


Abstract
The cdc25C phosphatase participates in regulating transition from the G2 phase of the cell cycle to mitosis by dephosphorylating cyclin-dependent kinase 1. The tumor suppressor p53 down-regulates expression of cdc25C as part of G2/M checkpoint control. Transcription of cdc25C oscillates during the cell cycle with no expression in resting cells and maximum transcription in G2. We had identified earlier a new mechanism of cell cycle-dependent transcription that is regulated by a cell cycle-dependent element (CDE) in conjunction with a cell cycle genes homology region (CHR). The human cdc25C gene was the first example. CDE/CHR tandem elements have since been found in promoters of many cell cycle genes. Here we show that the mouse cdc25C gene is regulated by a CHR but does not hold a CDE. Therefore, it is the first identified gene with CHR-dependent transcriptional regulation during the cell cycle not relying on a CDE located upstream of it. The CHR leads to repression of cdc25C transcription early in the cell cycle and directs a release of this repression in G2. Furthermore, we find that this CHR can cooperate in cell cycle-dependent transcription with elements placed directly upstream of it binding E2F, Sp1 or Sp3 transcription factors.
INTRODUCTION
The phase transitions of the cell division cycle are, in part, regulated through phosphorylation. The phosphatase cdc25C is a main regulator involved in transition from the G2 phase to mitosis in the cell division cycle (1,2). Microinjection of an antibody against cdc25C into HeLa cells leads to an inhibition of cell cycle progression before mitosis (3). Consistent with its function at the time of passage from G2 to mitosis, mRNA levels of cdc25C change periodically during the cell cycle. Only small amounts are found in G1 and increasing concentrations are observed in S phase with maximum levels in G2 (2).
Transcriptional regulation during the mammalian cell cycle has so far been mostly associated with the function of the E2F family of transcription factors and the retinoblastoma tumor suppressor protein pRb (4,5). Heterodimers formed by E2F and DP components are able to activate transcription of cell cycle promoters in late G1 and S phase. In G0 and early G1, complex formation of the retinoblastoma tumor suppressor pRb family of proteins with E2F/DP represses transcription. Transition from this repression in the early phase of the cell cycle to activation at later times is controlled by phosphorylation of pRb-family members (6,7).
More recently we have identified another mechanism by which gene transcription can be regulated in a cell cycle-dependent manner. This mechanism employs a tandem transcriptional element composed of the cell cycle-dependent element (CDE) and the cell cycle genes homology region (CHR). The fluctuations in mRNA levels from genes like cdc25C, cyclin A, cyclin B2 and cdc2 (cdk1) are dependent on differential transcription due to these tandem repressor elements in their promoters. CDE/CHR sites regulate the promoters by repression in G0 and G1 and release from repression later in the cell cycle (8–10). Both halves of the CDE/CHR element are required for transcriptional repression in G0 and G1. Inactivation of either the CDE or the CHR leads to deregulation of transcription with high expression levels already in early phases of the cell cycle. The proteins regulating through CDE/CHR elements have so far only been described poorly. A factor called CDF-1, which is different from E2F, has been shown by one group to bind to some CDE/CHR elements (11). However, this factor has resisted cloning and further molecular characterization. The cdc2 and cyclin A promoters were shown to bind E2F to the CDE half of the CDE/CHR site (12).
In the human cdc25C promoter a recognition site for the tumor suppressor p53 has been described. This site can bind p53 and is able to activate transcription in response to co-transfection of a p53-expressing plasmid, at least when artificially placed adjacent to the adenovirus E1b minimal promoter (13). We have demonstrated that transcription of cdc25C is repressed by p53. This down-regulation of cdc25C is not contingent upon the p53 site in its promoter but depends on CCAAT boxes. The down-regulation of cdc25C is part of the G2/M checkpoint control by p53 (14,15).
The trimeric nuclear transcription factor Y (NF-Y) binds to these CCAAT boxes and is the major activator for CDE/CHR-regulated promoters (8). NF-Y binding activity varies during the cell cycle. However, it only contributes as a general activator to the typical cell cycle-dependent expression of genes like cyclin B2. Concentration of its NF-YA subunit decreases only after the peak expression in G2 of the CDE/CHR genes has already been reached. NF-Y binding activity is also present in G1, at which time these cell cycle genes are not expressed (16). Another important transcriptional activator is Sp1. It binds to GC boxes and is the first identified member of a larger family of transcription factors (17). Sp1 was also recognized as a factor involved in transcription of the cell cycle genes, particularly through cooperation with E2F or pRb (18–20).
Here we describe the first CHR element that does not rely on intact CDE or E2F sites upstream of it to confer cell cycle-dependent transcription. The cdc25C CHR can cooperate with E2F and Sp1/3 sites in regulating promoter activity during the cell cycle.
MATERIALS AND METHODS
Cloning of the cdc25C promoter and plasmid constructs
The cdc25C genomic DNA upstream of the start codon was amplified from the adaptor-ligated Mouse GenomeWalker library (Clontech, Palo Alto, CA) by primer walking. The series of PCRs started with two nested gene-specific primers derived from the 5′ end of the cDNA (GenBank accession nos L16926 and L16994): CDCMO2-3′, 5′-ACAGAATGCTTAGGTTTGCCGAGTCG-3′; CDMO1-3′, 5′-GTCTAGGTACCTCTAGAGTGGGATAGGTCCTGTAGACATGAC-3′. After the last walking step the m25C-wt-luci construct was obtained by PCR amplification with the primers M25C-5′, 5′-GACCCAAGCTTCTGGTCTGCTGCCACCACCATCACTAAACCCGCC-3′, and M25C-3′, 5′-CTGTAGCCATGGCTTCAGAGTCTTCACCGAGGGAGGTGGG-3′, followed by cloning (HindIII/NcoI) in pGL3-Basic (Promega, Mannheim, Germany). In two independent experiments from different genomic DNA samples clones were obtained and sequencing of both strands yielded an identical sequence for both clones, which was submitted to the database (GenBank accession no. AF450244). The m25C-wt-luci 1302 bp promoter construct drives expression of firefly luciferase as reporter. The sequence of the human cdc25C promoter, including the segment close to the coding sequence (15), was described earlier (GenBank accession nos AY046902 and Z29077).
