

Summary
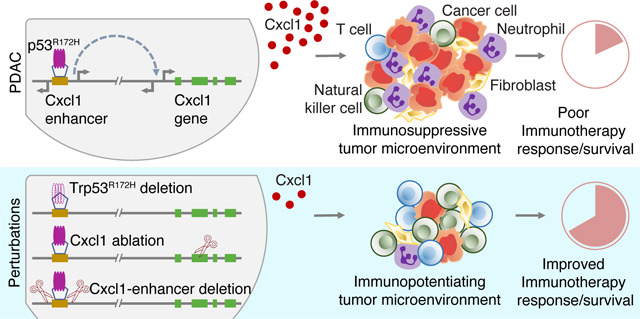
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive cancer without effective treatments. It is characterized by activating KRAS mutations and p53 alterations. However, how these mutations dysregulate cancer-cell-intrinsic gene programs to influence the immune landscape of the tumor microenvironment (TME) remains poorly understood. Here, we show that p53R172H establishes an immunosuppressive TME, diminishes the efficacy of immune checkpoint inhibitors (ICIs), and enhances tumor growth. Our findings reveal that the upregulation of the immunosuppressive chemokine Cxcl1 mediates these pro-tumorigenic functions of p53R172H. Mechanistically, we show that p53R172H associates with the distal enhancers of the Cxcl1 gene, increasing enhancer activity and Cxcl1 expression. p53R172H occupies these enhancers in an NF-κB-pathway-dependent manner, suggesting NF-κB’s role in recruiting p53R172H to the Cxcl1 enhancers. Our work uncovers how a common mutation in a tumor-suppressor transcription factor appropriates enhancers, stimulating chemokine expression and establishing an immunosuppressive TME that diminishes ICI efficacy in PDAC.
Keywords: enhancer, eRNA, mutant p53, p53R172H, p53R175H, chemokine, Cxcl1, PDAC, immunosuppression, immune checkpoint inhibitors, immunotherapy
Graphical Abstract
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the third leading cause of cancer deaths due to late diagnosis and limited response to existing therapies1. It is predicted to become the second leading cause of cancer-related deaths by 20302, highlighting the urgent need for new therapeutic strategies. One approach that could offer immediate benefits is enhancing PDAC’s susceptibility to existing therapeutic modalities, such as immune checkpoint inhibitors (ICIs). Despite their success in other cancers, ICIs have yet to show benefits for PDAC patients3–7. Recent strategies to harness the potential of ICIs by targeting additional factors such as DNA-repair proteins8–10, costimulatory receptors11,12, and immune modulators13–15 have shown promise in some cancers. Gaining deeper mechanistic insight into how oncogenic events shape cancer-cell-intrinsic gene programs to influence cell-extrinsic characteristics and sustain a pro-tumorigenic tumor microenvironment (TME) is crucial for unlocking this therapeutic approach.
About 90% of PDACs harbor activating KRAS mutations, suggesting it is a founding oncogenic event, and ~70% have alterations in the TP53 tumor suppressor gene, suggesting the abrogation of the genome-guarding role of p53 accelerates the malignant progression16. The majority of p53 alterations are missense mutations in the DNA binding domain, which abolishes recognition of canonical binding motifs17. The six most frequently mutated p53 residues in human cancer are broadly categorized as ‘contact’ mutations (R248 and R273) or ‘structural’ mutations (R175, G245, R249, R282) based on defects in binding or structural deformation of the DNA-binding domain, respectively18. Mutant p53 exerts pro-tumorigenic effects either by loss-of-function19,20, a dominant-negative effect of suppressing the wild-type p53 function21, or by acquiring additional gain-of-function22–24. These mutant proteins can associate with transcription factors and other effectors in augmenting the transactivation potential of the interacting partners25–33. However, the interacting partners, affected pathways, and altered gene programs are highly context-dependent34–38.
The TME of PDAC is highly desmoplastic, hypoxic, and considered immunologically cold39,40. The low tumor mutational burden and the resulting low neo-antigen burden further insulate PDAC from the effectiveness of ICIs41. Consequently, understanding how cell-intrinsic programs shape the PDAC TME could reveal novel therapeutic opportunities. Over the past decade, some progress has been made7. For example, cancer-cell-specific Pin1 establishes a desmoplastic and immunosuppressive TME, and its pharmacological targeting appears to synergize with immunochemotherapy in the treatment of PDAC42. Similarly, mutant p53 harboring pancreatic ductal cells accumulate neutrophils and polymorphonuclear myeloid-derived suppressor cells that are anti-inflammatory and resist immunotherapy43. Still, the mechanistic insights on how mutant p53 contributes to immunosuppressive TME remain poorly understood.
We hypothesized that mutant p53 - which is highly stabilized compared to its wild-type counterpart44,45 and retains a functional and potent transactivation domain46 could drive new gene programs through mechanisms distinct from those of p53WT. For example, the transcription factor KLF5, which is frequently mutated in many cancers, gains over 5,000 new binding sites in comparison to wild-type KLF5, including super-enhancers proximal to genes that promote tumor growth47. Similarly, the splice variant of the androgen receptor that lacks the ligand binding domain occupies unique genomic sites and displays novel DNA binding motifs48. In this study, we examined the transcriptional influence of p53 missense mutations. We focused on the most prevalent p53 mutation49 (R175H in humans and R172H in mice) in the context of almost universally co-occurring Kras activating mutation (G12D) in PDAC. To investigate the role of p53R172H in shaping the TME, we used cells derived from a genetically engineered mouse model (GEMM) of PDAC, created isogenic cell lines, and employed a host of techniques, including genome and transcriptome sequencing, CRISPR-Cas9 gene editing, immune profiling, and mouse survival studies with ICIs.
Our findings indicate that p53R172H drives tumor progression by modulating cancer-cell-specific gene expression, which reprograms the TME into an immunosuppressive milieu. Specifically, p53R172H is associated with lower T cell infiltration, higher myeloid-derived suppressor cell (MDSC) infiltration, and diminished efficacy of ICIs in mouse models of PDAC. At the molecular level, p53R172H exerts its immunosuppressive effects primarily by regulating chemokine genes, particularly Cxcl1. p53R172H associates with distal enhancers of Cxcl1 and amplifies its expression in an NF-κB-dependent manner. This interaction suggests a broader mechanism in which mutant p53 co-opts enhancers, which vastly outnumber gene promoters, through collaboration with other transcription factors. By interacting with the enhancer’s cognate binders, mutant p53 modulates cell-intrinsic gene expression that ultimately impacts the cell-extrinsic features of the TME. This study not only elucidates a mechanism of immune suppression driven by p53R172H but also highlights broader implications for p53 missense mutations and their cooperation with other transcription factors in pancreatic cancer, offering potential targets for therapeutic intervention.
Results
p53R172H regulates a subset of chemokines
To examine the transcriptional programs regulated by p53R172H, we used KPC cells derived from a GEMM of PDAC (LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre)50. Given the finding that more than 90% of cancer cases lose the Trp53WT allele in the presence of mutant p53 allele51,52, we used Exome-seq to check the status of Trp53 alleles and confirmed that the Trp53WT allele is indeed lost (Figure S1A). This Trp53R172H/− genotype enables the examination of the gain-of-function effects of p53R172H, distinguishing it from the dominant-negative confounding effect in the parental Trp53R172H/+ genotype.
To understand how p53R172H alters gene programs, we compared transcriptional programs between Trp53R172H/− and Trp53−/− cells. However, instead of comparing with cells derived from other GEMMs of PDAC (such as LSL-KrasG12D/+;LSL-Trp53−/−;Pdx-1-Cre)52–54, which likely harbor non-overlapping mutations arising from the genomic instability due to a lack of p53WT in both cases, we made isogenic Trp53−/− cells (KrasG12D/+;Trp53−/−) using CRISPR-Cas9 (Figure 1A) and selected single-cell clones and clonal mix population. Although all cells used in this study harbor KrasG12D/+, the genotypes hereafter are solely indicated by Trp53 status for simplicity. We confirmed the deletion of Exon-2 to Exon-10 - identical region deleted to generate Trp53−/− mice52 - at the DNA, RNA, and protein levels (Figure S1B-D). We performed RNA-seq to measure the p53R172H-mediated gene expression changes in the KrasG12D background and found several hundred genes under the control of p53R172H (Figure 1B). Notably, the expression of canonical p53 target genes55 remained relatively unchanged, confirming the lack of wild-type p53 function in both cells. We found the genes upregulated by p53R172H were enriched for chemokines and targets of the NF-κB transcription factor (Figure 1C). The genes suppressed by p53R172H were enriched for negative regulation of biosynthesis and metabolism, likely related to the control of proliferation (Figure S1E). We performed a chemokine array to measure the levels of secreted chemokines in the tissue culture media and found a subset of chemokines differentially regulated by p53R172H (Figure S1F). Most of the p53R172H-regulated chemokines belong to either CCL or ELR+CXCL chemokine subclass56.
Figure 1. p53R172H elevates the expression of a subset of chemokine genes.

A, Generation of Trp53−/− isogenic cells from the parental Trp53R172H/− cells. Trp53 gene with R172H mutation in exon-5 is deleted from intron-1 to intron-10 using CRISPR/Cas9.
B, Minus-average (MA) plot of RNA-seq transcripts showing the differentially expressed genes between Trp53R172H/− and Trp53−/− isogenic cells. Significantly upregulated and downregulated genes (adjusted p-value < 0.001 and four-fold change in normalized counts) are shown in blue and red, respectively. The wild-type p53-regulated genes are shown in yellow.
C, Pathways (left) and transcription factor targets (right) enriched in p53R172H-upregulated genes. Gene Ontology analysis was performed using Enrichr101 against the KEGG pathway database102 (left) and TRRUST database103 (right). Blue dots represent significant gene sets (p-value < 0.05), and the darker color represents higher significance.
D, Generation of Trp53R172H-restored isogenic cells in Trp53−/− cells using a Trp53R172H cDNA expression cassette in piggyback vector (Trp53−/− + pTrp53R172H). An empty vector without the Trp53R172H cDNA expression cassette (Trp53−/− + pEV) was inserted in Trp53−/− as a control.
E, mRNA levels of the expressed chemokine genes are shown as the z-score heatmap of RNA-seq transcripts per million (TPM) in the four isogenic cells.
F, Quantification of the three chemokine genes under p53R172H control by ELISA in the tissue culture media of the four isogenic cells. P-values are calculated from a one-way ANOVA test followed by a post hoc test with Benjamini-Hochberg correction. Panels B, C, E, and F use Trp53−/− clone-1 isogenic cells.
To exclude clonal variability as a confounding factor, we performed RNA-seq experiments with an additional single-cell clone of isogenic Trp53−/− cells, as well as a clonal-mix population generated by pooling three single-cell clones. Our results showed a similar number of p53R172H-regulated genes and the dependence of a similar set of chemokine genes on p53R172H across clonal and clonal-mix populations (Figure S2A). The genes upregulated and downregulated upon the deletion of Trp53R172H highly overlapped among clonal and clonal-mix populations (Figure S2B), and the overlapping genes showed enrichment for chemokine genes (Figure S2C), as observed previously with a representative clone. Similarly, enhanced secretion of Cxcl1, Cxcl5, and Ccl2 proteins was dependent on p53R172H in the clonal-mix population as measured by ELISA in tissue-culture media (Figure S2D).
We further substantiated the generality of p53R172H-mediated transcriptional regulation and its influence on chemokine gene expression using syngeneic Trp53−/− cells57, generated from a GEMM of PDAC (p48-Cre;KrasLSL-G12D/+;Trp53loxP/+)58. Similar to the parental KPC cells, we found that the Trp53WT allele is lost in these PDAC cells (Figure S3A), likely due to the loss-of-heterozygosity, which we confirmed at the mRNA and protein levels (Figure S3B-C). RNA-seq analysis of the syngeneic Trp53−/− cells, compared with the Trp53R172H/− cells, highlighted the dependency of similar chemokine genes on p53R172H (Figure S3D-E). Notably, specific chemokines—Cxcl1, Cxcl5, and Ccl2—showed reduced secretion, as measured by cytokine array (Figure S3F).
To determine whether p53R172H is sufficient to regulate chemokine expression, we ectopically expressed Trp53R172H cDNA in Trp53−/− using the PiggyBac expression vector (Figure 1D, see Methods). An empty vector (pEV) served as a control. We confirmed the expression of p53R172H (Figure S1G) and observed that mRNA levels of certain chemokine genes were partially restored in p53R172H-expressing Trp53−/− cells (Figure 1E). Further validation using ELISA confirmed that the reintroduction of p53R172H in Trp53−/− cells significantly restored the expression of specific chemokines—Cxcl1, Cxcl5, and Ccl2 (Figure 1F). Collectively, these findings show that p53R172H acquires a novel transcriptional regulatory function, driving the expression of a specific set of chemokine genes.
p53R172H creates an immunosuppressive TME and abrogates ICI efficacy
To test whether the loss of Trp53R172H suppresses the PDAC formation and growth, we orthotopically implanted Trp53R172H/− or Trp53−/− cells in the pancreas of immunocompetent wild-type mice. We profiled the immune landscape of the PDAC TME 21 days after implantation (Figure 2A). The tumors formed by Trp53R172H/− cells were significantly larger compared to the Trp53−/− cells (Figure 2B), indicating the role of p53R172H in PDAC formation and growth. Immune profiling of the tumors using fluorescence-activated cell sorting (FACS) showed that the Trp53−/− tumors had higher infiltration of T cells (CD45+CD3e+CD8+), activated T cells (CD45+CD3e+CD8+CD44+), cytotoxic T cells (CD45+CD3e+CD8+Gzmb+) and activated conventional CD4+ T cells (CD45+CD3e+CD8-CD4+CD44+Foxp3-) compared to the Trp53R172H/− tumors (Figure 2C). In contrast, Trp53−/− tumors had lower infiltration of MDSCs (CD45+CD11b+Gr-1+Arg-1+) than Trp53R172H/− tumors (Figure 2D). The tumors established with orthotopic implantation of the clonal-mix population of Trp53−/− cells were also smaller and showed higher infiltration of T cells, activated T cells, cytotoxic T cells, and activated conventional CD4+ T cells (Figure S4A-B). We used immunofluorescence to further examine the immune composition of the tumors. We found a higher number of cytotoxic T cells (CD8+) and a lower number of neutrophils (Gr-1+) and MDSCs (Gr-1+Arg-1+) in Trp53−/− cells (Figure 2E).
Figure 2. p53R172H creates immunosuppressive TME and abrogates ICIs efficacy.

A, Schematics and the experimental design of Trp53R172H/− and Trp53−/− isogenic cells’ orthotopic implantation in mouse pancreas, immune profiling of the tumors, ICIs treatment, and survival analysis.
B, Weights of the Trp53R172H/− and Trp53−/− tumors. The p-value is calculated using a two-tailed t-test.
C, Effect of the Trp53 status on T cell infiltration in PDAC tumors. The p-values are calculated using a two-tailed t-test.
D, Effect of the Trp53 status on MDSCs infiltration in PDAC tumors. The p-value is calculated using a two-tailed t-test.
E, Representative multiplex immunofluorescence staining of Trp53R172H/− and Trp53−/− tumors (left), and quantification of immune cells density from randomly selected 500 × 500 μm regions (right). The scale bars correspond to 200 μm and 50 μm in the main view and the magnified view, respectively. P-values are calculated using a two-tailed t-test.
F, Effect of the Trp53 status and ICIs on PDAC tumor growth. Control mice were treated with IgG. Tumor volumes from the last measurement with at least three mice left in the cohort were used to calculate p-values using a two-tailed t-test.
G, Kaplan-Meier survival curves showing the effect of Trp53 status and ICIs on the survival of mice implanted with either Trp53R172H/− or Trp53−/− cells. Control mice were treated with IgG. P-values are calculated using a log-rank (Mantel-Cox) test.
H, Experimental design and timeline of tumor challenge experiment in the long-term survivor mice implanted with Trp53−/− PDAC tumors and treated with ICIs.
