, this did not result in S6 phosphorylation in MEL cells.
Figure 4.

Calyculin A exposure of MEL cells results in S6 phosphorylation. (A) MEL cells were incubated for 4 h in phosphate-free medium with [32P]orthophosphate added for the final 2 h of incubation. The culture was then split into three equal aliquots and anisomycin (Anis) or calyculin A (Cal) added to two of the aliquots. The remaining aliquot served as the untreated control (Con). Following an additional 30 min of incubation, extracts were prepared from the cells and ribosomes were obtained by sedimentation through sucrose. The recovered ribosomal proteins were separated by electrophoresis in 15% polyacrylamide gels under denaturing conditions and visualized by autoradiography. Due to the increase in background phosphorylation in the calyculin A-treated cells, the amount of material loaded in this lane was reduced by 50%, to allow for a more balanced exposure of the autoradiograph. The mobility of the indicated molecular size markers are shown to the left of the autoradiograph. The two phosphoproteins near the 17.9 kDa molecular size marker are the acidic ribosomal phosphoproteins, P1 and P2. (B) MEL cell phosphoproteins were metabolically labeled with and without the addition of calyculin A (Cal). Acid-soluble cytosolic proteins were extracted, separated by electrophoresis in 12% polyacrylamide gels and phosphorylated proteins identified by autoradiography. To identify the position of migration of ribosomal protein S6, a partially purified ribosomal preparation from cells that had been metabolically labeled with [32P]orthophosphate in the the presence of calyculin A was run in an adjacent gel lane. (C) BaF3 proteins were labeled with [32P]orthophosphate. Calyculin A and erythropoietin were added for the final 30 min incubation, as indicated. The r-proteins were recovered as described above, except that the concentration of KCl in the sucrose buffer was increased to 500 mM to reduce background due to loosely adherent proteins. Under these conditions the acidic ribosomal proteins are lost in the supernatants. The recovered r-proteins were analyzed by electrophoresis and autoradiography.
The preceding results suggest that the inhibition of S6 phosphorylation in MEL cells occurred downstream of kinase activation. Since S6K is constitutively active in the absence of mitogen exposure, S6 itself might be dephosphorylated by a constitutively active phosphatase. In that case exposure to a phosphatase inhibitor during metabolic labeling should result in S6 phosphorylation. Therefore, MEL cell phosphoproteins were metabolically labeled in the absence of mitogens and exposed to an inhibitor of serine/threonine phosphatases (calyculin A) for the final 30 min labeling. As demonstrated in Figure 4A, S6 phosphorylation was readily detected when metabolic labeling was performed under these conditions.
The preceding data suggest that in MEL cells S6 phosphorylation is suppressed by the action of a phosphatase. However, since the preceding experiments utilized a prolonged sedimentation to partially purify the ribosomal proteins, it is possible that dephosphorylation occurred during preparation. To address this possibility, S6 phosphorylation was assessed in unfractionated cytosolic lysates, enriching for ribosomal proteins by extraction in 67% glacial acetic acid. As demonstrated in Figure 4B, no phosphoprotein was observed at the expected position of S6 in acid-extracted cytosolic proteins from control cells. However, in lysates prepared from cells that had been incubated with calyculin A for the final 30 min metabolic labeling, a phosphoprotein was observed that co-migrated with S6. In addition, while calyculin A generally increased protein phosphorylation, several proteins in addition to S6 also appeared to be specifically affected by this treatment. Thus, the inability to detect S6 phosphorylation in MEL cells was not simply an artifact of ribosome preparation. Rather, the preceding experiments provide evidence that the upstream pathways necessary for S6K activation are active in MEL cells and that the lack of S6 phosphorylation appears to be due to the action of a phosphatase that rapidly dephosphorylates S6.
