

Abstract
Introduction
Hemophagocytic lymphohistiocytosis characterized by hemophagocytosis leading to uncontrolled inflammation; the most common etiology in secondary cases of hemophagocytic lymphohistiocytosis is viral infections, especially Epstein–Barr virus. Visceral leishmaniasis is a vectorborne protozoal disease caused by Leishmania donovani complex. It is common in tropical and subtropical regions, with 50,000–90,000 new cases annually.
Case presentation
A 15-month-old Arab female was admitted to our hospital with 15 days of fever and decreased weight. On clinical examination, she had a markedly enlarged liver and spleen that were palpable 4 cm and 6 cm below the costal margin, respectively. The peripheral blood smear showed hypochromic microcytic anemia, poikilocytosis, reactive lymphocytosis, and mild thrombocytopenia. Bone marrow aspiration did not show malignancy or any other pathological findings. The patient was put on antibiotic therapy without improvement. Repeated bone marrow aspiration showed erythrophagocytosis; intracellular small round organisms looked like the amastigote form of Leishmania (Donovan bodies) with no evidence of malignancies. Her lab values showed ferritin greater than 500 ug/L, pancytopenia, and hypertriglyceridemia. The patient was diagnosed with hemophagocytic lymphohistiocytosis secondary to visceral leishmaniasis.
Conclusion
Hemophagocytic lymphohistiocytosis secondary to visceral leishmaniasis is an extensively rare phenomenon in the medical literature that causes challenges in diagnosis and management. Steroids should be used wisely to not cover the symptoms of infections or malignancy, and amphotericin B resistance should be kept in mind in unresponsive Leishmania cases.
Keywords: Visceral leishmaniasis, Hemophagocytic lymphohistiocytosis, Case report
Background
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome characterized by hemophagocytosis leading to uncontrolled inflammation. HLH could be familial or secondary to severe infections, malignancy, or rheumatologic diseases. Viral infections are the most common triggers in secondary HLH, and most commonly with Epstein–Barr virus [1].
Visceral leishmaniasis (VL) is a vectorborne protozoal disease caused by Leishmania donovani complex endemic to many countries in the tropical and subtropical regions, where it seen predominantly in the rural areas and among the “poorest of the poor.” Globally there are around 50,000–90,000 new cases per year, with 90% of these cases being reported from Brazil, China, Ethiopia, Eritrea, India, Kenya, Somalia, South Sudan, Sudan, and Yemen [2].
HLH secondary to VL is an extensively rare phenomenon in the medical literature that causes confusion in diagnosis and management.
Here we first report from Syria a case of visceral leishmaniasis that was complicated with hemophagocytic lymphohistiocytosis and resistant to amphotericin B in a 15-month-old female.
Case presentation
A 15-month-old Arab female was admitted to our hospital with 15 days of fever and decreased weight and activity. She had no dyspnea or cyanosis. There was no history of rash or diarrhea. She was up to date with her immunization, and the mother mentioned that her daughter was diagnosed with typhoid fever by a doctor 4 months before, and it had been successfully treated with oral cefixime for a week.
On clinical examination, she was alert and interactive, but appeared pale. She had a fever of up to 39.5 °C. Her blood pressure was 90/60 mmHg, pulse was 120/min, respiratory rate was 32/min, and weight was 8.5 kg. She had a markedly enlarged liver and spleen that were palpable 4 cm and 6 cm below the costal margin, respectively.
