

Abstract
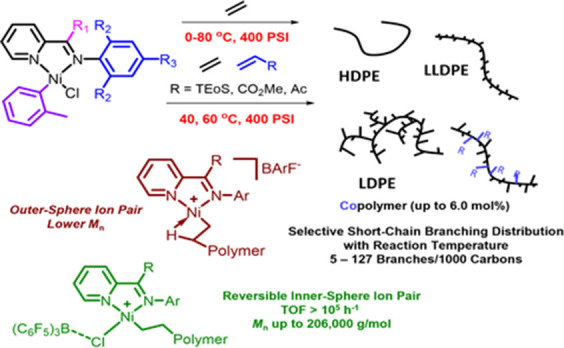
Design of catalysts for Ni-catalyzed olefin polymerization predominantly focuses on ligand design rather than the activation process when attempting to achieve a broader scope of polyolefin micro- and macrostructures. Air-stable alkyl-or aryl-functionalized NiII precatalysts were designed which eliminate the need of in situ alkylating processes and are activated solely by halide abstraction to generate the cationic complex for olefin polymerization. These complexes represent an emerging class of olefin polymerization catalysts, enabling the study of various cocatalysts forming either inner- or outer-sphere ion pairs. It is demonstrated that an organoboron cocatalyst activation produces a well-defined ion pair, which in contrast to ill-defined organoaluminum cocatalysts, can directly activate the complex by halide abstraction to yield comparatively higher molecular weight homo/copolymers. Under high ethylene pressure, broader branching densities and the gradual incorporation of short-chain branches were achieved, circumventing the need for elaborate ligand design and copolymerization with α-olefins. The underlying chain-walking mechanism and ion pair interactions were further elucidated by DFT calculations. A phenyl group on the bridging carbon functioned as a rotational barrier, producing higher molecular weight polymers compared to methyl-substituted analogs. Here, we provide a perspective to manipulate the iminopyridyl NiII system, leveraging ion pair interactions and ligand design to govern polyolefin molecular weights and microstructures.
Keywords: catalysis, ion pairs, olefin polymerization, iminopyridyl nickel complexes, organoboron cocatalyst
Introduction
The success of polymeric materials is rooted in their versatility, high performance, and affordable production.1−3 Among these materials, polyolefins continue to make up about half of the polymer production due to their exceptional chemical resistance and tunable material properties.4 In general, the physical properties of polyolefins are directly related to their molecular weight (MW) and microstructure, which is highly dependent on the involved catalytic system. Over the years, there has been a surge in the development of ligands aimed at the control of the polymer microstructure to influence material properties. However, effective microstructure control has required the use of increasingly complex ligands.5
Olefin polymerization systems rely not only on the transition metal complex but also on the cocatalyst used for activation.6−9 The ionic interaction between the metal complex and the counterion significantly impacts the catalytic activity and polymer architecture in early transition metal-catalyzed olefin polymerizations.10−14 For olefin polymerization reactions, this cation–anion interaction can exist as either inner- or outer-sphere ion pairs. Using well-defined alkyl-abstracting organoboron cocatalysts to activate zirconocene complexes, such as tris(pentafluorophenyl)borane (B(C6F5)3) and triphenylcarbenium tetraphenylborate (TBCF), showed dramatic differences in catalytic activity and stereoselectivity.15 The difference in the catalytic activity using these organoboron cocatalysts was attributed to the notion that smaller counterions favored inner-sphere ion pairs, which could compete with monomer coordination and reduce catalytic activity, and bulkier counterions preferred noncoordinating outer-sphere ion pairs, increasing catalytic activity (Figure 1A).10,16
Figure 1.
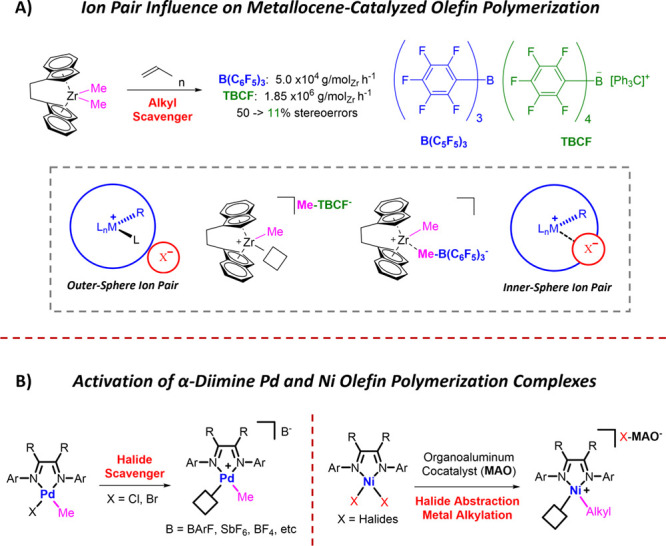
Ion pairs for olefin polymerization. (A) Activation of early transition metal complexes using alkyl abstracting inner- and outer-sphere counterions in comparison to (B) activation of α-diimine Pd complexes with halide abstracting activators and in-situ activation of α-diimine Ni complexes by halide abstracting/alkylating organoaluminum cocatalyst methylaluminoxane (MAO).
The development of the Brookhart-type catalysts launched the field of late-transition metal-catalyzed olefin polymerization (Figure 1B).17 While these α-diimine PdII and NiII complexes are highly tolerant of polar monomers and provide control over polymer microstructure through their unique “chain-walking” mechanism, investigations into the effect of the influence of the counterion for these catalytic systems are limited.18 Diimine PdII/NiII complexes can be designed with halide and carbyl ligands for activation by both organoaluminum and organoboron cocatalysts.11
Although alkylated diimine Ni complexes exist, they can suffer from poor stability.19 ,20 ,13 For example, in 1980, Dieck reported that diimine Ni dimethyl complexes are stable up to 0 °C.21,22 Switching to the sterically bulkier alkyl group, R = CH2SiMe3 exhibits higher thermal stability; however, comprehensive activation and polymerization studies have not been reported.23 Highly stable alkylated Ni complexes for polymerization remain elusive.
Diimine NiII analogs intended for olefin polymerization reactions were predominantly designed containing dihalide ligands limiting the activation to bifunctional organoaluminum cocatalysts such as methylaluminoxane (MAO), which functions through halide abstraction and metal alkylation for coordination insertion polymerization. Unfortunately, there are few catalytic systems that allow for the investigation of counterion effects in [N,N]-chelated NiII complexes. For instance, only two cationic carbyl diimine NiII complexes are reported for olefin polymerization due to lability of the Ni–C bond, rendering them thermally unstable (Figure 2A).10,17 Similarly, η3-coordinated cationic diimine Ni complexes isolated with a PF6– anion, are temperature sensitive and also require organoaluminum cocatalysts for olefin polymerization (Figure 2B).24 Alkylsilane substituted diimine Ni complexes which could copolymerize ethylene and vinyltrialkoxysilanes require an activation with a combination of B(C6F5)3 and Li(B(C6F5)4) and are also air-sensitive (Figure 2C).25 While thermally sensitive dimethyl diimine Ni complexes have been reported, they are limited to activation with organoboron cocatalysts which abstract an alkyl group to afford the cationic active species (Figure 2D).21
Figure 2.

Selected examples of [N,N]-chelated nickel olefin polymerization complexes. Carbyl [N,N]-chelated Ni complexes A–E and dihalide iminopyridyl Ni complexes with aryl imine (I–V), pyridine (VI), and bridging carbon (VII–IX) modifications. In this work, benchtop-stable carbyl iminopyridyl NiII complexes are reported.
Meanwhile, the activation through MAO limits the in-depth study on the influence of cocatalysts and reaction conditions to tailor polymer microstructures due to limited understanding of the anionic macrostructure of MAO.26 The ability to investigate inner- and outer-sphere ion pairs to govern polymeric properties is contingent upon a carbyl Ni precatalyst.11 Therefore, it is crucial to develop a robust carbyl NiII precatalyst activatable by a variety of cocatalysts to evaluate the impact of counterions on [N,N]-chelated NiII complexes, because the catalytic activity and the type of polymeric material produced can be influenced by the activation method and the choice of cocatalyst.27−30 The shift from intricate ligand design to utilization of ion pairs could offer control over polymer microstructure and MW.
Specifically, we seek to create an aryl-functionalized Ni precatalyst, activated solely by a halide abstraction, eliminating the need for an alkylation process to generate the cationic complex for olefin polymerization. These complexes would serve as an ideal system for a variety of activators, expanding beyond the limitation of organoaluminum cocatalysts. Thus, we selected iminopyridine ligands, with the [N,N]-ligand backbone serving as a diimine analog and the pyridine moiety acting as a stronger σ-donor to stabilize the labile Ni–C bond.31 Notably, benchtop-stable iminopyridyl mesityl NiII complexes have been prepared (Figure 2E) for use in Negishi cross-coupling reactions.32 Iminopyridine ligands have been extensively used in olefin polymerization, with the Ni complexes exhibited similar catalytic activity compared to their α-diimine analogues.