S6 phosphorylation is repressed in a lineage-specific manner in hematopoietic cells
The inability to phosphorylate S6 in MEL cells could reflect a lineage-specific effect, neoplastic transformation or a defect unique to these cells. Therefore, S6 phosphorylation was examined in other hematopoietic cell lines. As demonstrated in Figure 4C, S6 phosphorylation could not be detected in BaF3/EpoR cells following stimulation with erythropoietin. However, similarly to MEL cells, S6 phosphorylation was easily detected when these cells were exposed to calyculin A during metabolic labeling. Since erythropoietin exposure of BaF3/EpoR cells also resulted in PHAS I phosphorylation (data not shown), S6 and PHAS I phosphorylations were also uncoupled in these cells. Similarly, S6 phosphorylation was not observed in 32Dcl3 myeloid cells exposed to either a mitogen (IL3) or induced to differentiate with G-CSF (data not shown). Thus, repression of S6 phosphorylation is not unique to MEL cells.
The preceding data demonstrate that S6 phosphorylation is not an invariant result of mitogen receptor activation but is cell-type dependent. Assignment of a specific lineage is complicated for hematopoietic cell lines since establishment frequently results in aberrant expression of lineage-restricted gene products. Therefore, to determine if S6 phosphorylation was a normal response to erythropoietin receptor activation, S6 phosphorylation was assessed in primary cell cultures. Multipotential hematopoietic progenitor (CD34+) cells were obtained from normal donors and grown in the presence of SCF and erythropoietin to allow for expansion of erythroid cells. In the absence of their specific cytokines, primary hematopoietic cells undergo apoptosis (33) and thus the cells are enriched by both positive and negative selection. Myeloid cells were similarly expanded from the same CD34 population by growth in GM-CSF and G-CSF. Following 8 days in culture the cells were washed and then grown for an additional 24 h in either erythropoietin or G-CSF alone. Phenotype was determined by analysis of surface marker expression. The cells expanded in erythropoietin were ∼98% erythroid, while those expanded in G-CSF were essentially 100% myeloid (Fig. 5A). Thus, a highly enriched population of differentiating progenitors of restricted hematopoietic phenotype was obtained.
Figure 5.

S6 phosphorylation is suppressed in primary myeloid but not erythroid cells. Normal human CD34+ cells were expanded by incubation in medium containing either erythropoietin (EPO) or G-CSF, as described in the text. (A) Cell phenotype was assessed by FACS analysis using the following myeloid cell surface markers: CD13, CD14 and CD33. CD71 was used as an erythroid marker. (B) S6 phosphorylation was assessed by metabolic labeling as previously described. Erythropoietin (EPO) was added to an aliquot of the expanded erythroid cells and G-CSF added to the expanded myeloid cells for the final 30 min incubation, as indicated above each lane. Ribosomal proteins were recovered by sedimentation through sucrose containing 500 mM KCl, separated by electrophoresis and visualized by autoradiography. Ribosomal protein S6 is indicated by the arrow. As previously described, the acidic ribosomal phosphoproteins are not recovered during sedimentation at this concentration of KCl. (C) The recovered ribosomal proteins were visualized by silver staining.
Phosphorylation of S6 was assessed in the primary cell cultures as previously done for the cell lines. Phosphorylation was readily detectable in the primary erythroblasts (Fig. 5B). In contrast to the results previously obtained with Da3/EpoR cells, S6 phosphorylation was not acutely dependent on growth factor exposure in the primary erythroid cells. While we speculate that this may be due to the fact that overnight growth in low serum medium was omitted for the primary cell cultures, this possibility was not directly assessed in these experiments. However, this result provides evidence that suppression of S6 phosphorylation is not a general feature of erythroid cells.
In contrast to the results obtained with the primary erythroid cells, S6 phosphorylation was not detected in the cells expanded in GM-CSF and G-CSF. However, cell expansion in the presence of the myeloid cytokines was ∼2-fold less than for cells expanded in erythropoietin. Therefore, the recovered proteins were analyzed by staining to ensure that the inability to detect S6 phosphorylation in the myeloid cells was not due to significant differences in the amount of recovered protein. As shown in Figure 5C, the amount of protein recovered from the myeloid cells was slightly less than that recovered from the erythroid cells. However, this was insufficient to account for the lack of detectable phosphorylation in the myeloid cells. Further, even with more prolonged exposures S6 phosphorylation was not detected in primary myeloid cells.