The laboratory investigations showed that her hemoglobin (Hg) was 71 g/L, her platelets (PLTs) were 104 × 103/µL, and her white blood cells (WBCs) were 6.8 × 103/µL, with 16% neutrophils, 73% lymphocytes, and 7% monocytes. The erythrocyte sedimentation rate (ESR) was elevated, at 135 mm and 150 mm in the first and the second hour, respectively. The peripheral blood smear showed hypochromic microcytic anemia, poikilocytosis, reactive lymphocytosis, and mild thrombocytopenia. The Wright test was negative, and Widal was positive (S. typhi O: 1/320; S. typhi H: 1/640; S. paratyphi B: 1/320; and S. paratyphi A: 1/320). Chest x-ray was normal. Abdominal ultrasonography revealed no abnormalities except hepatomegaly and splenomegaly. Bone marrow aspiration (BMA) did not show malignancy or any other pathological findings. We initially considered typhoid fever relapse, but the patient did not improve with 7 days of intravenous ceftriaxone. Therefore, the antibiotic was changed to meropenem for 7 days as an empirical treatment but failed again to provide any clinical improvement.
Other differential diagnoses considered were protozoal infections such as leishmaniasis (kala-azar), infectious mononucleosis, neoplastic diseases such as acute leukemia and lymphoma, and other disorders such as rheumatic diseases. Leishmaniasis was the most suspected diagnosis, as these kinds of parasitic infections are common in our region; therefore, we conducted a direct agglutination test for visceral leishmaniasis, which was positive (1/128). BMA was performed again and showed erythrophagocytosis; intracellular small round organisms looked like the amastigote form of Leishmania (Donovan bodies) with no evidence of malignancies (Fig. 1). Then, we immediately started with conventional amphotericin B (AMB) 1 mg/kg/day. However, after 9 days of treatment, the symptoms did not regress and continued to progress, and the patient’s status continued to deteriorate, so blood units were transferred to her.
Fig. 1.
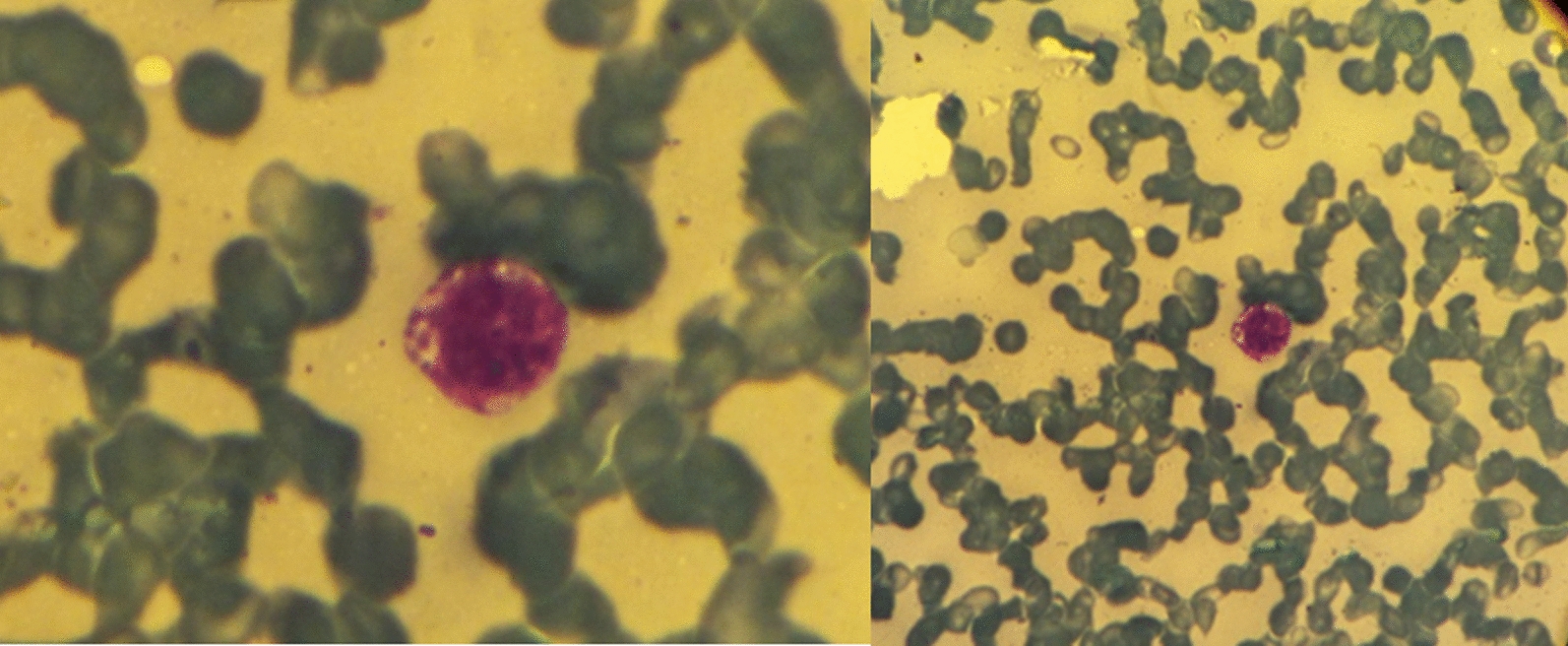
BMA showed erythrophagocytosis; intracellular small round organisms looked like the amastigote form of Leishmania (Donovan bodies) with no evidence of malignancies