Promoter mutants were produced by PCR-based mutagenesis on the basis of m25C-wt-luci. The mutant M1 was created employing the primers M1-for, 5′-TTGTTGAGctgtcGGAAGGTTTGAATGGTCAGACC-3′, and M1-rev, 5′-CAAACCTTCCgacagCTCAACAACCTATCACCAC-3′ (mutations are indicated by lower case letters). The other constructs mutated in the region of the CDE were obtained by PCR with equivalent primers. The construct M6 was also produced by PCR using primers M6-for, 5′-GGAAGGTgcatATGGTCAGACCTCTGGCTGTTG-3′, and M6-rev, 5′-CTGACCATatgcACCTTCCACTCTCTCAACAAC-3′. The other plasmids mutated in the region of the CHR were created with equivalent primers. The plasmid YA13m29 was provided by Roberto Mantovani and expresses a dominant-negative subunit A of NF-Y (16). pRL-null (Promega) contains a cDNA encoding Renilla luciferase. The mouse cyclin B2 promoter driving expression of firefly luciferase in B2-Luci plasmid (10) as well as reagents investigating the influence of p53 on cdc25C expression were described earlier (14,15).
A plasmid was created to express recombinant full-length human DP-1 (GenBank accession no. L23959) in Escherichia coli as a fusion protein with six histidine amino acids at the C-terminus. The cDNA was cloned as a NcoI/BglII fragment into pQE-60 (Qiagen, Hilden, Germany) after PCR amplification from a cDNA library (Clontech) with the following two primers: forward, 5′-TTCATGCCATGGCAAAAGATGCCGGTC-3′; reverse, 5′-ACCAGAAGATCTGTCGTCCTCGTCATTCTCG-3′.
All plasmid DNAs were purified through anion exchange columns (Qiagen) and confirmed by restriction analysis and sequencing.
Cell culture and transfections
NIH 3T3 cells were obtained from DSMZ (Braunschweig, Germany) and cultured in a humidified atmosphere with 10% CO2 at 37°C with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS; Biochrom, Berlin, Germany).
For cell cycle analyses, 4 × 104 cells were plated per well in 1 ml medium on 12-well plates and cultured overnight before transfection. Transfections were carried out in 4-fold using 6 µl Pfx-3 (Invitrogen, Leek, The Netherlands) with 2 µg luciferase reporter construct and 40 ng of pRL-null vector (Promega) in 1 ml Optimem transfection medium (Gibco, Eggenstein, Germany) per well. Cells were incubated for 4 h before replacing transfection medium with DMEM containing 10% FCS. Cells were then cultured for 1 day in this medium before starvation in DMEM without FCS for 60 h. After this time cells were harvested for 0 h time points. The mouse fibroblasts were stimulated with 20% FCS in DMEM to re-enter the cell cycle and subsequently analyzed at indicated times.
Transfections using the dominant-negative form of NF-YA were carried out in triplicate. In 24-well plates 5 × 104 cells were plated per well in 0.5 ml medium. Cells were cultured overnight before transfection. Transfection was carried out using 1 µl Fugene 6 (Roche, Mannheim, Germany) with 125 ng luciferase reporter construct, 2.5 ng of pRL-null vector and increasing amounts of construct YA13m29.
Co-transfection experiments with p53 wild-type or mutant expression plasmids and the mouse cdc25C reporter were carried out in SaOS-2 cells (DSMZ) (15,21). Cells were cultured in a humidified atmosphere with 5% CO2 at 37°C with McCoy’s 5A modified medium (Biochrom) supplemented with 15% FCS. Transfection was carried out in triplicate using lipofection with Fugene 6 (Roche) according to the manufacturer’s instructions. Exponentially growing cells were plated at a density of 5 × 104/well in 0.5 ml medium in 24-well plates. Cells were cultured overnight before transfection. m25C-wt-luci construct (250 ng) was co-transfected with 25 ng of constructs expressing wild-type or mutant p53 proteins and 25 ng of Renilla luciferase expression vector (pRL-null; Promega) as an internal control. DNA amount was held constant in all experiments by adjusting with pcDNA3.1/His C (Invitrogen).
Luciferase assays
Luciferase activities were assayed with the Dual Luciferase Assay System (Promega) as suggested by the manufacturer. Firefly luciferase activity was normalized to Renilla luciferase activity to compensate for variability in transfection efficiencies.
Fluorescence activated cell sorting (FACS) analyses
Cells were harvested, washed in PBS/EDTA (1 mM) and fixed with 75% ethanol in PBS/EDTA for ≥12 h at 4°C, and subsequently centrifuged and resuspended in 1 ml PBS/EDTA containing 50 µg/ml RNase A (Sigma, München, Germany). Propidium iodide (Sigma) staining followed at a final concentration of 40 µg/ml. Cells were filtered through a 35 µm pore size cell strainer (Falcon, Heidelberg, Germany). Flow cytometry was performed with a FACSCalibur instrument (Becton Dickinson, Heidelberg, Germany). A total of at least 10 000 cells per run was analyzed with the CELLQuest software (Becton Dickinson).
Electrophoretic mobility-shift assays (EMSAs)
Oligonucleotide probes were labeled by filling in 5′ overhangs using the Klenow fragment of DNA polymerase in the presence of [α-32P]deoxycytidine-5′-triphosphate. The labeled oligonucleotides were purified using the Nucleotide Removal Kit (Qiagen) according to the manufacturer’s instructions.
For Sp1/3 EMSAs HeLa nuclear extract with ∼5 µg of protein were incubated for 15 min at room temperature in 10 mM Tris pH 7.5, 0.5 mM EDTA, 1 mM MgCl2, 50 mM NaCl, 0.5 mM DTT, 6% glycerol and 0.75 µg of polydeoxyinosinic-deoxycytidylic acid, poly(dI-dC) (Sigma) in a total volume of 15 µl. Antibodies specific for Sp1 (sc-420) and Sp3 (sc-644) transcription factors were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and incubated with protein mixtures prior to addition of probe. For Sp1/3 shifts GC-box oligo_nucleotide was used as a positive control (22). After probe addition, reactions were incubated for 20 min at room temperature. The protein–DNA complexes were separated on native 4.5% polyacrylamide gels at 15°C as described (23). Gels were dried and visualized using a FLA-3000 Fujifilm phosphorimaging system.