Translation of TOP mRNAs is repressed in differentiating MEL cells in the absence of changes in S6 phosphorylation
We previously demonstrated that exposure of MEL cells to inducers of differentiation resulted in a decrease in the translation of rpmRNAs (18). Similarly, translational repression of rpmRNAs also occurs in 32D myeloid cells in response to a stimulus to differentiate (G-CSF) (S.Kroll and J.O.Hensold, unpublished observations) and S6 phosphorylation is also undetectable in these cells. This suggests that translational regulation of TOP mRNAs is independent of changes in S6 phosphorylation. To specifically address the role of the TOP in the translational regulation of rpmRNAs in MEL cells, a HGH reporter cDNA that was cloned downstream of the S16 promoter and 5′-UTR was utilized. As a control, a similar construct that differed only by introduction of a point mutation into the TOP was used. These cDNAs were introduced into MEL cells and stable clones selected. Translation of the wild-type and mutant TOP transcripts was assessed by northern blot hybridization of cytosolic RNA that had been fractionated by sedimentation in sucrose gradients. Since we had previously demonstrated that a significant percentage of MEL cell mRNAs accumulate in a salt-labile, translationally inactive 80S complex (18), the fractionations were performed in 500 mM KCl, to more clearly separate translating, polysomal mRNA from non-translating, subpolysomal mRNA. As demonstrated in Figure 6, under normal growth conditions the wild-type TOP mRNA was less efficiently translated than the mutant TOP mRNA (56 versus 88% of the mRNA in polysomes). Following exposure to the inducer of differentiation A23187, translation of the wild-type TOP mRNA was reduced by 43% (from 56 to 32% of the mRNA in polysomes). For the mutant TOP mRNA the decrease in translation in response to A23187 was 19% (from 88 to 71% of the mRNA in polysomes). Thus, while the mutant TOP mRNA was subject to translational repression in response to the inducer, this was >2-fold less than the repression observed for the wild-type TOP mRNA. As previously shown, this occurs in the absence of changes in S6 phosphorylation.
Figure 6.

Inducers of MEL cell differentiation repress translation of TOP mRNAs. A HGH cDNA that had been ligated 3′ to the rpS16 promoter and 5′-UTR was stably expressed in MEL cells. As a control a similar construct in which the TOP had been mutated (C→A) was similarly expressed. Cytosolic RNAs were fractionated by sedimentation through sucrose gradients and HGH mRNA identified by northern blot hybridization of the gradient fractions as described in Materials and Methods. Translation of each transcript was assessed under normal growth conditions and following 8 h exposure to the inducer of differentiation A23187. Results were quantified with a phosphorimager. The northern blots are displayed in the bottom panels and distribution of the mRNAs in gradient fractions is shown graphically at the top (plotted as a percentage of total HGH hybridization signal). Ethidium stained gels shown below each northern blot demonstrate the effect of the inducer on the amount of polyribosomes as well as providing a reference point for the sedimentation of 40S and 60S subunits (as indicated by the peak accumulation of 18S and 28S rRNA). The fractions corresponding to the position of sedimentation of individual 40S and 60S ribosomal subunits, as well as the disomes, trisomes and higher order polysomes (as determined by UV absorbance tracings of the fractionated material) are indicated above the northern blots. (Note that under these salt conditions the monosome peak is too small to be well visualized.) Arrows placed above the graphs similarly indicate positions of sedimentation of fractions containing 40S and 60S subunits as well as disomes, trisomes and higher order polysomes.