We then tested whether the increased CD8+ T cell infiltration in the TME of Trp53−/− tumors compared to Trp53R172H/− tumors would sensitize the tumors to ICIs. We treated the Trp53R172H/− or Trp53−/− cells-implanted mice with ICIs (anti-CTLA4 + anti-PD-1 combinatorial therapy) or IgG (control) three times in 3-day intervals and monitored the tumor growth and survival (Figure 2A). We used ultrasound to monitor the growth of PDAC tumors after ICIs administration until the mice reached a humane endpoint. In multiple replicate studies, we consistently observed a slower growth of Trp53−/− tumors than the Trp53R172H/− tumors (Figure 2F & Figure S4C). More importantly, the ICIs treatment dramatically increased the survival of mice implanted with Trp53−/− tumors (Figure 2G & Figure S4D). The ICIs treatment resulted in complete regression of Trp53−/− tumors in 6 out of 10 in the first cohort and 4 out of 8 in the second cohort, and these mice survived long-term. We challenged these long-term survivors with another orthotopic implantation of Trp53−/− cells a year after the first implantation and treatment. These mice were able to prevent tumor formation (Figure 2H), suggesting the establishment of immune memory. These in vivo studies indicate a role for p53R172H in establishing an immunosuppressive TME and facilitating evasion of anti-tumor immunity in PDAC.
Cxcl1 mediates an oncogenic role of p53R172H
We hypothesized that p53R172H establishes an immunosuppressive TME by regulating the expression of immune-modulating genes. To investigate this, we focused on p53R172H-dependent chemokines, particularly Ccl2, due to its strong association with p53R172H and its potential as a therapeutic target in PDAC59. Ccl2 is known to attract monocytes60–62 and promotes infiltration of natural killer (NK) cells63,64. Using CRISPR-Cas9, we generated isogenic Ccl2−/− cells while retaining KrasG12D/+ and Trp53R172H/− mutations (Figure S5A). The frameshift mutations in the Ccl2 gene resulted in the complete loss of Ccl2 expression (Figure S5B). Surprisingly, the absence of Ccl2 did not affect tumor growth upon orthotopic implantation (Figure S5C), nor did it enhance responsiveness to ICIs, unlike what was observed in Trp53−/− cells (Figure S5D). These findings led us to explore other p53R172H-dependent chemokines. We deleted the Cxcl1 or Cxcl5 gene in the Ccl2−/− cells and implanted the double knockout cells (Ccl2−/−;Cxcl1−/− or Ccl2−/−;Cxcl5−/−) in the pancreas of immunocompetent mice. The double knockout cells resulted in significantly smaller tumors (Figure S5E).
These results raised the question of whether the deletion of Cxcl1 or Cxcl5 alone could have impeded tumor growth. To address this, we made isogenic Cxcl1−/− or Cxcl5−/− cells using CRISPR-Cas9 while retaining the KrasG12D/+ and Trp53R172H/− mutations (Figure 3A). The frameshift mutation resulted in a complete loss of the Cxcl1 expression (Figure S5F). Orthotopic implantation of Trp53R172H/−;Cxcl1−/− or Trp53R172H/−;Cxcl5−/− cells in the pancreas of wild-type mice demonstrated that the loss of Cxcl1 significantly reduced tumor size, recapitulating the effects of p53R172H loss (Figure 3B). In contrast, Cxcl5 deletion alone did not impact tumor growth. Examination of the immune landscape of Cxcl1−/− tumors revealed a higher infiltration of T cells, activated T cells, cytotoxic T cells, and activated conventional CD4+ T cells, similar to the Trp53−/− tumors (Figure 3C). These results indicate that Cxcl1 contributes to p53R172H-mediated immunosuppression.
Figure 3. Cxcl1 mediates the immunosuppressive role of p53R172H and abrogates ICIs efficacy.

A, Generation of Trp53R172H/−;Cxcl1−/− and Trp53R172H/−;Cxcl5−/− isogenic cells from the parental Trp53R172H/− cells. A single guide-RNA-mediated genome editing using CRISPR/Cas9 resulted in a frameshift mutation.
B, Weights of the Trp53R172H/−;Cxcl1−/− and Trp53R172H/−;Cxcl5−/− tumors compared with the Trp53R172H/− tumors. P-values are calculated from a one-way ANOVA test followed by a post hoc test with Benjamini-Hochberg correction.
C, Effect of the Cxcl1 status in T cell infiltration in PDAC tumors. P-values are calculated using a two-tailed t-test.
D, Effect of the Cxcl1 status in leukocyte (CD45+) and neutrophil (CD45+CD11b+MHCII-Ly-6G+) infiltration in PDAC tumors. P-values are calculated using a two-tailed t-test.
E, Effect of the Cxcl1 status in lymphocyte (CD45+CD11b-CD90+NK1.1-) and NK cell (CD45+CD11b-CD90+NK1.1+) infiltration in PDAC tumors. P-values are calculated using a two-tailed t-test.
F, Effect of the Cxcl1 status in macrophage (CD45+CD11b+CD64+) infiltration in PDAC tumors. P-values are calculated using a two-tailed t-test.
G, Representative multiplex immunofluorescence staining of Trp53R172H/− and Trp53R172H/−;Cxcl1−/− tumors (left), and quantification of immune cells density from randomly selected 500 × 500 μm regions (right). The scale bars correspond to 200 μm and 50 μm in the main view and the magnified view, respectively. P-values are calculated using a two-tailed t-test.
H, Effect of the Cxcl1 status and ICIs in PDAC tumor growth. Control mice were treated with IgG. Trp53R172H/− tumor growth cohort is the same as in Figure S4C. Tumor volumes from the last measurement with at least three mice left in the cohort were used to calculate p-values using a two-tailed t-test.
I, Kaplan-Meier survival curves showing the effect of Cxcl1 status and ICIs in the survival of mice implanted with parental Trp53R172H/− or Trp53R172H/−;Cxcl1−/− cells. Control mice were treated with IgG. Trp53R172H/− mice survival cohort is the same as in Figure S4D. P-values are calculated using a log-rank (Mantel-Cox) test.
Cxcl1 is a chemotactic cytokine with a widely reported role in cancer65. The primary receptor for Cxcl1 is Cxcr2, which is mainly expressed in neutrophils66. Neutrophils can be recruited to the TME and differentiate into polymorphonuclear MDSCs (PMN-MDSCs), which contribute to a pro-tumorigenic environment67. We observed a reduced abundance of neutrophils among total CD45+ cells in Trp53R172H/−;Cxcl1−/− tumors (Figure 3D). We investigated other immune cell counterparts that could contribute to slower tumor growth. We observed an increase in infiltration of lymphocytes and innate lymphoid cells (CD45+CD11b-CD90+NK1.1-), natural killer (NK) cells (CD45+CD11b-CD90+NK1.1+), and macrophages (CD11b+CD64+) upon the loss of Cxcl1 (Figure 3E & Figure 3F), all of which have been reported to show anti-tumor activity68. Finally, we observed that the lymphocyte-to-neutrophil and NK-cell-to-neutrophil ratio was significantly higher in Cxcl1-ablated tumors, which suggests a TME with more robust anti-tumor activity (Figure S5G). We used immunofluorescence to further examine the immune composition of the tumors. We found a higher number of cytotoxic T cells (CD8+) and a lower number of immunosuppressive neutrophils (Gr-1+) and MDSCs (Gr-1+Arg-1+) in Trp53R172H/−;Cxcl1−/− tumors (Figure 3G).
We then tested whether the immune state of the TME in Trp53R172H/−;Cxcl1−/− tumors impedes tumor growth and elicits a response to ICIs. We found that Cxcl1 ablation slowed tumor growth (Figure 3H). More importantly, mice with Trp53R172H/−;Cxcl1−/− tumors exhibited better survival rates, with ICI treatment further improving survival (Figure 3I). Overall, our data suggest that one of the pro-tumorigenic pathways exploited by p53R172H involves the elevation of Cxcl1 expression, which contributes to the establishment of an immunosuppressive TME, promoting tumor growth and diminishing the efficacy of ICIs.
p53R172H occupies Cxcl1 enhancers and increases their transcription activation potential
The significance of Cxcl1 in shaping the PDAC TME prompted us to investigate the mechanism by which p53R172H regulates Cxcl1. p53 is a potent transcription factor with a robust transactivation domain, located in the N-terminus of the protein, that remains largely unaffected in mutant p53 proteins found in cancers. Most p53 mutations in cancers occur within the DNA-binding domain, resulting in the loss of the ability to bind the canonical DNA motifs recognized by wild-type p53.
Given its transcriptional activation potential, we investigated whether p53R172H associates with new genomic sites. After testing nine different antibodies, we identified two that performed well with CUT&RUN69. In Trp53R172H/− cells, we observed significant p53R172H occupancy at the Cxcl1 promoter (Figure 4A & Figure S6A) compared to the background signals from p53 CUT&RUN in Trp53−/− cells and the non-specific IgG signal in Trp53R172H/− cells. Peaks derived from two different p53R172H antibodies overlapped significantly (Figure S6B), primarily occupying gene promoters (Figure S6C). Even when analyzing unique peaks identified by each antibody, the CUT&RUN signals from both correlated well (Figure S6D), suggesting that the incomplete overlap may result from the thresholds used in peak calling. Interestingly, p53R172H binding was not confined to the Cxcl1 promoter; we identified even stronger peaks upstream of the Cxcl1 gene. Since these regions could function as transcriptional regulatory elements influencing Cxcl1 expression, we examined commonly used markers for such elements in Trp53R172H/− and Trp53−/− cells. We detected prominent signals for the transcription co-factor p300 and H3K27Ac (Figure 4B). Additionally, the lower levels of H3K4me3 and higher levels of H3K27Ac at these loci, compared to the Cxcl1 promoter, suggest that these regions are likely enhancers.
Figure 4. p53R172H occupies and modulates Cxcl1 enhancers.

A, p53R172H occupancy at and around the Cxcl1 gene in Trp53R172H/− and Trp53−/− cells using CUT&RUN. Non-specific IgG in Trp53R172H/− cells is used as a control.
B, p300, H3K27Ac, and H3K4me3 occupancy at and around the Cxcl1 gene in Trp53R172H/− and Trp53−/− cells using CUT&RUN.
C, Nascent RNA profiles at and around the Cxcl1 gene in Trp53R172H/−, Trp53−/−, or Trp53−/− + pTrp53R172H cells using PRO-seq. Nascent RNA profiles are used to call de novo enhancers using dREG71, and the annotated enhancers are shown at the bottom (names begin with “e”).
D, Quantification of enhancer RNA (eRNA) from PRO-seq in the three enhancers around the Cxcl1 gene.
E, Nascent RNA levels of the expressed Chemokine genes shown as the z-cores heatmap of PRO-seq RPM in the three isogenic cells.
F, Quantification of p300 and histone modification levels (left) and p53R172H occupancy (right) at the promoter and enhancers of the Cxcl1 gene using CUT&RUN. P-values are calculated excluding e8697 using a paired t-test.
A strong indicator of an active enhancer is the presence of short, unstable, non-polyadenylated enhancer RNA (eRNA) generated from the divergent transcription of enhancers70. To confirm that the p53R172H-occupied regions around the Cxcl1 gene are enhancers, we performed nascent RNA sequencing using PRO-seq on Trp53R172H/−, Trp53−/−, and Trp53−/− + pTrp53R172H cells (Figure 4C). We observed prominent divergent transcription at these putative enhancers. Using dREG71, which identifies de novo enhancers based on divergently transcribed nascent RNA profiles, we discovered three enhancers (e8695, e8696, and e8697, measuring 705 bp, 1065 bp, and 819 bp, respectively) within 15 kb of the Cxcl1 gene (Figure 4C). It is well-established that the level of enhancer transcription positively correlates with the transcription of target genes70. We quantified the eRNA at these enhancers and found that the deletion of Trp53R172H reduced their activity, while ectopic expression of p53R172H mostly restored it (Figure 4D), suggesting that p53R172H binding at these enhancers may significantly influence the transcription output of target genes. We measured chemokine gene transcription by quantifying nascent RNA in Trp53R172H/−, Trp53−/−, and Trp53−/− + pTrp53R172H cells. We observed increased nascent transcription of a similar subset of chemokine genes in the presence of p53R172H (Figure 4E), indicating transcriptional regulation by p53R172H.
To confirm the specificity of the p53R172H occupancy and to ensure that the p53R172H CUT&RUN signals are not a result of p53 antibodies non-specifically associating with chromatin-associated factors, we quantified p53R172H, p300, H3K27Ac, and H3K4me3 signals. At the promoters and enhancers of Cxcl1 (Figure 4F) and also at all p53R172H CUT&RUN peaks (n=7,721) (Figure S6E & Figure S6F), we found that the enrichment of p53R172H occupancy in Trp53R172H/− cells compared to Trp53−/− cells was significantly higher than the difference in the levels of chromatin-associated factors. Instead, the levels of p300 and histone modifications at the p53R172H-occupied regions were relatively similar between Trp53R172H/− and Trp53−/− cells. This suggests that p53R172H is not responsible for opening and priming the chromatin for transcription. Instead, p53R172H augments the transcription activation potential of these regions that seem to be already open and primed, likely by other transcription factors. It is plausible that p53R172H commutes to these enhancers through interaction with other transcription factors. This model is consistent with a lack of a p53R172H-specific DNA binding motif under the p53R172H CUT&RUN peaks. These observations also highlight that p300 binding and histone modifications are not the direct temporal determinants of the regulatory activity of promoters and enhancers72. Instead, transcription factor occupancy and divergent nascent transcription more precisely predict the activity of enhancers and the expression of their target gene73.
To confirm the specificity of p53R172H occupancy and ensure that the p53R172H CUT&RUN signals were not due to non-specific association of p53 antibodies with chromatin-associated factors, we quantified p53R172H, p300, H3K27Ac, and H3K4me3 signals. At both the promoters and enhancers of Cxcl1 (Figure 4F) and across all p53R172H CUT&RUN peaks (n=7,721) (Figure S6E & Figure S6F), we observed that the enrichment of p53R172H occupancy in Trp53R172H/− cells compared to Trp53−/− cells was significantly higher than the difference in the levels of chromatin-associated factor. The levels of p300 and histone modifications at p53R172H-occupied regions were relatively similar between Trp53R172H/− and Trp53−/− cells, suggesting that p53R172H is not responsible for opening and priming the chromatin for transcription. Instead, p53R172H appears to enhance the transcriptional activation potential of regions that are likely already open and primed by other transcription factors. It is plausible that p53R172H is recruited to these enhancers through interactions with other transcription factors. These observations also suggest that p300 binding and histone modifications are not direct temporal determinants of the regulatory activity of promoters and enhancers72. Instead, transcription factor occupancy and divergent nascent transcription more accurately predict enhancer activity and the expression of their target genes73.
Cxcl1 enhancers dictate Cxcl1 expression and its immunosuppressive function
To test whether and to what extent the Cxcl1 enhancers regulate the Cxcl1 expression, we systematically deleted them individually or in pairs using CRISPR-Cas9 (Figure 5A). We observed that the deletion of e8695 and e8696 but not e8697 (denoted as Δe8695, Δe8696, and Δe8697, respectively) significantly reduced the Cxcl1 expression (Figure 5B). These enhancer-deleted cells are KrasG12D/+, Trp53R172H/−, and Cxcl1+/+. The dual deletion of e8695 and e8696 (Δe8695 + Δe8696) further reduced the Cxcl1 expression, but the additional reduction was modest. To examine their contribution to the PDAC growth and TME, we orthotopically implanted the Δe8695, Δe8696, and Δe8697 cells in the pancreas of immunocompetent mice and extracted the tumors after 21 days. Δe8695 and Δe8696 tumors were significantly and consistently reduced in size as compared to their parental cell, and the extent of their effect was similar to their impact on Cxcl1 levels, whereas Δe8697 tumor size was partially reduced in one cohort but not detectably reduced in another cohort (Figure 5C & Figure S7A).
Figure 5. p53R172H-occupied Enhancers regulate Cxcl1 expression and dictate Cxcl1-mediated immunosuppression.

A, Generation of enhancer-deleted isogenic cells from the parental Trp53R172H/− cells. Two enhancers upstream of the Cxcl1 gene and one downstream of the Cxcl1 gene are deleted individually or in a pair using CRISPR/Cas9. The size of deleted regions is indicated in parentheses.