E2F EMSAs were carried out with ∼300 ng recombinant E2F1-GST and DP1-His per assay. Oligonucleotides employed to shift E2F as wild-type and mutant control were derived from the mouse B-myb promoter (24) and used in EMSAs as published earlier (25). The sequences of the mouse cdc25C gene specific EMSA oligonucleotides were as follows: Sp25C, 5′-GGAACATGACCCCGCCCCCTAACCAATC-3′ and 5′-GATTGGTTAGGGGGCGGGGTCATGTT-3′; Sp25Cmut, 5′-GGAACATGACTATATATCCTAACCAATC-3′ and 5′-GATTGGTTAGGATATATAGTCATGTT-3′; wt, 5′-GGTTGTTGAGAGAGTGGAAGGTTTGAATGGTCAGAC-3′ and 5′-GTCTGACCATTCAAACCTTCCACTCTCTCAACAA-3′. The sequences of the mutant EMSA probes can be derived on this basis from mutations indicated in the figures.
Nuclear extracts from HeLa cells were prepared as described and were a generous gift from Reinhard Lührmann (26). E2F-1 as glutathione-S-transferase (GST) fusion protein was expressed in E.coli from the plasmid pGST20TE2F-1 constructed and provided by Kristian Helin (27). The protein was expressed in E.coli using the QIAexpress System (Qiagen). DP-1-His was purified with Ni-NTA-agarose (Qiagen) according to manufacturer’s instructions. After isolation, DP-1 was dialyzed against 50 mM Tris pH 7.5, 5 mM DTT, 100 mM NaCl and supplemented with 50% glycerol.
RESULTS
Cell cycle-dependent transcription from the cdc25C promoter
A cdc25C promoter construct driving luciferase expression shows cell cycle dependence (Fig. 1). To obtain DNA sequence information necessary to create this construct the mouse cdc25C gene upstream of the coding region was amplified by primer walking starting from the 5′ end of the cDNA. With primers derived from the subsequent sequencing a DNA segment of 1302 bp upstream of the first coding ATG was amplified and cloned into a vector that could be used to drive luciferase reporter expression.
Figure 1.

Cell cycle-dependent expression from the mouse cdc25C promoter. NIH 3T3 mouse fibroblast cells were transfected with m25C-wt-luci or B2-luci plasmids. Cells were cultured for 24 h after transfection and then serum starved for 60 h. For the 0 h time points, resting cells after serum starvation were used, whereas the other time points are after addition of serum to the medium. (A) Samples from parallel experiments were analyzed for luciferase expression driven by the cdc25C or cyclin B2 promoters. Within a time point activities were normalized to expression of Renilla luciferase. The highest expression level for each plasmid measured by luciferase activity was set at 100% and other values were normalized to that. 890 relative light units (r.l.u.) for cdc25C and 2056 r.l.u. for cyclin B2 represent 100%. Assays were carried out four times. (B) In parallel, cells were analyzed for their DNA content by FACS at different times to determine their cycle distribution of the cell population used in the reporter assays.
Expression from the cdc25C promoter is low in resting cells compared with expression in the G2/M phase of the cell cycle (Fig. 1A). Stimulation of expression is ∼14-fold comparing minimum expression before addition of serum with maximum expression at 28 h after restimulation of cell division. Cell cycle distribution of cells was followed by FACS analysis of DNA stained cells with the maximum for S phase cells at ∼18 h after serum restimulation with a subsequent increase in G2/M cell population (Fig. 1B). Timing of cdc25C expression during the cell cycle is similar to expression from the cyclin B2 promoter, which is reasonable since products of both genes are needed in late G2 phase. cdc25C transcription starts earlier and reaches its peak level shortly after cyclin B2 (Fig. 1). Expression of the cdc25C reporter matches expression from the chromosomal gene in both human and mouse (2,28). Stimulation factor, timing and levels of expression of the mouse cdc25C reporter are similar to that of the human promoter (8,29).
Mouse and human cdc25C promoters share limited homology
The activating and cell cycle-regulatory sites in the human cdc25C promoter have been mapped (8). After finding that the mouse 1302 bp genomic DNA fragment upstream of the coding region is sufficient to drive transcription typical for the cdc25C gene we sought to identify the regulatory elements. We compared sequences of the mouse and human promoters (Fig. 2 and Supplementary Material). The overall homology between the two sequences is low. In the promoter region, 756 nt upstream of the first codon, only 357 bases or 47% are identical in the mouse and human promoters (Supplementary Material). However, a few segments are conserved in both promoters. For the human promoter, NF-Y and Sp1 sites had been identified as main activating elements (30). The CDE/CHR tandem element is responsible for cell cycle-dependent transcriptional regulation (8). Two of the NF-Y-binding CCAAT boxes are conserved, both with regard to sequence and distance from each other (Fig. 2). The third, most upstream of the CCAAT boxes, is not found in the mouse promoter. Another potentially activating site, the GC box with a consensus for binding Sp1 and Sp3 proteins, is found in both promoters. Most interestingly, the region of the CHR cell cycle element is conserved but the adjacent CDE is not found in the mouse promoter (Fig. 2).
Figure 2.

Limited homology between mouse and human cdc25C promoters indicates potential regulatory sites. The mouse cdc25C upstream region was cloned after amplification by primer walking and sequenced (GenBank accession no. AF450244). This sequence is compared with the human cdc25C promoter for which some regulatory sites have been mapped (GenBank accession nos AY046902 and Z29077). GC boxes binding Sp1 and Sp3 transcription factors, CCAAT boxes contacted by NF-Y trimeric proteins and the CDE and CHR cell cycle regulatory elements are indicated. Numbering starts at the first coding nucleotide triplet. A complete mouse promoter sequence with a longer comparison of mouse and human cdc25C promoters is given in the Supplementary Material.
NF-Y is the main transcriptional activator for cdc25C transcription
Transcription of many cell cycle genes is regulated by NF-Y as the predominant activator (8,16,30,31). A simple method to test for the activation by the trimeric NF-Y factor is to employ a dominant-negative subunit of it. We co-transfected, into normally growing NIH 3T3 cells, increasing amounts of a plasmid carrying a dominant-negative mutant of NF-YA (YA13m29) together with the wild-type cdc25C reporter construct (Fig. 3). More than 60% of the activity is lost at the highest concentration of the dominant-negative mutant. This indicates that more than half of the cdc25C promoter activity is dependent on activation by NF-Y.
Figure 3.

