

Summary
The current targeted therapy for BRAFV600E-mutant lung cancer consists of a dual blockade of RAF/MEK kinases often combining dabrafenib/trametinib (D/T). This regimen extends survival when compared to single-agent treatments, but disease progression is unavoidable. By using whole-genome CRISPR screening and RNA sequencing, we characterize the vulnerabilities of both persister and D/T-resistant cellular models. Oxidative stress together with concomitant induction of antioxidant responses is boosted by D/T treatment. However, the nature of the oxidative damage, the choice of redox detoxification systems, and the resulting therapeutic vulnerabilities display stage-specific differences. Persister cells suffer from lipid peroxidation and are sensitive to ferroptosis upon GPX4 inhibition in vivo. Biomarkers of lipid peroxidation are detected in clinical samples following D/T treatment. Acquired alterations leading to mitogen-activated protein kinase (MAPK) reactivation enhance cystine transport to boost GPX4-independent antioxidant responses. Similarly to BRAFV600E-mutant melanoma, histone deacetylase (HDAC) inhibitors decrease D/T-resistant cell viability and extend therapeutic response in vivo.
Keywords: lung adenocarcinoma, BRAF oncogene, targeted therapy, persister cells, ferroptosis, GPX4 inhibition, HDAC inhibition
Graphical abstract
Highlights
-
•
Unbiased genetic screen identifies ferroptosis as DTP vulnerability in BRAFV600E LUAD
-
•
GPX4 inhibitors extend survival when applied at early disease relapse
-
•
Relapsed tumors display biomarkers indicative of sensitivity to ferroptosis triggers
-
•
Acquired mutations restoring MAPK signaling sensitize tumors to HDAC inhibition
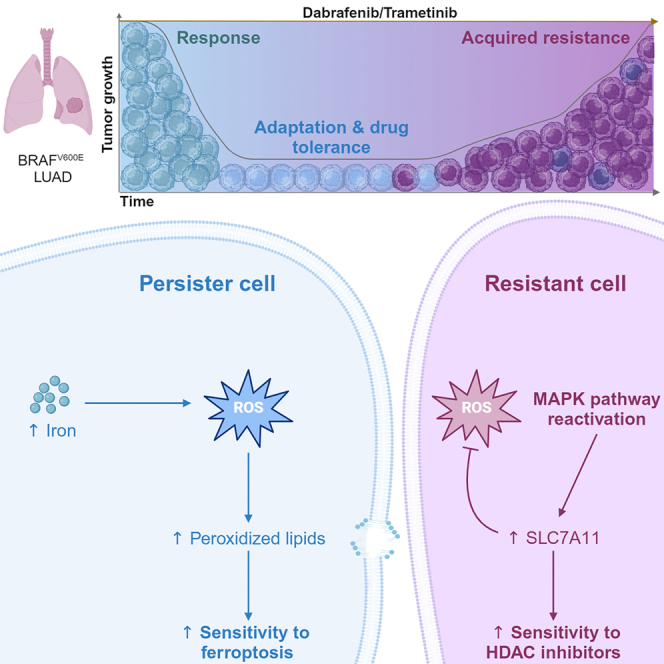
Nokin et al. identify two redox-related cellular states that involve an adaptive drug-tolerant stage followed by a genetic-defined resistance phase in BRAFV600E-driven lung cancer. Timely treatments with small molecules (GPX4 and HDAC inhibitors) that further enhance the resulting oxidative stress delay disease progression in vivo.
Introduction
The RAF family of serine/threonine kinases is comprised by three isoforms (A-, B-, and CRAF). They are known for mediating RAS activation to the downstream kinases MEK and ERK with few additional substrates. In spite of sharing such stringent substrate specificity, a similar activation mechanism, and the presence of three highly conserved regions, their implication in human cancer is strikingly different.1 While both mutations and oncogenic fusions implicating A- and CRAF have been identified in human tumors, they appear at a much lower frequency than those involving BRAF. Over 200 BRAF cancer-related mutations have been identified and are subdivided in three classes with different clinical properties.2,3 The most common substitution (V600E) belongs to the class 1 and mimics phosphorylation of the activation loop resulting in constitutive signaling as an active monomer in an RAS-independent manner. In lung adenocarcinoma (LUAD), BRAF mutations are detected in approximately 5% of cases with a balanced representation of V600E and non-V600E events. Yet, only V600E patients are treated with targeted agents.4 The standard of care consists of a dual blockade of RAF/MEK kinases often combining dabrafenib with trametinib (hereafter referred as D/T) resulting in higher response rate and extended progression-free survival when compared to single-agent treatments.5 However, as with other targeted treatments, tumor progression will eventually occur. A fraction of resistant tumors display mutations in other members of the RAS-ERK pathway resulting in reactivated signaling.6,7 These alterations co-exist with the original BRAFV600E driver, and it is unknown whether, as observed in patients with melanoma, the oncogenic combination could drive relapse at the expense of acquiring novel targetable vulnerabilities.8 Furthermore, there is limited information of how BRAFV600E-driven LUAD tumors adapt to D/T treatment and whether this is accompanied by actionable vulnerabilities.
Here, we combined naive BRAFV600E lung cancer cell lines as well as patient-derived xenograft (PDX) cell cultures with acquired D/T resistance in vivo, CRISPR-Cas9 screening, and pharmacological treatments to identify mechanisms of resistance and associated vulnerabilities that could potentially be implemented to fight disease relapse in the clinic.
Results
Genome-wide screen to identify mediators and molecular vulnerabilities upon D/T treatment
We used the LUAD BRAFV600E-mutant HCC364 human cell line to generate a Cas9 derivative by lentiviral delivery of the enzyme. The growth properties and the response to D/T of the Cas9-expressing cells were indistinguishable from the parental cell line (data not shown). To perform the screen, we selected the lentiviral CRISPRko sgRNA Brunello library.9 It was transduced into cells at a multiplicity of infection of 0.3 and 1,000× coverage and selected with puromycin during 1 week before being divided into control and D/T-treated groups. D/T treatment was performed using the IC50 values obtained for each individual compound (Figures S1A and S1B). This D/T combination induced an apoptotic response together with a fraction of surviving cells (Figure S1C) that were maintained during 3 weeks to enable positive and negative selection of single-guide RNAs (sgRNAs) compared to control conditions. At this point, the genomic DNA was harvested, and the sgRNA cassette was amplified and quantitated by Illumina sequencing (Figure 1A).
Figure 1.

Genome-wide CRISPR-Cas9 screening identifies determinants of dabrafenib/trametinib sensitivity in BRAF-mutant LUAD cells
(A) Schematic diagram illustrates the genome-wide CRISPR-Cas9 knockout library screening. Human Brunello sgRNA library was packed into lentiviral particles and transduced into Cas9-overexpressing HCC364 cells (HCC364-Cas9). The sgRNA-transduced cells were selected by puromycin and cultured in vehicle and dabrafenib/trametinib (D/T) (250/5 nM, refreshed every 3 days) for 3 weeks. Genomic DNA was extracted from the treated cells and the sgRNA fragment was amplified by PCR. Copy number of sgRNAs was determined by high-throughput sequencing analysis.
(B) Scatterplot depicting gene-level results for D/T negatively and positively selected hits in the CRISPR screen. A number of representative hits are shown in color and are detailed in C and D. n = 1 biological replicate.
(C and D) STRING protein network of the positively (C) and negatively (D) selected hits as defined in (B). Colored nodes highlight proteins enriched in certain signaling pathways. The edges represent protein-protein associations, and the line thickness indicates the strength of data support. n = 1 biological replicate.
(E) Scheme representing the major actors of ferroptosis, highlighting the central role of GPX4 in the detoxification of peroxidized lipids. See also Figure S1.
Bioinformatic comparisons using CRISPRAnalyzeR10 were performed to identify sgRNAs that appeared with differential representative scores (Figure 1B). The biological significance of the screening was confirmed by the identification of FBXW7, whose inactivation has been reported to elicit a multidrug resistance phenotype.11 The screen also identified several enriched and depleted sgRNAs targeting genes previously reported to mediate D/T resistance or to induce increased sensitivity, respectively. Among the most relevant enriched sgRNAs, we found those targeting negative regulators of the mitogen-activated protein kinase (MAPK) and PI3K pathways as well as inhibitors of the YAP transcriptional program (Figure 1B). In good agreement, sgRNAs directed against genes implicated in the activation of these processes or pathway effectors required for effective signaling appeared as drop-out hits. In sum, these results validate our genetic screen by confirming previous findings indicating that RTK/RAS-dependent hyperactivation of MAPK and PI3K signaling or elevated Hippo pathway activity results in D/T resistance6,12,13,14,15,16 (Figure 1C).
Lipid peroxidation as an early phenotype and potential vulnerability following D/T treatment
To identify therapeutic vulnerabilities associated with D/T adaptation, we further investigated the list of depleted sgRNAs following treatment. One of the most prominent candidates was GPX4 (ninth out of 736 significantly depleted sgRNAs) with a Z-ratio of −6.30 (Figure 1B). Interestingly, GPX4 has been reported to play an essential pro-survival function to sustain an early therapy-resistant cell state in a variety of tumor types and drug treatments.17,18 GPX4 is a selenocysteine-containing enzyme with lipid hydroperoxidase function essential to protect cells from ferroptosis, an iron-mediated oxidative form of cell death.19 In good agreement, we identified a collection of depleted guides converging on three biological processes functionally related to GPX4 and ferroptosis: selenocysteine synthesis, mevalonate pathway (e.g., PSTK, SEPSH2, MVK, and HMGCS1), and iron metabolism (e.g., FDX1L, HCSB, NFS1, and ISCU) (Figures 1D and 1E).
Drug treatments often induce a non-mutational drug-tolerant persister (DTP) transient state characterized by low proliferation rates. Over time, a subset of DTPs gradually resumes proliferation in the presence of the drug, originating a second population of cells known as drug-tolerant expanded persisters (DTEPs).20,21 Collectively, these populations provide a cellular reservoir from which acquired drug resistance mechanisms eventually unfold.21,22 We generated putative DTPs and DTEPs by maintaining BRAFV600E-mutant HCC364 cells on constant D/T treatment for 3 and 40 weeks, respectively. The cells at the 3-week time point—that coincided with the end of the CRISPR screen (Figure 1A)—were in a non or low-cycling stage. At the end of the screening process, the control population expanded 7,000-fold whereas the D/T-treated condition, potentially representing a DTP state, only increased by 1.5-fold. However, at 40 weeks on constant D/T treatment, the population displayed intermediate proliferation rates when compared to the parental controls and therefore potentially constituted a DTEP pool (Figure 2A). This different proliferation rate between DTP and DTEP cells was further confirmed by dissimilar enrichment scores of cell cycle-related terms (cell cycle checkpoints and e2F targets) using single-sample gene set enrichment analysis (ssGSEA) of RNA sequencing data (Figure 2B). In good agreement with previous reports,17,20 our DTEPs reverted to a drug-sensitive state upon D/T removal (drug holiday for 3 weeks), indicating that cell proliferation in the presence of the drugs was sustained by non-mutational adaptation (Figure 2A). Furthermore, induction of cellular senescence is also one of the underlying mechanisms by which cancer therapies exert anti-tumor activity and could be an early trigger to dormancy.23,24 We investigated the transcriptome of our DTP and DTEPs by applying SENCAN, a cancer senescence classifier.25 In accordance with their respective proliferation rates, we observed a 3-fold increase in the senescence score in the DTP state, reinforcing the concept that persistent D/T treatment triggers a gradual and adaptive evolution with increased proliferative properties resulting in a DTEP population (Figure 2C). More reassuringly, we applied a recently developed transcriptional signature defining the DTP state composed of 212 genes commonly up-regulated in lung DTP models.26 Following D/T treatment, our putative DTP and DTEP populations displayed an enrichment of the tolerance signature suggesting that their state is a bona fide persister phenotype (Figures 2D, 2E, and S1D).