However, traditional ligand design focusing on increasing steric bulk on the aryl imine produced lower MW polymers, seemingly contradicting the established trend reported for late-transition metal complexes where increasing axial steric bulk on the metal center favored chain-propagation and yielded higher MW polymers (Figure 2I–III).33−36 We hypothesize that the steric bulk of the single aryl imine moiety cannot sufficiently contribute to the effective blockage of the metal center to limit chain-transfer. These complexes could produce moderate MW polyethylene (PEs) (Mn < 26,000 g mol–1) at ambient conditions with the development of the “half-sandwich” iminopyridine ligands (Figure 2IV), where the “aryl cap” of the 8-arylnaphthyl imine aggressively shielded a single axial site.37 Guo and Dai later introduced a rotation-restricted strategy using rigid benzosuberyl substituents (Figure 2V), enhancing axial shielding and yielding moderate MW PEs (Mn < 24,000 g mol–1) and high MW PEs when combined with the “half-sandwich” scaffold.38−41 Alternatively, substituents at the 6-position of the pyridine (Figure 2VI) increased the mol % incorporation of polar monomers, but occupy the same coordination plane as ethylene and the growing polymer chain, promoting chain-transfer and the production of low MW PEs and oligomers. (Mn < 1700 g mol–1).42,43
Therefore, we sought to explore the impact of steric bulk on the substituent on the imino-carbon, commonly referred as bridging carbon, by introducing a phenyl group as a “rotational barrier”, which would restrict the aryl imine substituents in their mobility and increase axial bulk to limit chain-transfer. Surprisingly, few iminopyridyl Ni complexes have been designed with substituents other than a hydrogen or methyl group on the carbon.44 Laine et al. varied the bulk with hydrogen, methyl, and phenyl groups, but the MW was inconsistent with the steric bulk (Figure 2VII).45 Zohuri and co-workers found that replacing the methyl bridging substituent with a phenyl group led to a significant decrease in Mv, suggesting bulkier bridging carbon substituents negatively affect polymerization (Figure 2VIII).46 Zubris and co-workers alternatively explored alkyl groups on the bridging carbon but only produced low MW polymers (Mn < 1600 g mol–1) with an ethyl substituent (Figure 2IX).47
In this contribution, we disclose the synthesis of an emerging class of benchtop, air-stable iminopyridyl NiII complexes, C1–C8, which feature an o-tolyl ligand as the carbyl substituent as the aryl ligand has been found to produce benchtop-stable bipyridine NiII complexes.31 We investigate the significance and effect of the ion pair for ethylene polymerization, and its role in tailoring branching characteristics and density of the produced PE with respect to the reaction conditions. The complexes are designed with a focus on a rotational barrier provided by either a methyl or phenyl group on the bridging carbon and the ortho-substituents on the aryl imine. Various cocatalysts such as modified-MAO, sodium tetrakis[3,5-bis(trifluoromethyl) phenyl]borate (NaBArF), silver tetrakis[3,5-bis(trifluoromethyl) phenyl]borate (AgBArF), and organoboranes were evaluated for ethylene polymerization. Furthermore, we demonstrate the use of B(C6F5)3 as the primary cocatalyst for carbyl NiII iminopyridyl complexes, producing active complexes and yielding high MW PE (Mn > 100 kg mol–1) compared to known nonhalf sandwich and half-sandwich iminopydridyl complexes.
Density functional theory (DFT) studies support the establishment of a reversible inner-sphere anion coordination with ClB(C6F5)3–, which reduces the overall catalytic activity, but allows the production of higher MW polymers. Moreover, the activation of these complexes with B(C6F5)3 allows for the study that the substituent on the bridging carbon greatly influences the polymer MWs through a rotational restriction of the ortho-substituents on the aryl imine moiety. The chain-walking behavior leads to controlled branching densities and short-chain branching distributions, ranging from methyl to butyl, depending on the reaction temperature (0–80 °C), producing polymers with diverse macrostructures. DFT studies also support a proposed mechanistic pathway to elucidate the underlying chain-walking mechanism. Complex C7 is used to evaluate the efficacy of the carbyl iminopyridyl Ni complexes for copolymerization with polar monomers. In addition, a single catalyst can generate several types of PEs, such as high-density PE (HDPE), linear low-density PE (LLDPE), and low-density PE (LDPE) with controlled branching densities, circumventing the need for copolymerization with α-olefins. These carbyl NiII iminopyridyl complexes are competitive catalysts for olefin polymerization in which not only the MW but also the microstructure can be tuned by cocatalyst selection and ligand design, which is desired for a one-complex/one-monomer catalytic system.
Results and Discussion
Design and Development of Carbyl Iminopyridyl Nickel Catalysts
A series of iminopyridine ligands (L1–L8) were synthesized using a one-step condensation reaction of the aniline with the targeted ketone and were isolated in high yields (Figure 3a).34,45,46,48,49 Ligands L1, L3, L5, and L7 feature a methyl group on the bridging carbon atom and a varied steric bulk on the aryl imine moiety, ranging from isopropyl to benzosuberyl groups, whereas ligands L2, L4, L6, and L8 have a phenyl group on the bridging carbon. Notably, the m-xylyl substituents (L5 and L6) and the benzosuberyl substituent (L7 and L8) have only been investigated in iminopyridyl Pd complexes with a methyl group on the bridging carbon.50 The complexations were achieved either by ligand exchange using (PPh3)2Ni(o-Tol)Cl or by oxidative addition with Ni(COD)2 and 2-chlorotoluene (Figure 3b), forming iminopyridyl NiII catalysts C1–C8 in good yields (66–92% for C1–C7 and 10% for C8, Figure S2). We found that the more traditional route using Ni(COD)2 imposed challenges and resulted in low yielding reactions. In contrast, the alternative route using ligand exchange between a commercially available nickel precursor ((PPh3)2Ni(o-Tol)Cl) and the iminopyridine was more practical for the majority of the complexes with the exception of C4 and C6 which achieved higher yields using the Ni(COD)2 oxidative addition pathway.
Figure 3.
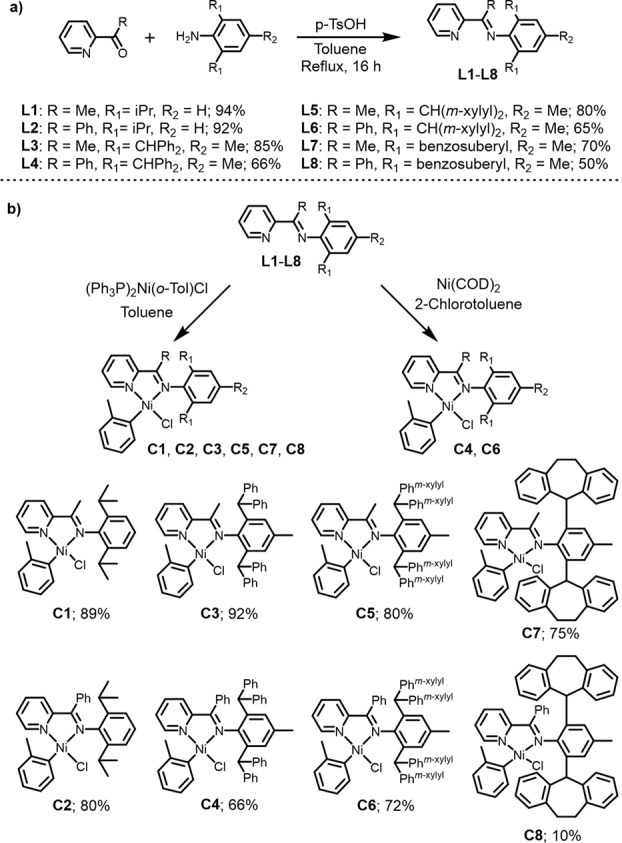
Synthesis of carbyl iminopyridyl NiII complexes. (a) Synthesis of iminopyridine ligands L1–L8. (b) Complexation of ligands to form NiII catalysts C1–C8.