NF-Y is the main activator for cdc25C transcription. Increasing amounts of dominant-negative NF-YA subunit down-regulate the wild-type cdc25C luciferase reporter construct. Firefly luciferase reporter m25C-wt-luci, together with pRL-null plasmid expressing Renilla luciferase control, was transfected in NIH 3T3 cells. Increasing amounts of a plasmid coding for a dominant-negative form of NF-YA, YA13m29, were co-transfected. Firefly luciferase activity was normalized to Renilla luciferase. The value for transfections without YA13m29 was set at 100% and other activities are given relative to that. Three independent experiments were carried out with three assays per experiment and standard deviations are shown.
p53 down-regulates transcription of cdc25C independent of a p53 consensus binding site
The mouse cdc25C promoter does not contain a p53 consensus binding site (Fig. 2 and Supplementary Material). The site, which can bind p53 in the human promoter, is located centered around nucleotide –787 relative to the translational start (13,15). This region is not homologous in human and mouse promoters (Fig. 2). However, transfection of SaOS-2 cells with a plasmid expressing p53 yields a 6.8-fold down-regulation of the mouse cdc25C promoter luciferase reporter when wild-type p53 is compared with a mutant form of p53 (Fig. 4). The mutant used as a control is an altered form with amino acid changes P72R and V143A, which lead to its inability to serve as a transcription factor (21,32,33). Down-regulation of transcription from the mouse cdc25C promoter by the p53 tumor suppressor proves that this regulation is independent of a p53 binding site in the promoter since this promoter lacks such a site (Fig. 2). This result is consistent with our previous observations and conclusions on the human cdc25C promoter (15).
Figure 4.
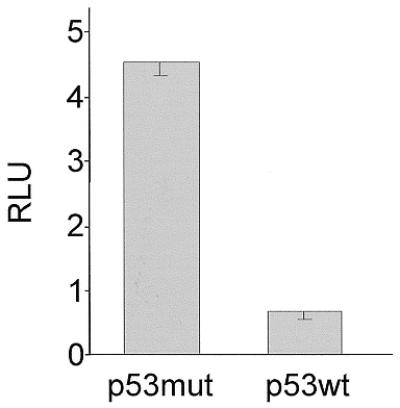
Wild-type p53 down-regulates transcription from the cdc25C promoter. SaOS-2 cells were co-transfected with the mouse cdc25C-promoter reporter and wild-type or transcriptionally inactive mutant p53-expressing plasmids. Luciferase activity was measured from lysates. Experiments were carried out in triplicate and standard deviations are shown .
The CHR is the major site responsible for regulating cell cycle-dependent transcription of mouse cdc25C
In order to elucidate the impact of potential cell cycle elements we mutated the putative CHR and the region upstream of it in reporter constructs and tested them for cell cycle-dependent transcription. These constructs were transfected in NIH 3T3 mouse fibroblasts and expression assayed by luciferase activity. Cells were starved by serum deprivation to test for G0 expression and restimulated by serum re-addition to the medium for 24 h to obtain promoter activities for a cell population enriched in G2. Stimulation of transcription of 17 mutant promoters from G0 to G2 was measured relative to the wild-type cdc25C promoter (Fig. 5). Average stimulation of 13.6-fold for wt-m25C-luci was set to 100%.
Figure 5.


Cell cycle-dependent transcription from wild-type and mutant cdc25C promoter luciferase constructs. Dominant importance of the CHR for cell cycle-dependent transcription. The CHR can cooperate with Sp1/3, CDE or E2F sites. Stimulation rates represent transcriptional regulation of wild-type cdc25C and mutant promoters during the cell cycle. NIH 3T3 cells were transfected with wild-type (wt) or mutant m25C-luci constructs, grown for 24 h, starved by serum deprivation and restimulated to enter the cell cycle. Cell lysates were assayed for luciferase activity. Wild-type and mutant constructs were tested in parallel experiments. Stimulation of promoter activity during the cell cycle was calculated by dividing luciferase activities 24 h after serum addition by activities before serum stimulation. Stimulation for wt-m25C-luci was set as 100% and percentages for mutants were calculated for every experiment relative to this stimulation. Average stimulation for wild-type was 13.6-fold. Experiments were repeated between two and six times for each individual mutant. Standard deviations are shown in the graphs. (A) Mutational analysis of the CHR and the region upstream of the CHR. (B) Analysis of bona fide CDE, E2F or Sp1/3 sites introduced upstream of the CHR. M12 represents a CDE and M13 represents an E2F site, M15 and M16 represent Sp1/3 sites, respectively. E2F/CHR or Sp1/3-CHR hybrid sites with additional mutations in the CHR, as in mutant M6, were analyzed with mutants M14 and M17, respectively.
Mutant promoters M6 to M11 have lost most of their ability to drive cell cycle-dependent transcription (Fig. 5A). These mutant promoters all contain changes in the putative CHR that lead to a lack of repression in G0. Activities in G2 stay at the same level as for the wild-type construct, whereas mutant reporter activities increase in resting cells. In mutants M6 to M11 this behavior results in a drop of cell cycle regulation of transcription to about one-fifth that of the wild-type cdc25C promoter. Even single nucleotide mutations as in M8 and M11 display a substantial loss of regulation. Therefore, this site is indeed a CHR.
Functional CHR elements have, until now, only been described in conjunction with CDEs. The respective sequence in the mouse cdc25C promoter upstream of the CHR is different from any CDE identified so far (8–10). Still, mutation of this region leads to a change in regulation. An alteration in cell cycle-dependent transcription is seen in mutants M1 and M4 with about half the regulation lost. Like regulation of mutants M6 to M11, changes in cell cycle transcription of M1 and M4 are also due to an increase of activity in G0 while transcriptional activation in G2 stays at the same level as activity for the wild-type promoter. In comparison with the wild-type cdc25C promoter, mutants M2 and M3 do not give a significant change in regulation; and the mutation introduced in M5 even shows an improved cell cycle-regulated transcription (Fig. 5A). Summing up the results from the mutations presented in Figure 5A, it is obvious that changes in the CHR result in a much stronger alteration of cell cycle-dependent transcription than mutations in the region upstream from it, where in other promoters a CDE is found. If the region upstream of the CHR in the mouse cdc25C promoter is changed to a CDE-like sequence, as in mutant M16, or even mutated to the CDE from the human cdc25C promoter, as in M12, cell cycle-regulated transcription is improved (Fig. 5B).