B, Quantification of the Cxcl1 chemokine level in enhancer-deleted isogenic cells and comparison with the Trp53R172H/− cells. P-values are calculated from a one-way ANOVA test followed by a post hoc test with Benjamini-Hochberg correction.
C, Weights of the enhancer-deleted isogenic tumors compared with the Trp53R172H/− tumors. Trp53R172H/−;Δe8695;Δe8696 cells were not implanted in mice due to the minimal effect over either Trp53R172H/−;Δe8695 or Trp53R172H/−;Δe8696 cells. P-values are calculated from a one-way ANOVA test followed by a post hoc test with Benjamini-Hochberg correction.
D, Effect of the e8696 and e8697 status in leukocyte and neutrophil infiltration in PDAC tumors. P-values are calculated from a one-way ANOVA test followed by a post hoc test with Benjamini-Hochberg correction.
E, Representative multiplex immunofluorescence staining of Trp53R172H/−;Δe8695 and Trp53R172H/−;Δe8696 tumors. The scale bars correspond to 200 μm and 50 μm in the main view and the magnified view, respectively.
F, Quantification of immune cell density from randomly selected 500 × 500 μm regions. P-values are calculated from a one-way ANOVA test followed by a post hoc test with Benjamini-Hochberg correction.
G, Kaplan-Meier survival curves showing the effects of e8696 or e8697 status and ICIs in the survival of mice implanted with parental Trp53R172H/− or Trp53R172H/−;Δe8696, or Trp53R172H/−;Δe8697 cells. Control mice were treated with IgG. Trp53R172H/− mice survival cohort is the same as in Figure S4D & Figure 3I. P-values are calculated using a log-rank (Mantel-Cox) test.
As shown in Figure 3, the ablation of Cxcl1 alone reprogrammed the immune landscape of the PDAC TME. Although Δe8695 and Δe8696 reduced Cxcl1 expression and tumor size, we sought to ensure that enhancer deletion recapitulated the Cxcl1-mediated immune reprogramming. To minimize batch effects, we processed all mice and samples together, which limited us to two enhancer-deleted cell lines. We selected Δe8696 and Δe8697 due to their equidistant positions from the Cxcl1 gene and their variability in inducing Cxcl1 expression. Moreover, based on Cxcl1 expression and tumor weight (Figure 5B & Figure 5C), we anticipated that Δe8695 would recapitulate the results of Δe8696. Consequently, we examined the immune landscape, tumor growth, and survival with Δe8696 and Δe8697 cells. As seen in the Cxcl1−/− tumors, we observed higher infiltration of immune cells, a lower fraction of neutrophils, and higher innate lymphoid cells, macrophages, and NK cells in Δe8696 tumors (Figure 5D & Figure S7B). We used immunofluorescence to further examine the immune composition of the tumors (Figure 5E). We found a higher density of cytotoxic T cells (CD8+) and a lower density of immunosuppressive neutrophils (Gr-1+) and MDSCs (Gr-1+Arg-1+) in Δe8696 tumors compared to Trp53R172H/− tumors (Figure 5F).
We then tested if the reprogrammed immune microenvironment influenced the efficacy of ICIs. We observed the mice with Δe8696 tumors responded well to ICIs (Figure 5G). The survival and ICIs benefits were not observed in Δe8697 despite its similar proximity to the Cxcl1 gene. e8697 is closer to the Cxcl2 gene, which is lowly expressed in these cells. Moreover, the transcription of e8697 is lower than that of e8696, and the CUT&RUN signal of p53R172H is not detected at this enhancer (Figure 4A & Figure S6A). These observations suggest that the p53R172H-occupied e8696 is the critical regulator of Cxcl1 expression and p53R172H-mediated immunosuppression in PDAC.
NF-κB occupies Cxcl1 promoter and enhancers
The arginine residue at position 172 of p53 stabilizes the DNA binding domain by contacting a zinc ion18. Therefore, the common R172H mutation alters p53’s conformation and abolishes its canonical DNA binding activity17. To further understand the mechanism underlying p53R172H DNA association, we investigated whether p53R172H associates with DNA independently or relies on other transcription factors. Our analysis of the DNA sequences under the p53R172H CUT&RUN peaks revealed no enrichment of the canonical wild-type p53 DNA binding motif or any novel motif. Instead, we observed an enrichment of several known transcription factors under the p53R172H peaks.
When we specifically examined the enhancers and promoters of the p53R172H-dependent chemokines, we found the NF-κB motif to be the most enriched, present in approximately 70% of examined sites (Figure 6A). To test if NF-κB binds these putative NF-κB sites, we used CUT&RUN with an antibody against the RelA (p65) subunit of NF-κB. We observed prominent NF-κB occupancy at the Cxcl1 promoter and enhancers (Figure 6B). Quantification showed a ~20–25% decrease in NF-κB occupancy in Trp53−/− cells compared to Trp53R172H/− cells (Figure 6C), suggesting a role of p53R172H in increasing NF-κB occupancy at these sites. Despite similar p300 occupancy and histone modifications (Figure S8A), the average NF-κB CUT&RUN signal was similarly lower in Trp53−/− cells compared to Trp53R172H/− cells (Figure 6D).
Figure 6. p53R172H occupancy in Cxcl1 Enhancers is dependent on NF-κB.

A, Enriched transcription factor motifs at the promoters and enhancers of the p53R172H-dependent chemokine genes identified using HOMER105.
B, NF-κB occupancy at the Cxcl1 gene and enhancers in Trp53R172H/− and Trp53−/− cells using CUT&RUN. Non-specific IgG is used as a control. The HOMER-identified NF-κB motifs are shown at the bottom (red bars).
C, Quantification of NF-κB occupancy at the promoter and enhancers of the Cxcl1 gene using CUT&RUN. P-values are calculated excluding e8697 using a paired t-test.
D, Quantification of NF-κB occupancy at the NF-κB CUT&RUN peaks using CUT&RUN.
E, Quantification of NF-κB phosphorylation in the Trp53R172H/− and Trp53−/− cells using ELISA on cell extracts. The antibody targets S536 in the p65 subunit of NF-κB.
F, Quantification of NF-κB localization in the cytoplasm vs nucleus using western blot (serial dilution of protein lysates). Vinculin and histone H3 were used as the markers of cytoplasmic and nuclear fractions, respectively. P-values are calculated from a two-way ANOVA test.
We observed a similar trend of lower NF-κB occupancy in Trp53−/− cells compared to Trp53R172H/− cells in the p53R172H CUT&RUN peaks (Figure S8B). This lower occupancy was not due to differential expression of NF-κB between the two cell types (Figure S8C). Instead, we found that the phosphorylation of the RelA subunit of NF-κB and its nuclear localization is reduced by ~25% in the absence of p53R172H (Figure 6E & Figure 6F). Phosphorylated NF-κB translocates into the nucleus, where it binds and regulates the expression of pro-inflammatory genes74. These data suggest that while NF-κB expression is unaffected by p53R172H, its phosphorylation, subsequent nuclear localization, and genome occupancy are moderately increased in the presence of p53R172H. However, this ~20–25% increase in NF-κB occupancy at the promoters and enhancers of Cxcl1 does not fully explain the four-fold higher expression of Cxcl1 in Trp53R172H/− cells compared to Trp53−/− cells (Figure 1F).
Our observation of both p53R172H and NF-κB occupancy at the Cxcl1 enhancers and promoters led us to examine the overlap between p53R172H and NF-κB CUT&RUN peaks. We found a significant overlap in their genomic occupancy, with most co-occupied regions coinciding with known promoters and enhancers (Figure S8D). We quantified p53R172H and NF-κB occupancy, p300 levels, histone modifications, and nascent transcription across three sets of CUT&RUN peaks: unique p53R172H peaks, common peaks, and unique NF-κB peaks (Figure S9A-C). In each case, the levels of chromatin-associated factor were similar between Trp53R172H/− and Trp53−/− cells. However, differences in p53R172H occupancy were reflected in nascent RNA profiles, indicating that nascent transcription is a better indicator of the transcriptional state of promoters and enhancers than chromatin-associated factors. These findings demonstrate that NF-κB binds to the promoters and enhancers of Cxcl1 largely independently of p53R172H, and its genome-wide occupancy significantly overlaps with that of p53R172H.
p53R172H depends on NF-κB for Cxcl1 enhancer binding
We next investigated whether p53R172H genome occupancy depends on NF-κB. To test this, we examined the impact of TPCA-1, a potent IKKβ inhibitor that prevents NF-κB activation, on NF-κB and p53R172H occupancy using CUT&RUN. As expected, TPCA-1 treatment significantly reduced NF-κB binding at the Cxcl1 enhancers in both Trp53R172H/− and Trp53−/− cells (Figure 7A). More importantly, p53R172H occupancy at the Cxcl1 enhancers was similarly reduced following TPCA-1 treatment, strongly suggesting that p53R172H binding is dependent on the NF-κB pathway.
Figure 7. NF-κB inhibition abrogates p53R172H occupancy.

A, Effect of inhibiting NF-κB activation (5 uM TPCA-1) in NF-κB and p53R172H occupancy at the Cxcl1 gene and enhancers. DMSO treatment serves as a control.
B, Change in p53R172H occupancy (left) and NF-κB occupancy (right) by NF-κB inhibition in the p53R172H and NF-κB CUT&RUN peaks, respectively.
C. Effects of TPCA-1 treatment in NF-κB (left) and p53R172H (right) occupancy in the two detectable enhancers of the Cxcl1 gene.
D, Effects of TPCA-1 treatment in Trp53R172H (left) and Cxcl1 (right) mRNA levels using RT-qPCR.
Our data showed that CUT&RUN signals for all p53R172H peaks and nearly all NF-κB peaks (except one) decreased following NF-κB inhibition by TPCA-1 (Figure 7B & Figure S10A), highlighting the broad impact of NF-κB on p53R172H chromatin binding. Specifically, the TPCA-1 treatment reduced the occupancy of both NF-κB and p53R172H to approximately 20% at the Cxcl1 enhancers, illustrating the critical role of NF-κB in facilitating p53R172H recruitment to these sites (Figure 7C). Additionally, we observed that TPCA-1 treatment led to a decrease in Cxcl1 gene expression, as expected, but did not affect Trp53R172H gene expression. This finding suggests that the reduced occupancy of p53R172H was not due to lower Trp53R172H expression but rather the direct effect of NF-κB pathway inhibition (Figure 7D).
Collectively, our findings reveal a novel mechanism by which the cancer-cell-intrinsic p53R172H occupies the enhancers of the Cxcl1 gene in an NF-κB-dependent manner. This appropriation of the NF-κB pathway by p53R172H amplifies Cxcl1 expression, contributing to the establishment of an immunosuppressive TME that, in turn, diminishes the efficacy of ICIs. These insights highlight potential therapeutic targets for enhancing ICI efficacy in PDACs harboring Trp53 missense mutations and elevated Cxcl1 expression.
Discussion
Our findings indicate that p53R172H promotes tumor growth by modulating cancer-cell-specific gene expression programs that shape the TME to suppress anti-tumor immunity. Specifically, Trp53R172H/− tumors exhibit fewer T cells, higher MDSC infiltration, and reduced ICI efficacy in mouse models of PDAC. At the molecular level, p53R172H exerts its immunosuppressive effects primarily through the regulation of chemokine genes, particularly Cxcl1. p53R172H binds to distal enhancers of Cxcl1 and amplifies its expression in an NF-κB-dependent manner. This interaction suggests a broader mechanism by which mutant p53 may co-opt enhancers—vastly outnumbering gene promoters—by interacting with the enhancer’s cognate binders to modulate cell-intrinsic gene expression, ultimately influencing cell-extrinsic features. This study not only elucidates the specific mechanisms of immunosuppression facilitated by p53R172H but also suggests broader implications for p53 missense mutations and their cooperation with other transcription factors in cancer progression.
Enhancers have emerged as key players in disease pathology. Over 90% of disease-associated variants are located in non-coding regions, predominantly within enhancers75. Despite this recognition, understanding the precise mechanisms by which enhancers influence disease remains a challenge. However, recent advances, exemplified by the FDA approval of Casgevy, the first CRISPR-Cas9-based therapy targeting the enhancer regulating the BCL11A transcription factor for sickle cell anemia76, underscore the therapeutic potential of manipulating enhancer activity. Our study further illuminates the significance of enhancers, revealing how oncogenic factors can appropriate them to drive the expression of immunosuppressive chemokines for PDAC progression and confer resistance to ICIs.
Our findings hold two other significant implications. First, they offer mechanistic insights into the therapeutic targeting of immunosuppressive chemokines and their receptors, as well as the upstream signaling pathways, in cancer treatment. High levels of Cxcl1 are associated with poor prognosis in various cancers, including PDAC77. A similar dependence of selected chemokine genes on p53R172H was observed in previous studies. shRNA-mediated knockdown of mutant p53 in human colon adenocarcinoma cell line SW480 (harboring R273H/P309S mutations) and pancreatic cancer cell line MIA-PaCa-2 (harboring R248W mutation) also reduced Cxcl1 expression78. This supports the promise of current clinical trials testing the blockade of tumor cell-derived Cxcl179, Cxcr2 deletion80, or its inhibition81. Similarly, the NF-κB signaling pathway that dictates Cxcl1 expression is activated in many cancers. However, despite its potential, no effective NF-κB inhibitor is available for clinical use in cancer treatment due to significant toxicities associated with blocking this pathway82,83. Our characterization of an enhancer targeting a key effector of NF-κB-mediated immunosuppression suggests a promising avenue for reducing toxicity while maintaining therapeutic efficacy.
Secondly, our findings establish a mechanistic link between a common mutation in the most frequently mutated gene and the ineffectiveness of ICIs in certain cancers. Mutations in transcription factors drive many cancers51, and enhancers vastly outnumber genes. The exploitation of enhancers by mutated transcription factors in our study highlights the importance of this mechanism and underscores the need for further investigation into tissue- and mutation-specific enhancer-gene regulatory circuits. It is conceivable that mutated transcription factors interact with abundant enhancers in the genome through liquid-liquid phase condensates, driving aberrant gene programs84.
More importantly, our observations align with clinical data. A mutant p53 RNA expression signature shows a significant correlation with reduced survival in 11 cancer types85,86. Similarly, PDAC with p53 missense mutations is associated with poor survival40, and compared to p53-null, they have enhanced fibrosis and lower lymphocyte and CD8+ T cell infiltration86,87. Degradation of mutant p53 and restoration of wild-type function are targets of several clinical trials25,88. Evidence suggests that the mutant p53-specific immune reprogramming in cancer is mediated by Cxcl1 overexpression. For example, ectopic expression of p53R175H, but not wild-type p53, whose level and activity are tightly suppressed in healthy cells89, increases Cxcl1 expression in colon cancer90. Similarly, overexpression of p53R175H, but not wild-type p53, specifically in the context of KRASG12D mutation, enhanced Cxcl1 expression in human pancreatic epithelial cells30. Another study switching p53WT expression to p53R172H revealed enrichment of cytokine-cytokine receptor genes, emphasizing the role of mutant p53 in cytokine regulation43. Moreover, in pancreatic ductal epithelial cells with KrasG12D mutation, chemokine genes Cxcl2 and Cxcl5 exhibited significantly higher expression in p53R172H cells compared to p53WT cells56. It is plausible that the p53R172H mutation interacts with NF-κB, utilizing it as a vehicle to reach specific enhancers. With a functional and potent transactivation domain, p53R172H could elevate the expression of the Cxcl1 gene beyond the baseline induction by NF-κB alone. Prior studies have indicated mutant p53’s propensity to prolong NF-κB signaling in cancer contexts91, while reciprocal inhibition between p53WT and NF-κB has also been documented92,93. Moreover, physical interaction between p53R172H and NF-κB has been reported19,20, possibly explaining the presence of p53R172H at Cxcl1 enhancers.
Our study suggests that a small fraction of PDAC patients who are p53NULL may respond to ICIs. Nonetheless, combination immuno-oncology therapy in pancreatic cancer yielded only a 3.1% response rate5. While it is a small fraction, the genotype of PDAC patients that respond to ICIs warrants attention. It’s plausible that tumors relying on p53R172H-mediated immunosuppression may become vulnerable to ICIs upon abrogation of this dependence. However, we lack evidence to suggest that p53R172H can serve as a biomarker for ICI responsiveness in PDAC at this stage. Instead, our data, along with evidence from the literature, suggest that overexpression of Cxcl1, implicated in various cancers through different mechanisms94–100, could be a better biomarker. Our work presents a novel and potent approach to abrogate Cxcl1 expression in PDAC by targeting enhancers, which generally have higher specificity and lower toxicity.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact (P.A.S.).