Reevaluation tests showed that Hg was 45 g/L, mean corpuscular volume (MCV) was 60.4 fL, PLTs were 8 × 103/µL, and WBCs were 3.72 × 103/µL, with 19% neutrophils, 71% lymphocytes, and 9% monocytes. Repeating the peripheral blood smear revealed severe erythrocytopenia, severe hypochromic agglutination, rouleaux formation, poikilocytosis (elliptocytes, few spherocytes, and few teardrop cells), slight leukocytopenia, and severe thrombocytopenia. Additional laboratory tests are presented in Table 1.
Table 1.
Summary of clinical investigations
| Investigation | Value | Normal |
|---|---|---|
| Cholesterol | 107 mg/dL | 130–230 mg/dL |
| Triglycerides | 529 mg\dL | 50–150 mg\dL |
| LDL | 73 mg/dL | 100–155 mg/dL |
| Ferritin | 1280.3 ng/mL | 15–150 ng/mL |
| Urea | 20 mg/dL | 10–20 mg/dL |
| Creatinine | 0.65 mg/dL | 0.60–1.30 mg/dL |
| Aspartate transaminase (AST) | 80 U/L | 5–50 U/L |
| Alanine aminotransferase (ALT) | 13 U/L | 5–45 U/L |
| Alkaline phosphates | 166 U/L | 90–320 U/L |
| Total protein | 8.0 g/dL | 6.0–8.0 g/dL |
| Albumin | 2.55 g/dL | 3.50–5.00 g/dL |
| C-reactive protein (CRP) | 123.2 mg/L | < 6 mg/L |
| Lactate dehydrogenase (LDH) | 948 U/L | < 480 U/L |
| Total bilirubin | 1.71 mg/dL | < 1.1 mg/dL |
In the light of the previous data, the patient had (1)pancytopenia, (2) hypertriglyceridemia, (3) splenomegaly, (4) fever, and (5) ferritin greater than 500 ug/L with (6) no evidence of malignancy, fulfilling the required criteria (which should be five out of eight or more) to confirm the diagnosis of HLH [4]. Therefore, we added prednisolone 2 mg/kg for 5 days without response, and then replaced it with intravenous immunoglobulins 2 g/kg for 2 days, also without response.
We replaced conventional AMB with meglumine antimoniate 60 mg/kg. After only 4 days, the fever disappeared, the hepatosplenomegaly began to recede, and blood count improved. After 8 days of the treatment in hospital, the patient continued 20 days with the same drug at home and finally completely recovered. At the follow-up 20 days later, the patient had good health, with complete resolution of VL and subsequently HLH.