All deregulated promoter mutants show an increase in luciferase activities in G0 instead of a loss in G2. Mutants such as M5 and M15, for which a stronger regulation than for the wild-type construct is observed, have an expression in G0 at a similar level to the wild-type promoter. Their change in stimulation compared with wild type results from an increase of promoter activity in G2. Mutant M16 is different in that it shows a lower than wild-type activity in resting cells, thereby displaying a increased fold induction in comparison with the wild-type cdc25C promoter. In contrast, the reduced cell cycle-dependent transcriptional regulation seen in mutants M1 and M4 is due to an increase in expression in G0 rather than lower levels of expression in G2 (Fig. 5 and data not shown). Each deregulated mutant promoter gives activities at the elevated level at which wild-type cdc25C promoter activity in G2 is observed. This implies a loss of repression rather than a reduction of activation when cell cycle elements are mutated.
The CHR is able to cooperate in regulating cell cycle-dependent transcription with CDE, E2F or Sp1/3 sites
All CHR elements identified so far to be relevant for cell cycle-dependent transcription are found in conjunction with a CDE site 4 nt upstream of the CHR (8–10). Therefore, we tested if the CDE from the human cdc25C promoter, introduced in the proper position with mutant M12 to create the CDE/CHR tandem element, would lead to cell cycle transcription in the context of the mouse promoter. The mutant M12 with an intact CDE/CHR is not only transcribed in a cell cycle-dependent manner, but displays an even stronger regulation with a lower activity in G0 compared with the wild-type promoter (Fig. 5B). As a control, mutation of the CHR in this CDE/CHR context has been described earlier (8). This indicates that the CHR from the mouse cdc25C promoter can cooperate with a CDE artificially placed in tandem with it. Yet the CDE alone cannot regulate cell cycle-dependent transcription in the human promoter (8).
An E2F site originating from the mouse B-myb promoter (25) was another element we chose to test in tandem with this CHR. Mutant promoter M13 with an E2F/CHR site shows a stronger cell cycle transcription than the wild-type promoter with just a CHR (Fig. 5B). As a control that the cell cycle-dependence of this new tandem element is contingent upon the CHR and not simply attributable solely to the introduced E2F site we changed the CHR in this context. M14 as the corresponding mutant has almost no ability to direct cell cycle-dependent transcription (Fig. 5B). Therefore, E2F and CHR sites cooperate in the M13 mutant promoter to regulate cell cycle-dependent transcription.
The ability of the mutant sites to bind E2F protein was tested in EMSAs. As a control, an oligonucleotide from the B-myb promoter carrying an E2F site was used by incubating it with E2F-1 and DP-1. This binding was compared with the same oligonucleotide with its E2F site mutated (Fig. 6, lanes 1 and 2). E2F does not bind to the wild-type cdc25C promoter oligonucleotide nor to probes mutated in the CHR and upstream from it (lanes 3–6). However, when an E2F site is introduced upstream of the CHR, binding similar to that of the B-myb probe is also observed in the context of the cdc25C promoter (M13 and lane 1). Binding to this E2F site is not dependent on an intact CHR since mutant oligonucleotide M14 is still able to bind E2F with similar affinity as M13 (Fig. 6). Taken together with functional assays this indicates that, on the one hand, an E2F site can bind E2F-1/DP-1 heterodimers even in the absence of an intact CHR in tandem with it. But, on the other hand, this isolated E2F site is not able to regulate cell cycle-dependent transcription (Figs 5B and 6).
Figure 6.

E2F/DP heterodimers cannot bind to the wild-type cdc25C promoter but form complexes on E2F/CHR sites. EMSAs with recombinant E2F-1-GST and DP-1-His proteins were carried out with probes from the B-myb promoter as controls (myb and myb mut). Sequences of wild-type and mutant oligonucleotides used to test binding to the cdc25C promoter can be derived from Figure 5.
Two other mutants were tested to see if changing the region upstream of the CHR to DNA binding sites for Sp1 and Sp3 would diminish cell cycle-dependent transcription, like a random mutation did in M1 and M4 mutants (Fig. 5A). Unexpectedly, both mutants M15 and M16, which were created to place Sp1/3 sites upstream of the CHR, do not show a reduced cell cycle-dependent transcription. Instead, cell cycle transcription is even more pronounced (Fig. 5B). Additional mutation of the CHR in the M16 construct to the M17 mutant leads to a total loss of cell cycle-dependent transcription (Fig. 5B). This proves that introduction of Sp1/3 sites alone is not sufficient to create a cell cycle element.
We tested protein binding to the Sp1/3 sites that were introduced in the cdc25C promoter and could support cell cycle-dependent transcription. Because of the similarity between E2F and Sp1/3 sites we used E2F-1/DP-1 proteins in EMSAs. The M16 and M17 mutants, both based on the cdc25C promoter and mutated to an Sp1/3 site, were able to shift a weak band compared with bona fide E2F sites (Fig. 6). However, mutant M15, also a cdc25C promoter segment with an Sp1/3 site, cannot yield a band with E2F (Fig. 6). Since mutants M15 and M16 showed cell cycle-dependent transcription and mutant M17 did not, and only mutants M16 and M17 but not M15 could bind E2F, it can be excluded that only E2F is responsible for cell cycle transcription through Sp1/3 sites introduced in the cdc25C promoter (Figs 5B and 6).
Sp1 and Sp3 protein binding to wild-type and mutant oligonucleotides was tested by EMSA from HeLa cell nuclear extract (Fig. 7). The GC control probe provides mobilities for the Sp1 and Sp3 protein–DNA complexes (Fig. 7A, lanes 1 and 2). A wild-type DNA fragment from the cdc25C promoter cell cycle regulatory region containing the CHR does not bind Sp1 or Sp3 (Fig. 7A, lane 3). Probes based on the same oligonucleotide with mutations in the CHR or the region upstream of it (M1, M4, M6 and M9) do not give an Sp1/3 shift (Fig. 7A). However, cdc25C promoter mutants engineered to contain Sp1/3 sites shift bands with the mobility of Sp1 and Sp3 (Fig. 7A, M15–M17). To confirm the identity of Sp1 and Sp3 shifts, we carried out competition EMSAs and experiments including specific antibodies (Fig. 7B). An oligonucleotide (GC) which can bind both Sp1 and Sp3 factors shifts two bands that can be self-competed (Fig. 7B, lanes 1 and 2). The bands are reduced in intensity or disappear and supershifted complexes become visible when antibodies specific for Sp1 or Sp3 are added (Fig. 7B, lanes 3 and 4). The same Sp1 and Sp3 complexes are shifted by the cdc25C promoter mutants M14 to M17. Competition with the Sp1/3-binding GC oligonucleotide abrogates binding of the two specific bands (Fig. 7B, lanes 5–12).