Materials availability
The lead contact can provide cell lines generated in this study upon request.
Data and code availability
RNA-seq, PRO-seq, Exome-seq, and CUT&RUN data have been deposited in GEO and will be publicly available as of the date of publication. Accession numbers and secure tokens for reviewer access are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse/human p1253 | Epicypher | SKU: 13-2015 |
| p53 (1C12) Mouse mAb | Cell Signaling | #2524 |
| anti-mouse/human p53 | Leica Biosystems | P53-DO7-L-CE |
| anti-mouse CD16/32;clone 93 | BioLegend | Cat# 101330;AB_2561482 |
| anti-mouse CD25 (APC-Cy7);clone PC61 | BioLegend | Cat# 102026;AB_830745 |
| anti-mouse CD279 (PD-1) (BV605);clone 29F.1A12 | BioLegend | Cat# 135220;AB_2562616 |
| anti-mouse CD3 (eFluor 450);clone 17A2 | eBioscience | Cat# 48-0032-80;AB_1272229 |
| anti-mouse CD366 (TIM3) (PE-Cy7);clone RMT3-23 | eBioscience | Cat# 25-5870-82;AB_2573483 |
| anti-mouse CD4 (BUV737);clone RM4-5 | BD Biosciences | Cat# 612844;AB_2732918 |
| anti-mouse CD45 (BUV395);clone 30-F11 | BD Biosciences | Cat# 564279;AB_2651134 |
| anti-mouse CD62L (BV711);clone MEL-14 | BioLegend | Cat# 104445;AB_2564215 |
| anti-mouse CD69 (APC);clone H1.2F3 | BioLegend | Cat# 104513;AB_492844 |
| anti-mouse CD8a (BV605);clone 53-6.7 | BioLegend | Cat# 100744;AB_2562609 |
| anti-mouse FoxP3 (PerCP-Cy5.5);clone FJK-16s | eBioscience | Cat# 45-5773-80;AB_914349 |
| anti-mouse/human CD11b (PE/Dazzle 594);clone M1/70 | BioLegend | Cat# 101255;AB_2563647 |
| anti-mouse/human CD44 (PE);clone IM7 | BioLegend | Cat# 103023;AB_493686 |
| anti-mouse/human Gzmb (AF700);clone QA16A02 | BioLegend | Cat# 372221;AB_2728388 |
| anti-mouse/human TCF1 (AF488);clone C63D9 | Cell Signaling Technology | Cat# 6444S |
| anti-Ly-6G (clone 18A) PerCP/Cyanine5.5 | BioLegend | Cat# 127615 |
| anti-CD90.2 (clone 30-H12) FITC | BioLegend | Cat# 105306 |
| anti-CX3CR1 (clone SA011F11) BV650 | BioLegend | Cat# 14903 |
| anti-NK1.1 (clone PK136) BV605 | BioLegend | Cat# 108753 |
| anti-Folate Receptor 2 (clone 10/FR2) APC | BioLegend | Cat# 153305 |
| anti-I-A/I-E (clone M5/114.15.2) BV711 | BioLegend | Cat# 107643 |
| anti-CD206 (clone C068C2) PE/Cyanine7 | BioLegend | Cat# 141719 |
| anti-CD64 (clone X54-5/7.1) PE/Dazzle | BioLegend | Cat# 139319 |
| anti-Ly6C (clone HK1.4) BV785 | BioLegend | Cat# 128041 |
| Streptavidin-PE | BioLegend | Cat# 405203 |
| anti-TIM-4 (clone RMT4-54) BV421 | BD Biosciences | Cat# 744874 |
| anti-CD200R3 (clone Ba13) BUV805 | BD Biosciences | Cat# 752653 |
| anti-CD45 (clone 30-F11) BUV395 | BD Biosciences | Cat# 565967 |
| anti-Siglec-F (clone E50-2440) APC-R700 | BD Biosciences | Cat# 565183 |
| anti-FCeR1a (clone MAR-1) BV750 | BD Biosciences | Cat# 751756 |
| anti-CD19 (clone 1D3) BUV615 | BD Biosciences | Cat# 751213 |
| anti-CD11c (clone N418) BUV737 | BD Biosciences | Cat# 749039 |
| anti-CD103 (clone M290) BUV563 | BD Biosciences | Cat# 741261 |
| anti-CD11b (clone M1/70) APC-eFluor 780 | Invitrogen | Cat# 47-0112-80 |
| anti-CD16/CD32 (clone 2.4G2) | Bio X Cell | Cat# BP0307 |
| Lyve1 Biotin (clone ALY7) 13-0443-82 | Thermo Scientific | Cat# |
| anti-CD8 alpha, clone: YTS169.4 | Abcam | Cat# ab22378 |
| anti-Neutrophil | Abcam | Cat# ab2557 |
| anti-Arginase-1 | Cell Signaling Technology | Cat# 93668T |
| VECTASHIELD Vibrance® Antifade Mounting Medium | VECTOR Laboratories | Cat# H-1700 |
| Chemicals, peptides, and recombinant proteins | ||
| Fixable Viability Dye eFluor 506 | eBioscience | Cat# 65-0866-18 |
| Fixable Viability Dye eFluor 780 | eBioscience | Cat# 65-0865-14 |
| DMEM | Gibco | Cat# 11995 |
| Heat-inactivated fetal bovine serum | Tissue Culture Biologics | Cat# 101HI |
| Tissue culture-treated 10 cm plates | Corning | Cat# CLS430167 |
| Trypsin-EDTA | Gibco | Cat# 25200 |
| α-PD-1 | BioXcell | Cat# BE0146 |
| α-CTLA4 | BioXcell | Cat# 9D9 |
| Collagenase IV | Thermo Scientific | Cat# 17104019 |
| DNase I, Grade II, from bovine pancreas | Sigma-Aldrich | Cat# 10104159001 |
| Liberase TL Research Grade | Sigma-Aldrich | Cat# 05401020001 |
| Lipofectamine 2000 transfection reagent | Thermo Scientific | Cat# 18324012 |
| PureFection | System Bioscience | Cat# LV750A |
| T4 Polynucleotide Kinase | New England Biolabs | Cat# M0201 |
| Tango Buffer | Thermo Scientific | Cat# BY5 |
| FastDigest BbsI restriction enzyme | Thermo Scientific | Cat# FD1014 |
| T7 DNA Ligase | New England Biolabs | Cat# M0318 |
| Alkaline Phosphatase | Thermo Scientific | Cat# EF0654 |
| ACK Lysing Buffer | Thermo Scientific | Cat# A1049201 |
| Plasmid-Safe ATP-Dependent DNase | Biosearch Technologies | Cat# E3101K |
| QIAprep Spin Miniprep Kit | Qiagen | Cat# 27104 |
| S.O.C. Medium | Thermo Scientific | Cat# 15544034 |
| Carbenicillin Disodium Salt | Thermo Scientific | Cat# 10177012 |
| anti-mouse CXCL1 | Leinco Technologies | Cat# K125 |
| anti-mouse CCL2 | Bio-Techne | Cat# MAB479 |
| Recombinant Mouse CXCL1 | Bio-Techne | Cat# 453-KC |
| Recombinant Mouse CCL2 | Bio-Techne | Cat# 452-M2 |
| anti-Mouse CXCL1- Biotin | Leinco Technologies | Cat# K124 |
| anti-Mouse CXCL1- Biotin | Bio-Techne | Cat# BAF479 |
| HRP Streptavidin | BioLegend | Cat# 405210 |
| TMB Substrate Solution | BioLegend | Cat# 421101 |
| Tissue-Tek® O.C.T. Compound | Sakura Finetek | Cat# 62550 |
| Antigen Unmasking Solution | VECTOR Laboratories | Cat# H-3300 |
| Stop Solution | BioLegend | Cat# 423001 |
| Critical commercial assays | ||
| FoxP3 Transcription Factor Staining Buffer Set | eBioscience | Cat# 00-5523-00 |
| Chemically competent Stbl3 E. coli cells | Thermo Scientific | Cat# C737303 |
| One Shot TOP10 Chemically Competent E. coli cells | Thermo Scientific | Cat# C404006 |
| Cytofix/Cytoperm Fixation/Permeabilization buffer | BD Biosciences | Cat# 554714 |
| Precision Count Beads | BioLegend | Cat# 424902 |
| UltraComp eBeads Compensation Beads | Invitrogen | Cat# 01-2222-42 |
| Mouse Cytokine Array C3 | RayBiotech | Cat# AAM-CYT-3 |
| Mouse LIX/ CXCL5 ELISA kit | Thermo Scientific | Cat# EMCXCL5 |
| 96-well ELISA plates | BioLegend | Cat# 423501 |
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4368577 |
| ChIC/CUT&RUN Kit | EpiCypher | Cat# 14-1048 |
| TPCA-1 | Selleckchem | Cat# S2824 |
| Deposited data | ||
| RNA-seq | This study | GSE275105 Secure token: ujstwuygpfsbpkz |
| PRO-seq | This study | GSE275106 Secure token: ytwtwwqkzvgjtgp |
| Exome-seq | This study | GSE275107 Secure token: sbcrgsaqndcplgx |
| CUT&RUN | This study | GSE275108 Secure token: ctcryaimjtefzav |
| Experimental models: Cell lines | ||
| KrasG12D/+;Trp53R172H/− | Dr. Ashok Saluja lab, University of Miami | N/A |
| KrasG12D/+;Trp53−/− (syngenic) | Dr. Ronald DePinho lab, MD Anderson Cancer Center |
N/A |
| KrasG12D/+;Trp53−/− (isogenic) | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Ccl2−/− | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Ccl2−/−;Cxcl1−/− | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Ccl2−/−;Cxcl5−/− | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Cxcl1−/− | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Cxcl5−/− | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Δe8695 | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Δe8695;Δe8696 | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Δe8696 | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Δe8697 | This study | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | Jackson Laboratory | Strain# 000664 |
| Plasmids | ||
| PB-CMV-MCS-EF1α-Puro cDNA cloning and expression vector | System Biosciences | Cat# PB510B-1 |
| Trp53 (NM_011640) Mouse Untagged Clone | Origene | Cat# MC205636 |
| pU6-(BbsI)_CBh-Cas9-T2A-BFP (gift from Ralf Kuehn) | Addgene | Cat# 64323 |
| Oligonucleotides | ||
| See Table_1_Primer_Oligos.xlsx | ||
| Software and algorithms | ||
| FlowJo v10.8.1 | BD Biosciences | https://www.flowjo.com |
| Prism v10.3.0 | Graphpad | https://www.graphpad.com/features |
| CRISPick Design Tool | Broad Institute’s Genetic Perturbation Platform | https://portals.broadinstitute.org/gppx/crispick/public |
| Gene Set Enrichment Analysis (GSEA) | Subramanian et al., 2005 | http://www.broad.mit.edu/gsea/downloads.jsp |
| ImageJ Fiji | ImageJ | https://imagej.net/ij/ |
| QuPath software v.0.5.1 | QuPath | https://qupath.github.io/ |
| Adobe Illustrator CC2023 | Adobe | https://www.adobe.com |
| Bowtie2 v2.2.9 | Langmead et al., 2019 | https://github.com/BenLangmead/bowtie2 |
| Picard v2.8.0 | Broad Institute | https://github.com/broadinstitute/picard |
| SAMtools v0.1.19 | Danecek et al.57 | https://github.com/samtools/samtools |
| R v4.3.3 | R Foundation | https://cran.r-project.org/bin/ |
| DESeq2 v1.40.2 | Love et al.59 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ggplot2 v3.5.0 | Hadley Wickham | https://cran.r-project.org/web/packages/ggplot2/index.html |
| msigdbr v7.5.1 | Igor Dolgalev | https://cran.r-project.org/web/packages/msigdbr/index.html |
| clusterProfiler v4.8.3 | Yu et al., 2021 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| Enrichr | Kuleshov et al., 2016 | http://amp.pharm.mssm.edu/Enrichr/ |
| UCSC Genome Browser | Kent et al. 2002 | https://genome.ucsc.edu/ |
| HOMER (version v4.11.1) | Heinz et al. | http://homer.ucsd.edu/homer/index.html |
| SAMtools (version 1.9) | Danecek et al. | https://www.htslib.org/doc/1.9/samtools.html |
| SEACR | Meers et al. | https://github.com/FredHutch/SEACR?tab=readme-ov-file |
RNA-seq - GSE275105. Secure token: ujstwuygpfsbpkz
PRO-seq - GSE275106. Secure token: ytwtwwqkzvgjtgp
Exome-seq - GSE275107. Secure token: sbcrgsaqndcplgx
CUT&RUN - GSE275108. Secure token: ctcryaimjtefzav
Table_1_Primer_Oligos.xlsx provides the primers and DNA oligos used to clone the guide RNA sequence in the Cas9 and gRNA-expressing plasmids.
The lead contact can provide any additional information required to reanalyze the data reported in this paper upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mice
We obtained C57Bl/6 mice from the Edwin L. Steele Laboratories, Massachusetts General Hospital, and The Jackson Laboratory (Bar Harbor, Maine). C57Bl/6 mice were bred and maintained in Cox-7 gnotobiotic animal facility at Edwin L Steele Laboratories. We housed the mice under specific pathogen-free conditions at the Koch Institute animal facility and the Massachusetts General Hospital animal facility. We gender-matched and age-matched the mice to be 6–10 weeks old at the time of experimentation. All experimental use of animals followed the Public Health Service Policy on Humane Care of Laboratory Animals and was approved by the Committee on Animal Care at MIT, the Institutional Animal Care and Use Committees (Massachusetts General Hospital/Harvard Medical School), and the Association for Assessment and Accreditation of Laboratory Animal Care International.
PDAC cell lines
We received the KPC cells (KrasG12D/+;Trp53R172H/+) generated from the GEMMs of PDAC (LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre) from Dr. Ashok Saluja from the University of Miami. We received the HY19636 cells (KrasG12D/+;Trp53−/−) generated from the GEMMs of PDAC (p48-Cre;KrasLSL-G12D/+;Trp53loxP/+ from Dr. Ronald DePinho, MD Anderson Cancer Center.
We cultured the PDAC cells in Dulbecco’s Modified Eagle Medium supplemented with 4.5 g/L D-glucose, L-glutamine, and 110 mg/L sodium pyruvate (Gibco), along with 10% heat-inactivated fetal bovine serum (Tissue Culture Biologics), on tissue culture-treated 10 cm plates (Corning). The cells were maintained at 37°C with 5% CO2 and passaged using 0.25% Trypsin-EDTA (Gibco) upon reaching ~70% confluency. For tumor implantation, we trypsinized the cells and resuspended them in PBS at a concentration of 2.5 million cells/ml.
Isogenic Trp53−/−, Ccl2−/−, Cxcl1−/−, Cxcl5−/−, and Cxcl1-enhancers-deleted (Δe8695, Δe8696, and Δe8697) PDAC cell lines
We generated CRISPR-deleted cells by first cloning specific sgRNAs into the Cas9 and Blue Fluorescent Protein (BFP) expressing pU6-(BbsI)_CBh-Cas9-T2A-BFP vector (Addgene). We designed the sgRNA sequences using the CRISPick Design Tool from the Broad Institute’s Genetic Perturbation Platform (see Table_1_Primer_Oligos.xlsx). The complementary top and bottom strands of the sgRNA oligos were synthesized with overhangs compatible with Type II restriction enzymes. We then phosphorylated the 5’ ends of these oligos with T4 Polynucleotide Kinase (NEB) at 37°C for 30 minutes.