The structures of C1, C3, C4, C5, C7, and C8 were determined by single-crystal X-ray diffraction, revealing in all cases square planar geometries (Figure 4). In addition, the square planar configuration allowed for NiII complexes C1-C7 to be fully characterized by 1H NMR and 13C NMR (Figures S3–S20). The topographic steric maps quantify the steric effect on the metal center imposed by the rotational barrier of the bridging carbon on the aryl imine substituents.51 Catalysts C1, C3, C5 and C7 illustrate the steric influence provided by the aryl imine moiety on the blockage of the metal center. However, the inclusion of the phenyl group on the bridging carbon atom increases the buried volume percentage to 47.0% for C4 and 54.5% for C8 compared to their methyl analogues (45.1% for C3 and 47.6% for C7). These data support our hypothesis that the substituent on the bridging carbon atom can introduce a rotational barrier, enhancing the shielding of the metal center. This is achieved by constraining the rotation of the ortho-aryl imine substituents, thereby imposing greater axial bulk and potentially retarding chain-transfer mechanisms.
Figure 4.
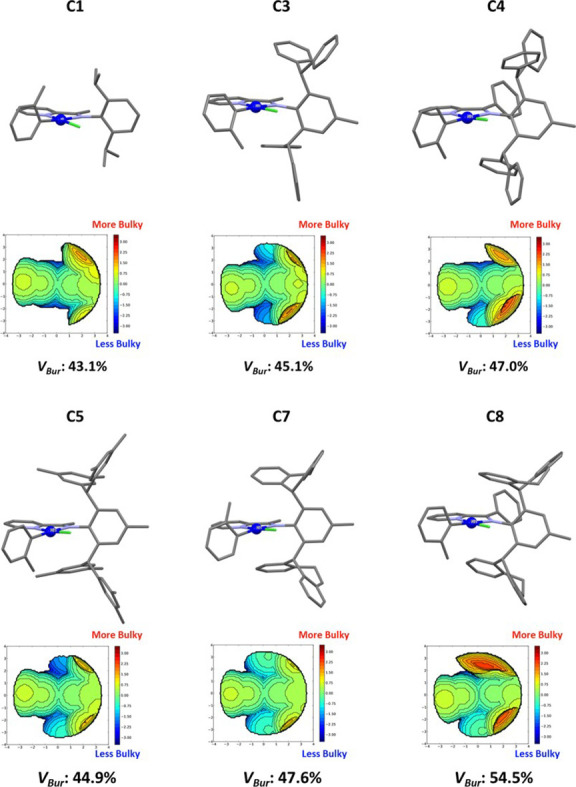
X-ray structures of carbyl iminopyridyl NiII complexes. Stick models, steric maps, and VBur% of C1, C3, C4, C5, C7, and C8.
Air- and Moisture Stability
During the synthesis of the complex series, we noticed a stability and robustness in atmospheric environments. Therefore, we thought to conduct a more detailed study to confirm their benchtop stability. Complexes C1 and C3 were selected as representative complexes for air- and moisture stability tests. Here, we subjected the complexes to open air and were analyzed by 1H NMR spectroscopy for any signs of degradation. After 3 weeks of taking aliquots, we did not find any decomposition or degradation of complexes (Figures S150 and S151). A vigorous moisture sensitivity test was conducted by treating the complexes with excessive amounts of pure water. The 1H NMR analysis of the dried crude samples showed a decomplexation of C1 of only 13.4% and 18% for C3 (Figures S152 and S153). These studies illustrate the high stability of the complexes and were therefore referred to as “benchtop” stable.
Cocatalyst Study
We selected C1 as a model to study the counterion effect for ethylene polymerization by using various cocatalysts. Preliminary polymerizations with C1 were conducted by in situ activation with modified-MAO (MMAO) and diethylaluminum chloride (Et2AlCl) at 20 °C (Table 1, entry 1, 2). The MW of the polymers was analyzed using high-temperature gel permeation chromatography (HT-GPC) and was found to be 6.5 and 4.0 kg mol–1, respectively. Although the organoaluminum cocatalysts produced a highly active species (TOF > 535,700 h–1), the polymerization reaction resulted in a higher dispersity (Đ = 2.47 and 2.11) for the isolated polymers. The in situ activation of C1 with NaBArF produced PE with higher MW (Mn = 8.5 kg mol–1, Table 1, entry 3). However, the catalytic activity was poor (TOF = 12,900 h–1). We suggest that the sodium cation is unable to efficiently abstract the halide from the metal center, leading to lower productivity. The poor activity was addressed with AgBArF, which produces AgCl with greater lattice energy than NaCl, implying a stronger halide abstraction.52 Activating with AgBArF produced a 12 times more active catalyst (TOF = 162,900 h–1), but reduced the polymer MW to 6.4 kg mol–1 (Table 1, entry 4). Other commonly used cocatalysts, such as NaBF4 and AgPF6, were investigated but were found to produce no polymer, illustrating the significant impact the ion pair has on olefin polymerization. To further enhance catalytic activity, (B(C6F5)3) was used as an additive, with reports showing that the organoborane promotes ethylene polymerizations with diimine NiII catalysts.53 Although the sequential addition of both B(C6F5)3 and AgBArF significantly increased the activity of C1 (TOF = 274,300 h–1), the MW of the resulting PE decreased to 4.6 kg mol–1 (Table 1, entry 5). While the dispersity remained consistent when only using AgBArF, it increased to 2.17 with the addition of B(C6F5)3. This increase in dispersity suggested a simultaneous addition of AgBArF and B(C6F5)3 could generate active species with differing ion pairs. Remarkably, activating C1 with only B(C6F5)3 produced PE with the highest MW for the tested cocatalysts (Mn = 11.4 kg mol–1) and exhibited good catalytic activity and lowest dispersity (TOF = 102,900 h–1, Đ = 1.93, Table 1, entry 6). We hypothesized that the B(C6F5)3 can abstract the halide and generate the B(C6F5)3-Cl anion. For instance, Erker and Le Gendre54 showed that BCF selectively abstracts a chloride from titanecene complexes generating a frustrated Lewis pair with a B(C6F5)3-Cl anion which has an inner-sphere coordination. This report motivated us to investigate if a similar phenomenon occurs with our complexes. Therefore, a 19F NMR was conducted and a shift in all three peaks of the fluorenes of B(C6F5)3 was observed which implies the formation of B(C6F5)3-Cl (Figure S149). Additionally, we believe that aryl ligand abstraction by B(C6F5)3 is unlikely for two reasons: Ni(I) halides complexes are not known to initiate polymerization, and Ni(I) halides tend to dimerize, and such dimers are very stable and therefore catalytically inactive. For these reasons, we strongly believe that the B(C6F5)3 activates the complex by halide abstraction.
Table 1. Ethylene Polymerization of C1 with Different Cocatalystsa.
| entry | cocatalyst | time (min) | yield (g) | productivity (kg mol–1Ni h) | TOF (103)(h–1) | B/1000Cb | Mnc (kg mol–1) | Đc | Tmd (°C) |
|---|---|---|---|---|---|---|---|---|---|
| 1e | MMAO (200 equiv) | 5 | 3.0 | 18,000 | 642.9 | 27 | 6.5 | 2.47 | 109.3 |
| 2e | Et2AlCl (200 equiv) | 5 | 2.5 | 15,000 | 535.7 | 40 | 4.0 | 2.11 | 102.7 |
| 3 | NaBArF (1.2 equiv) | 10 | 0.3 | 360 | 12.9 | 26 | 8.5 | 1.98 | 108.3 |
| 4 | AgBArF (1.2 equiv) | 10 | 3.8 | 4560 | 162.9 | 32 | 6.4 | 2.00 | 105.2 |
| 5f | AgBArF (1.2 equiv) | 5 | 3.2 | 7680 | 274.3 | 41 | 4.6 | 2.17 | 104.6 |
| 6 | B(C6F5)3 (10 equiv) | 10 | 2.4 | 2880 | 102.9 | 25 | 11.4 | 1.93 | 108.6 |
Conditions: 5 μmol of C1, 400 psi ethylene, 45 mL of toluene, 20 °C.
Determined by 1H NMR in D2-tetrachloroethane at 125 °C.
Determined by GPC in 1,2,4-trichlorobenzene at 160 °C using polystyrene calibration.
Determined by differential scanning calorimetry.
2 μmol of C1.
Included 10 equiv of B(C6F5)3. TOF = Turnover Frequency, Mn = Number-Average Molecular Weight, Đ = Dispersity, Tm = Melting Temperature.
In transition metal polymerization catalysis, it is known that inner- or outer-sphere ion pairing is attributed to different catalytic activity of the metal center. During polymerization, monomer insertion occurs at the open site of the active catalyst; however this coordination site can be occupied by the cocatalyst.55 Weakly coordinating anions, such as BArF–, should prefer outer-sphere ion pairs, facilitating monomer coordination and higher catalytic activity.56 However, this leads to the more β-CH agostic species, which can lead to chain-transfer to monomer by β-H elimination and lower MWs, as observed experimentally with the activation of C1 with AgBArF. Activation with B(C6F5)3 produces ClB(C6F5)3– as the counterion and more favorably lead to inner-sphere ion pairs, blocking the vacant coordination site and competing with monomer coordination and the formation of β-CH agostic species.16 This inner-sphere coordination would reduce the overall catalytic activity, but would also lead to a lower impact of chain-transfer reactions and to higher MW polymers,57 as observed with the activation of C1 with B(C6F5)3. This study suggests that the use of B(C6F5)3 for Ni-catalyzed ethylene polymerization has a profound influence on the chain-propagation to access high MW polymers via an inner-sphere ion pair interaction.