Figure 7.


Binding of Sp1 and Sp3 proteins to the mutants of the cdc25C promoter tested by EMSA. Nuclear extracts from HeLa cells were employed to test for binding of Sp1 and Sp3 proteins to wild-type and mutant oligonucleotides derived from the mouse cdc25C promoter. The GC probe was used as positive control. cdc25C mutant probes follow the same nomenclature given in Figure 5. (A) EMSA with GC control oligonucleotide, wild-type and mutant cdc25C promoter probes. (B) EMSA bands representing Sp1 or Sp3 shifts are identified by reaction with specific antibodies. (C) The Sp25C oligonucleotide represents a segment from the mouse cdc25C promoter upstream of the CHR and NF-Y binding sites probing for Sp1/3 binding (Fig. 2). GC probe and Sp1-specific antibody serve as reagents to identify Sp1/3 proteins.
Sp1 and Sp3 can bind to an activator site in the cdc25C promoter
There is one putative Sp1/3 site in the cdc25C promoter centered around nucleotide –709 in the mouse promoter (Fig. 2). The homologous region in the human promoter, containing a sequence for a classical Sp1 site (34), had been deleted with a loss of about one-third of the activity and thereby shown to be relevant for activation (30), but protein binding to this site remained to be tested. In EMSAs the GC oligonucleotide and a specific antibody were used as positive controls. The Sp25C probe containing the putative Sp1/3 site from the mouse cdc25C promoter was employed to compete with the two complexes specific for Sp1 and Sp3 (Fig. 7C, lanes 1–4). The cdc25C promoter segment used as a probe was able to shift Sp1 and Sp3 factors whereas a mutant of the Sp1/3 site (Sp25Cmut) did not (Fig. 7C, lanes 5 and 9). Supershift with the antibody against Sp1 and competition with self and the GC oligonucleotide (Fig. 7C, lanes 6–8) are consistent with the notion that the putative Sp1/3 site in the cdc25C promoter is indeed a binding site for Sp1 and Sp3.
DISCUSSION
All CHR elements identified until now need a CDE in tandem with them to be functional (8–10). We describe here for the first time a CHR that does not have a CDE 4 nt upstream. The CHR in the mouse cdc25C promoter is homologous to the CHR in the human promoter (Fig. 2). However, the human cdc25C CHR can only repress and, thereby, regulate transcription during the cell cycle while the CDE 5′ from it is intact (8). The nucleotide sequence 5′ from the mouse cdc25C CHR is different from any known CDE (8–10). Nevertheless, mutation of this segment reduces cell cycle-dependent transcription in mutants M1 to M4 to some extent, but it is clearly not as pronounced as with mutations in the CHR (Fig. 5A). In contrast, for cyclin A, cdc2 and human cdc25C promoters mutations in CDEs and CHRs had equally strong effects on deregulation of cell cycle-dependent transcription. Furthermore, an extensive and detailed mutational analysis of the CDE in the human cdc25C promoter shows that a mutation from 5′-GGCGG-3′ to 5′-AGTGG-3′, as found in the mouse promoter described here, would clearly result in a loss of cell cycle-dependent transcriptional regulation (8). Comparison of the CHR elements in the human and mouse cdc25C promoters reveals that the homology extends beyond the CHR core (Fig. 2). However, a detailed study testing various nucleotides in and around the CHR core in the human promoter has shown that nucleotides outside the core 5′-TTTGAA-3′ are not needed for cell cycle-dependent transcriptional regulation in this assay (11). The difference in CDE/CHR combinations of mouse and human cdc25C promoters is not likely the result of a general species difference since CDE/CHR-regulated promoters from mouse have been described with CDEs that conform the CDE consensus derived from human cdc25C CDE, e.g. as seen in the mouse cyclin B2 promoter (10). Another point indicating that it is not just the difference in organism that dictates the combination of CDEs and CHRs results from the fact that in most transfections of reporter constructs for cell cycle assays mouse NIH 3T3 cells are used irrespective of the origin of the gene.
CDE sequences display some homology to E2F sites. In fact, for cyclin A and cdc2 it has been described that E2F can bind to CDEs in the promoters (12). Furthermore, in the B-myb promoter a CHR-like element, named the downstream repression site, was identified to regulate cell cycle-dependent transcription together with an E2F site (35,36). Therefore, we tested if the isolated CHR in the mouse cdc25C promoter can cooperate with an E2F site. Transcriptional up-regulation during the cell cycle by this hybrid E2F/CHR is even more pronounced than with wild-type CHR element alone (Fig. 5B). Still, this regulation is dependent on the CHR since its mutation leads to a drop of cell cycle transcription despite an intact E2F site (Fig. 5B, mutant M14). We conclude that the cdc25C CHR can cooperate with an E2F site.
Originally thought of as a negative control we created two constructs that yielded Sp1/3 sites upstream from the CHR (mutants M15 and M16). Surprisingly, the two mutant promoters did slightly better in up-regulating cell cycle-dependent stimulation of transcription (Fig. 5B), different from promoters with other mutations in this region which show a moderate loss of regulation (Fig. 5A, mutants M1 to M4). Both Sp1/3 mutants of the cdc25C promoter (M15 and M16) are able to bind Sp1 and Sp3 proteins. Also the E2F mutants M13 and M14 bind well to Sp1/3 (Fig. 7A and B). This suggests that Sp1 and Sp3 proteins can also bind in vivo to this region adjacent to the central regulator site CHR, implying that E2F as well as Sp1/3 sites can cooperate with the CHR in cell cycle-regulated transcription.