Figure 2.

Transcriptomic analysis of drug-tolerant persister and drug-tolerant expanded persister HCC364 cells points to redox homeostasis as a potential vulnerability in these cell populations
(A) Drug-tolerant persisters (DTPs) and drug-tolerant expanded persisters (DTEPs) were generated by maintaining BRAFV600E-mutant HCC364 cells on constant D/T (250/5 nM) treatment for 3 and 40 weeks, respectively. Growth curves of parental, DTP, and DTEP HCC364 cells in the presence of 250/5 nM D/T. DTEPs underwent a drug holiday for 3 weeks and were rechallenged with D/T to further confirm the non-genetic adaptation of these cells to the treatment (DTEP + drug hol + D/T). Data were analyzed using two-way ANOVA followed by Tukey’s multiple comparisons post-test and are shown as the mean values ± SEM. n = 3 biological replicates.
(B) RNA sequencing analyses were performed on HCC364 parental, DTP, and DTEPs cells. E2F targets and cell cycle checkpoints ssGSEA scores in parental, DTP, and DTEP HCC364 cells. n = 1 biological replicate.
(C) Senescence score in parental (PAR), DTP, and DTEP HCC364 cells. n = 1 biological replicate.
(D) Running enrichment scores of GSEA (left panel) in DTP vs. PAR, DTEP vs. PAR, and DTEP vs. DTP using the drug tolerance signature and the E2F targets and cell cycle checkpoints gene sets. Normalized enrichment scores (NESs) against the log10 of adjusted p value are shown (right panel). n = 1 biological replicate.
(E) Heatmap of the significantly differentially expressed genes from the 3 gene sets (drug tolerance signature, E2F targets, and cell cycle checkpoints) in PAR, DTP, and DTEP cells. n = 1 biological replicate. See also Figure S1.
In addition, gene set enrichment analysis (GSEA) pathway showed that modulation of seleno-aminoacid metabolism, cholesterol homeostasis, and oxidative stress were found in both DTPs and DTEPs (data not shown), thus pointing to redox homeostasis as a potential vulnerability in these cell populations. These data further support the potential dependency to GPX4-mediated detoxification of peroxidized lipids (highlighted by the CRISPR screen). Therefore, we next wanted to assess whether GPX4 function could be an essential feature during the entire period of drug-dependent cellular evolution and could consequently be a common vulnerability. Following D/T treatment, we observed a significant reduction in the redox-buffer potential measured by glutathione (GSH) cellular content with a concomitant increase in reactive oxygen species (ROS) in both populations (Figures 3A and 3B). Interestingly, this phenotype was accompanied by an elevation of both intracellular iron levels and peroxidized lipids that could explain their dependency on the GPX4 antioxidant function (Figures 3C and 3D). Importantly, D/T-treated cells displayed increased sensitivity to the GPX4 inhibitor RSL3 as well as to the ferroptosis activators erastin and FIN56 (Figures 3E, 3F, and 3G). In all cases, DTPs and DTEPs displayed enhanced sensitivity suggesting a higher GPX4 dependency. In agreement with the screen result, this sensitivity was further confirmed by CRISPR-mediated GPX4 depletion in the DTEP cells (Figure S2). Furthermore, the toxicity induced by RSL3 and erastin and the concomitant increase in lipid peroxidation were suppressed by co-treatment with the antioxidants N-acetylcysteine (NAC) and Trolox or with various ferroptosis inhibitors (liproxstatin, ferrostatin, and deferoxamine) reinforcing their increased sensitivity to ferroptosis (Figures 3H, 3I, and S3)
Figure 3.

DTP and DTEP HCC364 cells are hypersensitive to ferroptosis induction in vitro
(A) Intracellular GSH concentration in parental, DTP, and DTEP HCC364 cells. n = 3 biological replicates.
(B) ROS level in parental, DTP, and DTEP HCC364 cells. n = 6 biological replicates.
(C) Iron level in parental, DTP, and DTEP HCC364 cells. n = 4 biological replicates.
(D) Peroxidized lipids content in parental, DTP, and DTEP HCC364 cells. n = 6 biological replicates.
(E–G) Cell viability assessment by MTT assay of parental, DTP, and DTEP HCC364 cells treated with serial dilutions of RSL3 (E), erastin (F), or FIN56 (G) for 72 h (upper panel). IC50 values (lower panel) are represented for each condition. n = 4 biological replicates.
(H–J) Cell viability assessment by MTT assay of parental, DTP, and DTEP HCC364 cells treated with serial dilutions of RSL3 in the presence of 2.5 mM N-acetylcysteine (NAC, H), 10 μM Trolox (I), or 200 nM selenium (Se, J) for 72 h n = 3 biological replicates. All data were analyzed using one-way ANOVA followed by Dunnett’s multiple comparisons post-test and are shown as the mean values ± SEM. See also Figures S2–S4 and S10.
As mentioned earlier, the genetic screen and the RNA sequencing analysis identified biological processes that are mechanistically linked to GPX4 detoxification functions. These included components of the mevalonate pathway and selenoprotein biosynthesis as well as iron metabolism that could represent additional therapeutic vulnerabilities. The relevance of the selenoprotein biosynthetic pathway in this context was illustrated by the complete rescue of RSL3 sensitivity upon selenium supplementation (Figure 3J). Similarly, the mevalonate pathway provides essential precursors to selenoprotein biosynthesis, through the isopentenylation of selenocysteine-tRNA (Figure 1E).27,28 In good agreement, targeting HMG-CoA reductase by several statins also affected DTEPs, although we failed to observe an additive effect when combined with GPX4 inhibition (Figure 4).
Figure 4.

GPX4 inhibition reduces the tumor growth of D/T-resistant cells in vivo
(A) DTPs and DTEPs were generated by maintaining BRAFV600E-mutant 1D-PDX cells on increasing concentrations of D/T treatment for 3 and 40 weeks, respectively. Growth curves of parental, DTP, and DTEP 1D cells in the presence of 250/5 nM D/T. DTEPs underwent a drug holiday for 3 weeks and were rechallenged with D/T to further confirm the non-genetic adaptation of these cells to the treatment (DTEP + drug hol + D/T). Data were analyzed using two-way ANOVA followed by Tukey’s multiple comparisons post-test and are shown as the mean values ± SEM. n = 3 biological replicates.
(B) Cell viability assessment by MTT assay of parental, DTP, and DTEP 1D cells treated with serial dilutions of RSL3 for 72 h. Data are shown as the mean values ± SEM. IC50 values ± SD are indicated for each condition. n = 3 biological replicates.
(C) Cell viability assessment by MTT assay of parental and DTEP 1D cells treated with 50 nM RSL3 in the presence of 10 μM Trolox, 200 nM ferrostatin, 100 nM liproxstatin, 1 μM deferoxamine, 200 nM Se, or 2.5 mM NAC for 72 h. Data are normalized to untreated cells and shown as the mean values ± SEM. Data were analyzed using two-way ANOVA followed by Tukey’s multiple comparisons post-test. n = 3 biological replicates.
(D) Schematic outline of the 1D intrapulmonary xenograft model. After lung orthotopic injection, mice were treated with dabrafenib (30 mg/kg/day) and trametinib (0.2 mg/kg/day) by oral gavage 5 days per week. At disease progression, mice were treated with RSL3 (10 mg/kg/day) by intraperitoneal injection 5 days per week. Mice were sacrificed and lungs were collected for further analysis.
(E) Follow-up of intralung 1D tumor growth by bioluminescence imaging. Mice were treated as mentioned in D. Data are represented as mean signal intensity (p/s/cm2/sr) ± SEM, n = 6 or 7 mice for each group.
(F) Representative images of bioluminescence signal in mice treated with RSL3.
(G) Fold change in tumor growth compared to baseline (the start of RSL3 treatment, day 40 post-implantation) for individual cell xenografts treated for 14 days with vehicle or with 10 mg/kg/day RSL3. Fold changes were calculated based on the values presented in Figure 4E (p/s/cm2/sr). Statistical significance was tested using unpaired two-tailed t test. n = 6 or 7 mice for each group.
(H) Representative 4-HNE and GPX4 immunostaining in lung sections of mice treated with RSL3. Quantification of 4-HNE staining scores in 1D tumors from mice treated with vehicle or with RSL3. Data are shown as mean scores (average of three independent stainings) per mice and were analyzed using Mann-Whitney t test. n = 6 or 7 mice for each group. See also Figures S1 and S9.
As a whole, these results demonstrate that following D/T treatment, BRAFV600E-mutant LUAD persister cells display a disabled antioxidant program that renders them vulnerable to ferroptosis triggers.
Sensitivity of D/T persister cells to GPX4 inhibition in vivo
Evidence presented so far has identified the GPX4-antioxidant role as an essential pro-survival function during tumor adaptation upon D/T treatment and its inhibition as a potential therapy to target disease relapse. To further validate this hypothesis using a clinically relevant model, we utilized a BRAFV600E-mutant cell line (clone 1D) generated from a PDX of lung cancer that progressed following treatment with an RAF inhibitor.29 These cells were cultured with increasing concentrations of D/T starting from their respective IC50 values (Figures S1A and S1B) until they partially regained proliferative capacity and therefore reached a putative DTEP state (Figure 4A). Indeed, the sensitivity to D/T was partly restored following a drug holiday period suggesting that it was due to an adaptive mechanism. Remarkably, 1D-DTPs and 1D-DTEPs showed up to 30-fold increase in their sensitivity to RSL3 when compared to the parental controls (Figure 4B). 1D-DTEPs also presented an increased sensitivity to erastin (Figure S5A). In both contexts, cellular viability was restored by co-treatment with ferroptosis inhibitors, antioxidants, and selenium (Figures 4C and S5B). Similarly to HCC364 cells, 1D-DTEPs displayed increased ROS, peroxidized lipids, and intracellular iron levels (Figures S5C–S5E).
To further evaluate the role of GPX4 in the adaptation to D/T in a more translational setting, we performed in vivo assays. To this end, we introduced the luciferase gene into the parental 1D-PDX cells before their implantation in the lungs of immunocompromised mice via direct lung injection (Figure 4D). D/T treatment was started when overt progression was observed resulting in significant disease control with approximately a 10-fold tumor burden reduction. Yet, the therapeutic response was transient and D/T-treated tumors slowly regained proliferative capacity indicative of progressive disease (Figure 4E). This feature was calculated following clinical criteria and was determined to occur when D/T-treated tumors reached a volume 20% higher to that estimated at the beginning of the drug treatment.30 At this point, the disease progression cohort was split in two groups with one receiving the GPX4 inhibitor RSL3 as a next-line therapy. Remarkably, GPX4 inhibition of D/T-resistant tumors in vivo resulted in a 10-fold tumor growth reduction compared to the untreated cohort (Figures 4E–4G). Immunohistochemical staining of 4-hydroxynonenal (4-HNE) further confirmed the accumulation of lipid peroxides in lung tumors upon RSL3 treatment while GPX4 expression was comparable between the two groups (Figure 4H). These results suggest that the non-genetic adaptation to D/T therapy in vivo renders BRAFV600E-mutant tumors vulnerable to ferroptosis triggers.