Next, we employed the Golden Gate cloning method to ligate the annealed sgRNA cassettes into the BbsI-digested pU6-(BbsI)_CBh-Cas9-T2A-BFP vector. The reaction mixture consisted of the annealed sgRNA DNA cassette, the digested vector, 1X Tango Buffer (Thermo Scientific), 0.5 mM DTT, 0.5 mM ATP, FastDigest BbsI restriction enzyme (Thermo Scientific), and T7 DNA Ligase (NEB). We performed six cycles of digestion and ligation, alternating between 37°C for restriction digestion (5 minutes) and 21°C for ligation (5 minutes), for a total incubation time of 60 minutes. To eliminate excess unligated linear DNA, we treated the reaction with Plasmid-Safe ATP-Dependent DNase (Biosearch Technologies).
Following the ligation, we transformed the gRNA-cloned pU6-(BbsI)_CBh-Cas9-T2A-BFP plasmid into chemically competent Stbl3 E. coli cells (Thermo Scientific). We heat-shocked the cells at 42°C, incubated them on ice, and then recovered them in S.O.C. medium (Thermo Scientific) before plating them on LB agar containing Carbenicillin (Thermo Scientific). After overnight incubation, we selected single colonies, grew them in LB medium with Carbenicillin, and stored them as glycerol stocks at −80°C. Plasmid DNA was isolated from the selected colonies using the QIAprep Spin Miniprep Kit (Qiagen), and successful sgRNA insertion was confirmed by sequencing.
To achieve CRISPR-mediated gene deletion, we transfected 200 ng of the sequence-verified gRNA-cloned plasmid into 100,000 cells and seeded the previous day in a 24-well plate to reach 70–90% confluency. We prepared the transfection complexes by combining the purified plasmid DNA with Lipofectamine 2000 transfection reagent (ThermoFisher) in Opti-MEM I Reduced Serum Medium, incubated the mixture for 20 minutes, and then added it to the cells. Seventy-two hours post-transfection, we used FACS to select BFP-expressing single-cell clones, which we then isolated into 96-well plates. Finally, we sequenced these single-cell clones to confirm the presence of the intended gene edits - the deletion of the target region using a pair of gRNAs or the occurrence of frameshift or nonsense mutations in cases where a single gRNA was employed.
Ectopic Expression of p53R172H in Trp53−/− Cells
To test the sufficiency of p53R172H, we ectopically expressed Trp53R172H in Trp53−/− cells using a PiggyBac cDNA Cloning and Expression Vector. We PCR-amplified the Trp53R172H open reading frame (ORF) from the MC205636 plasmid using primers designed to add EcoRI and NotI restriction sites at both ends (see Table_1_Primer_Oligos.xlsx). The amplified product and the PiggyBac vector (System Biosciences) were digested with EcoRI and NotI at 37°C for 15 minutes. We then ran the digested products on a 1% agarose gel at 150 V for 2 hours and gel-purified the desired fragments: the vector backbone (6430 bp) and Trp53R172H insert (1819 bp). To prevent self-ligation, we dephosphorylated the vector using Alkaline Phosphatase (Thermo Scientific) and phosphorylated the restriction-digested Trp53R172H ORF with T4 Polynucleotide Kinase at 37°C for 30 minutes. We ligated the insert into the vector using T7 DNA Ligase at room temperature for 30 minutes.
The ligated plasmid construct was then transformed into One Shot TOP10 Chemically Competent E. coli cells (Thermo Scientific) via heat shock at 42°C, followed by recovery in SOC medium. After overnight incubation on LB agar plates containing Carbenicillin, we picked single colonies, cultured them in LB medium with Carbenicillin, and stored the cultures as glycerol stocks at −80°C. We isolated plasmid DNA from selected colonies using the Qiagen Miniprep kit and confirmed the successful insertion of the Trp53R172H ORF by sequencing.
For stable expression of Trp53R172H in Trp53−/− cells, we transfected 500 ng of the sequence-verified PiggyBac-Trp53R172H plasmid into one million Trp53−/− cells grown in a six-well plate, which had been seeded the previous day to achieve 60–80% confluency. We prepared the transfection complexes by combining the PiggyBac-Trp53R172H plasmid with PureFection (System Bioscience) in serum-free DMEM, vortexed the mixture for 30 seconds, incubated it for 15 minutes at room temperature and then added it dropwise to the cells. After allowing transposase activity for 72 hours, we applied puromycin selection. Although most cells died, a few colonies appeared after 10 days. We isolated individual colonies and verified the stable integration of the Trp53R172H construct. This process successfully generated Trp53−/− cells with stable, ectopic expression of the p53R172H mutant protein.
To answer whether the Cxcl1 enhancer binding is specific to mutant p53, we tried expressing Trp53WT in Trp53−/− cells using multiple approaches, such as ectopic expression using piggyback plasmid, Dox-inducible system, or base-editing. Despite our efforts, we were unable to generate viable Trp53WT-expressing cells, consistent with previous findings that restoration of p53WT induces senescence87, a therapeutic strategy under investigation in cancer clinical trials77.
METHOD DETAILS
Orthotopic pancreatic tumor implantation
We made a ~1 cm incision in the skin and abdominal wall directly over the spleen to expose the pancreas. Using forceps, we gently spread the pancreas and injected 50,000 cells in a 20 μL volume into the pancreas, forming a bubble. We then closed the abdominal wall with absorbable sutures and used staples to close the skin. The staples were removed 7 to 10 days later to ensure proper skin healing and apposition.
Tumor growth measurements and treatment
We measured tumor growth using high-frequency ultrasound. Every three days, we employed a Visualsonics Vevo 2100 ultrasound device equipped with a high-frequency 550S probe to noninvasively monitor tumor growth longitudinally under anesthesia. Once the tumors reached approximately 5 mm in diameter, we randomized the mice into different groups. Following randomization, we treated the mice with either IgG (control) or a cocktail consisting of 200 μg of α-PD-1 (BioXcell) and 100 μg of α-CTLA4 (BioXcell) in a 100 μl volume, administered every third day for a total of three doses.
Tumor extraction and processing
We dissected tumors from the pancreas of mice, weighed them, and then minced and digested them in RPMI buffer containing 1 mg/ml Collagenase IV (Thermo Scientific), 1 mg/ml DNase I (Thermo Scientific), and 0.25 mg/ml Liberase (Sigma-Aldrich) at 37C for 20 minutes. We then strained the tumors through 100-micron filters using the back-end of a 1 ml syringe plunger into 50 ml conical Falcon tubes to create a single-cell suspension. We lysed red blood cells in 1 ml of ACK lysing buffer (Thermo Scientific) on ice for 2 minutes, followed by two washes with pre-chilled FACS buffer (PBS containing 1% FBS and 2 mM EDTA).
Staining for flow cytometry
For intracellular protein staining, we added Brefeldin A at 1x to all reagents until the fixation/permeabilization step. We stained cells for 15 minutes on ice with Fixable Viability Dye eFluor 780 (eBioscience) or Fixable Viability Dye eFluor 506 (eBioscience) to differentiate between live and dead populations and ɑCD16/CD32 (BioLegend) to prevent non-specific antibody binding. After incubation, we washed the cells with FACS buffer and stained them for surface proteins using fluorophore-conjugated antibodies resuspended in FACS buffer at the specified dilutions for 20 minutes at 4°C. Following surface staining, we washed the cells twice with FACS buffer and fixed them using the Foxp3 Transcription Factor Fixation/Permeabilization buffer (eBioscience). Samples processed for myeloid cell staining and flow cytometry were not fixed. After fixation, we washed the cells twice with FACS buffer, then stained them for intracellular proteins in FACS buffer overnight at 4°C. To obtain the absolute cell count, we added Precision Count Beads (BioLegend) to the samples according to the manufacturer’s instructions. We washed the cells twice with FACS buffer before proceeding to acquiring data. We performed flow cytometry sample acquisition on an LSR Fortessa cytometer or FACSymphony A3 Cell Analyzer (BD Biosciences) and analyzed the collected data using FlowJo v10.8.1 software (BD Biosciences).
Multiplex immunofluorescent staining of mouse tumors
We fixed tissues harvested from mice with 4% paraformaldehyde and embedded them in either paraffin or Tissue-Tek® O.C.T. Compound (Sakura Finetek). We sectioned the paraffin-embedded tissues at 4 μm and the O.C.T.-embedded tissues at 10 μm thickness. We deparaffinized and hydrated the paraffin-embedded slides, then retrieved epitopes using citrate-based Antigen Unmasking Solution (VECTOR Laboratories) at 98°C for 20 minutes. We blocked the slides with 5% normal donkey serum and 0.3% Triton-X in phosphate-buffered saline (PBS) for 1 hour at room temperature, followed by incubation overnight at 4°C with primary antibodies: 1:200 anti-CD8ɑ (Abcam), 1:250 anti-Neutrophil (Abcam), and 1:100 Arginase-1 (Cell Signaling Technology). After washing with PBS, we incubated the slides with a solution containing 4’,6-diamidino-2-phenylindole (DAPI) and secondary antibodies at a 1:200 dilution (711–166-152, Cy™3 AffiniPure™ F(ab’)₂ Fragment Donkey Anti-Rabbit IgG (H+L), Jackson ImmunoResearch; 712–606-150, Alexa Fluor® 647 AffiniPure™ F(ab’)₂ Fragment Donkey Anti-Rat IgG (H+L), Jackson ImmunoResearch) for 1 hour at room temperature, then mounted them with cover glass using VECTASHIELD Vibrance® Antifade Mounting Medium (VECTOR Laboratories). We imaged the stained slides at 20x magnification using a Zeiss Axio Scan Z1 (Carl Zeiss) and analyzed the captured image data to quantify CD8-positive cells or Gr-1 + Arginase-1-positive cells using QuPath software v.0.5.1106. For quantification, we randomly selected 5–8 regions of interest (ROIs) (500 × 500 μm) in each viable tumor tissue, calculated the average proportion and density of cells with the corresponding protein expression in every ROI, and reported these values as the representative value for each case.
Cytokine and chemokine array
For the Trp53R172H/− and Trp53−/− cells, we plated 25,000 cells in 6-well tissue culture plates on day 1. To account for the background signal from the serum, we incubated media alone in an empty well. When the cells reached 10–20% confluency, we refreshed the media. After 48 hours, we collected the supernatants and centrifuged them to remove cellular debris. We then incubated the supernatants with the Mouse Cytokine Array C3 kit (RayBiotech) according to the manufacturer’s instructions. The membranes were imaged using the ChemiDoc MP Imaging System from Bio-Rad. We measured and analyzed cytokine intensities using the Protein Array Analyzer for Image J software, normalizing the results with control spots to determine relative cytokine intensities.
ELISA
To measure the protein concentration of chemokine production across all cell lines, we coated 96-well ELISA plates (BioLegend) with 50 μl of 1 μg/ml anti-mouse CXCL1 (Leinco Technologies) or CCL2 (Bio-Techne) for the CCL2-specific ELISA. The coating was done overnight at 4°C in 50 mM Carbonate-Bicarbonate buffer (pH 9.5). After coating, we washed the plates three times with PBS containing 0.05% Tween 20 (wash solution) and blocked them with PBS containing 1% bovine serum albumin. We then prepared recombinant mouse CXCL1 protein (Bio-Techne) or recombinant mouse CCL2 protein (Bio-Techne) in a blocking buffer to generate a standard curve. Culture supernatants were centrifuged to remove cellular debris before being incubated on the pre-coated ELISA plates at room temperature for four hours, followed by thorough washing.
Next, we incubated the plates with 50 μl of 1 μg/ml anti-mouse CXCL1-biotin (Leinco Technologies) or anti-mouse CCL2-biotin (Bio-Techne) diluted in blocking buffer for two hours at room temperature, followed by additional washes. We then incubated the plates for 30 minutes with 50 μl of HRP Streptavidin (BioLegend) diluted at 1:1000 in blocking buffer at room temperature, followed by three washes. To detect the reaction, we added 50 μl of TMB Substrate Solution (BioLegend) for 4 minutes before stopping the reaction with 25 μl of Stop Solution (BioLegend). We analyzed the plates on an Infinite m200 microplate reader at a wavelength of 450 nm (Tecan). Both standards and culture supernatants were tested in duplicate.
We used commercially available ELISA kits for mouse CXCL5 (Thermo Scientific) and followed the manufacturer’s instructions to complete the assays.
Quantitative real-time PCR (qRT-PCR)
We quantified the expression levels of specific genes using quantitative reverse transcription polymerase chain reaction (qRT-PCR). We extracted total RNA from KrasG12D/+;Trp53R172H/− and KrasG12D/+;Trp53−/− cells in tissue culture with the RNeasy Mini Kit (Qiagen), adhering to the manufacturer’s protocol. We determined the concentration and purity of the extracted RNA using Qubit Fluorometric Quantification (Thermo Scientific) and NanoDrop spectrophotometer (Thermo Scientific), respectively. We synthesized cDNA from 1 μg of total RNA with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems), following the supplier’s instructions. We used the Power SYBR Green PCR Master Mix (Applied Biosystems) to perform qRT-PCR using a LightCycler® 480 System (Roche). Each reaction included 2 μL of cDNA template, 10 μL of SYBR Green master mix, and 0.5 μM of each forward and reverse primer (see Table_1_Primer_Oligos.xlsx), totaling 20 μL. The thermal cycling conditions were: initial denaturation at 95°C for 10 minutes, 40 cycles of 95°C for 15 seconds, and 60°C for 1 minute. We conducted a melting curve analysis to verify the specificity of the amplification products. We calculated relative gene expression normalizing to the housekeeping gene GAPDH. To ensure reproducibility and reliability, we performed all reactions in triplicate.
RNA-seq
We extracted total RNA from KrasG12D/+;Trp53R172H/− cells, isogenic and syngenic KrasG12D/+;Trp53−/− cells, and Trp53−/− + pTrp53R172H and Trp53−/− + pEV cells in tissue culture using the RNeasy Mini Kit (Qiagen) following the manufacturer’s protocol. We assessed RNA quality and concentration with an Agilent 2100 Bioanalyzer (Agilent Technologies). We prepared the library from RNA samples with an RNA integrity number (RIN) greater than 7.0 using the TruSeq Stranded mRNA Library Prep Kit (Illumina), according to the recommended protocol. Sequencing was performed on an Illumina NovaSeq 6000 platform, generating paired-end reads of 150 base pairs. We processed the raw sequence data for quality control using FastQC and trimmed adapters with Cutadapt. We aligned high-quality reads to the reference genome (mm10) using bowtie2 and performed transcript quantification using rsem. We conducted differential expression analysis with DESeq2 to identify significant changes in gene expression between conditions.
Exome-seq
We performed Exome sequencing (Exome-seq) to confirm the mutations in the Trp53 and Kras genes and to comprehensively analyze the coding regions of the genome. We extracted genomic DNA from the samples using a standard phenol-chloroform extraction method and quantified it with a Qubit fluorometer. Using the Agilent SureSelect mouse All Exon V5 kit, we captured the exonic regions according to the manufacturer’s protocol. We then subjected the captured libraries to paired-end sequencing on the Illumina NovaSeq 6000 platform. We processed the raw sequence data using the BWA-MEM algorithm to align it to the mouse reference genome (mm10). We performed subsequent variant calling using the GATK Best Practices pipeline and filtered the variants from BAM files using Mutect2. We annotated the identified variants with ANNOVAR and filtered them based on quality metrics, allele frequency, and predicted functional impact. Finally, we used CNVkit to determine the copy number status of the mutated positions from BAM files.
PRO-seq
We adapted precision run-on sequencing (PRO-seq) from a previously published paper107. We prepared tissue culture cells for the nuclear run-on reaction by cell permeabilization. We removed the tissue culture medium, rinsed the cells with PBS, and placed the plates on ice. We then scraped the cells while still on ice, collected them into a 15 ml conical tube, and centrifuged them at 1,000g for 5 minutes. For nuclei isolation, we resuspended the pellet in ice-cold douncing buffer (10 mM Tris-Cl pH 7.4, 300 mM sucrose, 3 mM CaCl2, 2 mM MgCl2, 0.1% Triton X-100, 0.5 mM DTT, 0.1X Halt protease inhibitor, and 0.02 U/μl RNase inhibitor). We transferred it to a 7 ml dounce homogenizer (Wheaton, cat # 357542). After incubating on ice for 5 minutes, we dounced the cells 25 times with a tight pestle, transferred them back to the 15 ml conical tube, and centrifuged to pellet the nuclei. We washed the pellet twice in a douncing buffer. Then we resuspended the washed pellet in storage buffer (10 mM Tris-Cl pH 8.0, 5% glycerol, 5 mM MgCl2, 0.1 mM EDTA, 5 mM DTT, 1× Halt protease inhibitor, and 0.2 U/μl RNase inhibitor) at a concentration of 5 million nuclei per 50 μl of storage buffer. We flash-froze the suspension in liquid nitrogen and stored it at −80°C.