DFT Study for Counter Ion Interaction
To validate the above proposition of a stronger inner-sphere interaction for the ClB(C6F5)3– anion, a computational investigation was conducted. The DFT calculations aimed at elucidating the structural and energetic differences in the interaction of the cationic nickel catalyst with the BArF– and ClB(C6F5)3– anions. The ligand was initially simplified to the unsubstituted pyridine-imine, NC5H4-2-CH=NH, and the PE chain was truncated to a propyl group, generated by insertion of an ethylene molecule into the Ni-CH3 bond, for a full quantum mechanical (QM) approach. However, later calculations also addressed the system with the real ligand (L1) at the QM/MM level (Figure 5, Top). The geometry optimizations were conducted in the presence of a polarizable continuum with the permittivity of dichloromethane and thermal corrections were applied to obtain standard Gibbs energy values in condensed medium (298 K, 1 mol/L). The cation–anion interaction was investigated only for the most stable Ni–Pr isomer, namely the β-agostic complex with the propyl group trans to the pyridine ring (Figure S140). At the full QM level on the simplified system, for both anions, the addition with displacement of the β-agostic CH ligand is exoergic. The addition of ethylene lowers the Gibbs energy of the system by −12.8 kcal mol–1, whereas the anion coordination lowers it by −13.2 kcal mol–1 for ClB(C6F5)3– and −5.1 kcal mol–1 for BArF– (Figure 5, Bottom). The coordination site left open by dissociation of the propyl C–H function is saturated by interaction with a Cl lone pair for the former anion and an F lone pair for the latter one. The suitability of the QM/MM approach was first tested by reoptimizing handling the three noninteracting C6H3-3,5-(CF3)2 groups of the BArF anion at the molecular mechanics level, yielding comparable stabilization (−5.5 vs −5.1 kcal mol–1) and metric parameters (Figure S141). The results of the QM/MM calculations for the model system with ligand L1 are shown in Figure 5. Views of the optimized molecules are in Figure S143.
Figure 5.
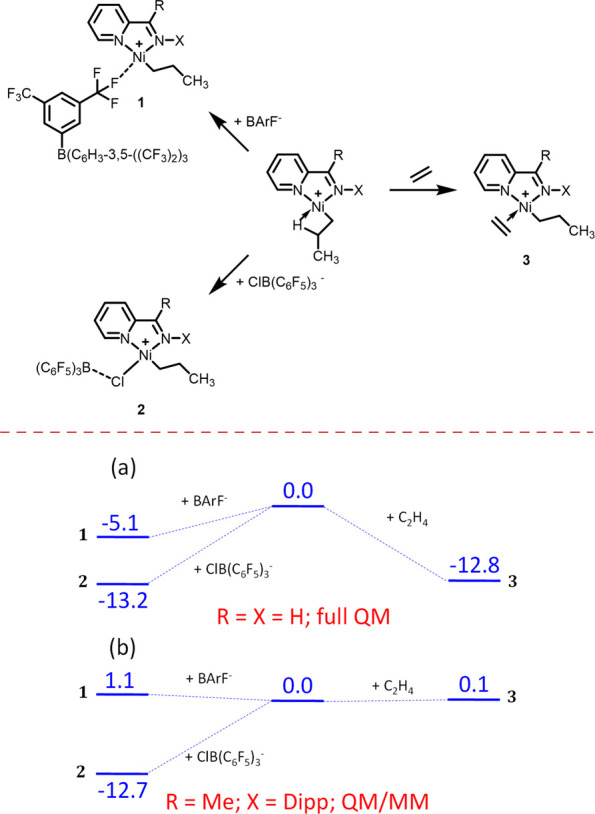
Cation–anion ion pair interaction with C1. Top: Interaction between the [Ni(NC5H4CRNX)(Pr)]+ cation (center) and the BArF– and ClB(C6F5)3– anions (left) and ethylene monomer (right). Bottom: Gibbs energy profile (values in kcal mol–1) for the (a) simplified ligand (X = R = H) at the full QM level; (b) ligand L1 (X = Dipp, R = Me) at the QM/MM level (the Dipp substituent in the cation, the three C6F5 groups in the Cl-BCF anion and the three noninteracting C6H3-3,5-(CF3)2 groups in the BArF anion were treated at the MM level).
The greater stabilization associated with the ClB(C6F5)3– coordination is obviously related to the greater donor power of the Cl lone pair in the B–Cl bond, relative to the F lone pair in the C–F bond of the BArF– anion. In the structure of the [Ni(NC5H4CHNH)(Pr)]+ClB(C6F5)3– adduct, the Ni–Cl distance (2.250 Å) is slightly longer than in the optimized [Ni(NC5H4CHNH)(Pr)Cl] complex (2.225 Å, a lengthening by only 0.025 Å), but the B–Cl distance (2.255 Å) is much longer than in the free ClB(C6F5)3– anion (2.003 Å, a lengthening by 0.252 Å).58 For the QM/MM calculation on [Ni(NC5H4CMeNDipp)(Pr)]+ClB(C6F5)3–, the corresponding distances are 2.264 Å (Ni–Cl) and 2.695 vs 2.135 Å for B–Cl in the adduct and free ClB(C6F5)3– anion, respectively (Figure S143). Furthermore, the B atom is pyramidalized (sum of the three CBC angles = 350.7° in the QM model, 353.2° in the QM/MM L1 model) to a much lesser extent than in the free ClB(C6F5)3– anion (QM: 340.8°; QM/MM: 341.0°), whereas the neutral B(C6F5)3 Lewis acid has a planar B atom (360°). These structural parameters suggest that the Ni–Cl–B bonding is better considered as a weak dative interaction from the Cl ligand in the neutral [Ni(NC5H4CHNH)(Pr)Cl] complex to the B atom in the B(C6F5)3 Lewis acid (Ni–Cl → B), rather than as a Ni⃖Cl–B dative interaction from the ClB(C6F5)3– anion to the [Ni(NC5H4CHNH)(Pr)]+ cation. The Gibbs energy of the [Ni(NC5H4CHNH)(Pr)Cl] + B(C6F5)3 system is −12.2 kcal mol–1 relative to the β-agostic [Ni(NC5H4CHNH)(Pr)]+ cation + ClB(C6F5)3– anion, namely only +1.0 kcal mol–1 relative to the inner-sphere ion pair (Figure S142). Thus, according to these calculations, the Lewis acid–base interaction between [Ni(NC5H4CHNH)(Pr)Cl] and B(C6F5)3 is weak but can furnish the β-agostic alkyl complex as a kinetically competent intermediate and then the ethylene adduct can form rather easily. In the structure of the inner-sphere [Ni(NC5H4CRNX)(Pr)]+BArF– ion pairs (QM: X = R = H; QM/MM; R = Me, X = Dipp), the Ni–F distance is 2.082 (QM) or 2.091 (QM/MM) Å and the C–F distance of the Ni-coordinated CF bond is 1.432 (QM) or 1.429 (QM/MM) Å, whereas all other noninteracting CF bonds are a bit shorter (QM: 1.35–1.36 Å; QM/MM: 1.382–1.384 Å), attesting the non-negligible effect of the F → Ni interaction on the C–F bond. Comparison of the energetic parameters for the simplified system (full QM, Figure 5, Bottom a) and for the better L1 model (QM/MM, Figure 5, Bottom b) suggests that the steric effect of the Dipp substituent plays an important role particularly on the monomer coordination and on the coordination of the bulkier BArF– anion, whereas it has a very minor destabilizing effect on the addition of the ClB(C6F5)3– anion.