Discussion
Visceral leishmaniasis is one of the fatal diseases caused by Leishmania donovani and L. chagasi that affects several internal organs such as the spleen, liver, and bone marrow and is transmitted by phlebotomine sandflies [3]. According to the World Health Organization (WHO), around 50,000–90,000 of VL cases are reported per year, with 90% of these cases being reported from Brazil, China, Ethiopia, Eritrea, India, Kenya, Somalia, South Sudan, Sudan, and Yemen. It is associated with chronic fever, malaise, weight loss, and splenomegaly. [2, 3].
VL is diagnosed using histopathology, culture, molecular techniques, and serologic tests. Histopathology uses splenic aspiration or bone marrow aspirates. Bone marrow aspirates are less sensitive compared with splenic aspirates. Direct agglutination has a sensitivity of 97–100% and specificity of 86–92% [4, 5].
Secondary hemophagocytic lymphohistiocytosis may develop because of overwhelming immune system activation secondary to severe infections, malignancy, or rheumatologic diseases. The presentation of HL is variable and may present with fever, hemophagocytosis, splenomegaly, hypertriglyceridemia and/or hypofibrinogenemia, and cytopenias [1].
Diagnosis of hemophagocytic lymphohistiocytosis is done using the Revised Diagnostic Guidelines for HLH [6] (Table 2)
Table 2.
Revised Diagnostic Guidelines for HLH[6]
|
A diagnosis of HLH can be established if one of either 1 or 2 below is fulfilled. (1) A molecular diagnosis consistent with HLH. (2) Diagnostic criteria for HLH are fulfilled (five out of the eight criteria below). (A) Initial diagnostic criteria (to be evaluated in all patients with HLH) Fever Splenomegaly Cytopenias (affecting ≥ 2 of 3 lineages in the peripheral blood): Hemoglobin < 90 g/L (in infants < 4 weeks: hemoglobin < 100 g/L) Platelets < 100 × 109/L Neutrophils < 1.0 × 109/L Hypertriglyceridemia and/or hypofibrinogenemia: Fasting triglycerides ≥ 3.0 mmol/L (that is, ≥ 265 mg/dL) Fibrinogen ≤ 1.5 g/L Hemophagocytosis in bone marrow or spleen or lymph nodes No evidence of malignancy (B) New diagnostic criteria Low or absent NK-cell activity (according to local laboratory reference) Ferritin ≥ 500 mg/L Soluble CD25 (that is, soluble IL-2 receptor) ≥ 2400 U/mL |
NK, natural killer
HLH secondary to VL is an extensively rare phenomenon in the medical literature, and it is a challenge to diagnose. The first case of VL/HLH was reported in 1984. The vast majority of patients are children, with a higher incidence among males than females [7, 8].
Patients with VL/HLH may present with fever, splenomegaly, hepatomegaly, pancytopenia, lymphadenopathy, jaundice, ascites, petechiae, papilloedema, bleeding petechiae, ecchymosis, hemorrhagic suffusions, gingival bleeding, and enterorrhagia [7, 8]. This clinical presentation of VL overlaps with HLH, and could cause a delay in diagnosis with a high rate of morbidities without ideal treatment for HLH [9].
Systematic review of published VL/HLH cases in 2008 showed that the mean delay in diagnosis of the reported cases is 9 weeks, and the mortality is 14% because of bleeding or infections.
The ideal treatment of HLH is still unknown; Henter et al. [6] recommended using etoposide, dexamethasone, cyclosporine A in selected patients, intrathecal therapy with methotrexate, and corticosteroids. For patients with familial disease or molecular diagnosis and patients with severe and persistent, or reactivated, disease, they recommend subsequent hematopoietic stem cell transplantation (HSCT).
Common agents used to treat VL are antimony sodium stibogluconate, amphotericin, paromomycin, and oral miltefosine. Although AMB is highly efficacious, there is a high risk of resistance in laboratory experimentation [5, 10].