MAPK reactivation-mediated resistance and associated ROS vulnerabilities
Persister cells represent a reservoir from which acquired resistance eventually arises. In the context of D/T treatment, various bypass mutations including activating substitutions in K- and NRAS have been described in BRAFV600E lung tumors as potential mediators of clinical resistance.4 In order to investigate whether RAS-mediated MAPK reactivation would also result in sensitivity to GPX4 inhibition and induction of ferroptosis, we took advantage of a D/T-resistant cell line (DFCI471) derived from a fresh biopsy at disease progression (Figures S6A–D). Sequencing of both the tumor and plasma revealed an NRASQ61K mutation that co-existed with the BRAFV600E oncogene with no additional genomic alterations potentially linked to D/T resistance.14 Indeed, short hairpin RNA-mediated NRAS knockdown decreased MAPK signaling and reverted the resistant phenotype (Figures S6E–S6H). These DFCI471 cells, unlike DTEPs from both HCC364 and 1D cells, turned out to be resistant to ferroptosis trigger erastin or the GPX4 inhibitor RSL3 (Figures 5A and 5B). To further confirm this result using another NRAS-dependent cellular model, we rescued the HCC364 DTPs by exogenous expression of NRASQ61K. Lentiviral delivery of the oncogene restored cell proliferation and induced D/T resistance increasing the IC50 of both compounds up to 50-fold (Figures S7A–S7C). This was correlated with the restoration of MAPK signaling and downstream transcriptional output to levels comparable to those of parental HCC364 cells (Figures S7D and S7E). In good agreement with the response of DFCI471 cells, the HCC364NRAS cells were insensitive to RSL3 or to erastin and displayed drug sensitivity values similar to those of the parental cells (Figures 5A and 5B). Interestingly, these phenotypes were replicated in another cellular setting in which D/T resistance occurred independently of acquired RAS mutations. We derived a spontaneous D/T-resistant HCC364 clone that displayed EGFR amplification (CNV 6.3), resulting in MAPK signaling and proliferation levels under D/T treatment similar to those of the untreated parental cells (Figures S7F–S7H). Furthermore, these HCC364EGFRa cells were insensitive to ferroptosis triggers (Figures 5A and 5B). These results suggest that in contrast to the persister state, acquired resistance to D/T driven by MAPK reactivation does not result in sensitivity to ferroptosis inducers.
Figure 5.

NRAS-mediated resistance leads to increased sensitivity to oxidative stress but not to ferroptosis
(A and B) DFCI471 cells derived from a biopsy of a patient with BRAFV600E LUAD at disease progression upon D/T treatment. HCC364NRAS cells were generated by exogenous expression of NRASQ61K in HCC364-DTPs and HCC364EGFRa cells were derived spontaneously from HCC364-DTEPs after continuous D/T (250/5 nM) treatment for 70 weeks. Cell viability assessment by MTT assay of parental and DTEP HCC364, HCC364EGFRa, HCC364NRAS, and DFCI471 cells treated with serial dilutions of RSL3 (A) or erastin (B) for 72 h. Data are shown as the mean values ± SEM. n = 3 biological replicates.
(C) Quantitative reverse-transcription PCR (RT-PCR) analysis of relative SLC7A11 mRNA level in parental and DTEP HCC364, HCC364EGFRa, HCC364NRAS, and DFCI471 cells. Data were analyzed using one-way ANOVA followed by Tukey’s multiple comparisons post-test and are shown as the mean values ± SEM. n = 5 biological replicates.
(D) Immunoblot of SLC7A11, SLC3A2, and GPX4 in parental and DTEP HCC364, HCC364EGFRa, HCC364NRAS, and DFCI471 cells. HSP90 was used as a loading control. n = 3 biological replicates.
(E) Intracellular ROS level in DFCI471 cells co-treated with 1 μM vorinostat or 10 nM panobinostat and 2.5 mM NAC for 72 h. Data were analyzed using two-way ANOVA followed by Tukey’s multiple comparisons post-test and are shown as the mean values ± SEM. n = 3 or 5 biological replicates.
(F) Clonogenic assay of DFCI471, HCC364, HCC364EGFRa, and HCC364NRAS cells co-treated with 1 μM vorinostat or 10 nM panobinostat and 2.5 mM NAC. Data were analyzed using two-way ANOVA followed by Tukey’s multiple comparisons post-test and are shown as the mean values ± SEM. n = 3 biological replicates. See also Figures S2, S6, S7, and S10.
We speculated that in this context of acquired resistance due to MAPK reactivation, GPX4 might not be a limiting redox factor since RAS/MAPK signaling has been shown to increase cellular GSH production by up-regulating the cystine/glutamate antiporter xCT.31 Indeed, we found that DFCI471, HCC364NRAS, and HCC364EGFRa cell lines displayed increased SLC7A11 mRNA and protein levels (Figures 5C and 5D). Accordingly, we went on to identify alternative actionable vulnerabilities that could be effective in the context of acquired resistance. A functionally equivalent NRAS mutation (Q61H) has been reported to mediate D/T insensitivity in BRAFV600E-mutant melanoma cells.8 In this tumor type, the resistant cells were treated with the histone deacetylase inhibitor (HDACi) vorinostat leading to reduced expression of SLC7A11 and elevation of oxidative stress to toxic levels.8 Similarly, we found that DFCI471 cells treated with vorinostat or the related HDACi panobinostat triggered ROS production with a concomitant decrease in cell viability (Figures 5E and 5F). Treatment with the antioxidant NAC diminished the ROS production induced by both HDAC inhibitors and partly restored cell proliferation (Figures 5E and 5F). This illustrated the central role of oxidative stress as an acquired vulnerability following NRAS-mediated D/T resistance in BRAFV600E-mutant LUAD. Similarly, HDACi treatment decreased the proliferation rate of both HCC364NRAS and HCC364EGFRa cells. This was again linked to ROS-dependent stress as co-treatment with NAC partly restored the growth rate of HDACi-treated cells (Figures 5F and S7I). In agreement with the pharmacologic approach, CRISPR-mediated ablation of SLC7A11 decreased the ability of these cells to grow in the presence of D/T (Figure S2).
Finally, we investigated whether HDACi could be applicable in a more clinically relevant setting. To this end, we took advantage of a second PDX-derived cell line (clone 1E).29 In our hands, 1E-PDX cells became insensitive to D/T and displayed reactivated MAPK signaling as well as proliferation rates similar to those of the parental controls (Figures 6A–6C). Unlike the 1D-DTEPs (Figure 4A), D/T-resistant 1E cells (1E-R) maintained their high proliferative capacity following a drug holiday, suggesting that it may be mediated by a genetic alteration. Targeted sequencing analysis identified a BRAF gene amplification (CNV 15.4) compared to parental controls (CNV 2). This mechanism of genetic resistance has been previously described in BRAFV600E-mutant melanoma.32 Similarly to the previous examples of MAPK reactivation, 1E-R cultures displayed elevated SLC7A11 protein and mRNA levels (Figures 6B and 6D) as well as cystine uptake (Figure 6E). Likewise, 1E-R cells were insensitive to the ferroptosis triggers RSL3 and erastin (Figures S8A and S8B). Importantly, they showed increased sensitivity to HDACi treatment when compared to 1E-PDX parental cells in an oxidative stress-dependent manner (Figures 6F, S8C, and S8D).
Figure 6.

MAPK pathway reactivation through BRAF amplification is associated with ROS vulnerabilities and in vivo targeting of NRAS-mediated D/T resistance in BRAFV600E LUAD cells with HDAC inhibitors
(A) DTPs and 1E-R cells were generated by maintaining BRAFV600E-mutant 1E-PDX cells on increasing concentrations of D/T treatment for 3 and 30 weeks, respectively. Growth curves of parental, DTP, and -R 1E cells in the presence of 250/5 nM D/T. 1E-R cells underwent a drug holiday for 3 weeks and were rechallenged with D/T to confirm the genetic adaptation of these cells to the treatment (1E-R + drug hol + D/T). Data were analyzed using two-way ANOVA followed by Tukey’s multiple comparisons post-test and are shown as the mean values ± SEM. n = 3 biological replicates.
(B) Immunoblot of BRAF, phospho-ERK (p-ERK), and SLC7A11 in 1E and 1E-R cells cultured in the presence of 250/5 nM D/T for 48 h. HSP90 was used as a loading control. n = 2 biological replicates.
(C) Quantitative RT-PCR analysis of relative MAPK pathway target genes (DUSP4, DUSP6, ETV4, ETV5, SPRY2, and PHLDA1) mRNA levels in 1E and 1E-R cells cultured in the presence of 250/5 nM D/T for 48 h. Data were analyzed using one-way ANOVA followed by Tukey’s multiple comparisons post-test and are shown as the mean values ±SEM. n = 3 biological replicates.
(D) Quantitative RT-PCR analysis of SLC7A11 mRNA level in 1E and 1E-R cells. Data were analyzed using unpaired t test with Welch’s correction and are shown as the mean values ± SEM. n = 3 biological replicates.
(E) Intracellular cystine uptake in HCC364 (parental and DTEP) and 1E (parental and -R) cells. Data were analyzed using two-way ANOVA followed by Tukey’s multiple comparisons post-test and are shown as the mean values ± SEM. n = 4 biological replicates.
(F) Clonogenic assay of 1E and 1E-R cells co-treated with 1 μM vorinostat or 10 nM panobinostat and 2.5 mM NAC. Data were analyzed using two-way ANOVA followed by Tukey’s multiple comparisons post-test and are shown as the mean values ± SEM. n = 4 biological replicates.
(G) Schematic outline of the DFCI471 subcutaneous xenograft model. Mice were treated with vorinostat (100 mg/kg/day) or panobinostat (10 mg/kg/day) by intraperitoneal injection 5 days per week. After 14 days, mice were sacrificed and tumors were collected for further analysis.
(H) Follow-up of tumor growth of the indicated conditions. Data are represented as mean tumor volume ±SEM. n = 8 or 10 mice for each group.
(I) Fold change in tumor growth compared to baseline.
(J) Immunoblot of acetyl and phospho-histone H3 (Ac and p-histone H3), histone H3, and SLC7A11 in 4 representative tumors per condition. Tubulin was used as a loading control. n = 4 biological replicates. See also Figure S8.
Altogether, these results suggest that acquired resistance-mediated MAPK signaling reactivation results in both ROS toxicities and associated vulnerabilities that are mechanistically different from those observed during the non-genetic adaptation observed in DTEPs.
Targeting NRAS-dependent acquired resistance in vivo
We next investigated the therapeutic potential of the HDACi vorinostat and panobinostat on D/T-resistant LUAD in vivo. To this end, we used the DFCI471 model since RAS-mediated D/T resistance is frequent in the clinical setting.4 DFCI471 xenografts were implanted and allowed to grow before treatment initiation (Figure 6G). Remarkably, exposure to the HDACi compounds induced a significant decrease in DFCI471 tumor growth that correlated with decreased levels of the proliferation marker phosphorylated histone H3 (Figures 6H and 6I). The higher therapeutic benefit observed with panobinostat may be due to increased HDAC inhibition in vivo as measured by acetylated H3 levels in tumor extracts (Figure 6J). In good agreement, panobinostat more efficiently reduced the expression levels of SLC7A11 potentially explaining its higher therapeutic effect (Figure 6J).
In sum, our results suggest that enhanced oxidative stress caused by HDACi might be a therapeutic vulnerability to treat BRAFV600E D/T-resistant tumors due to acquired alterations leading to MAPK reactivation.
Discussion
Most targeted therapies have limited efficacy when applied as single-agent treatments due to the rapid onset of drug resistance. When possible, improved regimens have been implemented in the clinic by applying rational drug combinations. BRAF-mutant LUAD has been one of these cases as demonstrated by a recent study reporting superior 5-year survival rates in a non-comparative phase 2 trial evaluating single vs. combined D/T therapy.5 Unfortunately, even if administered as combination regimens, tumor progression eventually unfolds, leaving drug-insensitive patients with limited therapeutic alternatives.4 Several mechanisms driving resistance to D/T have been identified in relapsing BRAF-mutant patients (mostly in the context of melanoma), often resulting in MAPK reactivation or increased signaling through parallel and downstream pathways such as PI3K or Hippo/Yap, respectively. Regarding BRAF-mutant LUAD, these resistance mechanisms have only been documented in a few case reports.6,7,12,13,16 Such heterogeneous clinical landscape complicates the anticipation of potential treatments to overcome disease progression. In this context, the identification of targetable molecular vulnerabilities that could be utilized to fight the onset of acquired resistance is a pressing medical need.