DISCUSSION
The results presented here provide evidence for a novel form of regulation of S6 phosphorylation, whereby S6 phosphorylation is dissociated from activation of its kinase, S6K, by the apparent action of a phosphatase that acts downstream of S6K. This activity is subject to developmental regulation as S6 phosphorylation is repressed in normal myeloid, but not erythroid, cells. However, despite lack of detectable S6 phosphorylation, TOP mRNAs are translationally regulated in response to induction of cell differentiation. This latter result demonstrates that translational regulation of 5-TOP mRNAs is not absolutely dependent upon phosphorylation of ribosomal protein S6 and thus provides evidence for the existence of alternative pathways for regulating the expression of these proteins.
S6K and PHAS I share similar upstream activation pathways and the constitutive phosphorylation of PHAS I and activation of S6K in MEL cells is presumably due to constitutive erythropoietin receptor activation in these cells (23). However, S6 phosphorylation is not detectable in MEL cells unless the cells are exposed to a phosphatase inhibitor during metabolic labeling. This provides evidence for the existence of a phosphatase that acts downstream of S6K. This appears distinct from the PP2A phosphatase regulation of S6K recently reported in TGFβ-treated cells (34) or shown to associate with S6K in Jurkat I cells (35). In both of these reports the phosphatase plays a role in S6K activation. In contrast, the data presented here provides evidence for a phosphatase that acts independently of S6K activation. In the absence of evidence to suggest an intervening step between kinase activation and S6 phosphorylation, the most likely target of this phosphatase is S6 itself.
Substrate specificity for serine/threonine phosphatases is typically imparted by different regulatory subunits that associate with these enzymes (36). The data presented here suggest that such a subunit is expressed in MEL, BaF3/EpoR and 32D cells, but not in Da3/EpoR cells. Conversely, phosphorylation of S6 is detected in primary cultures of erythroid, but not myeloid, cells. The most reasonable explanation for these observations is that suppression of S6 phosphorylation is a lineage-specific event that is repressed in a transformation-dependent manner in MEL cells. In this regard it is noteworthy that one of the key transforming events in MEL cells is overexpression of PU.1 (37,38), which is a primary regulator of myeloid gene expression (39). This may account for the lack of S6 phosphorylation in MEL cells while it is easily detectable in normal erythroid cells.
Phosphorylation of ribosomal protein S6 has been proposed to regulate translation of mRNAs that contain a TOP (4,6,14). Consistent with this, in the absence of rpS6 phosphorylation in MEL cells, translation of a wild-type TOP mRNA is repressed relative to the identical mRNA containing a mutant TOP. However, translation of the wild-type TOP mRNA is further repressed relative to the mutant TOP mRNA in inducer-exposed cells and this also occurs in the absence of S6 phosphorylation. Thus, translational regulation of TOP mRNAs in differentiating MEL cells is independent of changes in phosphorylation of S6. While this does not exclude the possibility that S6 phosphorylation regulates translation of TOP mRNAs, it does demonstrate that independent mechanisms exist for regulating their translation. Since we have not assessed S6K activation under all conditions analyzed, we cannot exclude the possibility that regulation of TOP translation is mediated by S6K, via phosphorylation of alternative substrates. However, this appears to be unlikely since in MEL cells PHAS I is phosphorylated in response to inducers and this phosphorylation shares activation pathways with S6K. In addition, the only other protein that has been suggested to be a substrate of S6K is eIF4B (F.Peiretti and J.Hershey, University California-Davis, Davis, CA, personal communication) and this protein is constitutively phosphorylated in MEL cells both under normal growth conditions and in inducer-exposed cells (J.O.Hensold, R.Duncan and J.Hershey, unpublished observations). Consistent with this, translational repression of TOP mRNAs in response to amino acid deprivation has been determined to be regulated independently of S6K (40).