In conclusion, the B(C6F5)3 Lewis acid is able to activate the Ni–Cl bond to remove the chloride ion from the nickel coordination sphere, but the cation interaction with ClB(C6F5)3– competes efficiently with the ethylene coordination, whereas the BArF– anion has essentially no retardation effect. The inner-sphere [Ni(NC5H4CMeNDipp)(Pr)]+ClB(C6F5)3– ion pair, better described as a [Ni(NC5H4CMeNDipp)(Pr)(Cl)]···B(C6F5)3 Lewis acid–base adduct, is predicted to be the resting state of the propagation process, rather than the outer-sphere [Ni(NC5H4CMeNDipp)(Pr)(C2H4)]+ClB(C6F5)3– ion pair, and the energy span of the propagation process is greater, resulting in a slower polymerization.58
Ethylene Polymerization Study with C1–C8
Since B(C6F5)3 has been found to be an exceptional activator for this system yielding higher MW materials, we sought to investigate the substituent effect using B(C6F5)3 for all iminopyridyl complexes. Ethylene polymerizations were conducted at 20 °C with 400 psi of ethylene pressure, yielding the results summarized in Table 2. Complexes with the methyl group on the bridging carbon atom (C1, C3, C5, and C7) highlight the effect of increasing the steric bulk on the aryl imine moiety. No significant difference in MWs was observed for the polymers produced by C1, C3 and C5 (Mn = 11.4, 7.8, and 11.7 kg mol–1), in line with the marginal increase of the calculated VBur%. Although the VBur% increases to only 47.6% in C7, the polymers produced have the highest MWs of the methyl bridging carbon series (Mn = 153.7 kg mol–1) with lower dispersity (Đ = 1.77) at 20 °C. This is over 10 times higher than any previously tested complex with a methyl-substituted carbon bridging and 5 times higher than similar catalysts activated with organoaluminum (Table 2, entry 4).41 The data suggests that the benzosuberyl ligand provides a rigid steric environment that orients the aryl moiety closed to the metal center and further supports the hypothesis that higher MW materials can also be achieved with a more rigid ligand design.
Table 2. Ethylene Polymerization of C1–C8 at 20 °Ca.
| entry | catalyst | time (min) | yield (g) | productivity (kg mol–1Ni h) | TOF (103)(h–1) | B/1000Cb | Mnc (kg mol–1) | Đc | Tmd (°C) |
|---|---|---|---|---|---|---|---|---|---|
| 1e | C1 | 10 | 2.4 | 2880 | 102.9 | 25 | 11.4 | 1.93 | 108.6 |
| 2 | C3 | 10 | 1.7 | 1020 | 36.4 | 30 | 7.8 | 1.94 | 101.0 |
| 3 | C5 | 10 | 1.6 | 960 | 34.3 | 37 | 11.7 | 1.97 | 99.7 |
| 4 | C7 | 10 | 0.8 | 480 | 16.8 | 26 | 153.7 | 1.77 | 101.5 |
| 5e | C2 | 60 | 1.5 | 300 | 10.7 | 36 | 6.9 | 1.92 | 102.1 |
| 6 | C4 | 60 | 0.9 | 90 | 3.2 | 49 | 23.8 | 1.75 | 112.0 |
| 7 | C6 | 60 | 1.2 | 120 | 4.3 | 45 | 37.0 | 1.62 | 82.9 |
| 8 | C8 | 180 | 0.5 | 167 | 0.6 | 42 | 71.3 | 2.06 | 91.5 |
Conditions: 10 μmol of Ni catalyst, 10 equiv of B(C6F5)3, 400 psi ethylene, 45 mL of toluene.
Determined by 1H NMR in D2-tetrachloroethane at 125 °C.
Determined by GPC in 1,2,4-trichlorobenzene at 160 °C using polystyrene calibration.
Determined by differential scanning calorimetry.
5 μmol of Ni. TOF = Turnover Frequency, Mn = Number-Average Molecular Weight, Đ = Dispersity, Tm = Melting Temperature.
Complexes with the phenyl group on the bridging carbon (C2, C4, C6, and C8) should increase the rotational restriction of the aryl imine substituents. C2 exhibited low catalytic activity (TOF = 10,700 h–1) and lower in MW (Mn = 6.9 kg mol–1) compared with C1. Surprisingly, C4 exhibited a completely different result and was able to produce PE with MW more than triple compared to C3 (Mn = 23.8 kg mol–1) (Table 2, entry 6). The polymerization results of C4 supported our hypothesis that the phenyl substituent on the bridging carbon atom acts as a rotational barrier and is key for producing higher MW polymers. Like C4, C6 produced much higher MW PE (Mn = 37.0 kg mol–1) with lower dispersity (Table 2, entry 7) and further confirmed our hypothesis, where bulkier bridging carbon substituents can provide higher shielding to the metal center. As for C2, the isopropyl group is not bulky enough even with the phenyl group acting as a rotational barrier, thus resulting in low MW PEs. Synthetic challenges in preparing pure and isomer-free ligands, low complexation, and turnover yields, classified C8 as a complex with undesirable properties. The data suggests that emphasis on the design of ligands with a rotational barrier provided by the bridging carbon atom substituent may be more effective at producing higher MW polymers.
Copolymerization
The copolymerization of ethylene with a small percent incorporation of polar monomers (2–5 mol %) can introduce functionalities that combine the high strength and durability of PE with improved chemical resistance, adhesion, polarity, and mechanical properties. Numerous Ni complexes have been reported for the copolymerization of ethylene with polar monomers, utilizing various ligands such as α-diimine, phosphine-sulfonate, salicylaldimine, and N-heterocyclic carbene, among others.59 However, iminopyridine ligands have predominantly been reported for ethylene copolymerization in the form of Pd complexes, capable of producing copolymers with both high molecular weights and percentages of methyl acrylate (MA) and acrylic acid incorporation.40,50,60,61 In comparison, dihalide iminopyridyl NiII complexes can only produce low MW copolymers with moderate MA incorporation (4.8 kg mol–1 with 2.4 mol %).43 We selected C7 for ethylene/polar monomer copolymerization as the complex can produce high MW PE at ambient conditions. Interestingly, C7 produced ethylene/MA copolymers (Table 3, entries 5 and 6) with higher MW (35.8 kg mol–1) and 2.2 mol % incorporation. Additionally, we tested vinyltriethoxysilane (VTEoS) and vinyl acetate (VAc), which have never been reported as comonomers for iminopyridyl NiII complexes. With 0.5 M VTEoS loading, C7 produced copolymer with moderate MW (66.3 kg mol–1) and 3 mol % incorporation (Table 3, entry 2). Increasing the loading to 1.0 M nearly doubled the mol % incorporation with a slight decrease in MW to 48.1 kg mol–1 (Table 3, entry 4). For copolymerization with VAc, C7 produced copolymers with MWs of 53.3 kg mol–1 and incorporation of 0.26 mol % (Table 3, entry 7).
Table 3. Ethylene/Polar Monomer Copolymerization of C7a.
| entry | monomer (M) | temp. (°C) | yield (g) | TOF (103)(h–1) | B/1000Cb | Mnc (kg mol–1) | Đc | incorp.b (mol %) | Tmd (°C) | Tgd (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1e | 40 | 3.2 | 11.4 | 57 | 111.6 | 1.71 | 68.7 | –36.0 | ||
| 2 | VTEoS [0.5] | 40 | 0.8 | 0.7 | 52 | 66.3 | 1.60 | 3.1 | 56.2 | –55.0 |
| 3f | VTEoS [0.5] | 60 | 1.2 | 2.1 | 69 | 33.0 | 1.68 | 4.6 | 27.2 | –64.7 |
| 4 | VTEoS [1.0] | 40 | 0.45 | 0.4 | 50 | 48.1 | 1.79 | 5.9 | 46.8 | –63.6 |
| 5g | MA [0.1] | 40 | 0.15 | 0.06 | 61 | 55.6 | 1.57 | 0.34 | 73.1 | –39.2 |
| 6g | MA [0.1] | 60 | 0.3 | 0.1 | 83 | 35.8 | 1.66 | 2.2 | 26.4 | –51.9 |
| 7g | VAc [0.25] | 60 | 0.5 | 0.2 | 78 | 53.3 | 1.65 | 0.26 | 46.9 | –49.8 |
Conditions: 20 μmol of C7, 1.2 equiv of AgBArF, 400 psi ethylene, 45 mL of toluene, 120 min.
Determined by 1H NMR in D2-tetrachloroethane at 125 °C.
Determined by GPC in 1,2,4-trichlorobenzene at 160 °C using polystyrene calibration.
Determined by differential scanning calorimetry.
10 μmol of C7, 60 min.
10 umol of C7
240 min. [M] = molar concentration, TOF = Turnover Frequency, Mn = Number-Average Molecular Weight, Đ = Dispersity, Tm = Melting Temperature, VTEoS = vinyltriethoxysilane, MA = methyl acrylate, VAc = vinyl acetate.
Branching Density and Distribution
To confirm the thermal stability of the carbyl complexes, C1 was assessed at reaction temperatures ranging from 0 to 80 °C. The complex is still highly active in polymerization reactions up to 60 °C, which is indicative of good thermal stability (TOF = 94,300 h–1). However, at 80 °C, the activity dramatically decreases to 30,000 h–1 which suggests catalyst degradation. We observed that the MW of PEs from C1 is highly dependent on the reaction temperature. With decreasing temperatures, the MW of PEs increased to 65.1 kg mol–1 at 0 °C (Table 4, entry 1). Only oligomers were generated at temperatures above 40 °C (Table 4, entry 4, 5). A similar trend was observed in the iminopyridyl NiII catalysts C2–C7, with C4 producing high MW polymers at 0 °C (Mn = 164.6 kg mol–1) and 5.6 kg mol–1 at 60 °C with the addition of the rotational barrier provided by the bridging carbon (Table S1).