Accumulating evidence suggests that DTPs are the cellular reservoir that eventually leads to fully resistant disease upon additional acquired modifications.21,22,33 Irrespective of whether DTPs are rare pre-existing entities in the primary tumor, drug-induced, or the result of stochastic reprogramming, eradicating DTPs could be of great therapeutic value. Yet, while efficacious therapeutic approaches have yet to be established, a number of potential vulnerabilities have been identified in DTPs.33 Among those, up-regulation of antioxidant responses is linked with the persister phenotype in multiple cancer types.34,35,36,37 Also, a recent report demonstrated that NPC1L1-mediated uptake of vitamin E was increased in DTPs and its inhibition by ezetimibe had a significant anti-tumor effect in a pre-clinical cancer model.38 Interestingly, metabolic reprogramming through adaptation of both mitochondrial respiration and lipid metabolism has been shown to be a key mediator of resistance to MAPK inhibitors in BRAF-mutant melanoma.39,40,41,42 Further investigations will determine whether, as suggested by the results presented here, targeting these altered metabolic pathways (only identified in the context of melanoma) could also represent an effective therapeutic strategy to overcome resistance in BRAF-mutated LUAD.
In this context, our genome-wide genetic screen identified the lipid hydroperoxidase GPX4 as a top candidate required by BRAF-mutant lung cancer cells to survive D/T treatment (Figure 1). Indeed, GPX4 has been previously identified as a key mediator to sustain the DTP cell state in various tumor types.17,18,43 Our findings using BRAFV600E PDX-derived lung cancer cell lines suggest that GPX4 inhibition in vivo at the time of early relapse on D/T treatment (based on RECIST criteria) results in significant disease control (Figures 4E–4G). Importantly, a pleural effusion sample obtained from a patient with metastatic LUAD who initially responded to D/T displayed extensive signs of lipid peroxidation (identified by 4-HNE staining) at the time of early disease progression that was absent in the pre-treatment diagnostic samples (Figure 7). This result suggests that GPX4 inhibitors and/or other ferroptosis-inducing treatments could represent an alternative line of treatment when early signs of disease progression are detected. Admittedly, it is currently unclear at what point of disease progression the acquired resistance takes over adaptive drug tolerance. Recent results at the single-cell level have demonstrated that the DTP state is in part conserved at the early resistant setting.26 Our transcriptional data in vitro and, more importantly, the response of PDX to GPX4 inhibitors and the identification of lipid peroxidation markers in a patient biopsy reinforce this hypothesis. Altogether, we believe that this evidence is clinically relevant since patients with BRAFV600E mutant progressing on D/T would undergo treatment discontinuation followed by chemoimmunotherapy or immunotherapy alone, if PD-L1 expression is equal or higher than 50%, with relatively modest efficacy.44 Unfortunately, GPX4 inhibition failed to up-regulate immune checkpoint proteins both in vitro and in vivo (Figure S9), as this might have provided a potential synergistic effect with immunotherapy. Nevertheless, novel compounds with reduced toxicity should be developed in order to consider GPX4 inhibition or alternative ferroptosis triggers a realistic clinical option. In any case, our data, together with pre-existing evidence, reinforce the existence of targetable redox vulnerabilities during the DTP state that could have future clinical applicability.
Figure 7.

Clinical evidence of the occurrence of lipid peroxidation at disease progression following D/T treatment
Representative 4-HNE immunostaining in patient samples pre- and post-D/T treatment. Immunostaining for the epithelial marker CK7 was performed in consecutive sections as control. Scale bars represent 75 μm.
Further supporting the increased susceptibility to ferroptosis following D/T treatment, iron levels are also elevated in DTP and DTEP populations as evidenced by increased ferritin and stable transferrin receptor (TFRC) levels (Figure S10). Although it may seem counterintuitive that cells undergoing oxidative cells stress would up-regulate iron metabolism, this is consistent with the increased dependency of DTP cells on iron-sulfur cluster (ISC) biosynthesis. Indeed, ISC biosynthesis is a significant characteristic of sensitivity in DTPs, as evidenced by their dependency on various ISC assembly factors that appeared as drop-out hits in the genetic screen (Figure 1). Importantly, ISC availability is emerging as a major regulator of ferroptosis sensitivity, with ISC triggering an iron starvation response,45 leading to increased iron uptake and iron mobilization via ferritin degradation, thereby predisposing to ferroptosis.46 At the mitochondrial level, ISC availability is also required for proper regulation of GSH availability, suggesting a potential interaction between ISC availability and GSH dynamics. Further analyses will be required to identify the molecular mechanisms that result in increased ISC dependency.
Finally, our results also demonstrate that ferroptosis activators, that show therapeutic efficacy in the persister stage, display reduced anti-tumor efficacy once resistance mechanisms that reactivate MAPK signaling have been acquired. This may be due to the fact that RAS/MAPK signaling directly activates the expression and activity of the cystine/glutamate antiporter xCT.31 Of note, the SLC7A11 subunit of the antiporter is overexpressed in patients with KRAS-mutant LUAD and its inhibition leads to prolonged survival.47 Imported cystine is then reduced to cysteine providing a rate-limiting substrate for the cellular GSH synthesis and increased antioxidant power. In addition, RAS signaling also inhibits ferroptotic death by up-regulating the ferroptosis suppressor protein 1.48 In sum, in the context of MAPK signaling reactivation, GPX4 is unlikely to be a limiting redox factor thus reducing the therapeutic effect of direct inhibitors or other ferroptosis triggers as described in this manuscript. Indeed, recent evidence suggests that redox metabolism is a crucial function in DTP and DTEP lung cancer populations and its control is exerted at multiple levels.49 This complex and still incompletely characterized network of anti-oxidative functions potentially offers various options with therapeutic potential to increase ROS levels to a lethal threshold in these cellular populations. Some of these options, such as the recently reported BET inhibition,49 may be better tolerated than the ferroptosis triggers described here.
Nevertheless, we have observed that acquired resistance due to MAPK reactivation also results in ROS-dependent vulnerabilities mechanistically different from those observed in the DTP stage. We have confirmed elevated SLC7A11 expression levels and cystine uptake in PDX-derived lung cancer D/T-resistant cell lines with either an NRAS-activating mutation, EGFR amplification, or copy-number gain of the BRAFV600E driver (Figures 5C, 5D, 6B, 6D, and 6E). Treatment of these resistant cells with HDAC inhibitors significantly reduced their viability in an ROS-dependent manner (Figures 5F and 6G–6I). Furthermore, this treatment resulted in a 3-fold decrease in tumor progression in vivo. This is in good agreement with previous findings in D/T-resistant BRAFV600E-mutant melanoma harboring an NRAS mutation.8 However, as previously reported in this tumor context, HDAC inhibitors display ROS-independent effects on cancer cells,8 and it is therefore likely that the therapeutic benefit described here in LUAD is not entirely dependent on the control of the XCt antiporter. Yet, supported by several pre-clinical works,8,50,51,52,53 a phase 1/2 clinical trial (NCT02836548) has been initiated to evaluate vorinostat for the treatment of patients with BRAFV600E-mutant advanced melanoma progressing on D/T treatment.54 Preliminary results were recently reported demonstrating that intermittent vorinostat treatment elicited durable anti-tumor responses in a subset of patients accompanied by the elimination of MAPK reactivating mutations when evaluated by ctDNA.55 The termination of this study will potentially confirm its anti-tumor efficacy previously anticipated in the pre-clinical setting.35 It may also inform of its potential applicability to other BRAF-mutant tumor types progressing on D/T inhibitor therapy due to acquired mutations leading to MAPK reactivation.
Limitations of the study
Our work identifies two redox-related cellular states as sequential treatment vulnerabilities to enhance the response of BRAFV600E-mutant lung cancer to D/T combination therapy. Defining the most sensitive windows to GPX4 and HDAC inhibitors that would maximize therapeutic benefit remains a critical point. Evidence from our in vivo models and from an index patient undergoing D/T treatment suggests some extent of overlapping that could in part be due to the transition nature of the DTEPs. The absence of bona fide DTP and DTEP markers in vivo together with the lack of robust longitudinal sampling during D/T treatment further complicates the definition of precise temporary regimens based on these findings. Furthermore, novel GPX4 inhibitors with enhanced pharmacodynamics properties should be developed. In sum, future studies are mandatory to explore the effectiveness of these stage-specific treatments across a broader spectrum of BRAFV600E-mutant lung cancer models.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-PD-L1 (clone 28-8) | Abcam | Cat#ab205921; RRID: AB_2687878 |
| Rabbit monoclonal anti-PD-L2 (clone D7U8C) | Cell signaling Technology | Cat#82723; RRID:AB_2799999 |
| Rabbit monoclonal anti-CD47 (clone D307P) | Cell signaling Technology | Cat#63000; RRID:AB_2799637 |
| Rabbit monoclonal anti-VISTA (clone D5L5T) | Cell signaling Technology | Cat#54979; RRID:AB_2799474 |
| Rabbit monoclonal anti-CD200 (clone E519V) | Cell signaling Technology | Cat#23451; RRID:AB_3102027 |
| Rabbit polyclonal anti-B7H3 (CD276) | Thermo Fisher Scientific | Cat#PA5-114525; RRID:AB_2890419 |
| Rabbit monoclonal anti-keratin 7 (clone SP52) | Roche Diagnostics | Cat# 05986818001; RRID: AB_3102028 |
| Rabbit polyclonal anti-GPX4 | Abcam | Cat# ab231174; RRID:AB_3073732 |
| Rabbit polyclonal anti-4 Hydroxynonenal | Abcam | Cat# ab46545; RRID:AB_722490 |
| Rabbit monoclonal anti-xCT/SLC7A11 (clone D2M7A) | Cell signaling Technology | Cat#12691; RRID:AB_2687474 |
| Rabbit monoclonal anti-4F2hc/SLC3A2 (clone D3F9D) | Cell signaling Technology | Cat#47213; RRID:AB_2799323 |
| Rabbit polyclonal anti-GPX4 | Cell signaling Technology | Cat#52455; RRID:AB_2924984 |
| Rabbit monoclonal anti-BRAF (clone 55C6) | Cell signaling Technology | Cat#9433; RRID:AB_2259354 |
| Rabbit monoclonal anti-Phospho-MEK1/2 Ser217/221 (clone 41G9) | Cell signaling Technology | Cat#9154; RRID:AB_2138017 |
| Rabbit monoclonal anti-MEK1/2 (clone D1A5) | Cell signaling Technology | Cat#8727; RRID:AB_10829473 |
| Rabbit polyclonal anti-Phospho-p44/42 MAPK (Erk1/2) Thr202/Tyr204 | Cell signaling Technology | Cat#9101; RRID:AB_331646 |
| Mouse monoclonal anti-p44/42 MAPK (Erk1/2) (clone L34F12) | Cell signaling Technology | Cat#4696; RRID:AB_390780 |
| Rabbit monoclonal anti-Phospho-Akt Ser473 (clone D9E) | Cell signaling Technology | Cat#4060; RRID:AB_2315049 |