Sp1/3 had earlier been implicated in cell cycle transcription but never in conjunction with CDE/CHR regulation. For example, binding of Sp1/3 to a GC element has been shown for the hamster thymidine kinase promoter which is necessary for growth-related transcription from this promoter (37). Furthermore, it has been shown that Sp1 and E2F sites overlap in the mouse cyclin E promoter and have an antagonistic effect on transcriptional activity (38). It has been found that pRb can form complexes with Sp1 during all phases of the cell cycle. This complex activates the hamster dihydrofolate reductase promoter (19). Karlseder et al. showed that cooperation between E2F and Sp1 is necessary for general activity of growth-dependent transcription of the mouse thymidine kinase promoter (18). Sp1 binding sites are necessary for E2F-1-dependent repression of the human cyclin D1 promoter. E2F-1 can act through an Sp1 site (20). To a limited extent this could also be the case for cdc25C transcription since we observe a weak binding of E2F also to the Sp1/3 mutants M16 and M17 (Fig. 6). Recently, cooperation between Sp1 and E2F sites has been reported in a promoter regulating DNA polymerase expression (39). In summary, it appears to be a novel finding that Sp1/3 sites can substitute for the CDE in a CDE/CHR tandem element.
Generally, standard deviations in experiments presented in Figure 5 are relatively large. The reason is that we have performed several completely independent experiments and included them in these results. Therefore, variations stemming from, for example, synchronization of cells, transfections and luciferase measurements are the cause for these standard deviations. In particular, quality of synchronization varies to a large extent from one experiment to another. Because of these strong potential variations we refrained from just presenting several measurements from experiments carried out in parallel with just one representative synchronization.
The regulatory region of other cdc25 genes, like that of mouse cdc25B, are different from that of cdc25C. The cdc25B promoter contains a TATA box and its transcription during the cell cycle is, in part, mediated by a cell cycle-regulated repressor (40). The two sites overlap and are different from the CHR described for the cdc25C promoter (Fig. 2).
In summary, it becomes evident that the CHR can act as a single element in cell cycle-dependent transcription or function in tandem with CDE, E2F or Sp1/3 sites. It appears that E2F, Sp1 and Sp3 proteins can cooperate with the protein binding to the CHR. So far this factor has eluded identification. From the work described here it is obvious that a priority of future work will be to elucidate the identity of this transcriptional regulator and analyze its interactions with other factors.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Kristian Helin, Reinhard Lührmann, Roberto Mantovani and Bert Vogelstein for generously providing reagents, Jana Lorenz for expert technical assistance and Christine Lange-zu Dohna for help in the initial phase of this project. K.R. is a recipient of a graduate fellowship awarded by the Freistaat Sachsen. K.E. is supported by grants from the Bundesministerium für Bildung und Forschung through the IZKF and the Deutsche Forschungsgemeinschaft.
DDBJ/EMBL/GenBank accession no. AF450244
REFERENCES
- 1.Millar J.B. and Russell,P. (1992) The cdc25 M-phase inducer: an unconventional protein phosphatase. Cell, 68, 407–410. [DOI] [PubMed] [Google Scholar]
- 2.Sadhu K., Reed,S.I., Richardson,H. and Russell,P. (1990) Human homolog of fission yeast cdc25 mitotic inducer is predominantly expressed in G2. Proc. Natl Acad. Sci. USA, 87, 5139–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Millar J.B., Blevitt,J., Gerace,L., Sadhu,K., Featherstone,C. and Russell,P. (1991) p55CDC25 is a nuclear protein required for the initiation of mitosis in human cells. Proc. Natl Acad. Sci. USA, 88, 10500–10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weinberg R.A. (1995) The retinoblastoma protein and cell cycle control. Cell, 81, 323–330. [DOI] [PubMed] [Google Scholar]
- 5.Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev., 12, 2245–2262. [DOI] [PubMed] [Google Scholar]
- 6.Muller H. and Helin,K. (2000) The E2F transcription factors: key regulators of cell proliferation. Biochim. Biophys. Acta, 1470, M1–M12. [DOI] [PubMed] [Google Scholar]
- 7.Bernards R. (1997) E2F: a nodal point in cell cycle regulation. Biochim. Biophys. Acta, 1333, M33–M40. [DOI] [PubMed] [Google Scholar]
- 8.Zwicker J., Lucibello,F.C., Wolfraim,L.A., Gross,C., Truss,M., Engeland,K. and Müller,R. (1995) Cell cycle regulation of the cyclin A, cdc25C and cdc2 genes is based on a common mechanism of transcriptional repression. EMBO J., 14, 4514–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fajas L., Le Cam,L., Polanowska,J., Fabbrizio,E., Servant,N., Philips,A., Carnac,G. and Sardet,C. (2000) A CDE/CHR-like element mediates repression of transcription of the mouse RB2 (p130) gene. FEBS Lett., 471, 29–33. [DOI] [PubMed] [Google Scholar]
- 10.Lange-zu Dohna C., Brandeis,M., Berr,F., Mössner,J. and Engeland,K. (2000) A CDE/CHR-tandem element regulates cell cycle-dependent repression of cyclin B2 transcription. FEBS Lett., 484, 77–81. [DOI] [PubMed] [Google Scholar]
- 11.Liu N., Lucibello,F.C., Korner,K., Wolfraim,L.A., Zwicker,J. and Müller,R. (1997) CDF-1, a novel E2F-unrelated factor, interacts with cell cycle-regulated repressor elements in multiple promoters. Nucleic Acids Res., 25, 4915–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu N., Lucibello,F.C., Engeland,K. and Müller,R. (1998) A new model of cell cycle-regulated transcription: repression of the cyclin A promoter by CDF-1 and anti-repression by E2F. Oncogene, 16, 2957–2963. [DOI] [PubMed] [Google Scholar]
- 13.Resnick-Silverman L., St Clair,S., Maurer,M., Zhao,K. and Manfredi,J.J. (1998) Identification of a novel class of genomic DNA-binding sites suggests a mechanism for selectivity in target gene activation by the tumor suppressor protein p53. Genes Dev., 12, 2102–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manni I., Mazzaro,G., Gurtner,A., Mantovani,R., Haugwitz,U., Krause,K., Engeland,K., Sacchi,A., Soddu,S. and Piaggio,G. (2001) NF-Y mediates the transcriptional inhibition of the cyclin B1, cyclin B2, and cdc25C promoters upon induced G2 arrest. J. Biol. Chem., 276, 5570–5576. [DOI] [PubMed] [Google Scholar]