Conclusion
Hemophagocytic lymphohistiocytosis secondary to visceral leishmaniasis is an extensively rare phenomenon in the medical literature that causes challenges in diagnosis and management. The clinical presentation of VL overlaps with HLH, and could cause delay in diagnosis with a high rate of morbidities without ideal treatment for HLH. Steroids should be used wisely to not cover the symptoms of infections or malignancy, and AMB resistance should be kept in mind in unresponsive Leishmania cases.
Acknowledgements
Not applicable.
Abbreviations
- HLH
Hemophagocytic lymphohistiocytosis
- VL
Visceral leishmaniasis
- LD
Leishmania donovani
- Hg
Hemoglobin
- PLT
Platelet
- WBC
White blood cell
- ESR
Erythrocyte sedimentation rate
- BMA
Bone marrow aspiration
- AMB
Amphotericin B
- MCV
Mean corpuscular volume
- HSCT
Hematopoietic stem cell transplantation
Author contributions
YR, KA, MS, and BB: analysis and interpretation of the patient data, writing of the manuscript, and revision. FAS and AD: supervision and patient care. MZBA: critical review and revision. Please direct correspondence to the corresponding author Muhamad Zakaria Brimo Alsaman. All authors read and approved the final manuscript.
Funding
There are no sources of funding.
Availability of data and materials
All data are included in this published article and its supplementary information files.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient’s legal guardian for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hines M, Bhatt N, Talano J-AM. Diagnosis, treatment, and management of hemophagocytic lymphohistiocytosis in the critical care unit. Critical Care of the Pediatric Immunocompromised Hematology/Oncology Patient: Springer; 2019. p. 159–82.
- 2.Leishmaniasis world health organization 2022 https://www.who.int/news-room/fact-sheets/detail/leishmaniasis.
- 3.Guerin PJ, Olliaro P, Sundar S, Boelaert M, Croft SL, Desjeux P, et al. Visceral leishmaniasis: current status of control, diagnosis, and treatment, and a proposed research and development agenda. Lancet Infect Dis. 2002;2(8):494–501. 10.1016/S1473-3099(02)00347-X [DOI] [PubMed] [Google Scholar]
- 4.Da Silva MRB, Stewart JM, Costa CHN. Sensitivity of bone marrow aspirates in the diagnosis of visceral leishmaniasis. Am J Trop Med Hyg. 2005;72(6):811–4. 10.4269/ajtmh.2005.72.811 [DOI] [PubMed] [Google Scholar]
- 5.Maxfield L, Crane JS. Leishmaniasis. StatPearls. Treasure Island (FL): StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC.; 2022.
- 6.Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–31. 10.1002/pbc.21039 [DOI] [PubMed] [Google Scholar]
- 7.Rajagopala S, Dutta U, Chandra KP, Bhatia P, Varma N, Kochhar R. Visceral leishmaniasis associated hemophagocytic lymphohistiocytosis–case report and systematic review. J Infect. 2008;56(5):381–8. 10.1016/j.jinf.2008.02.013 [DOI] [PubMed] [Google Scholar]
- 8.Carvalho FHGD, Lula JF, Teles LDF, Caldeira AP, Carvalho SFGD. Hemophagocytic lymphohistiocytosis secondary to visceral leishmaniasis in an endemic area in the north of Minas Gerais, Brazil. Revista da Sociedade Brasileira de Medicina Trop. 2020. 10.1590/0037-8682-0491-2019. 10.1590/0037-8682-0491-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tunç B, Ayata A. Hemophagocytic syndrome: a rare life-threatening complication of visceral leishmaniasis in a young boy. Pediatr Hematol Oncol. 2001;18(8):531–6. 10.1080/088800101753328501 [DOI] [PubMed] [Google Scholar]
- 10.Ponte-Sucre A, Gamarro F, Dujardin J-C, Barrett MP, López-Vélez R, García-Hernández R, et al. Drug resistance and treatment failure in leishmaniasis: a 21st century challenge. PLoS Negl Trop Dis. 2017;11(12): e0006052. 10.1371/journal.pntd.0006052 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are included in this published article and its supplementary information files.