| Rabbit monoclonal anti-Phospho-Akt Thr308 (clone D25E6) | Cell signaling Technology | Cat#13038; RRID:AB_2629447 |
| Rabbit polyclonal anti-Akt | Cell signaling Technology | Cat#9272; RRID:AB_329827 |
| Rabbit monoclonal anti-Phospho-EGF Receptor Tyr1068 (clone D7A5) | Cell signaling Technology | Cat#3777; RRID:AB_2096270 |
| Rabbit monoclonal anti-EGF Receptor (clone D38B1) | Cell signaling Technology | Cat#4267; RRID:AB_2246311 |
| Rabbit monoclonal anti-Phospho-Histone H3 Ser10 (clone D2C8) | Cell signaling Technology | Cat#3377; RRID:AB_1549592 |
| Rabbit monoclonal anti-Histone H3 (clone D1H2) | Cell signaling Technology | Cat#4499; RRID:AB_10544537 |
| Rabbit monoclonal anti-CD71/TFRC (clone D7G9X) | Cell signaling Technology | Cat#13113; RRID:AB_2715594 |
| Rabbit monoclonal anti-FTH1 (clone D1D4) | Cell signaling Technology | Cat#4393; RRID:AB_11217441 |
| Rabbit monoclonal anti-HSP90 (clone C45G5) | Cell signaling Technology | Cat#4877; RRID:AB_2233307 |
| Goat polyclonal anti-NRAS | Abcam | Cat#ab77392; RRID:AB_1524048 |
| Rabbit polyclonal anti-acetyl-Histone H3 | Merck | Cat#06–599; RRID:AB_2115283 |
| Rabbit polyclonal anit-Actin | Merck | Cat#A2066; RRID:AB_476693 |
| Mouse monoclonal anti-α-Tubulin (clone B-5-1-2) | Merck | Cat#T6074; RRID:AB_477582 |
| Anti-goat IgG, HRP-linked antibody | Thermo Fisher Scientific | Cat#A15999; RRID:AB_2534673 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell signaling Technology | Cat#7074; RRID:AB_2099233 |
| Anti-mouse IgG, HRP-linked Antibody | Cell signaling Technology | Cat#7076; RRID:AB_330924 |
| Anti-rabbit IgG EnVision+ HRP | Agilent | Cat# K400311-2 |
| Biological samples | ||
| Pleural effusions at baseline and after treatment from a BRAFV600E NSCLC patient | DanaFarber Cancer Institute (DF/HCC 02–180) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Dabrafenib mesylate | TargetMol | Cat#T8474 |
| Trametinib | TargetMol | Cat#T125 |
| RSL3 | TargetMol | Cat#T3646 |
| Erastin | TargetMol | Cat#T1765 |
| FIN56 | TargetMol | Cat#T4066 |
| Trolox | TargetMol | Cat#T1710 |
| Ferrostatin-1 | TargetMol | Cat#T6500 |
| Liproxstatin-1 | TargetMol | Cat#T2376 |
| Deferoxamine mesylate | TargetMol | Cat#T8474 |
| Vorinostat | TargetMol | Cat#T1583 |
| Panobinostat | TargetMol | Cat#T2383 |
| Lovastatin | TargetMol | Cat#T1207 |
| Simvastatin | TargetMol | Cat#T0687 |
| Atorvastatin | TargetMol | Cat#T20765 |
| Fluvastatin sodium | TargetMol | Cat#T1487 |
| N-acetyl-L-cysteine (NAC) | Merck | Cat#A7250 |
| Sodium selenite | Merck | Cat#214485 |
| Critical commercial assays | ||
| GSH-Glo™ Glutathione Assay | Promega | Cat#V6911 |
| CellROX™ Green Flow Cytometry Assay kit | Thermo Fisher Scientific | Cat#C10492 |
| FerroOrange Assay Kit | Dojindo Laboratories | Cat#F374 |
| BODIPY™ 665/676 (Lipid Peroxidation Sensor) | Thermo Fisher Scientific | Cat#B3932 |
| FITC Annexin V Apoptosis Detection Kit I | BD Biosciences | Cat#556547 |
| Deposited data | ||
| RNA-seq data | This paper | GEO:GSE271247 |
| Experimental models: Cell lines | ||
| HCC364 | Dana-Farber Cancer Institute, authenticated by Eurofins Genomics using the Applied Biosystems AmpFLSTR Identifier Plus | RRID:CVCL_5134 |
| DFCI471 | Dana-Farber Cancer Institute14 | N/A |
| 1D and 1E PDX-derived cell lines | Prof. P. Lito29 | N/A |
| Experimental models: Organisms/strains | ||
| NSG mice | Charles River | Strain code 614 |
| Oligonucleotides | ||
| qRT-PCR sequences | This paper | See Table S1 |
| sgRNA sequences | This paper | See Table S2 |
| Recombinant DNA | ||
| pLVX-HA-NRASQ61K | This paper | N/A |
| pLKO.1-TRC shRNA control | MISSION (Merck) | N/A |
| pLKO.1-TRCN0000033256 (shNRAS#1) | MISSION (Merck) | N/A |
| pLKO.1-TRCN0000033257 (shNRAS#2) | MISSION (Merck) | N/A |
| HP138-puro | Dr. I. Cheeseman | Addgene Plasmid #134246 |
| TLCV2 | Dr. A. Karpf | Addgene Plasmid #87360 |
| lenti-sgRNA blast | Dr. B. Stringer | Addgene Plasmid #104993 |
| Human sgRNA library Brunello in lentiGuide-Puro | Dr. D. Root & J. Doench | Addgene Plasmid #73178 |
| pJS10-Cas9 blast | This paper | N/A |
| pLenti6-CMV-luciferase | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism8 | GraphPad | https://www.graphpad.com |
| QuPath 0.4.3 | QuPath | https://qupath.github.io/ |
| CRISPRAnalyzeR | https://github.com/boutroslab/CRISPRAnalyzeR | |
| MAGeCK | https://sourceforge.net/projects/mageck/ | |
| R version 4.1 | R Core Team 2017 | https://www.r-project.org |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David Santamaría (d.santamaria@usal.es).
Materials availability
Further requests for cell lines generated in this study should be directed to and will be fulfilled by the lead contact, David Santamaría (d.santamaria@usal.es), with the Materials Transfer Agreement.
Data and code availability
Original/source data of RNA-sequencing (accession number: GSE271247) have been deposited at https://www.ncbi.nlm.nih.gov/geo/and are publicly available as of the date of publication. This study did not report new original code. Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Cell lines and cell culture
Human BRAFV600E-mutant lung adenocarcinoma cell line HCC364 was obtained from the Dana Farber Cancer Institute. DFCI471 cells were derived from a fresh biopsy of a BRAFV600E-mutated LUAD patient who progressed following D/T treatment.14 These cells were maintained in vitro in presence of a high concentration of dabrafenib (1μM). BRAFV600E-mutant PDX-derived cell lines (clone 1D and 1E) generated from a patient at disease progression after treatment with a RAF inhibitor29 at Prof. Lito’s laboratory (Memorial Sloan Kettering Cancer Center, New York, NY, USA). All cell lines were regularly checked for mycoplasma contamination using MycoAlert Mycoplasma Detection Kit (Lonza). Cells were either cultured in standard DMEM (HCC364 and derivatives, 1D and 1E cells) or in RPMI-1640 (DFCI471 cells) medium both containing 10% fetal bovine serum (FBS, ThermoFisher Scientific), 2 mM L-glutamine (ThermoFisher Scientific) and 1% penicillin-streptomycin (ThermoFisher Scientific).
Generation of cell line derivatives: HCC364-DTPs, DTEPs and EGFRa were generated by treating HCC364 cells with 250 nM dabrafenib and 5 nM trametinib (D/T 250/5 nM) for 3, 40 and 70 weeks, respectively. 1D-DTPs and 1E-DTPs were obtained by treating parental 1D and 1E PDX-derived cells with D/T 250/5 nM for 3 weeks 1D-DTEPs were generated by maintaining the D/T 250/5 nM treatment for 40 weeks 1E-DTEPs were generated by maintaining the D/T 250/5 nM treatment for 30 weeks. From this point onwards a cell line derivative containing a BRAF gene amplification (CNV 15.4) was generated and was referred to as 1E-R. In all cases, the drugs were refreshed every 3 days. In order to determine if long-term D/T treated cells acquired a genetic alterations as resistance mechanism, these cells were cultured in absence of D/T (drug holiday) for 3 weeks and, then, treated again with the drugs. The proliferation of these rechallenged cells was followed during 1 week and compared to long term cells constantly cultured with D/T (250/5 nM). HCC364NRAS cells were generated by lentiviral transduction of pLVX-HA-NRASQ61K in HCC364 DTPs, allowing resistance to D/T by exogenous expression of NRASQ61K mutant.
Animals and in vivo studies
All animal experimental procedures were performed according to the Federation of European Laboratory Animal Sciences Associations (FELASA). The procedures were reviewed and approved either by the Ethic Committee of the University of Bordeaux (France) under the number APAFIS #15445 or by the Institutional Animal Care and Ethics Committee of the University of Liège (Belgium) under the number 22–2422. Female 6-week-old NSG mice were purchased from Charles River (strain code 614). All animals were housed either in the Animal Facility A2 of the University of Bordeaux (institutional agreement number: A33063916) or in the GIGA-accredited animal facility of the University of Liège (institutional agreement number: LA1610002).
For subcutaneous xenografts, 2.5x106 DFCI471 cells were resuspended in 100 μL RPMI supplemented with 20% (v/v) matrigel (BD Biosciences) and were inoculated subcutaneously in one flank of 8-week-old female NSG mice. Tumor volume was measured with a caliper using the formula W x W x L/2, where W and L denote width and length, respectively. When tumor volume reached 150 mm3, mice were randomized in 3 groups and treated with vehicle, vorinostat (100 mg/kg/day) or panobinostat (10 mg/kg/day) by intraperitoneal injection 5 days per week (n = 10 per condition). Drugs were dissolved in 5% DMSO, 30% PEG-300 and 5% Tween-80 solution. After 2 weeks, mice were sacrificed and tumors were collected. One piece was embedded in paraffin for IHC and the rest was frozen in liquid nitrogen for total protein extraction.
For lung orthotopic implantation, PDX-1D cells were first stably transduced with pLenti6-CMV-luciferase and selected with 10 μg/mL blasticidin. One million 1D cells were resuspended in 50 μL DMEM with 20% (v/v) matrigel and were implanted by transcutaneous intralung injection in 8-week-old female NSG mice. Tumor growth was monitored using bioluminescence imaging once a week. When tumors started to grow (18 days post-implantation), mice were treated with a combination of dabrafenib (30 mg/kg/day) and trametinib (0.2 mg/kg/day) by oral gavage 5 days per week (n = 15 D/T and 3 vehicle mice). When D/T treated tumors reached a volume 20% higher to that estimated at the beginning of the drug treatment (disease progression), mice were randomized in 2 groups and treated with RSL3 (10 mg/kg/day) by intraperitoneal injection 5 days per week (n = 6–7 per condition). All drugs were dissolved in 5% DMSO, 30% PEG-300 and 2% Tween-80 solution. After 2 weeks, mice were sacrificed and lungs were collected for further analysis.
Patient sample
Pre and post D/T treatment samples were obtained from a 74 years old male patient with a BRAFV600E mutated NSCLC, who gave his written informed consent to the correlative protocol DF/HCC 02–180. Two samples were included in this study: a baseline pleural effusion and a recurrent pleural effusion. Tumor DNA only sequencing using the DFCI OncoPanel platform, and immunohistochemistry (described in methods details) were performed on both these samples. The protocol DF/HCC 02–180 was approved by the Dana-Farber Cancer Institute IRB.
Method details
shRNA-mediated NRAS knock-down
The following vectors from the MISSION library were used: MISSION pLKO.1- TRC shRNA control, TRCN0000033256 and TRCN0000033257 targeting human NRAS. Cells were infected with lentiviral particles and selected with Puromycin 1 μg/ml in order to obtain stable populations.
GPX4 and SLC7A11 inducible CRISPR-Cas9 Knock-out
Parental cell lines (HCC364-DTPs and HCC364-EGFRa) were co-transfected with transposase and hp138-puro (a gift from Iain Cheeseman –Addgene plasmid # 134246–) and selected with puromycin to generate doxycycline-inducible Cas9 derivatives. HCC364-NRAS were infected with lentiviral particles generated using TLCV2 (a gift from Adam Karpf –Addgene plasmid # 87360–) and FACS-sorted using the GFP reporter. In all cases expression of Cas9 was confirmed by western blotting. Cells were subsequently infected with lentiviral particles generated using plenti-sgRNA blast (a gift from Brett Stringer –Addgene plasmid # 104993–) and selected with blasticidin. The specific sgRNA sequences are listed in Tables S2.