These results raise questions regarding the function of cell-specific suppression of S6 phosphorylation. Cytokine receptors in different hematopoietic cells transmit functionally equivalent signals (41). Thus, differential expression of an S6 phosphatase provides a means to achieve cell-specific differences in gene expression despite the activation of similar upstream pathways. Additional evidence that translation is differentially regulated in hematopoietic cells is provided by the observation that ribosomal protein S19 mutations have been found in 25% of patients with Diamond–Blackfan anemia (DBA) (42). Since the DBA defect is largely confined to erythroid cells, this suggests that these cells may be uniquely dependent upon a function of this ribosomal protein. Thus, it appears likely that specific differences in translational regulation contribute to lineage-specific differences in gene expression by affecting the translation of mRNAs with specific structural or sequence characteristics.
Acknowledgments
ACKNOWLEDGEMENTS
The authors would like to acknowledge Susann Brady-Kalny and Adam Goldfarb for helpful suggestions. This work was supported by the Office of Research and Development, Medical Research Service, Department of Veterans’ Affairs (J.H.) and grant DK43414 from the National Institutes of Health (J.H.) and grant IBN-28317 from the National Science Foundation (G.L.).
REFERENCES
- 1.Schmidt E. (1999) The role of c-myc in cellular growth control. Oncogene, 18, 2988–2996. [DOI] [PubMed] [Google Scholar]
- 2.Warner J. (1999) The economics of ribosomal biosynthesis in yeast. Trends Biochem. Sci., 24, 437–440. [DOI] [PubMed] [Google Scholar]
- 3.Meyuhas O. and Hornstein,E. (2000) Translational control of TOP mRNAs. In Sonenberg,N., Hershey,J. and Mathews,M. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 671–693.
- 4.Jefferies H., Reinhard,C., Kozma,S. and Thomas,G. (1994) Rapamycin selectively represses translation of the “polypyrimidine tract” mRNA family. Proc. Natl Acad. Sci. USA, 91, 4441–4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Terada N., Patel,H., Takase,K., Kohno,K., Nairn,A. and Gelfand,E. (1994) Rapamycin selectively inhibits translation of mRNAs encoding elongation factors and ribosomal proteins. Proc. Natl Acad. Sci. USA, 91, 11477–11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fumagalli S. and Thomas,G. (2000) S6 phosphorylation and signal transduction. In Sonenberg,N., Hershey,J. and Mathews,M. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 695–717.
- 7.Shima H., Pende,M., Chen,Y., Fumagalli,S., Thomas,G. and Kozma,S. (1998) Disruption of the p70s6k/p85s6k gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J., 17, 6649–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saitoh M., ten Dijke,P., Miyazono,K. and Ichijo,H. (1998) Cloning and characterization of p70(S6K beta) defines a novel family of p70 S6 kinases. Biochem. Biophys. Res. Commun., 253, 470–476. [DOI] [PubMed] [Google Scholar]
- 9.Brown E., Beal,P., Keith,C., Chen,J., Shin,T. and Schreiber,S. (1995) Control of p70 S6 kinase by kinase activity of FRAP in vivo. Nature, 377, 441–446. [DOI] [PubMed] [Google Scholar]
- 10.Cheathum B., Vlahos,C., Cheathum,L., Wang,L., Blenis,J. and Khan,C. (1994) Phosphatidylinositol 3-kinase is required for insulin stimulation of pp70 S6 kinase, DNA synthesis and glucose transporter translocation. Mol. Cell. Biol., 14, 4902–4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Downward J. (1994) Regulating S6 kinase. Nature, 371, 378–379. [DOI] [PubMed] [Google Scholar]
- 12.Burnett P., Barrow,R., Cohen,N., Snyder,S. and Sabatini,D. (1998) RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl Acad. Sci. USA, 95, 1432–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearson T., Dennis,P., Han,J., Williamson,N., Kozma,S., Wettenhall,R. and Thomas,G. (1995) The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J., 21, 5279–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawasome H., Papst,P., Webb,S., Keller,G., Johnson,G., Gelfand,E. and Terada,N. (1998) Targeted disruption of p70s6k defines its role in protein synthesis and rapamycin sensitivity. Proc. Natl Acad. Sci. USA, 95, 5033–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brooks R. (1977) Continuous protein synthesis is required to maintain the probability of entry into S-phase. Cell, 12, 311–317. [DOI] [PubMed] [Google Scholar]
- 16.Sherton C. and Kabat,D. (1976) Changes in RNA and protein metabolism preceding onset of hemoglobin synthesis in cultured Friend leukemia cells. Dev. Biol., 48, 118–131. [DOI] [PubMed] [Google Scholar]
- 17.Bilello J., Kuhne,J., Warnecke,G. and Koch,G. (1980) Amplification of translational control as a trigger of in vitro differentiation. In Rossi,G.B. (ed.), In Vivo and In Vitro Erythropoiesis: The Friend System. Elsevier/North-Holland Biomedical Press, Amsterdam, The Netherlands, pp. 229–238.