- 15.Krause K., Haugwitz,U., Wasner,M., Wiedmann,M., Mössner,J. and Engeland,K. (2001) Expression of the cell cycle phosphatase cdc25C is down-regulated by the tumour suppressor protein p53 but not by p73. Biochem. Biophys. Res. Commun., 284, 743–750. [DOI] [PubMed] [Google Scholar]
- 16.Bolognese F., Wasner,M., Lange-zu Dohna,C., Gurtner,A., Ronchi,A., Muller,H., Manni,I., Mössner,J., Piaggio,G., Mantovani,R. and Engeland,K. (1999) The cyclin B2 promoter depends on NF-Y, a trimer whose CCAAT-binding activity is cell-cycle regulated. Oncogene, 18, 1845–1853. [DOI] [PubMed] [Google Scholar]
- 17.Philipsen S. and Suske,G. (1999) A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res., 27, 2991–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlseder J., Rotheneder,H. and Wintersberger,E. (1996) Interaction of Sp1 with the growth- and cell cycle-regulated transcription factor E2F. Mol. Cell Biol., 16, 1659–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noe V., Alemany,C., Chasin,L.A. and Ciudad,C.J. (1998) Retinoblastoma protein associates with SP1 and activates the hamster dihydrofolate reductase promoter. Oncogene, 16, 1931–1938. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe G., Albanese,C., Lee,R.J., Reutens,A., Vairo,G., Henglein,B. and Pestell,R.G. (1998) Inhibition of cyclin D1 kinase activity is associated with E2F-mediated inhibition of cyclin D1 promoter activity through E2F and Sp1. Mol. Cell Biol., 18, 3212–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krause K., Wasner,M., Reinhard,W., Haugwitz,U., Lange-zu Dohna,C., Mössner,J. and Engeland,K. (2000) The tumour suppressor protein p53 can repress transcription of cyclin B. Nucleic Acids Res., 28, 4410–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagen G., Muller,S., Beato,M. and Suske,G. (1994) Sp1-mediated transcriptional activation is repressed by Sp3. EMBO J., 13, 3843–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Engeland K., Andrews,N.C. and Mathey-Prevot,B. (1995) Multiple proteins interact with the nuclear inhibitory protein repressor element in the human interleukin-3 promoter. J. Biol. Chem., 270, 24572–24579. [DOI] [PubMed] [Google Scholar]
- 24.Lam E.W. and Watson,R.J. (1993) An E2F-binding site mediates cell-cycle regulated repression of mouse B-myb transcription. EMBO J., 12, 2705–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zwicker J., Liu,N., Engeland,K., Lucibello,F.C. and Müller,R. (1996) Cell cycle regulation of E2F site occupation in vivo. Science, 271, 1595–1597. [DOI] [PubMed] [Google Scholar]
- 26.Makarova O.V., Makarov,E.M. and Lührmann,R. (2001) The 65 and 110 kDa SR-related proteins of the U4/U6.U5 tri-snRNP are essential for the assembly of mature spliceosomes. EMBO J., 20, 2553–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helin K., Harlow,E. and Fattaey,A. (1993) Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol. Cell. Biol., 13, 6501–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nargi J.L. and Woodford-Thomas,T.A. (1994) Cloning and characterization of a cdc25 phosphatase from mouse lymphocytes. Immunogenetics, 39, 99–108. [DOI] [PubMed] [Google Scholar]
- 29.Lucibello F.C., Truss,M., Zwicker,J., Ehlert,F., Beato,M. and Müller,R. (1995) Periodic cdc25C transcription is mediated by a novel cell cycle-regulated repressor element (CDE). EMBO J., 14, 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zwicker J., Gross,C., Lucibello,F.C., Truss,M., Ehlert,F., Engeland,K. and Müller,R. (1995) Cell cycle regulation of cdc25C transcription is mediated by the periodic repression of the glutamine-rich activators NF-Y and Sp1. Nucleic Acids Res., 23, 3822–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mantovani R. (1998) A survey of 178 NF-Y binding CCAAT boxes. Nucleic Acids Res., 26, 1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas M., Kalita,A., Labrecque,S., Pim,D., Banks,L. and Matlashewski,G. (1999) Two polymorphic variants of wild-type p53 differ biochemically and biologically. Mol. Cell Biol., 19, 1092–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Como C.J. and Prives,C. (1998) Human tumor-derived p53 proteins exhibit binding site selectivity and temperature sensitivity for transactivation in a yeast-based assay. Oncogene, 16, 2527–2539. [DOI] [PubMed] [Google Scholar]
- 34.Jones K.A. and Tjian,R. (1985) Sp1 binds to promoter sequences and activates herpes simplex virus ‘immediate-early’ gene transcription in vitro. Nature, 317, 179–182. [DOI] [PubMed] [Google Scholar]
- 35.Bennett J.D., Farlie,P.G. and Watson,R.J. (1996) E2F binding is required but not sufficient for repression of B-myb transcription in quiescent fibroblasts. Oncogene, 13, 1073–1082. [PubMed] [Google Scholar]
- 36.Liu N., Lucibello,F.C., Zwicker,J., Engeland,K. and Müller,R. (1996) Cell cycle-regulated repression of B-myb transcription: cooperation of an E2F site with a contiguous corepressor element. Nucleic Acids Res., 24, 2905–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sorensen P. and Wintersberger,E. (1999) Sp1 and NF-Y are necessary and sufficient for growth-dependent regulation of the hamster thymidine kinase promoter. J. Biol. Chem., 274, 30943–30949. [DOI] [PubMed] [Google Scholar]
- 38.Vogt B., Zerfass-Thome,K., Schulze,A., Botz,J.W., Zwerschke,W. and Jansen-Dürr,P. (1999) Regulation of cyclin E gene expression by the human papillomavirus type 16 E7 oncoprotein. J. Gen. Virol., 80, 2103–2113. [DOI] [PubMed] [Google Scholar]
- 39.Huang D., Jokela,M., Tuusa,J., Skog,S., Poikonen,K. and Syvaoja,J.E. (2001) E2F mediates induction of the Sp1-controlled promoter of the human DNA polymerase epsilon B-subunit gene POLE2. Nucleic Acids Res., 29, 2810–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Korner K., Jerome,V., Schmidt,T. and Müller,R. (2001) Cell cycle regulation of the murine cdc25B promoter: essential role for nuclear factor-Y and a proximal repressor element. J. Biol. Chem., 276, 9662–9669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.