Genome-wide sgRNA library construction
In this study, the human Brunello CRISPR knockout sgRNA library (Addgene #73178), a gift from David Root and John Doench (Broad Institute, Boston, USA), was used to identify genes responsible for D/T resistance in HCC364 cells. This pooled library contains 76,441 unique sgRNA sequences targeting 19114 protein-coding genes (4 sgRNAs/gene) and 1000 control non-targeting sgRNAs. Amplification of pooled sgRNA library, lentivirus production and titer determination were performed as previously described.56
CRISPR/Cas9 screening for determinants of dabrafenib/trametinib sensitivity
The workflow of this screen is illustrated in Figure 1A. First, we generated a stable Cas9-expressing HCC364 cells (HCC364-Cas9) by lentiviral transduction of pJS10-Cas9 plasmid (MOI = 1) and selection with blasticidin. Both the expression and the activity of Cas9 were validated by western blot and FACS analysis using control sgRNAs, respectively. Then we transduced HCC364-Cas9 cells with the Brunello library at a MOI of 0.3 aiming for coverage of, on average, 1,000 cells per sgRNA. The positively transduced cells were selected with 1 μg/mL of puromycin for 7 days to generate a mutant cell pool. 8x107 cells, representing the baseline of sgRNAs representation, were collected for further analysis. This latter was then treated with vehicle (DMSO) or 250 nM dabrafenib and 5 nM trametinib for 3 weeks (medium was replaced every 3–4 days). After the treatment, at least 8x107 cells were harvested for genomic DNA extraction using Quick-DNA Midiprep plus kit (Zymo Research D4075). Illumina sequencing libraries were generated using PCR amplification with primers specific to the genome integrated lentiviral vector backbone sequence, as previously described.56 These libraries were sequenced with a MiSeq instrument (Illumina). The number of reads was adjusted to cover each sgRNA with approximately 1000 reads.
CRISPR/cas9 screening data analysis
Fastq files were processed (screen quality, read cleaning and mapping) and analyzed (hit calling) using CRISPRAnalyzeR.10 Hit calling was performed using MaGECK and a Z-Ratio was calculated between the pre-treated and treated cells.
RNA-sequencing and data analysis
Transcriptome sequencing (RNA-seq) was performed in parental, DTP and DTEP-HCC364 cells. Fastq files from different lanes were merged and sequencing quality was analyzed using FastQC version 0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were trimmed using trimmomatic version 0.3957 using parameters LEADING:30 TRAILING:30 MINLEN:36. Read mapping and gene quantification were performed using RSEM version 1.3.258 with STAR aligner and default parameters.
Gene Set Enrichment Analysis (GSEA) was performed with fgsea R package version 1.20.059 on estimated Transcript Per Million (TPM) log fold-changes with KEGG, Reactome, Wikipathway and Hallmark gene sets from MSig database version 760 as well as drug tolerance gene signature.26 The score computation to evaluate the activity of E2F targets (Hallmarks) and cell cycle checkpoints (Reactome) pathways was performed following single sample GSEA method by using GSVA R package version 1.42.061 on log2 transformed TPM with parameters method = "ssgsea", min.sz = 10, ssgsea.norm = T. Senescence score was computed from RNA-seq raw counts with the online tool SENCAN - cancer senescence classifier (https://rhpc.nki.nl/sites/senescence/classifier.php) reached on the August the 10th 2022.
GSH measurement
Cells were seeded in 96-well white plates and cultured overnight at 37°C in a 5% CO2 incubator. Glutathione (GSH) intracellular concentration was assessed using GSH-Glo Glutathione Assay (V6911, Promega) following the manufacturer’s instructions.
ROS measurement
The cells were treated in presence of drugs for 72 h. ROS production by cancer cell lines was measured using CellROX Green Flow Cytometry assay kit (C10492, ThermoFisher Scientific) according to the manufacturer’s protocol.
Intracellular iron measurement
Intracellular ferrous ion (Fe2+) levels were determined by flow cytometry using FerroOrange probe (F374-10, Dojindo). Briefly, 24 h after seeding, cells were washed with HBSS three times. After trypsinization, the cell suspension in complete medium was transferred to a FACS tube and was centrifuged at 1,500 rpm for 3 min. The cell pellet was washed once with HBSS and then, was resuspended in serum-free DMEM medium with 0.2 μM FerroOrange. A negative control was performed by adding 1 μM of deferoxamine. The cells were incubated for 15–30 min in a 37°C incubator equilibrated with 5% CO2. The samples were analyzed using a LSRFortessa flow cytometer (BD Biosciences).
Peroxidized lipids content measurement
Peroxidized lipids content was measured using the lipid peroxidation probe BODIPY 665/676 (B3932, ThermoFisher Scientific). The cells were treated in the presence of drugs (Erastin, RSL3 and Trolox) for 72 h. After trypsinization, the cell pellet was resuspended in 4 μM BODIPY staining solution in PBS and incubated in the dark for 20 min at 37°C. The cells were washed with PBS 3 times and analyzed using a FACSCanto II flow cytometer (BD Biosciences).
Cell growth assay
For IC50 determination assays, 12-points serial dilutions of the different drugs/compounds (Dabrafenib, Trametinib, Erastin, RSL3, FIN56, Trolox, Ferrostatin-1, Liproxstatin-1, Deferoxamine, Vorinostat, Panobinostat, Lovastatin, Simvastatin, Atorvastatin, Fluvastatin, N-acetyl-L-cysteine and sodium selenite) were added to 96-well plates containing cells (HCC364, 3,000 cells per well; DFCI471, 1D and 1E, 5,000 cells per well). After 72 h incubation, cell viability was determined using MTT reagent and absorbance was read at 595 nm on a spectrophotometer plate reader. For proliferation curve assays, cell viability was measured at day 1, 3, 5 and 7 after treatment with D/T (refreshed every 3 days).
Clonogenic assay
Cells were seeded into 6-well plates (10,000 cells per well) and cultured with the indicated drugs (Dabrafenib, Trametinib, Vorinostat, Panobinostat and N-acetyl-L-cysteine) for 10–15 days. At the end of the assay, cells were fixed with 4% of formaldehyde and stained with 2% of crystal violet. After photo scanning, crystal violet was released by cell lysis using 5% SDS solution and quantified by absorbance measurement using a spectrophotometer plate reader.
Apoptosis assay
HCC364 cells were treated with Dabrafenib/Trametinib (D/T 250/5 nM) for 24, 48 and 72 h. Cells were collected and apoptosis was measured with the FITC Annexin V Apoptosis Detection Kit I (BD Biosciences) using a FACSCanto II flow cytometer (BD Biosciences).
Immunohistochemistry
Following the deparaffinization step, tissue sections were autoclaved for 11 min at 126°C in 10 mM citrate buffer (pH6), Target Retrieval Solution (S1699, Dako) or in Tris/EDTA buffer (pH9). Endogenous peroxidases were blocked by incubation in 3% H2O2 solution for 20 min and slides were incubated with animal free blocking solution (15019L, Cell Signaling) for 20 min at room temperature (RT). The following antibodies were used for the primary reaction [1 h at room temperature in diluent solution (S2022, Dako)]: anti-PD-L1 (1/300, clone 28-8, Abcam), anti-PD-L2 (1/200, clone D7U8C, Cell Signaling), anti-CD47 (1/400, clone D307P, Cell Signaling), anti-VISTA (1/400, clone D5L5T, Cell Signaling), anti-CD200 (1/200, clone E519V, Cell Signaling), anti-B7-H3 (1/1500, polyclonal, #PA5-114525, ThermoFisher Scientific), anti-keratin 7 (ready to use, clone SP52, Ventana Medical Systems), anti-GPX4 (1/500, ab231174, Abcam) and anti-4-hydroxynonenal (4-HNE, 1/400, ab46545, Abcam). After washes in PBS, the secondary reaction was performed using the rabbit Envision+/HRP (Agilent Technologies) according to the manufacturer’s recommendations. Positive cells were visualized using SignalStain DAB Substrate Kit (Cell Signaling). Samples were counterstained with hematoxylin and mounted with EUKITT (Kindler GmbH). For anti-GPX4 and anti-4-HNE antibodies, the intensity of the staining (0, 1+, 2+, 3+) in tumor cells was assessed in three independent immunolabeled tissue sections and results are expressed as mean of these three scores per lung. Regarding the expression of immune checkpoints by tumor cells, the number of positive cells per mm2 was precisely determined by computerized counts (QuPath 0.4.3 software for digital pathology image analysis).
Somatic variants analysis
DNA samples were screened for somatic variants by using the ‘Solid Tumor Solution’ (Sophia Genetics SA, Switzerland) according to the manufacturer’s protocol. Sequencing was performed on a MiSeq platform using v3 chemistry (Illumina, USA). Data were analyzed using the Sophia DDM Platform with Sophia Genetics proprietary algorithms.
Cystine uptake
Cells were seeded in 96-well back plates and cultured overnight at 37°C in a 5% CO2 incubator. Cystine uptake by cancer cells was measured using the Cystine uptake assay kit (UP05-12, Dojindo) according to the manufacturer’s protocol.
Western blotting
Cells were extracted in RIPA buffer (150 mM NaCl, 0.5% Na+-deoxycholate, 1% Triton X-100, 0.1% SDS, 50 mM Tris-HCl pH 7.5 and protease/phosphatase inhibitors). Protein concentrations were determined using the DC Protein Assay kit (Bio-Rad). Twenty μg of proteins were separated by 7.5–12.5% SDS-PAGE and transferred to PVDF or nitrocellulose membranes. Membranes were saturated for 1h with blocking buffer [5% skim milk in tris-buffered saline with 0.1% Tween 20 (TBS-T)] for 1 h at room temperature (RT) and probed overnight in primary antibody at 4°C. The following primary antibodies raised against xCT/SLC7A11 (#12691), 4F2hc/CD98/SLC3A2 (#47213), GPX4 (#52455), BRAF (#9433), phospho-MEK1/2 S217/221 (#9154), MEK1/2 (#8727), phospho-ERK1/2 T202/Y204 (#9101), ERK1/2 (#4696), P-AKT S473 (#4060), P-AKT T308 (#13038), AKT (#9272), phospho-EGFR Y1068 (#3777), EGFR (#4267), phospho-Histone H3 S10 (#3377), histone H3 (#4499), TFRC (#13113), FTH1 (#4393) and HSP90 (#4877) were purchased from Cell Signaling Technology. Antibodies also included those raised against NRAS (ab77392, Abcam), acetyl-Histone H3 (06–599, Sigma-Aldrich), actin (A2066, Sigma-Aldrich) and alpha-tubulin (T6074, Sigma-Aldrich). After three washes, the membranes were exposed to appropriate secondary HRP-linked antibody (anti-rabbit IgG #7074 or anti-mouse IgG #7076 antibodies from Cell Signaling Technology or anti-goat IgG #A15999 from Thermo Fisher Scientific) at RT for 1 h. The immunoreactive bands were visualized using Clarity Western ECL Substrate (Bio-Rad).
RNA extraction, reverse transcription and quantitative PCR (RT-qPCR)
Total cellular RNA was extracted reverse-transcribed and then subjected to PCR amplification and quantification, as previously described.62 Briefly, one microgram of total cellular RNA, extracted using the Quick-RNA MiniPrep Plus Kit (Zymo Research), was reverse-transcribed utilizing the OneScript cDNA Synthesis Kit (Abm). The resulting cDNA was then amplified by PCR with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). The PCR signals were captured using a CFX384 Touch Real-Time PCR System and analyzed with Bio-Rad’s CFX Manager software. Primer sequences are listed in Table S1.