We mixed 10 × 106 nuclei in 100 μl of storage buffer with 100 μl of 2x nuclear run-on buffer (10 mM Tris-HCl pH 8.0, 5 mM MgCl2, 1 mM DTT, 300 mM KCl, 1% Sarkosyl, 50 μM biotin-11-A/C/G/UTP, 0.2 units/μl RNase inhibitor) and incubated at 37°C for three minutes. We extracted total RNA, which includes biotin-labeled nascent RNA, using Trizol following the manufacturer’s protocol. We isolated and base-hydrolyzed the RNAs with a final concentration of 200 nM NaOH to an average size between 100–150 nucleotides. We isolated nascent RNAs with magnetic beads coated with streptavidin, followed by 3’ adapter ligation. After another round of biotin-streptavidin affinity purification, we removed the mRNA cap and ligated the 5’ adapter. Following the third biotin-streptavidin affinity purification, we generated cDNA by reverse transcription. We prepared PRO-seq libraries for sequencing using Illumina TruSeq small-RNA adaptors with twelve cycles of PCR.
To map the PRO-seq sequencing reads, we clipped adapters from PRO-seq reads using Cutadapt. We discarded reads shorter than 15 bp as they map to multiple sites in the genome. We mapped the reads to the mouse genome mm10 using bowtie2, requiring them to map uniquely to the genome and allowing up to 2 mismatches.
Enhancer calling
We identified enhancers in KrasG12D/+;Trp53R172H/− cells as described previously108. We used dREG, a computational tool designed for genome-wide detection of regulatory elements from nascent transcription data, which leverages bidirectional transcription signals from PRO-seq to provide a high-resolution map of enhancers.
CUT&RUN
We performed the Cleavage Under Targets and Release Using Nuclease (CUT&RUN) assay using the CUTANA™ ChIC/CUT&RUN Kit (EpiCypher, 14–1048) according to the manufacturer’s protocol. We prepared CUT&RUN with two different antibodies against p53, NF-κB subunit p65, p300, H3K4me3, and H3K27Ac. We prepared 500,000 cells per reaction and used concanavalin A-coated magnetic beads to capture cells. We added 0.5 ug antibody to permeabilized cells and incubated overnight at 4ºC. We permeabilized the cells after overnight incubation with digitonin and introduced pAG-MNase to the cell-bead complexes to facilitate targeted chromatin digestion. We initiated the digestion by adding calcium chloride and incubating the samples at 4ºC for 2 hours. To stop the reaction, we added EGTA and collected the supernatants containing the CUT&RUN-enriched chromatin. We purified the released DNA fragments using SPRIselect reagent and eluted the DNA. We quantified the eluted DNA using a Qubit fluorometer and prepared sequencing libraries using the NEBNext Ultra II DNA Library Prep Kit (New England Biolabs) according to the manufacturer’s instructions. The resulting libraries were sequenced on an Illumina NextSeq 500.
To map the CUT&RUN sequencing reads, we clipped adapters from CUT&RUN reads using Cutadapt. We discarded reads shorter than 15 bp as they map to multiple sites in the genome. We mapped the reads to the mouse genome mm10 in the --very-sensitive-local mode using bowtie2, requiring them to uniquely map to the genome. We then removed the PCR duplicates using the unique molecular identifiers (UMIs). We subsampled reads in the treatment group to match the reads in the control (IgG) group and called peaks using SEACR_1.3 (https://github.com/FredHutch/SEACR). We generated a union of p53R172H peaks (n= 7,721) from two sets of peaks called with two p53 antibodies (Epicypher p53 antibody and Leica p53 antibody). As a control, we selected 100,000 random regions in the genome matched for p53R172H CUT&RUN peak widths and filtered for expression levels of more than 1 CUT&RUN reads.
We used the Hypergeometric Optimization of Motif EnRichment (HOMER) software to identify enriched DNA sequences within CUT&RUN peaks. We extracted genomic sequences from these peaks and analyzed de novo motif enrichment using the HOMER motif discovery algorithm. As a background control, we generated 100,000 random genomic regions, each 1,000 nucleotides in length.
TPCA-1 treatment
We dissolved 10 mg of TPCA-1 in 1.193 ml of DMSO to create a 30 mM stock solution. We plated cells in 15 cm tissue culture-treated plates and allowed them to adhere. 24 hours after plating them, we treated the cells with TPCA-1 (final concentration of 5 μM). As a control, we treated cells with 0.1% DMSO. We harvested cells 24 hours after the treatment and proceeded with either qRT-PCR or CUT&RUN.
Supplementary Material
ACKNOWLEDGMENTS
This research was funded by Program Project Grant P01-CA042063 from the NCI (P.A.S.), the United States Public Health Service grants R01-GM034277 from the NIH (P.A.S.), R35-CA197743, U01-CA224348, R01-CA259253, R01-CA208205, R01-NS118929, U01-CA261842, and grants from the Ludwig Cancer Center at Harvard, Nile Albright Research Foundation, Jane’s Trust Foundation, National Foundation for Cancer Research to (R.K.J.), NCI R00CA204595-05 and Pew-Stewart Scholarship to (S.S.), and Howard Hughes Medical Institute to (H.M.S.). The Emerald Foundation Postdoctoral Transition Award currently supports D.B.M. The Gertrude B. Elion Research Fellowship from GSK and the Ludwig Cancer Institute at MIT previously supported him. We thank our colleagues Tyler Jacks, William Freed-Pastor, the Late Brandon Horton, Shayla Nguyen, Skyler Kauffman, and Nathan Han for their helpful discussion. We thank the Swanson Biotechnology Center Flow Cytometry Facility for help with FACS and Stuart Levine and the staff at BioMicro Center at the Koch Institute for Integrative Cancer Research for their help with NGS. We also thank the Animal Facility at MGH and the FACS core at the Ragon Institute.
DECLARATION OF INTERESTS
R.K.J. is a Consultant for Accurius, DynamiCure, and SynDevRx; owns equity in Accurius, Enlight, and SynDevRx; served on the Board of Trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, Tekla Healthcare Opportunities Fund, Tekla World Healthcare Fund and received a research Grant from Sanofi. No funding or reagents from these organizations were used in this study. S.S. is an SAB member for Related Sciences, Arcus Biosciences, Ankyra Therapeutics, Prox Bio, and Repertoire Immune Medicines. S.S. is a co-founder of Danger Bio. S.S. is a consultant for TAKEDA and Merck and receives funding for unrelated projects from Leap Therapeutics and iTeos Therapeutics. S.S.’s interests are reviewed and managed under MIT’s policies for potential conflicts of interest.
References:
- 1.Park W., Chawla A., and O’Reilly E.M. (2021). Pancreatic Cancer. Jama 326, 851–862. 10.1001/jama.2021.13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel R.L., Giaquinto A.N., and Jemal A. (2024). Cancer statistics, 2024. CA: A Cancer J. Clin. 74, 12–49. 10.3322/caac.21820. [DOI] [PubMed] [Google Scholar]
- 3.Royal R.E., Levy C., Turner K., Mathur A., Hughes M., Kammula U.S., Sherry R.M., Topalian S.L., Yang J.C., Lowy I., et al. (2010). Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. Journal of immunotherapy (Hagerstown, Md. : 1997) 33, 828–833. 10.1097/cji.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma P., Dirix L., Vos F.Y.F.L.D., Allison J.P., Decoster L., Zaucha R., Park J.O., Vanderwalde A.M., Kataria R.S., Ferro S., et al. (2018). Efficacy and tolerability of tremelimumab in patients with metastatic pancreatic ductal adenocarcinoma. J. Clin. Oncol. 36, 470–470. 10.1200/jco.2018.36.4_suppl.470. [DOI] [Google Scholar]
- 5.O’Reilly E.M., Oh D.-Y., Dhani N., Renouf D.J., Lee M.A., Sun W., Fisher G., Hezel A., Chang S.-C., Vlahovic G., et al. (2019). Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncology 5, 1431–1438. 10.1001/jamaoncol.2019.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brahmer J.R., Tykodi S.S., Chow L.Q.M., Hwu W.-J., Topalian S.L., Hwu P., Drake C.G., Camacho L.H., Kauh J., Odunsi K., et al. (2012). Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. New Engl J Medicine 366, 2455–2465. 10.1056/nejmoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng R., Liu X., Zhang Y., Liu Y., Wang Y., Guo S., Jin X., Zhang J., Guan Y., and Liu Y. (2024). Frontiers and future of immunotherapy for pancreatic cancer: from molecular mechanisms to clinical application. Front. Immunol. 15, 1383978. 10.3389/fimmu.2024.1383978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaufman B., Shapira-Frommer R., Schmutzler R.K., Audeh M.W., Friedlander M., Balmaña J., Mitchell G., Fried G., Stemmer S.M., Hubert A., et al. (2015). Olaparib Monotherapy in Patients With Advanced Cancer and a Germline BRCA1/2 Mutation. J Clin Oncol 33, 244–250. 10.1200/jco.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higuchi T., Flies D.B., Marjon N.A., Mantia-Smaldone G., Ronner L., Gimotty P.A., and Adams S.F. (2015). CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 3, 1257–1268. 10.1158/2326-6066.cir-15-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reiss K.A., Mick R., Teitelbaum U., O’Hara M., Schneider C., Massa R., Karasic T., Tondon R., Onyiah C., Gosselin M.K., et al. (2022). Niraparib plus nivolumab or niraparib plus ipilimumab in patients with platinum-sensitive advanced pancreatic cancer: a randomised, phase 1b/2 trial. Lancet Oncol. 23, 1009–1020. 10.1016/s1470-2045(22)00369-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freed-Pastor W.A., Lambert L.J., Ely Z.A., Pattada N.B., Bhutkar A., Eng G., Mercer K.L., Garcia A.P., Lin L., Rideout W.M., et al. (2021). The CD155/TIGIT axis promotes and maintains immune evasion in neoantigen-expressing pancreatic cancer. Cancer Cell 39, 1342–1360.e14. 10.1016/j.ccell.2021.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giampazolias E., Costa M.P.da, Lam K.C., Lim K.H.J., Cardoso A., Piot C., Chakravarty P., Blasche S., Patel S., Biram A., et al. (2024). Vitamin D regulates microbiome-dependent cancer immunity. Science 384, 428–437. 10.1126/science.adh7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hidalgo M., Semenisty V., Bockorny B., Borazanci E., Hoff D.D.von, Feliu J., Sarvise M.P., Abad D.G., Peled A., Bohana-Kashtan O., et al. (2019). 91O A Multi-Center Phase IIA Trial to Assess the Safety and Efficacy of BL-8040 (a CXCR4 Inhibitor) in Combination with Pembrolizumab and Chemotherapy in Patients with Metastatic Pancreatic Adenocarcinoma (PDAC). Ann. Oncol. 30, xi33. 10.1093/annonc/mdz451. [DOI]
- 14.Zemek R.M., Jong E.D., Chin W.L., Schuster I.S., Fear V.S., Casey T.H., Forbes C., Dart S.J., Leslie C., Zaitouny A., et al. (2019). Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Sci. Transl. Med. 11. 10.1126/scitranslmed.aav7816. [DOI] [PubMed] [Google Scholar]
- 15.Aggarwal R.R., Zhang J., Zhu X., Monk P., Jones R.J., Linch M.D., Costin D., Bono J.S.D., Karsh L.I., Petrylak D.P., et al. (2023). First-in-class oral innate immune activator BXCL701 combined with pembrolizumab in patients with metastatic, castration-resistant prostate cancer (mCRPC) of small cell neuroendocrine (SCNC) phenotype: Phase 2a final results. J. Clin. Oncol. 41, 176–176. 10.1200/jco.2023.41.6_suppl.176. [DOI] [Google Scholar]
- 16.Raphael B.J., Hruban R.H., Aguirre A.J., Moffitt R.A., Yeh J.J., Stewart C., Robertson A.G., Cherniack A.D., Gupta M., Getz G., et al. (2017). Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer cell 32, 185–203.e13. 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu J., Sammons M.A., Donahue G., Dou Z., Vedadi M., Getlik M., Barsyte-Lovejoy D., Alawar R., Katona B.W., Shilatifard A., et al. (2015). Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 525, 206–211. 10.1038/nature15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho Y., Gorina S., Jeffrey P.D., and Pavletich N.P. (1994). Crystal Structure of a p53 Tumor Suppressor-DNA Complex: Understanding Tumorigenic Mutations. Science 265, 346–355. 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 19.Wang Z., Burigotto M., Ghetti S., Vaillant F., Tan T., Capaldo B.D., Palmieri M., Hirokawa Y., Tai L., Simpson D.S., et al. (2024). Loss-of-Function but Not Gain-of-Function Properties of Mutant TP53 Are Critical for the Proliferation, Survival, and Metastasis of a Broad Range of Cancer Cells. Cancer Discov. 14, OF1–OF18. 10.1158/2159-8290.cd-23-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boettcher S., Miller P.G., Sharma R., McConkey M., Leventhal M., Krivtsov A.V., Giacomelli A.O., Wong W., Kim J., Chao S., et al. (2019). A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365, 599–604. 10.1126/science.aax3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willis A., Jung E.J., Wakefield T., and Chen X. (2004). Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 23, 2330–2338. 10.1038/sj.onc.1207396. [DOI] [PubMed] [Google Scholar]
- 22.Shaulsky G., Goldfinger N., and Rotter V. (1991). Alterations in tumor development in vivo mediated by expression of wild type or mutant p53 proteins. Cancer Res. 51, 5232–5237. [PubMed] [Google Scholar]
- 23.Olive K.P., Tuveson D.A., Ruhe Z.C., Yin B., Willis N.A., Bronson R.T., Crowley D., and Jacks T. (2004). Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860. 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 24.Lang G.A., Iwakuma T., Suh Y.-A., Liu G., Rao V.A., Parant J.M., Valentin-Vega Y.A., Terzian T., Caldwell L.C., Strong L.C., et al. (2004). Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119, 861–872. 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 25.Cooks T., Pateras I.S., Tarcic O., Solomon H., Schetter A.J., Wilder S., Lozano G., Pikarsky E., Forshew T., Rozenfeld N., et al. (2013). Mutant p53 Prolongs NF-κB Activation and Promotes Chronic Inflammation and Inflammation-Associated Colorectal Cancer. Cancer cell 23, 634–646. 10.1016/j.ccr.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexandrova E.M., Yallowitz A.R., Li D., Xu S., Schulz R., Proia D.A., Lozano G., Dobbelstein M., and Moll U.M. (2015). Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 523, 352–356. 10.1038/nature14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alexandrova E.M., Mirza S.A., Xu S., Schulz-Heddergott R., Marchenko N.D., and Moll U.M. (2017). p53 loss-of-heterozygosity is a necessary prerequisite for mutant p53 stabilization and gain-of-function in vivo. Cell Death Dis. 8, e2661–e2661. 10.1038/cddis.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanel W., Marchenko N., Xu S., Yu S.X., Weng W., and Moll U. (2013). Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell death and differentiation 20, 898–909. 10.1038/cdd.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu J., Reumers J., Couceiro J.R., Smet F.D., Gallardo R., Rudyak S., Cornelis A., Rozenski J., Zwolinska A., Marine J.-C., et al. (2011). Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 7, 285–295. 10.1038/nchembio.546. [DOI] [PubMed] [Google Scholar]