- 18.Hensold J., Barth-Baus,D. and Stratton,C. (1996) Inducers of erythroleukemic differentiation cause mRNAs that lack poly(A)-binding protein to accumulate in translationally inactive, salt-labile 80S ribosomal complexes. J. Biol. Chem., 271, 23246–23254. [DOI] [PubMed] [Google Scholar]
- 19.Petryshyn R., Chen,J.-J. and London,I. (1984) Growth-related expression of a double-stranded RNA-dependent protein kinase. J. Biol. Chem., 259, 14736–14742. [PubMed] [Google Scholar]
- 20.Palatnik C., Wilkins,C. and Jacobson,A. (1984) Translational control during early Dictylostelium development: possible involvement of poly(A) sequences. Cell, 36, 1017–1025. [DOI] [PubMed] [Google Scholar]
- 21.Kroll S.L., Barth-Baus,D. and Hensold,J.L. (2001) The carboxy-terminal domain of the granulocyte colony-stimulating factor receptor uncouples ribosomal biogenesis from cell cycle progression in differentiating 32D myeloid cells. J. Biol. Chem., 276, 49410–49418. [DOI] [PubMed] [Google Scholar]
- 22.Raught B., Gingras,A.-C. and Sonenberg,N. (2000) Regulation of ribosomal recruitment in eukaryotes. In Sonenberg,N., Hershey,J. and Mathews,M. (eds), Translational Control of Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 245–294.
- 23.Li J.-P., D’Andrea,A., Lodish,H. and Baltimore,D. (1990) Activation of cell growth by binding of Friend spleen focus-forming virus gp55 glycoprotein to the erythropoietin receptor. Nature, 343, 762–764. [DOI] [PubMed] [Google Scholar]
- 24.Avni D., Biberman,Y. and Meyuhas,O. (1997) The 5′ terminal oligopyrimidine tract confers translational control of TOP mRNAs in a cell type- and sequence context-dependent manner. Nucleic Acids Res., 25, 995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shama S., Avni,D., Frederickson,R., Sonenberg,N. and Meyuhas,O. (1995) Overexpression of initiation factor eIF-4E does not relieve the translational repression of ribosomal protein mRNAs in quiescent cells. Gene Expr., 4, 241–252. [PMC free article] [PubMed] [Google Scholar]
- 26.Yi T., Zhang,J., Miura,O. and Ihle,J. (1994) Hematopoietic cell phosphatase associates with erythropoietin (Epo) receptor after Epo-induced tyrosine phosphorylation: identification of two potential binding sites. Blood, 85, 87–95. [PubMed] [Google Scholar]
- 27.Hensold J., Dubyak,G. and Housman,D. (1991) Calcium ionophore, A23187, induces commitment to differentiation but inhibits the subsequent expression of erythroid genes in murine erythroleukemia cells. Blood, 77, 1362–1370. [PubMed] [Google Scholar]
- 28.Lin T.-A., Kong,X., Haystead,T., Pause,A., Belsham,G., Sonenberg,N. and Lawrence,J. (1994) PHAS-I as a link between mitogen-activated protein kinase and translation initation. Science, 266, 653–656. [DOI] [PubMed] [Google Scholar]
- 29.Levy S., Avni,D., Hariharan,N., Perry,R. and Meyuhas,O. (1991) Oligopyrimidine tract at the 5′ end of mammalian ribosomal protein mRNAs is required for their translational control. Proc. Natl Acad. Sci. USA, 88, 3319–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shama S. and Meyuhas,O. (1996) The translational cis-regulatory element of mammalian ribosomal protein mRNAs is recognized by the plant translational apparatus. Eur. J. Biochem., 236, 383–388. [DOI] [PubMed] [Google Scholar]