Quantification and statistical analysis
All experiments were performed as several independent biological replicates. All results were reported as means with standard deviation (SD) or Standard Error Mean (SEM) as indicated in figure legends. The number of biological replicates as well as the number of animals in treatment groups are indicated in the figure legends. Two group comparisons were performed using unpaired Student’s t test with or without Welsch’s correction according to homoscedasticity. When an experiment required comparisons between more than two groups, statistical analysis was performed using one-way or two-way ANOVA depending on the number of grouping factors. Dunnett’s or Tukey’s post-test was applied for simple or multiple comparisons, respectively. In the case of discrete variables (IHC scores), the comparison between groups was performed by Mann-Whitney’s U test. In all cases, a bilateral p < 0.05 was considered as statistically significant with a 95% confidence interval. Statistical tests are indicated in figure legends and exact p values are indicated in the figures. All analyses were conducted with GraphPad Prism 8 (GraphPad Software, USA).
Acknowledgments
Research in D.S. laboratory was funded by Siric-BRIO (Bordeaux Recherche Intégrée en Cancérologie, INCa convention 2018-006 and INSERM convention 18CI05500) and a Senior Chair Idex-Université de Bordeaux (2016-0396) and grants from Institut National Du Cancer (INCA_13911), Ministerio de Ciencia e Innovación (Retos PID2020-116824RB-I00), and by AECC Excellence Program 2022 (EPAEC222641CICS). M.-J.N. was supported by postdoctoral fellowships from Fondation ARC pour la recherche contre le cancer and from FNRS-Télévie. This work was also supported by the Fund André Daners, managed by the King Baudouin Foundation and by the Foundation Leon Fredericq. A.V. received funding from ISCIII (grant PI22-00548, co-funded by FEDER). E.N. received funding from ISCIII (grant PI21/00789, co-funded by FEDER). C.A. received funding by the Giovanni Armenise-Harvard Foundation, the Italian Ministry of University and Research (FARE-R207ENY9KZ), the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant 101001288), and AIRC under IG 2021-ID. 25737 project. The authors are thankful to Mrs. F. Perin and A. Demanche for expert technical assistance. We acknowledge the technology platforms of the GIGA at University of Liege: animal, imaging/flow cytometry, and viral vector facilities. The authors also thank the Animalerie A2 (Bordeaux University) for excellent assistance and CRISP’edit and Vect’UB facilities (TBMCore UAR CNRS3427, Inserm US05, Bordeaux University) for help with library packaging into lentiviral particles and CRISPR experiments and analysis. The schemes were created with BioRender.com.
Author contributions
M.-J.N., C.A., and D.S. conceived this project and designed the study. M.-J.N., E.D., E.R., S.S.J., V.P.-M., B.T., L.G., R.C., V.V., B.K., F.L., J.H., A.V., and M.H. performed the experiments and analyzed the data. S.d.H., J.Y.X., B.S., E.Z., A.B., D.C., O.C., P.L., P.A.J., B.R., and M.A. provided materials, methodological support, and/or conceptual advice. M.-J.N., P.P., E.N., B.R., C.A., and D.S. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Declaration of interests
D.S. received research fees from Aelin Therapeutics. C.A. received research fees from Revolution Medicines, Aelin Therapeutics, Verastem, Roche, and Boehringer Ingelheim. E.N. reports research funding from Pfizer and Roche. P.L. is listed as an inventor on patent applications filed by MSKCC that describe approaches to treat KRAS or BRAF-mutant tumors. P.A.J. has received consulting fees from AstraZeneca, Boehringer Ingelheim, Pfizer, Roche/Genentech, Takeda Oncology, ACEA Biosciences, Eli Lilly and Company, Araxes Pharma, Ignyta, Mirati Therapeutics, Novartis, Loxo Oncology, Daiichi Sankyo, Sanofi Oncology, Voronoi, SFJ Pharmaceuticals, Takeda Oncology, Transcenta, Silicon Therapeutics, Syndax, Nuvalent, Bayer, Eisai, Biocartis, Allorion Therapeutics, Accutar Biotech, Monte Rosa, Scorpion Therapeutics, Merus, Frontier Medicines, Hongyun Biotechnology, Duality, and AbbVie; post-marketing royalties from DFCI-owned intellectual property on EGFR mutations licensed to Lab Corp; sponsored research agreements with AstraZeneca, Daiichi Sankyo, Puma, Boehringer Ingelheim, Eli Lilly and Company, Revolution Medicines, and Astellas Pharmaceuticals; and stock ownership in Gatekeeper Pharmaceuticals. M.M.A. reports grants and personal fees from Genentech, grants and personal fees from Bristol Myers Squibb, personal fees from Merck, grants and personal fees from AstraZeneca, grants from Lilly, and personal fees from Maverick, Blueprint Medicine, Syndax, Ariad, Nektar, Gritstone, ArcherDX, Mirati, NextCure, Novartis, EMD Serono, and Panvaxal/NovaRx, outside the submitted work.
Published: August 1, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2024.101663.
Contributor Information
Marie-Julie Nokin, Email: mjnokin@uliege.be.
Chiara Ambrogio, Email: chiara.ambrogio@unito.it.
David Santamaría, Email: d.santamaria@usal.es.
Bolero Consortium:
Marie-Julie Nokin, Ernest Nadal, Chiara Ambrogio, and David Santamaría
Supplemental information
References
- 1.Lavoie H., Therrien M. Structural keys unlock RAS-MAPK cellular signalling pathway. Nature. 2022;609:248–249. doi: 10.1038/d41586-022-02189-7. [DOI] [PubMed] [Google Scholar]
- 2.Wiesweg M., Preuss C., Roeper J., Metzenmacher M., Eberhardt W., Stropiep U., Wedeken K., Reis H., Herold T., Darwiche K., et al. BRAF mutations and BRAF mutation functional class have no negative impact on the clinical outcome of advanced NSCLC and associate with susceptibility to immunotherapy. Eur. J. Cancer. 2021;149:211–221. doi: 10.1016/j.ejca.2021.02.036. [DOI] [PubMed] [Google Scholar]
- 3.Dagogo-Jack I., Martinez P., Yeap B.Y., Ambrogio C., Ferris L.A., Lydon C., Nguyen T., Jessop N.A., Iafrate A.J., Johnson B.E., et al. Impact of BRAF Mutation Class on Disease Characteristics and Clinical Outcomes in BRAF-mutant Lung Cancer. Clin. Cancer Res. 2019;25:158–165. doi: 10.1158/1078-0432.CCR-18-2062. [DOI] [PubMed] [Google Scholar]
- 4.Tabbo F., Pisano C., Mazieres J., Mezquita L., Nadal E., Planchard D., Pradines A., Santamaria D., Swalduz A., Ambrogio C., et al. How far we have come targeting BRAF-mutant non-small cell lung cancer (NSCLC) Cancer Treat Rev. 2022;103 doi: 10.1016/j.ctrv.2021.102335. [DOI] [PubMed] [Google Scholar]
- 5.Planchard D., Besse B., Groen H.J.M., Hashemi S.M.S., Mazieres J., Kim T.M., Quoix E., Souquet P.J., Barlesi F., Baik C., et al. Phase 2 Study of Dabrafenib Plus Trametinib in Patients With BRAF V600E-Mutant Metastatic NSCLC: Updated 5-Year Survival Rates and Genomic Analysis. J. Thorac. Oncol. 2022;17:103–115. doi: 10.1016/j.jtho.2021.08.011. [DOI] [PubMed] [Google Scholar]
- 6.Facchinetti F., Lacroix L., Mezquita L., Scoazec J.Y., Loriot Y., Tselikas L., Gazzah A., Rouleau E., Adam J., Michiels S., et al. Molecular mechanisms of resistance to BRAF and MEK inhibitors in BRAF(V600E) non-small cell lung cancer. Eur. J. Cancer. 2020;132:211–223. doi: 10.1016/j.ejca.2020.03.025. [DOI] [PubMed] [Google Scholar]
- 7.Ortiz-Cuaran S., Mezquita L., Swalduz A., Aldea M., Mazieres J., Leonce C., Jovelet C., Pradines A., Avrillon V., Chumbi Flores W.R., et al. Circulating Tumor DNA Genomics Reveal Potential Mechanisms of Resistance to BRAF-Targeted Therapies in Patients with BRAF-Mutant Metastatic Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020;26:6242–6253. doi: 10.1158/1078-0432.CCR-20-1037. [DOI] [PubMed] [Google Scholar]
- 8.Wang L., Leite de Oliveira R., Huijberts S., Bosdriesz E., Pencheva N., Brunen D., Bosma A., Song J.Y., Zevenhoven J., Los-de Vries G.T., et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell. 2018;173:1413–1425.e14. doi: 10.1016/j.cell.2018.04.012. [DOI] [PubMed] [Google Scholar]
- 9.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R., et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winter J., Schwering M., Pelz O., Rauscher B., Zhan T., Heigwer F., Boutros M. CRISPRAnalyzeR: Interactive analysis, annotation and documentation of pooled CRISPR screens. bioRxiv. 2017 doi: 10.1101/109967. Preprint at. [DOI] [Google Scholar]
- 11.Sanchez-Burgos L., Navarro-Gonzalez B., Garcia-Martin S., Sirozh O., Mota-Pino J., Fueyo-Marcos E., Tejero H., Anton M.E., Murga M., Al-Shahrour F., Fernandez-Capetillo O. Activation of the integrated stress response is a vulnerability for multidrug-resistant FBXW7-deficient cells. EMBO Mol. Med. 2022;14 doi: 10.15252/emmm.202215855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nokin M.J., Ambrogio C., Nadal E., Santamaria D. Targeting Infrequent Driver Alterations in Non-Small Cell Lung Cancer. Trends Cancer. 2021;7:410–429. doi: 10.1016/j.trecan.2020.11.005. [DOI] [PubMed] [Google Scholar]
- 13.Wang V.E., Xue J.Y., Frederick D.T., Cao Y., Lin E., Wilson C., Urisman A., Carbone D.P., Flaherty K.T., Bernards R., et al. Adaptive Resistance to Dual BRAF/MEK Inhibition in BRAF-Driven Tumors through Autocrine FGFR Pathway Activation. Clin. Cancer Res. 2019;25:7202–7217. doi: 10.1158/1078-0432.CCR-18-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abravanel D.L., Nishino M., Sholl L.M., Ambrogio C., Awad M.M. An Acquired NRAS Q61K Mutation in BRAF V600E-Mutant Lung Adenocarcinoma Resistant to Dabrafenib Plus Trametinib. J. Thorac. Oncol. 2018;13:e131–e133. doi: 10.1016/j.jtho.2018.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niemantsverdriet M., Schuuring E., Elst A.T., van der Wekken A.J., van Kempen L.C., van den Berg A., Groen H.J.M. KRAS Mutation as a Resistance Mechanism to BRAF/MEK Inhibition in NSCLC. J. Thorac. Oncol. 2018;13:e249–e251. doi: 10.1016/j.jtho.2018.07.103. [DOI] [PubMed] [Google Scholar]
- 16.Lin L., Sabnis A.J., Chan E., Olivas V., Cade L., Pazarentzos E., Asthana S., Neel D., Yan J.J., Lu X., et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015;47:250–256. doi: 10.1038/ng.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hangauer M.J., Viswanathan V.S., Ryan M.J., Bole D., Eaton J.K., Matov A., Galeas J., Dhruv H.D., Berens M.E., Schreiber S.L., et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–250. doi: 10.1038/nature24297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Viswanathan V.S., Ryan M.J., Dhruv H.D., Gill S., Eichhoff O.M., Seashore-Ludlow B., Kaffenberger S.D., Eaton J.K., Shimada K., Aguirre A.J., et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–457. doi: 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stockwell B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401–2421. doi: 10.1016/j.cell.2022.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharma S.V., Lee D.Y., Li B., Quinlan M.P., Takahashi F., Maheswaran S., McDermott U., Azizian N., Zou L., Fischbach M.A., et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin H., Wang L., Bernards R. Rational combinations of targeted cancer therapies: background, advances and challenges. Nat. Rev. Drug Discov. 2023;22:213–234. doi: 10.1038/s41573-022-00615-z. [DOI] [PubMed] [Google Scholar]