- 30.Kim M.P., Li X., Deng J., Zhang Y., Dai B., Allton K.L., Hughes T.G., Siangco C., Augustine J.J., Kang Y., et al. (2021). Oncogenic KRAS Recruits an Expansive Transcriptional Network through Mutant p53 to Drive Pancreatic Cancer Metastasis. Cancer Discov. 11, 2094–2111. 10.1158/2159-8290.cd-20-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agostino S.D., Strano S., Emiliozzi V., Zerbini V., Mottolese M., Sacchi A., Blandino G., and Piaggio G. (2006). Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 10, 191–202. 10.1016/j.ccr.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 32.Inga A., Monti P., Fronza G., Darden T., and Resnick M.A. (2001). p53 mutants exhibiting enhanced transcriptional activation and altered promoter selectivity are revealed using a sensitive, yeast-based functional assay. Oncogene 20, 501–513. 10.1038/sj.onc.1204116. [DOI] [PubMed] [Google Scholar]
- 33.Adorno M., Cordenonsi M., Montagner M., Dupont S., Wong C., Hann B., Solari A., Bobisse S., Rondina M.B., Guzzardo V., et al. (2009). A Mutant p53/Smad Complex Opposes p63 to Empower TGFβ-Induced Metastasis. Cell 137, 87–98. 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 34.Mello S.S., and Attardi L.D. (2013). Not all p53 gain-of-function mutants are created equal. Cell Death Differ. 20, 855–857. 10.1038/cdd.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amelio I., and Melino G. (2020). Context is everything: extrinsic signalling and gain-of-function p53 mutants. Cell Death Discov. 6, 16. 10.1038/s41420-020-0251-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oren M., and Prives C. (2024). p53: A tale of complexity and context. Cell 187, 1569–1573. 10.1016/j.cell.2024.02.043. [DOI] [PubMed] [Google Scholar]
- 37.Baugh E.H., Ke H., Levine A.J., Bonneau R.A., and Chan C.S. (2018). Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ 25, 154–160. 10.1038/cdd.2017.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hassin O., Nataraj N.B., Shreberk-Shaked M., Aylon Y., Yaeger R., Fontemaggi G., Mukherjee S., Maddalena M., Avioz A., Iancu O., et al. (2022). Different hotspot p53 mutants exert distinct phenotypes and predict outcome of colorectal cancer patients. Nat. Commun. 13, 2800. 10.1038/s41467-022-30481-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orth M., Metzger P., Gerum S., Mayerle J., Schneider G., Belka C., Schnurr M., and Lauber K. (2019). Pancreatic ductal adenocarcinoma: biological hallmarks, current status, and future perspectives of combined modality treatment approaches. Radiat Oncol Lond Engl 14, 141. 10.1186/s13014-019-1345-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maddalena M., Mallel G., Nataraj N.B., Shreberk-Shaked M., Hassin O., Mukherjee S., Arandkar S., Rotkopf R., Kapsack A., Lambiase G., et al. (2021). TP53 missense mutations in PDAC are associated with enhanced fibrosis and an immunosuppressive microenvironment. Proc National Acad Sci 118, e2025631118. 10.1073/pnas.2025631118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balsano R., Zanuso V., Pirozzi A., Rimassa L., and Bozzarelli S. (2023). Pancreatic Ductal Adenocarcinoma and Immune Checkpoint Inhibitors: The Gray Curtain of Immunotherapy and Spikes of Lights. Curr. Oncol. 30, 3871–3885. 10.3390/curroncol30040293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koikawa K., Kibe S., Suizu F., Sekino N., Kim N., Manz T.D., Pinch B.J., Akshinthala D., Verma A., Gaglia G., et al. (2021). Targeting Pin1 renders pancreatic cancer eradicable by synergizing with immunochemotherapy. Cell 184, 4753–4771.e27. 10.1016/j.cell.2021.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siolas D., Vucic E., Kurz E., Hajdu C., and Bar-Sagi D. (2021). Gain-of-function p53R172H mutation drives accumulation of neutrophils in pancreatic tumors, promoting resistance to immunotherapy. Cell Reports 36, 109578. 10.1016/j.celrep.2021.109578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Terzian T., Suh Y.-A., Iwakuma T., Post S.M., Neumann M., Lang G.A., Pelt C.S.V., and Lozano G. (2008). The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 22, 1337–1344. 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suh Y.-A., Post S.M., Elizondo-Fraire A.C., Maccio D.R., Jackson J.G., El-Naggar A.K., Pelt C.V., Terzian T., and Lozano G. (2011). Multiple Stress Signals Activate Mutant p53 In Vivo. Cancer Res. 71, 7168–7175. 10.1158/0008-5472.can-11-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sullivan K.D., Galbraith M.D., Andrysik Z., and Espinosa J.M. (2018). Mechanisms of transcriptional regulation by p53. Cell Death Differ. 25, 133–143. 10.1038/cdd.2017.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X., Choi P.S., Francis J.M., Gao G.F., Campbell J.D., Ramachandran A., Mitsuishi Y., Ha G., Shih J., Vazquez F., et al. (2017). Somatic super-enhancer duplications and hotspot mutations lead to oncogenic activation of the KLF5 transcription factor. Cancer Discov. 8, CD-17–0532. 10.1158/2159-8290.cd-17-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basil P., Robertson M.J., Bingman W.E., Dash A.K., Krause W.C., Shafi A.A., Piyarathna B., Coarfa C., and Weigel N.L. (2022). Cistrome and transcriptome analysis identifies unique androgen receptor (AR) and AR-V7 splice variant chromatin binding and transcriptional activities. Sci. Rep. 12, 5351. 10.1038/s41598-022-09371-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiang Y.-T., Chien Y.-C., Lin Y.-H., Wu H.-H., Lee D.-F., and Yu Y.-L. (2021). The Function of the Mutant p53-R175H in Cancer. Cancers 13, 4088. 10.3390/cancers13164088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hingorani S.R., Wang L., Multani A.S., Combs C., Deramaudt T.B., Hruban R.H., Rustgi A.K., Chang S., and Tuveson D.A. (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer cell 7, 469–483. 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 51.Donehower L.A., Soussi T., Korkut A., Liu Y., Schultz A., Cardenas M., Li X., Babur O., Hsu T.-K., Lichtarge O., et al. (2019). Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 28, 1370–1384.e5. 10.1016/j.celrep.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacks T., Remington L., Williams B.O., Schmitt E.M., Halachmi S., Bronson R.T., and Weinberg R.A. (1994). Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 4, 1–7. 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 53.Gopinathan A., Morton J.P., Jodrell D.I., and Sansom O.J. (2015). GEMMs as preclinical models for testing pancreatic cancer therapies. Dis. Model. Mech. 8, 1185–1200. 10.1242/dmm.021055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morton J.P., Timpson P., Karim S.A., Ridgway R.A., Athineos D., Doyle B., Jamieson N.B., Oien K.A., Lowy A.M., Brunton V.G., et al. (2010). Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc National Acad Sci 107, 246–251. 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fischer M. (2017). Census and evaluation of p53 target genes. Oncogene 36, 3943–3956. 10.1038/onc.2016.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Giuliano S., Guyot M., Grépin R., and Pagès G. (2014). The ELR+CXCL chemokines and their receptors CXCR1/CXCR2. OncoImmunology 3, e28399. 10.4161/onci.28399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deng Y., Xia X., Zhao Y., Zhao Z., Martinez C., Yin W., Yao J., Hang Q., Wu W., Zhang J., et al. (2021). Glucocorticoid receptor regulates PD-L1 and MHC-I in pancreatic cancer cells to promote immune evasion and immunotherapy resistance. Nat. Commun. 12, 7041. 10.1038/s41467-021-27349-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bardeesy N., Aguirre A.J., Chu G.C., Cheng K., Lopez L.V., Hezel A.F., Feng B., Brennan C., Weissleder R., Mahmood U., et al. (2006). Both p16Ink4a and the p19Arf-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. 103, 5947–5952. 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanford D.E., Belt B.A., Panni R.Z., Mayer A., Deshpande A.D., Carpenter D., Mitchem J.B., Plambeck-Suess S.M., Worley L.A., Goetz B.D., et al. (2013). Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin. Cancer Res. 19, 3404–3415. 10.1158/1078-0432.ccr-13-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gschwandtner M., Derler R., and Midwood K.S. (2019). More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front Immunol 10, 2759. 10.3389/fimmu.2019.02759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kalbasi A., Komar C., Tooker G.M., Liu M., Lee J.W., Gladney W.L., Ben-Josef E., and Beatty G.L. (2017). Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 23, 137–148. 10.1158/1078-0432.ccr-16-0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nywening T.M., Belt B.A., Cullinan D.R., Panni R.Z., Han B.J., Sanford D.E., Jacobs R.C., Ye J., Patel A.A., Gillanders W.E., et al. (2018). Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 67, 1112. 10.1136/gutjnl-2017-313738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Metelitsa L.S., Wu H.-W., Wang H., Yang Y., Warsi Z., Asgharzadeh S., Groshen S., Wilson S.B., and Seeger R.C. (2004). Natural Killer T Cells Infiltrate Neuroblastomas Expressing the Chemokine CCL2. J. Exp. Med. 199, 1213–1221. 10.1084/jem.20031462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morrison B.E., Park S.J., Mooney J.M., and Mehrad B. (2003). Chemokine-mediated recruitment of NK cells is a critical host defense mechanism in invasive aspergillosis. J. Clin. Investig. 112, 1862–1870. 10.1172/jci18125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Korbecki J., Barczak K., Gutowska I., Chlubek D., and Baranowska-Bosiacka I. (2022). CXCL1: Gene, Promoter, Regulation of Expression, mRNA Stability, Regulation of Activity in the Intercellular Space. Int J Mol Sci 23, 792. 10.3390/ijms23020792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Korbecki J., Kupnicka P., Chlubek M., Gorący J., Gutowska I., and Baranowska-Bosiacka I. (2022). CXCR2 Receptor: Regulation of Expression, Signal Transduction, and Involvement in Cancer. Int J Mol Sci 23, 2168. 10.3390/ijms23042168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bruni D., Angell H.K., and Galon J. (2020). The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 20, 662–680. 10.1038/s41568-020-0285-7. [DOI] [PubMed] [Google Scholar]
- 68.Fritz J.H., and Stoiber D. (2022). Editorial: Innate lymphocytes in tumor surveillance. Front. Immunol. 13, 1093318. 10.3389/fimmu.2022.1093318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Skene P.J., and Henikoff S. (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, e21856. 10.7554/elife.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim T.-K., Hemberg M., Gray J.M., Costa A.M., Bear D.M., Wu J., Harmin D.A., Laptewicz M., Barbara-Haley K., Kuersten S., et al. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187. 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Danko C.G., Hyland S.L., Core L.J., Martins A.L., Waters C.T., Lee H.W., Cheung V.G., Kraus W.L., Lis J.T., and Siepel A. (2015). Identification of active transcriptional regulatory elements from GRO-seq data. Nature Publishing Group 12, 433–438. 10.1038/nmeth.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morgan M.A.J., and Shilatifard A. (2020). Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 52, 1271–1281. 10.1038/s41588-020-00736-4. [DOI] [PubMed] [Google Scholar]
- 73.Tippens N.D., Liang J., Leung A.K.-Y., Wierbowski S.D., Ozer A., Booth J.G., Lis J.T., and Yu H. (2020). Transcription imparts architecture, function and logic to enhancer units. Nat Genet 52, 1067–1075. 10.1038/s41588-020-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu T., Zhang L., Joo D., and Sun S.-C. (2017). NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2, 17023. 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lappalainen T., and MacArthur D.G. (2021). From variant to function in human disease genetics. Science 373, 1464–1468. 10.1126/science.abi8207. [DOI] [PubMed] [Google Scholar]
- 76.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.-S., Domm J., Eustace B.K., Foell J., Fuente J. de la, Grupp, S., Handgretinger, R., et al. (2020). CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 384, 252–260. 10.1056/nejmoa2031054. [DOI] [PubMed] [Google Scholar]
- 77.Korbecki J., Bosiacki M., Barczak K., Łagocka R., Chlubek D., and Baranowska-Bosiacka I. (2023). The Clinical Significance and Role of CXCL1 Chemokine in Gastrointestinal Cancers. Cells 12, 1406. 10.3390/cells12101406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yan W., and Chen X. (2009). Identification of GRO1 as a Critical Determinant for Mutant p53 Gain of Function*. J Biol Chem 284, 12178–12187. 10.1074/jbc.m900994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li J., Byrne K.T., Yan F., Yamazoe T., Chen Z., Baslan T., Richman L.P., Lin J.H., Sun Y.H., Rech A.J., et al. (2018). Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 49, 178–193.e7. 10.1016/j.immuni.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Verzella D., Cornice J., Arboretto P., Vecchiotti D., Nolfi M.D.V., Capece D., Zazzeroni F., and Franzoso G. (2022). The NF-κB Pharmacopeia: Novel Strategies to Subdue an Intractable Target. Biomedicines 10, 2233. 10.3390/biomedicines10092233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ijichi H., Chytil A., Gorska A.E., Aakre M.E., Bierie B., Tada M., Mohri D., Miyabayashi K., Asaoka Y., Maeda S., et al. (2011). Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest 121, 4106–4117. 10.1172/jci42754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dimitrakopoulos F.-I.D., Kottorou A.E., Kalofonou M., and Kalofonos H.P. (2020). The Fire Within: NF-κB Involvement in Non–Small Cell Lung Cancer. Cancer Res. 80, 4025–4036. 10.1158/0008-5472.can-19-3578. [DOI] [PubMed] [Google Scholar]
- 83.Aggarwal B.B., and Sung B. (2011). NF-κB in Cancer: A Matter of Life and Death. Cancer Discov. 1, 469–471. 10.1158/2159-8290.cd-11-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ahn J.H., Davis E.S., Daugird T.A., Zhao S., Quiroga I.Y., Uryu H., Li J., Storey A.J., Tsai Y.-H., Keeley D.P., et al. (2021). Phase separation drives aberrant chromatin looping and cancer development. Nature, 1–5. 10.1038/s41586-021-03662-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leroy K., Haioun C., Lepage E., Métayer N.L., Berger F., Labouyrie E., Meignin V., Petit B., Bastard C., Salles G., et al. (2002). p53 gene mutations are associated with poor survival in low and low-intermediate risk diffuse large B-cell lymphomas. Ann. Oncol. 13, 1108–1115. 10.1093/annonc/mdf185. [DOI] [PubMed] [Google Scholar]
- 86.Chen X., Zhang T., Su W., Dou Z., Zhao D., Jin X., Lei H., Wang J., Xie X., Cheng B., et al. (2022). Mutant p53 in cancer: from molecular mechanism to therapeutic modulation. Cell Death Dis. 13, 974. 10.1038/s41419-022-05408-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Peuget S., Zhou X., and Selivanova G. (2024). Translating p53-based therapies for cancer into the clinic. Nat. Rev. Cancer, 1–24. 10.1038/s41568-023-00658-3. [DOI] [PubMed]