Table 4. Ethylene Polymerization of C1 at 0–80 °Ca.
| entry | temp (°C) | time (min) | yield (g) | productivity (kg mol–1Ni h) | TOF (103)(h–1) | Mnb (kg mol–1) | Đb | Tmc (°C) | B/1000Cd | branching distribution (%)f | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Me | Et | Pr | Bu | Am | Lg | ||||||||||
| 1 | 0 | 30 | 1.1 | 440 | 15.7 | 65.1 | 1.65 | 127.8 | 5 | 100 | 0 | 0 | 0 | 0 | 0 |
| 2 | 20 | 10 | 2.4 | 2880 | 102.9 | 11.4 | 1.93 | 108.6 | 25 | 94.3 | 3.5 | 2.2 | 0 | 0 | 0 |
| 3 | 40 | 10 | 1.8 | 2160 | 77.1 | 3.7 | 1.66 | 64.4 | 56(46)e | 80.0 | 4.7 | 2.9 | 2.6 | 2.1 | 7.6 |
| 4 | 60 | 10 | 2.2 | 2640 | 94.3 | 2.0 | 1.51 | 9.3 | 95(72)e | 74.0 | 6.2 | 4.1 | 2.6 | 3.4 | 9.7 |
| 5 | 80 | 10 | 0.7 | 840 | 30.0 | 1.0 | 1.74 | –23.6 | 127(81)e | 66.4 | 7.5 | 4.5 | 2.8 | 6.6 | 12.1 |
Conditions: 5 μmol of Ni catalyst, 10 equiv of B(C6F5)3, 400 psi ethylene, 45 mL of toluene.
Determined by GPC in 1,2,4-trichlorobenzene at 160 °C using polystyrene calibration.
Determined by differential scanning calorimetry.
Determined by 1H NMR in D2-tetrachloroethane at 125 °C.
Branching density corrected for saturated end groups.
Determined by 13C NMR in D2-tetrachloroethane at 125 °C. Percentages of branch length determined from relative intensity ratios of methyl (1B1, 1B2, 1B3), methylene (2B4, 3B5) or methine (brB4+) signals of the respective branch. TOF = Turnover Frequency, Mn = Number-Average Molecular Weight, Đ = Dispersity, Tm = Melting Temperature, Me = Methyl, Et = Ethyl, Pr = Propyl, Bu = Butyl, Am = Amyl, Lg = Large.
Surprisingly, the branching density of PEs obtained from C1 exhibited a linear relationship with respect to the reaction temperature under high pressure conditions where chain-walking should be suppressed (Figure S1).621H NMR and differential scanning calorimetry (DSC) showed that the PEs obtained from C1 at 0 °C had 5 branches/1000 carbon atoms and a Tm of 127.8 °C, classifying the polymer as HDPE. At 20 °C, the branching density increased to 25 branches/1000 carbon atoms and a decrease in Tm to 108.6 °C, characteristic of LLDPE. A wax-like LLDPE was obtained at 40 °C, with a branching density of 56 branches/1000 carbon atoms and a Tm of 64.4 °C. An LDPE-like material was generated at 60 °C with a branching density of 95 branches/1000 carbon atoms and a Tm of 9.3 °C, and an oligomeric oil was formed at 80 °C (Table 3, entries 1–5). A similar linear branching trend was observed with complexes C2–C7 in the same reaction temperature range. For example, C4 ranged from 18 branches/1000 carbon atoms at 0 °C to 104 branches/1000 carbon atoms at 60 °C. We suggest that the unique control of the branching density exhibited by the carbyl NiII complexes is influenced by the iminopyridine ligand in combination with the inner-sphere organoboron/Ni ion pair and reaction temperature. In comparison to “half-sandwich” iminopyridyl NiII complexes, their branching density is limited to a range of 79 to 125 branches/1000 carbon atoms at 25 and 70 °C.63 In summary, a single catalyst can produce several types of PEs with broad branching densities. This versatility is achieved without the need for expensive comonomers, making it highly desirable in polyolefin syntheses and processes. We point out that it is unusual for these complexes to produce a wide variety of PE at high pressures, where chain-walking should be retarded.
The ability to produce a wide range of branching densities resulted in the production of diverse materials and prompted further investigation into their unique microstructures. The material properties of the PE suggest a branching microstructure commonly achieved through α-olefin copolymerization, which introduces short-chain branches (SCBs). It has been understood that the controlled incorporation of SCBs, where the branch length is 5 carbon atoms or less, can dramatically impact the mechanical properties of PE.64 Therefore, the analysis of the branching microstructure of polymers produced by C1 under high ethylene pressure was conducted using quantitative 13C NMR spectroscopy (Figures S58–S63) and is included in Table 4.65,66
At 0 °C, only methyl branches are observed. Increasing the temperature to 20 °C induces the incorporation of ethyl, and propyl branches. Additional ethyl and propyl branches were observed in the polymer at 40 °C, with butyl, amyl, and long-chain branches (LCBs) being detected. At 60 and 80 °C, incorporation of short- and long-chain branches gradually increased at the expense of methyl branches. Current imine-chelated NiII complexes must utilize low pressure conditions and/or elaborate ligand designs to promote chain-walking and tailor the polymer microstructure.67,68 However, this strategy is not effective as the incorporation of SCBs is negligible. Since our system produces SCBs under high pressure conditions and is not supported by established mechanistic understandings, we sought to further investigate the underlying chain-walking mechanism through computational studies. Therefore, additional DFT calculations were conducted for C1 with respect to temperatures ranging from 0 to 80 °C.
DFT Calculations for Chain-Walking Mechanism
The polymerization is modeled from the β-CH agostic species 1, the resting state, with a propagating polymer chain (Figure S144). The metal can either chain-walk across the polymer or produce branches at key intermediates 1, 3, 5, 7, 9, and 11 via ethylene coordination (Figure 6a). The Gibbs energy profile at 60 °C serves as a representative data set to discuss the branching distribution and free energies (Figure 6b). The remaining Gibbs energy profiles are included as Figures S145–S148. From these profiles, it can be determined that higher reaction temperatures enable the gradual incorporation of longer branches due to increased thermal energy (kbT), which is consistent with the branching distribution reported in Table 3. The first pathway is a coordination/insertion of ethylene to produce a linear polymer (Linear), where monomer coordination must overcome an energy barrier (TS1MC) of 16.3 kcal mol–1 and is exergonic by 3.9 kcal mol–1. Alternatively, the agostic species 1 can undergo β-hydride elimination (TS2BHE) to form the olefin π-complex 2, with an energy barrier of 12.2 kcal mol–1. This complex can then undergo 2,1-reinsertion (TS23RI), with an energy barrier of 5.1 kcal mol–1, generating in the new β-CH agostic species 3, a process exergonic by −12.6 kcal mol–1 and more favorable than direct ethylene coordination–insertion by 4.7 kcal mol–1. Species 3 can again either undergo monomer coordination (TS3MC) with an energy barrier of 8.1 kcal mol–1 to produce a methyl branch (Methyl) or β-hydride elimination (TS4BHE) for further chain-walking. However, with a 13.0 kcal mol–1 energy barrier for chain-walking, methyl branch formation is more favorable, and is the predominant branch experimentally observed at all temperatures.
Figure 6.

Mechanism and Gibbs energy diagram for chain-walking mechanism. (a) Chain-walking and propagation mechanism with Gibbs energies for reaction temperatures from 0 to 80 °C. (b) Free energy diagram for chain-walking and propagation for C1 calculated from DFT at 60 °C. TS: transition state; BHE: β-hydride elimination; RI: reinsertion; MC: monomer coordination.