- 22.Ramirez M., Rajaram S., Steininger R.J., Osipchuk D., Roth M.A., Morinishi L.S., Evans L., Ji W., Hsu C.H., Thurley K., et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016;7 doi: 10.1038/ncomms10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saleh T., Gewirtz D.A. Considering therapy-induced senescence as a mechanism of tumour dormancy contributing to disease recurrence. Br. J. Cancer. 2022;126:1363–1365. doi: 10.1038/s41416-022-01787-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L., Lankhorst L., Bernards R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer. 2022;22:340–355. doi: 10.1038/s41568-022-00450-9. [DOI] [PubMed] [Google Scholar]
- 25.Jochems F., Thijssen B., De Conti G., Jansen R., Pogacar Z., Groot K., Wang L., Schepers A., Wang C., Jin H., et al. The Cancer SENESCopedia: A delineation of cancer cell senescence. Cell Rep. 2021;36 doi: 10.1016/j.celrep.2021.109441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Figarol S., Delahaye C., Gence R., Asslan R., Pagano S., Tardy C., Colinge J., Villemin J.-P., Maraver A., Ferrer I., et al. Farnesyltransferase inhibition overcomes the adaptive resistance to osimertinib in EGFR-mutant NSCLC. Nat Commun. 2024;15:5345. doi: 10.1038/s41467-024-49360-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moosmann B., Behl C. Selenoprotein synthesis and side-effects of statins. Lancet. 2004;363:892–894. doi: 10.1016/S0140-6736(04)15739-5. [DOI] [PubMed] [Google Scholar]
- 28.Zheng J., Conrad M. The Metabolic Underpinnings of Ferroptosis. Cell Metabol. 2020;32:920–937. doi: 10.1016/j.cmet.2020.10.011. [DOI] [PubMed] [Google Scholar]
- 29.Xue Y., Martelotto L., Baslan T., Vides A., Solomon M., Mai T.T., Chaudhary N., Riely G.J., Li B.T., Scott K., et al. An approach to suppress the evolution of resistance in BRAF(V600E)-mutant cancer. Nat. Med. 2017;23:929–937. doi: 10.1038/nm.4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eisenhauer E.A., Therasse P., Bogaerts J., Schwartz L.H., Sargent D., Ford R., Dancey J., Arbuck S., Gwyther S., Mooney M., et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur. J. Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 31.Lim J.K.M., Delaidelli A., Minaker S.W., Zhang H.F., Colovic M., Yang H., Negri G.L., von Karstedt S., Lockwood W.W., Schaffer P., et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. Natl. Acad. Sci. USA. 2019;116:9433–9442. doi: 10.1073/pnas.1821323116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson D.B., Menzies A.M., Zimmer L., Eroglu Z., Ye F., Zhao S., Rizos H., Sucker A., Scolyer R.A., Gutzmer R., et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer. 2015;51:2792–2799. doi: 10.1016/j.ejca.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikubo M., Inoue Y., Liu G., Tsao M.S. Mechanism of Drug Tolerant Persister Cancer Cells: The Landscape and Clinical Implication for Therapy. J. Thorac. Oncol. 2021;16:1798–1809. doi: 10.1016/j.jtho.2021.07.017. [DOI] [PubMed] [Google Scholar]
- 34.Raha D., Wilson T.R., Peng J., Peterson D., Yue P., Evangelista M., Wilson C., Merchant M., Settleman J. The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer Res. 2014;74:3579–3590. doi: 10.1158/0008-5472.CAN-13-3456. [DOI] [PubMed] [Google Scholar]
- 35.Dhimolea E., de Matos Simoes R., Kansara D., Al'Khafaji A., Bouyssou J., Weng X., Sharma S., Raja J., Awate P., Shirasaki R., et al. An Embryonic Diapause-like Adaptation with Suppressed Myc Activity Enables Tumor Treatment Persistence. Cancer Cell. 2021;39:240–256.e11. doi: 10.1016/j.ccell.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oren Y., Tsabar M., Cuoco M.S., Amir-Zilberstein L., Cabanos H.F., Hutter J.C., Hu B., Thakore P.I., Tabaka M., Fulco C.P., et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature. 2021;596:576–582. doi: 10.1038/s41586-021-03796-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahu N., Stephan J.P., Cruz D.D., Merchant M., Haley B., Bourgon R., Classon M., Settleman J. Functional screening implicates miR-371-3p and peroxiredoxin 6 in reversible tolerance to cancer drugs. Nat. Commun. 2016;7 doi: 10.1038/ncomms12351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Z., Qin S., Chen Y., Zhou L., Yang M., Tang Y., Zuo J., Zhang J., Mizokami A., Nice E.C., et al. Inhibition of NPC1L1 disrupts adaptive responses of drug-tolerant persister cells to chemotherapy. EMBO Mol. Med. 2022;14 doi: 10.15252/emmm.202114903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen S., Faouzi S., Souquere S., Roy S., Routier E., Libenciuc C., Andre F., Pierron G., Scoazec J.Y., Robert C. Melanoma Persister Cells Are Tolerant to BRAF/MEK Inhibitors via ACOX1-Mediated Fatty Acid Oxidation. Cell Rep. 2020;33 doi: 10.1016/j.celrep.2020.108421. [DOI] [PubMed] [Google Scholar]
- 40.Hong X., Roh W., Sullivan R.J., Wong K.H.K., Wittner B.S., Guo H., Dubash T.D., Sade-Feldman M., Wesley B., Horwitz E., et al. The Lipogenic Regulator SREBP2 Induces Transferrin in Circulating Melanoma Cells and Suppresses Ferroptosis. Cancer Discov. 2021;11:678–695. doi: 10.1158/2159-8290.CD-19-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aloia A., Mullhaupt D., Chabbert C.D., Eberhart T., Fluckiger-Mangual S., Vukolic A., Eichhoff O., Irmisch A., Alexander L.T., Scibona E., et al. A Fatty Acid Oxidation-dependent Metabolic Shift Regulates the Adaptation of BRAF-mutated Melanoma to MAPK Inhibitors. Clin. Cancer Res. 2019;25:6852–6867. doi: 10.1158/1078-0432.CCR-19-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang G., Frederick D.T., Wu L., Wei Z., Krepler C., Srinivasan S., Chae Y.C., Xu X., Choi H., Dimwamwa E., et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Invest. 2016;126:1834–1856. doi: 10.1172/JCI82661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang M.T., Tsai L.C., Nakagawa-Goto K., Lee K.H., Shyur L.F. Phyto-sesquiterpene lactones DET and DETD-35 induce ferroptosis in vemurafenib sensitive and resistant melanoma via GPX4 inhibition and metabolic reprogramming. Pharmacol. Res. 2022;178 doi: 10.1016/j.phrs.2022.106148. [DOI] [PubMed] [Google Scholar]
- 44.Dudnik E., Peled N., Nechushtan H., Wollner M., Onn A., Agbarya A., Moskovitz M., Keren S., Popovits-Hadari N., Urban D., et al. BRAF Mutant Lung Cancer: Programmed Death Ligand 1 Expression, Tumor Mutational Burden, Microsatellite Instability Status, and Response to Immune Check-Point Inhibitors. J. Thorac. Oncol. 2018;13:1128–1137. doi: 10.1016/j.jtho.2018.04.024. [DOI] [PubMed] [Google Scholar]
- 45.Alvarez S.W., Sviderskiy V.O., Terzi E.M., Papagiannakopoulos T., Moreira A.L., Adams S., Sabatini D.M., Birsoy K., Possemato R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551:639–643. doi: 10.1038/nature24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terzi E.M., Sviderskiy V.O., Alvarez S.W., Whiten G.C., Possemato R. Iron-sulfur cluster deficiency can be sensed by IRP2 and regulates iron homeostasis and sensitivity to ferroptosis independent of IRP1 and FBXL5. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abg4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu K., Li K., Lv J., Feng J., Chen J., Wu H., Cheng F., Jiang W., Wang J., Pei H., et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Invest. 2020;130:1752–1766. doi: 10.1172/JCI124049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muller F., Lim J.K.M., Bebber C.M., Seidel E., Tishina S., Dahlhaus A., Stroh J., Beck J., Yapici F.I., Nakayama K., et al. Elevated FSP1 protects KRAS-mutated cells from ferroptosis during tumor initiation. Cell Death Differ. 2022;30:442–456. doi: 10.1038/s41418-022-01096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen M., Mainardi S., Lieftink C., Velds A., de Rink I., Yang C., Kuiken H.J., Morris B., Edwards F., Jochems F., et al. Targeting of vulnerabilities of drug-tolerant persisters identified through functional genetics delays tumor relapse. Cell Rep. Med. 2024;5 doi: 10.1016/j.xcrm.2024.101471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Madorsky Rowdo F.P., Baron A., Gallagher S.J., Hersey P., Emran A.A., Von Euw E.M., Barrio M.M., Mordoh J. Epigenetic inhibitors eliminate senescent melanoma BRAFV600E cells that survive long-term BRAF inhibition. Int. J. Oncol. 2020;56:1429–1441. doi: 10.3892/ijo.2020.5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maertens O., Kuzmickas R., Manchester H.E., Emerson C.E., Gavin A.G., Guild C.J., Wong T.C., De Raedt T., Bowman-Colin C., Hatchi E., et al. MAPK Pathway Suppression Unmasks Latent DNA Repair Defects and Confers a Chemical Synthetic Vulnerability in BRAF-NRAS-and NF1-Mutant Melanomas. Cancer Discov. 2019;9:526–545. doi: 10.1158/2159-8290.CD-18-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith M.P., Wellbrock C. Molecular Pathways: Maintaining MAPK Inhibitor Sensitivity by Targeting Nonmutational Tolerance. Clin. Cancer Res. 2016;22:5966–5970. doi: 10.1158/1078-0432.CCR-16-0954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jonas O., Oudin M.J., Kosciuk T., Whitman M., Gertler F.B., Cima M.J., Flaherty K.T., Langer R. Parallel In Vivo Assessment of Drug Phenotypes at Various Time Points during Systemic BRAF Inhibition Reveals Tumor Adaptation and Altered Treatment Vulnerabilities. Clin. Cancer Res. 2016;22:6031–6038. doi: 10.1158/1078-0432.CCR-15-2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huijberts S., Wang L., de Oliveira R.L., Rosing H., Nuijen B., Beijnen J., Bernards R., Schellens J., Wilgenhof S. Vorinostat in patients with resistant BRAFV600E mutated advanced melanoma: a proof of concept study. Future Oncol. 2020;16:619–629. doi: 10.2217/fon-2020-0023. [DOI] [PubMed] [Google Scholar]
- 55.Embaby A., Huijberts S., Wang L., Leite de Oliveira R., Rosing H., Nuijen B., Sanders J., Hofland I., van Steenis C., Kluin R.J.C., et al. A proof-of-concept study of sequential treatment with the HDAC inhibitor vorinostat following BRAF and MEK inhibitors in BRAFV600mutated melanoma. Clin. Cancer Res. 2024 doi: 10.1158/1078-0432.CCR-23-3171. [DOI] [PubMed] [Google Scholar]
- 56.Joung J., Konermann S., Gootenberg J.S., Abudayyeh O.O., Platt R.J., Brigham M.D., Sanjana N.E., Zhang F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017;12:828–863. doi: 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2021 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- 60.Liberzon A., Birger C., Thorvaldsdottir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nokin M.J., Darbo E., Travert C., Drogat B., Lacouture A., San Jose S., Cabrera N., Turcq B., Prouzet-Mauleon V., Falcone M., et al. Inhibition of DDR1 enhances in vivo chemosensitivity in KRAS-mutant lung adenocarcinoma. JCI insight. 2020;5 doi: 10.1172/jci.insight.137869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original/source data of RNA-sequencing (accession number: GSE271247) have been deposited at https://www.ncbi.nlm.nih.gov/geo/and are publicly available as of the date of publication. This study did not report new original code. Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.