- 88.Weisz L., Damalas A., Liontos M., Karakaidos P., Fontemaggi G., Maor-Aloni R., Kalis M., Levrero M., Strano S., Gorgoulis V.G., et al. (2007). Mutant p53 Enhances Nuclear Factor κB Activation by Tumor Necrosis Factor α in Cancer Cells. Cancer research 67, 2396–2401. 10.1158/0008-5472.can-06-2425. [DOI] [PubMed] [Google Scholar]
- 89.Freed-Pastor W.A., and Prives C. (2012). Mutant p53: one name, many proteins. Genes & Development 26, 1268–1286. 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bianchi A., Silva I.D.C., Deshpande N.U., Singh S., Mehra S., Garrido V.T., Guo X., Nivelo L.A., Kolonias D.S., Saigh S.J., et al. (2023). Cell-autonomous Cxcl1 sustains tolerogenic circuitries and stromal inflammation via neutrophil-derived TNF in pancreatic cancer. Cancer Discov. 10.1158/2159-8290.cd-22-1046. [DOI] [PMC free article] [PubMed]
- 91.Webster G.A., and Perkins N.D. (1999). Transcriptional Cross Talk between NF-κB and p53. Mol. Cell. Biol. 19, 3485–3495. 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schneider G., Henrich A., Greiner G., Wolf V., Lovas A., Wieczorek M., Wagner T., Reichardt S., Werder A. von, Schmid, R.M., et al. (2010). Cross talk between stimulated NF-κB and the tumor suppressor p53. Oncogene 29, 2795–2806. 10.1038/onc.2010.46. [DOI] [PubMed] [Google Scholar]
- 93.Rahnamoun H., Lu H., Duttke S.H., Benner C., Glass C.K., and Lauberth S.M. (2017). Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nature communications 8, 754. 10.1038/s41467-017-01117-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Man X., Yang X., Wei Z., Tan Y., Li W., Jin H., and Wang B. (2022). High expression level of CXCL1/GROα is linked to advanced stage and worse survival in uterine cervical cancer and facilitates tumor cell malignant processes. BMC Cancer 22, 712. 10.1186/s12885-022-09749-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yu S., Yi M., Xu L., Qin S., Li A., and Wu K. (2020). CXCL1 as an Unfavorable Prognosis Factor Negatively Regulated by DACH1 in Non-small Cell Lung Cancer. Front. Oncol. 9, 1515. 10.3389/fonc.2019.01515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Korbecki J., Bosiacki M., Barczak K., Łagocka R., Brodowska A., Chlubek D., and Baranowska-Bosiacka I. (2023). Involvement in Tumorigenesis and Clinical Significance of CXCL1 in Reproductive Cancers: Breast Cancer, Cervical Cancer, Endometrial Cancer, Ovarian Cancer and Prostate Cancer. Int. J. Mol. Sci. 24, 7262. 10.3390/ijms24087262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang N., Liu W., Zheng Y., Wang S., Yang B., Li M., Song J., Zhang F., Zhang X., Wang Q., et al. (2018). CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-κB/SOX4 signaling. Cell Death Dis. 9, 880. 10.1038/s41419-018-0876-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhuo C., Ruan Q., Zhao X., Shen Y., and Lin R. (2022). CXCL1 promotes colon cancer progression through activation of NF-κB/P300 signaling pathway. Biol. Direct 17, 34. 10.1186/s13062-022-00348-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cao Z., Fu B., Deng B., Zeng Y., Wan X., and Qu L. (2014). Overexpression of Chemokine (C-X-C) ligand 1 (CXCL1) associated with tumor progression and poor prognosis in hepatocellular carcinoma. Cancer Cell Int. 14, 86. 10.1186/s12935-014-0086-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hu J., Zhao Q., Kong L.-Y., Wang J., Yan J., Xia X., Jia Z., Heimberger A.B., and Li S. (2021). Regulation of tumor immune suppression and cancer cell survival by CXCL1/2 elevation in glioblastoma multiforme. Sci. Adv. 7, eabc2511. 10.1126/sciadv.abc2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., and Ma’ayan A. (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 14, 128. 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kanehisa M., Furumichi M., Sato Y., Kawashima M., and Ishiguro-Watanabe M. (2022). KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592. 10.1093/nar/gkac963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Han H., Cho J.-W., Lee S., Yun A., Kim H., Bae D., Yang S., Kim C.Y., Lee M., Kim E., et al. (2018). TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 46, D380–D386. 10.1093/nar/gkx1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., and Mesirov J.P. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., and Glass C.K. (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bankhead P., Loughrey M.B., Fernández J.A., Dombrowski Y., McArt D.G., Dunne P.D., McQuaid S., Gray R.T., Murray L.J., Coleman H.G., et al. (2017). QuPath: Open source software for digital pathology image analysis. Sci. Rep. 7, 16878. 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mahat D.B., Kwak H., Booth G.T., Jonkers I.H., Danko C.G., Patel R.K., Waters C.T., Munson K., Core L.J., and Lis J.T. (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nature Protocols 11, 1455–1476. 10.1038/nprot.2016.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mahat D.B., Tippens N.D., Martin-Rufino J.D., Waterton S.K., Fu J., Blatt S.E., and Sharp P.A. (2024). Single-cell nascent RNA sequencing unveils coordinated global transcription. Nature 631, 216–223. 10.1038/s41586-024-07517-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq, PRO-seq, Exome-seq, and CUT&RUN data have been deposited in GEO and will be publicly available as of the date of publication. Accession numbers and secure tokens for reviewer access are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse/human p1253 | Epicypher | SKU: 13-2015 |
| p53 (1C12) Mouse mAb | Cell Signaling | #2524 |
| anti-mouse/human p53 | Leica Biosystems | P53-DO7-L-CE |
| anti-mouse CD16/32;clone 93 | BioLegend | Cat# 101330;AB_2561482 |
| anti-mouse CD25 (APC-Cy7);clone PC61 | BioLegend | Cat# 102026;AB_830745 |
| anti-mouse CD279 (PD-1) (BV605);clone 29F.1A12 | BioLegend | Cat# 135220;AB_2562616 |
| anti-mouse CD3 (eFluor 450);clone 17A2 | eBioscience | Cat# 48-0032-80;AB_1272229 |
| anti-mouse CD366 (TIM3) (PE-Cy7);clone RMT3-23 | eBioscience | Cat# 25-5870-82;AB_2573483 |
| anti-mouse CD4 (BUV737);clone RM4-5 | BD Biosciences | Cat# 612844;AB_2732918 |
| anti-mouse CD45 (BUV395);clone 30-F11 | BD Biosciences | Cat# 564279;AB_2651134 |
| anti-mouse CD62L (BV711);clone MEL-14 | BioLegend | Cat# 104445;AB_2564215 |
| anti-mouse CD69 (APC);clone H1.2F3 | BioLegend | Cat# 104513;AB_492844 |
| anti-mouse CD8a (BV605);clone 53-6.7 | BioLegend | Cat# 100744;AB_2562609 |
| anti-mouse FoxP3 (PerCP-Cy5.5);clone FJK-16s | eBioscience | Cat# 45-5773-80;AB_914349 |
| anti-mouse/human CD11b (PE/Dazzle 594);clone M1/70 | BioLegend | Cat# 101255;AB_2563647 |
| anti-mouse/human CD44 (PE);clone IM7 | BioLegend | Cat# 103023;AB_493686 |
| anti-mouse/human Gzmb (AF700);clone QA16A02 | BioLegend | Cat# 372221;AB_2728388 |
| anti-mouse/human TCF1 (AF488);clone C63D9 | Cell Signaling Technology | Cat# 6444S |
| anti-Ly-6G (clone 18A) PerCP/Cyanine5.5 | BioLegend | Cat# 127615 |
| anti-CD90.2 (clone 30-H12) FITC | BioLegend | Cat# 105306 |
| anti-CX3CR1 (clone SA011F11) BV650 | BioLegend | Cat# 14903 |
| anti-NK1.1 (clone PK136) BV605 | BioLegend | Cat# 108753 |
| anti-Folate Receptor 2 (clone 10/FR2) APC | BioLegend | Cat# 153305 |
| anti-I-A/I-E (clone M5/114.15.2) BV711 | BioLegend | Cat# 107643 |
| anti-CD206 (clone C068C2) PE/Cyanine7 | BioLegend | Cat# 141719 |
| anti-CD64 (clone X54-5/7.1) PE/Dazzle | BioLegend | Cat# 139319 |
| anti-Ly6C (clone HK1.4) BV785 | BioLegend | Cat# 128041 |
| Streptavidin-PE | BioLegend | Cat# 405203 |
| anti-TIM-4 (clone RMT4-54) BV421 | BD Biosciences | Cat# 744874 |
| anti-CD200R3 (clone Ba13) BUV805 | BD Biosciences | Cat# 752653 |
| anti-CD45 (clone 30-F11) BUV395 | BD Biosciences | Cat# 565967 |
| anti-Siglec-F (clone E50-2440) APC-R700 | BD Biosciences | Cat# 565183 |
| anti-FCeR1a (clone MAR-1) BV750 | BD Biosciences | Cat# 751756 |
| anti-CD19 (clone 1D3) BUV615 | BD Biosciences | Cat# 751213 |
| anti-CD11c (clone N418) BUV737 | BD Biosciences | Cat# 749039 |
| anti-CD103 (clone M290) BUV563 | BD Biosciences | Cat# 741261 |
| anti-CD11b (clone M1/70) APC-eFluor 780 | Invitrogen | Cat# 47-0112-80 |
| anti-CD16/CD32 (clone 2.4G2) | Bio X Cell | Cat# BP0307 |
| Lyve1 Biotin (clone ALY7) 13-0443-82 | Thermo Scientific | Cat# |
| anti-CD8 alpha, clone: YTS169.4 | Abcam | Cat# ab22378 |
| anti-Neutrophil | Abcam | Cat# ab2557 |
| anti-Arginase-1 | Cell Signaling Technology | Cat# 93668T |
| VECTASHIELD Vibrance® Antifade Mounting Medium | VECTOR Laboratories | Cat# H-1700 |
| Chemicals, peptides, and recombinant proteins | ||
| Fixable Viability Dye eFluor 506 | eBioscience | Cat# 65-0866-18 |
| Fixable Viability Dye eFluor 780 | eBioscience | Cat# 65-0865-14 |
| DMEM | Gibco | Cat# 11995 |
| Heat-inactivated fetal bovine serum | Tissue Culture Biologics | Cat# 101HI |
| Tissue culture-treated 10 cm plates | Corning | Cat# CLS430167 |
| Trypsin-EDTA | Gibco | Cat# 25200 |
| α-PD-1 | BioXcell | Cat# BE0146 |
| α-CTLA4 | BioXcell | Cat# 9D9 |
| Collagenase IV | Thermo Scientific | Cat# 17104019 |
| DNase I, Grade II, from bovine pancreas | Sigma-Aldrich | Cat# 10104159001 |
| Liberase TL Research Grade | Sigma-Aldrich | Cat# 05401020001 |
| Lipofectamine 2000 transfection reagent | Thermo Scientific | Cat# 18324012 |
| PureFection | System Bioscience | Cat# LV750A |
| T4 Polynucleotide Kinase | New England Biolabs | Cat# M0201 |
| Tango Buffer | Thermo Scientific | Cat# BY5 |
| FastDigest BbsI restriction enzyme | Thermo Scientific | Cat# FD1014 |
| T7 DNA Ligase | New England Biolabs | Cat# M0318 |
| Alkaline Phosphatase | Thermo Scientific | Cat# EF0654 |
| ACK Lysing Buffer | Thermo Scientific | Cat# A1049201 |
| Plasmid-Safe ATP-Dependent DNase | Biosearch Technologies | Cat# E3101K |
| QIAprep Spin Miniprep Kit | Qiagen | Cat# 27104 |
| S.O.C. Medium | Thermo Scientific | Cat# 15544034 |
| Carbenicillin Disodium Salt | Thermo Scientific | Cat# 10177012 |
| anti-mouse CXCL1 | Leinco Technologies | Cat# K125 |
| anti-mouse CCL2 | Bio-Techne | Cat# MAB479 |
| Recombinant Mouse CXCL1 | Bio-Techne | Cat# 453-KC |
| Recombinant Mouse CCL2 | Bio-Techne | Cat# 452-M2 |
| anti-Mouse CXCL1- Biotin | Leinco Technologies | Cat# K124 |
| anti-Mouse CXCL1- Biotin | Bio-Techne | Cat# BAF479 |
| HRP Streptavidin | BioLegend | Cat# 405210 |
| TMB Substrate Solution | BioLegend | Cat# 421101 |
| Tissue-Tek® O.C.T. Compound | Sakura Finetek | Cat# 62550 |
| Antigen Unmasking Solution | VECTOR Laboratories | Cat# H-3300 |
| Stop Solution | BioLegend | Cat# 423001 |
| Critical commercial assays | ||
| FoxP3 Transcription Factor Staining Buffer Set | eBioscience | Cat# 00-5523-00 |
| Chemically competent Stbl3 E. coli cells | Thermo Scientific | Cat# C737303 |
| One Shot TOP10 Chemically Competent E. coli cells | Thermo Scientific | Cat# C404006 |
| Cytofix/Cytoperm Fixation/Permeabilization buffer | BD Biosciences | Cat# 554714 |
| Precision Count Beads | BioLegend | Cat# 424902 |
| UltraComp eBeads Compensation Beads | Invitrogen | Cat# 01-2222-42 |
| Mouse Cytokine Array C3 | RayBiotech | Cat# AAM-CYT-3 |
| Mouse LIX/ CXCL5 ELISA kit | Thermo Scientific | Cat# EMCXCL5 |
| 96-well ELISA plates | BioLegend | Cat# 423501 |
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4368577 |
| ChIC/CUT&RUN Kit | EpiCypher | Cat# 14-1048 |
| TPCA-1 | Selleckchem | Cat# S2824 |
| Deposited data | ||
| RNA-seq | This study | GSE275105 Secure token: ujstwuygpfsbpkz |
| PRO-seq | This study | GSE275106 Secure token: ytwtwwqkzvgjtgp |
| Exome-seq | This study | GSE275107 Secure token: sbcrgsaqndcplgx |
| CUT&RUN | This study | GSE275108 Secure token: ctcryaimjtefzav |
| Experimental models: Cell lines | ||
| KrasG12D/+;Trp53R172H/− | Dr. Ashok Saluja lab, University of Miami | N/A |
| KrasG12D/+;Trp53−/− (syngenic) | Dr. Ronald DePinho lab, MD Anderson Cancer Center |
N/A |
| KrasG12D/+;Trp53−/− (isogenic) | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Ccl2−/− | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Ccl2−/−;Cxcl1−/− | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Ccl2−/−;Cxcl5−/− | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Cxcl1−/− | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Cxcl5−/− | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Δe8695 | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Δe8695;Δe8696 | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Δe8696 | This study | N/A |
| KrasG12D/+;Trp53R172H/−;Δe8697 | This study | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | Jackson Laboratory | Strain# 000664 |
| Plasmids | ||
| PB-CMV-MCS-EF1α-Puro cDNA cloning and expression vector | System Biosciences | Cat# PB510B-1 |
| Trp53 (NM_011640) Mouse Untagged Clone | Origene | Cat# MC205636 |
| pU6-(BbsI)_CBh-Cas9-T2A-BFP (gift from Ralf Kuehn) | Addgene | Cat# 64323 |
| Oligonucleotides | ||
| See Table_1_Primer_Oligos.xlsx | ||
| Software and algorithms | ||
| FlowJo v10.8.1 | BD Biosciences | https://www.flowjo.com |
| Prism v10.3.0 | Graphpad | https://www.graphpad.com/features |
| CRISPick Design Tool | Broad Institute’s Genetic Perturbation Platform | https://portals.broadinstitute.org/gppx/crispick/public |
| Gene Set Enrichment Analysis (GSEA) | Subramanian et al., 2005 | http://www.broad.mit.edu/gsea/downloads.jsp |
| ImageJ Fiji | ImageJ | https://imagej.net/ij/ |
| QuPath software v.0.5.1 | QuPath | https://qupath.github.io/ |
| Adobe Illustrator CC2023 | Adobe | https://www.adobe.com |
| Bowtie2 v2.2.9 | Langmead et al., 2019 | https://github.com/BenLangmead/bowtie2 |
| Picard v2.8.0 | Broad Institute | https://github.com/broadinstitute/picard |
| SAMtools v0.1.19 | Danecek et al.57 | https://github.com/samtools/samtools |
| R v4.3.3 | R Foundation | https://cran.r-project.org/bin/ |
| DESeq2 v1.40.2 | Love et al.59 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ggplot2 v3.5.0 | Hadley Wickham | https://cran.r-project.org/web/packages/ggplot2/index.html |
| msigdbr v7.5.1 | Igor Dolgalev | https://cran.r-project.org/web/packages/msigdbr/index.html |
| clusterProfiler v4.8.3 | Yu et al., 2021 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| Enrichr | Kuleshov et al., 2016 | http://amp.pharm.mssm.edu/Enrichr/ |
| UCSC Genome Browser | Kent et al. 2002 | https://genome.ucsc.edu/ |
| HOMER (version v4.11.1) | Heinz et al. | http://homer.ucsd.edu/homer/index.html |
| SAMtools (version 1.9) | Danecek et al. | https://www.htslib.org/doc/1.9/samtools.html |
| SEACR | Meers et al. | https://github.com/FredHutch/SEACR?tab=readme-ov-file |
RNA-seq - GSE275105. Secure token: ujstwuygpfsbpkz
PRO-seq - GSE275106. Secure token: ytwtwwqkzvgjtgp
Exome-seq - GSE275107. Secure token: sbcrgsaqndcplgx
CUT&RUN - GSE275108. Secure token: ctcryaimjtefzav
Table_1_Primer_Oligos.xlsx provides the primers and DNA oligos used to clone the guide RNA sequence in the Cas9 and gRNA-expressing plasmids.
The lead contact can provide any additional information required to reanalyze the data reported in this paper upon request.