Once the agostic species 3 crosses the barrier TS4BHE, it forms olefin π-complex 4, which can undergo 3,2-reinsertion (TS45RI) with a minimal barrier of only 3.0 kcal mol–1, resulting in the β-CH agostic complex 5 in an exergonic step (−6.1 kcal mol–1). Following the formation of complex 5, β-elimination must overcome a barrier of 23.2 kcal mol–1 (TS6BHE). In contrast, the barrier for ethylene insertion to produce ethyl branched (Ethyl) is significantly lower at 5.2 kcal mol–1 (TS5MC) and occurs in a mildly exergonic step (−0.3 kcal mol–1). After β-elimination from complex 5, the subsequent reinsertion step is barrierless for all reaction temperatures, leading to the spontaneous formation of β-CH agostic complex 7. The overall chain-walking step from 5 to 7 is endergonic by 5.3 kcal mol–1. The large energy barrier from 5 to 7 confirms the preference for higher precent incorporation of ethyl branches over propyl branches across all reaction temperatures. From complex 7, the barrier for propyl branch formation (TS7MC) is 6.5 kcal mol–1, which is slightly lower than the barrier of 9.2 kcal mol–1 (TS8BHE) for β-elimination leading to complex 8, which then spontaneously converts to complex 9 at temperatures above 40 °C. Following this, β-elimination to olefin π-complex 10, followed by the 6,5-reinsertion to β-CH agostic complex 11 is the precursor to form amyl branches. The β-elimination step is endergonic by 13.0 kcal mol–1 with an activation barrier of 16.6 kcal mol–1 (TS10BHE), yet the reinsertion step is exergonic and thermodynamically favorable. Although the high activation barrier value of 22.3 kcal mol–1 for reinsertion (TS1011RI) suggests a low amount of amyl branches, reactions above 60 °C have greater percent incorporation (Table 3, entries 3–5) due to the increase in the total thermal energy in the system. This increase in thermal energy subsequently also produces larger chain branches (Lg).
Conclusions
In this work, benchtop-stable iminopyridyl Ni(II) complexes were prepared bearing an o-tolyl carbyl substituent which allows for the study of the effect that ion pairs have on olefin polymerization. Early transition metal complexes demonstrate the importance of the counterion in controlling polyolefin microstructures, but the influence and impact of counterion effects in late transition complexes, specifically for [N,N]-chelated NiII complexes have been largely unexplored, due to the absence of a suitable thermodynamically stable complexes. We demonstrated that the well-defined, inner-sphere ClB(C6F5)3– counterion provides an altered resting state for propagation, inhibiting key intermediates responsible for chain-walking and chain-termination, thus producing higher MW PE with controlled short-chain branching distributions. This branching control provided materials with properties comparable to HDPE, LLDPE, and LDPE. Additionally, we demonstrated that the substituents on the bridging carbon can act as a rotational barrier imposing more axial bulk around the metal center which favors chain-propagation. This emphasizes the importance of substituent placement on the bridging carbon moiety, instead of the more traditional ligand design focusing on the aryl imine. We anticipate that this study will invigorate the exploration of a broader scope of activators and inner-sphere counterions with the extensive iminopyridine or related diimine ligand catalog for the synthesis of polyolefins with tailored branching characteristics and properties.
Acknowledgments
The authors H.S.R., Y.-S.L., M.A., and E.H. thank the Robert A. Welch Foundation for funding (H-E-0041 and E-2066-202110327) through the Center of Excellence in Polymer Chemistry and acknowledge the National Science Foundation (CHEM-2108576) for supporting parts of this work. R.P. and E.D. thank the CALMIP Mesocenter of the University of Toulouse for the allocation of computational resources. S.S.S. and L.C.G. were supported by NSF EFRI Award # 2029359, and acknowledge the use of the Carya, Opuntia, and Sabine Clusters and the advanced support from the Research Computing Data Core at the University of Houston. Jacobo Strong is acknowledged for his contributions to early B(C6F5)3 activation studies, insightful and engaging discussions, and for a critical review and editing of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.4c02708.
Author Contributions
H.S.R. and Y.-S.L. conceived the project. H.S.R. and Y.-S.L. performed experimental studies including catalyst preparation and characterization polymer synthesis and analysis. M.A. supported experimental studies, catalyst preparation, polymerization studies, and analysis. H.S.R., Y.-S.L., and E.H. drafted the initial manuscript. H.S.R., Y.-S.L., and E.H. designed and edited figures. H.S.R., Y.-S.L., M.A., S.S.S., L.C.G., R.P., and E.H. edited and revised the text. E.D. and R.P. conducted DFT simulations for the counterion study and made the respective figures. S.S.S. and L.C.G. performed DFT simulations for the chain-walking mechanism and made the respective figures. L.C.G., R.P., and E.H. supervised the study. The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Hustad P. D. Frontiers in Olefin Polymerization: Reinventing the World’s Most Common Synthetic Polymers. Science 2009, 325 (5941), 704–707. 10.1126/science.1174927. [DOI] [PubMed] [Google Scholar]
- Mülhaupt R. Green Polymer Chemistry and Bio-based Plastics: Dreams and Reality. Macromol. Chem. Phys. 2013, 214 (2), 159–174. 10.1002/macp.201200439. [DOI] [Google Scholar]
- Stürzel M.; Mihan S.; Mülhaupt R. From Multisite Polymerization Catalysis to Sustainable Materials and All-Polyolefin Composites. Chem. Rev. 2016, 116 (3), 1398–1433. 10.1021/acs.chemrev.5b00310. [DOI] [PubMed] [Google Scholar]
- Sauter D. W.; Taoufik M.; Boisson C. Polyolefins, a Success Story. Polymers 2017, 9 (6), 185. 10.3390/polym9060185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Zhang Y.; Hu X.; Wang C.; Jian Z. Advances on Controlled Chain Walking and Suppression of Chain Transfer in Catalytic Olefin Polymerization. ACS Catal. 2022, 12 (22), 14304–14320. 10.1021/acscatal.2c04272. [DOI] [Google Scholar]
- Chen A.; Ma Z.; Pan Y.; Chen M.; Zou C. Cocatalyst Effect in Transition Metal Catalyzed Ethylene Polymerization and Copolymerization. ChemCatChem. 2022, 14 (19), e202200578 10.1002/cctc.202200578. [DOI] [Google Scholar]
- Li L.; Stern C. L.; Marks T. J. Bis(Pentafluorophenyl)(2-perfluorobiphenylyl)borane. A New Perfluoroarylborane Cocatalyst for Single-Site Olefin Polymerization. Organometallics 2000, 19 (17), 3332–3337. 10.1021/om0002780. [DOI] [Google Scholar]
- Urciuoli G.; Zaccaria F.; Zuccaccia C.; Cipullo R.; Budzelaar P. H. M.; Vittoria A.; Ehm C.; Macchioni A.; Busico V. Cocatalyst effects in Hf-catalysed olefin polymerization: taking well-defined Al–alkyl borate salts into account. Dalton Trans. 2024, 53 (5), 2286–2293. 10.1039/D3DT04081J. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Zhang Z.; Zou C.; Chen C. A general cocatalyst strategy for performance enhancement in nickel catalyzed ethylene (co)polymerization. Chin. Chem. Lett. 2022, 33 (9), 4363–4366. 10.1016/j.cclet.2021.12.036. [DOI] [Google Scholar]
- Chen E. Y.-X.; Marks T. J. Cocatalysts for Metal-Catalyzed Olefin Polymerization: Activators, Activation Processes, and Structure–Activity Relationships. Chem. Rev. 2000, 100 (4), 1391–1434. 10.1021/cr980462j. [DOI] [PubMed] [Google Scholar]
- Chen Y.-X.; Metz M. V.; Li L.; Stern C. L.; Marks T. J. Sterically Encumbered (Perfluoroaryl) Borane and Aluminate Cocatalysts for Tuning Cation–Anion Ion Pair Structure and Reactivity in Metallocene Polymerization Processes. A Synthetic, Structural, and Polymerization Study. J. Am. Chem. Soc. 1998, 120 (25), 6287–6305. 10.1021/ja973769t. [DOI] [Google Scholar]