- 31.Merrick W. and Hensold,J. (2000) Analysis of eukaryotic translation in purified and semipurified systems. Curr. Protocols Cell Biol., 11.9.1–11.9.26. [DOI] [PubMed] [Google Scholar]
- 32.Chou M. and Blenis,J. (1995) The 70 kDa S6 kinase: regulation of a kinase with multiple roles in mitogenic signaling. Curr. Opin. Cell Biol., 7, 806–814. [DOI] [PubMed] [Google Scholar]
- 33.Arai K., Lee,F., Miyajima,A., Miyatake,S., Arai,N. and Yokota,T. (1990) Cytokines: coordinators of immune and inflammatory reponses. Annu. Rev. Biochem., 59, 783–836. [DOI] [PubMed] [Google Scholar]
- 34.Petritsch C., Beug,H., Balmain,A. and Oft,M. (2000) TGF-β inhibits p70 S6 kinase via protein phosphatase 2A to induce G1 arrest. Genes Dev., 14, 3093–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peterson R., Desai,B., Hardwick,J. and Schreiber,S. (1999) Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycin associated protein. Proc. Natl Acad. Sci. USA, 96, 4438–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Depaoli-Roach A., Park,I., Cerovsky,V., Csortos,C., Drubin,S., Kuntz,M., Sitikov,A., Tang,P., Verin,A. and Zolnierowicz,S. (1994) Serine/threonine protein phosphatases in the control of cell function. Adv. Enzyme Regul., 34, 199–224. [DOI] [PubMed] [Google Scholar]
- 37.Moreau-Gachelin F., Tavitian,A. and Tambourin,P. (1988) Spi-1 is a putative oncogene in virally-induced murine erythroleukemias. Nature, 331, 277–280. [DOI] [PubMed] [Google Scholar]
- 38.Moreau-Gachelin F., Ray,D., Mattei,M.-G., Tambourin,P. and Tavitian,A. (1989) The putative oncogene, Spi-1: murine chromosomal localization and transcriptional activation in murine erythroleukemias. Oncogene, 4, 1449–1456. [PubMed] [Google Scholar]
- 39.Shivdesani R. and Orkin,S. (1996) The transcriptional control of hematopoiesis. Blood, 87, 4025–4039. [PubMed] [Google Scholar]
- 40.Tang H., Hornstein,E., Stolovich,M., Levy,G., Livingstone,M., Templeton,D., Avruch,J. and Meyuhas,O. (2001) Amino acid-induced translation of TOP mRNAs is fully dependent on phosphatidylinositol 3-kinase-mediated signaling, is partially inhibited by rapamycin and is independent of S6K1 and rpS6 phosphorylation. Mol. Cell. Biol., 21, 8671–8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Socolovsky M., Lodish,H. and Daley,G. (1998) Control of hematopoietic differentiation: lack of specificity in signaling by cytokine receptors. Proc. Natl Acad. Sci. USA, 95, 6573–6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Draptchinskaia N., Gustavsson,P., Andersson,B., Pettersson,M., Willig,T.-N., Dianzani,I., Ball,S., Tchernia,G., Klar,J., Matsson,H., Tentler,D., Mohandas,N., Carlsson,B. and Dahl,N. (1999) The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nature Genet., 21, 169–175. [DOI] [PubMed] [Google Scholar]