- Mohammed M.; Nele M.; Al-Humydi A.; Xin S.; Stapleton R. A.; Collins S. Counterion Effects on Propylene Polymerization Using Two-State ansa-Metallocene Complexes. J. Am. Chem. Soc. 2003, 125 (26), 7930–7941. 10.1021/ja0207706. [DOI] [PubMed] [Google Scholar]
- Li H.; Li L.; Marks T. J.; Liable-Sands L.; Rheingold A. L. Catalyst/Cocatalyst Nuclearity Effects in Single-Site Olefin Polymerization. Significantly Enhanced 1-Octene and Isobutene Comonomer Enchainment in Ethylene Polymerizations Mediated by Binuclear Catalysts and Cocatalysts. J. Am. Chem. Soc. 2003, 125 (36), 10788–10789. 10.1021/ja036289c. [DOI] [PubMed] [Google Scholar]
- Zaccaria F.; Sian L.; Zuccaccia C.; Macchioni A.. Chapter One - Ion pairing in transition metal catalyzed olefin polymerization. In Adv. Organomet. Chem.; Pérez P. J. Ed.; Academic Press: 2020; Vol. 73, pp 1–78. [Google Scholar]
- Chien J. C.; Song W.; Rausch M. D. Effect of counterion structure on zirconocenium catalysis of olefin polymerization. Macromolecules 1993, 26 (12), 3239–3240. 10.1021/ma00064a044. [DOI] [Google Scholar]
- Chen M.-C.; Roberts J. A. S.; Marks T. J. Marked Counteranion Effects on Single-Site Olefin Polymerization Processes. Correlations of Ion Pair Structure and Dynamics with Polymerization Activity, Chain Transfer, and Syndioselectivity. J. Am. Chem. Soc. 2004, 126 (14), 4605–4625. 10.1021/ja036288k. [DOI] [PubMed] [Google Scholar]
- Johnson L. K.; Killian C. M.; Brookhart M. New Pd(II)- and Ni(II)-Based Catalysts for Polymerization of Ethylene and.alpha.-Olefins. J. Am. Chem. Soc. 1995, 117 (23), 6414–6415. 10.1021/ja00128a054. [DOI] [Google Scholar]
- Ittel S. D.; Johnson L. K.; Brookhart M. Late-Metal Catalysts for Ethylene Homo- and Copolymerization. Chem. Rev. 2000, 100 (4), 1169–1204. 10.1021/cr9804644. [DOI] [PubMed] [Google Scholar]
- Schleis T.; Spaniol T. P.; Okuda J.; Heinemann J.; Mülhaupt R. Ethylene polymerization catalysts based on nickel(II) 1,4-diazadiene complexes: the influence of the 1,4-diazadiene backbone substituents on structure and reactivity1Dedicated to Professor Akira Nakamura with best personal wishes.1. J. Organomet. Chem. 1998, 569 (1), 159–167. 10.1016/S0022-328X(98)00784-0. [DOI] [Google Scholar]
- Souther K. D.; Leone A. K.; Vitek A. K.; Palermo E. F.; LaPointe A. M.; Coates G. W.; Zimmerman P. M.; McNeil A. J. Trials and tribulations of designing multitasking catalysts for olefin/thiophene block copolymerizations. J. Polym. Sci., Part A: Polym. Chem. 2018, 56 (1), 132–137. 10.1002/pola.28885. [DOI] [Google Scholar]
- Xu M.; Liu Y.; Pang W.; Pan Y.; Chen M.; Zou C.; Tan C. Cocatalyst effects in α-diimine nickel catalyzed ethylene polymerization. Polymer 2022, 255, 125116 10.1016/j.polymer.2022.125116. [DOI] [Google Scholar]
- Svoboda M.; Dieck H. t. Diazadien-nickel-alkyle. J. Organomet. Chem. 1980, 191 (1), 321–328. 10.1016/S0022-328X(00)88576-9. [DOI] [Google Scholar]
- Cámpora J.; del Mar Conejo M. a.; Mereiter K.; Palma P.; Pérez C.; Reyes M. L.; Ruiz C. Synthesis of dialkyl, diaryl and metallacyclic complexes of Ni and Pd containing pyridine, α-diimines and other nitrogen ligands: Crystal structures of the complexes cis-NiR2py2 (R = benzyl, mesityl). J. Organomet. Chem. 2003, 683 (1), 220–239. 10.1016/S0022-328X(03)00691-0. [DOI] [Google Scholar]
- De Souza R.-F.; Mauler R. S.; Simon L. C.; Nunes F. F.; Vescia D. V. S.; Cavagnolli A. [η3-methallyl-nickel-dad]PF6 complex: new catalyst precursor for ethylene polymerization. Macromol. Rapid Commun. 1997, 18 (9), 795–800. 10.1002/marc.1997.030180907. [DOI] [Google Scholar]
- Johnson L. K.; McLain S. J.; Sweetman K. J.; Wang Y.; Bennett A. M. A.; Wang L.; McCord E. F.; Lonkin A.; Ittel S. D.; Radzewich C. E.. WO 2003044066, 2003.
- Negureanu L.; Hall R. W.; Butler L. G.; Simeral L. A. Methyaluminoxane (MAO) Polymerization Mechanism and Kinetic Model from Ab Initio Molecular Dynamics and Electronic Structure Calculations. J. Am. Chem. Soc. 2006, 128 (51), 16816–16826. 10.1021/ja064545q. [DOI] [PubMed] [Google Scholar]
- Bonnet M. C.; Dahan F.; Ecke A.; Keim W.; Schulz R. P.; Tkatchenko I. Synthesis of cationic and neutral methallyl nickel complexes and applications in ethene oligomerisation. J. Chem. Soc., Chem. Commun. 1994, 5, 615–616. 10.1039/c39940000615. [DOI] [Google Scholar]
- Wang C.; Friedrich S.; Younkin T. R.; Li R. T.; Grubbs R. H.; Bansleben D. A.; Day M. W. Neutral Nickel(II)-Based Catalysts for Ethylene Polymerization. Organometallics 1998, 17 (15), 3149–3151. 10.1021/om980176y. [DOI] [Google Scholar]
- Liang T.; Goudari S. B.; Chen C. A simple and versatile nickel platform for the generation of branched high molecular weight polyolefins. Nat. Commun. 2020, 11 (1), 372. 10.1038/s41467-019-14211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiebel E.; Voccia M.; Falivene L.; Caporaso L.; Mecking S. The Impact of Charge in a Ni(II) Polymerization Catalyst. ACS Catal. 2021, 11 (9), 5358–5368. 10.1021/acscatal.1c00778. [DOI] [Google Scholar]
- Shields B. J.; Kudisch B.; Scholes G. D.; Doyle A. G. Long-Lived Charge-Transfer States of Nickel(II) Aryl Halide Complexes Facilitate Bimolecular Photoinduced Electron Transfer. J. Am. Chem. Soc. 2018, 140 (8), 3035–3039. 10.1021/jacs.7b13281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biewer C.; Hamacher C.; Kaiser A.; Vogt N.; Sandleben A.; Chin M. T.; Yu S.; Vicic D. A.; Klein A. Unsymmetrical N-Aryl-1-(pyridin-2-yl)methanimine Ligands in Organonickel(II) Complexes: More Than a Blend of 2,2’-Bipyridine and N,N-Diaryl-α-diimines?. Inorg. Chem. 2016, 55 (24), 12716–12727. 10.1021/acs.inorgchem.6b01874. [DOI] [PubMed] [Google Scholar]
- Laine T. V.; Lappalainen K.; Liimatta J.; Aitola E.; Löfgren B.; Leskelä M. Polymerization of ethylene with new diimine complexes of late transition metals. Macromol. Rapid Commun. 1999, 20 (9), 487–491. . [DOI] [Google Scholar]
- Sun W.-H.; Song S.; Li B.; Redshaw C.; Hao X.; Li Y.-S.; Wang F. Ethylene polymerization by 2-iminopyridylnickel halide complexes: synthesis, characterization and catalytic influence of the benzhydryl group. Dalton Transactions 2012, 41 (39), 11999–12010. 10.1039/c2dt30989k. [DOI] [PubMed] [Google Scholar]
- Yue E.; Zhang L.; Xing Q.; Cao X.-P.; Hao X.; Redshaw C.; Sun W.-H. 2-(1-(2-Benzhydrylnaphthylimino)ethyl)pyridylnickel halides: synthesis, characterization, and ethylene polymerization behavior. Dalton Transactions 2014, 43 (2), 423–431. 10.1039/C3DT52234B. [DOI] [PubMed] [Google Scholar]
- Wang F.; Chen C. A continuing legend: the Brookhart-type α-diimine nickel and palladium catalysts. Polym. Chem-Uk 2019, 10 (19), 2354–2369. 10.1039/C9PY00226J. [DOI] [Google Scholar]
- Chen Z.; Allen K. E.; White P. S.; Daugulis O.; Brookhart M. Synthesis of Branched Polyethylene with “Half-Sandwich” Pyridine-Imine Nickel Complexes. Organometallics 2016, 35 (11), 1756–1760. 10.1021/acs.organomet.6b00165. [DOI] [Google Scholar]
- Lu W.; Liao Y.; Dai S. Facile access to ultra-highly branched polyethylenes using hybrid “sandwich” Ni(II) and Pd(II) catalysts. J. Catal. 2022, 411, 54–61. 10.1016/j.jcat.2022.05.003. [DOI] [Google Scholar]
- Ge Y.; Cai Q.; Wang Y.; Gao J.; Chi Y.; Dai S. Synthesis of High-Molecular-Weight Branched Polyethylene Using a Hybrid “Sandwich” Pyridine-Imine Ni(II). Catalyst. Front. Chem. 2022, 10, 886888 10.3389/fchem.2022.886888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y.-D.; Cai Q.; Dai S.-Y. Synthesis of High Molecular Weight Polyethylene and E-MA Copolymers Using Iminopyridine Ni(II) and Pd(II) Complexes Containing a Flexible Backbone and Rigid Axial Substituents. Chin. J. Polym. Sci. 2023, 41 (2), 233–239. 10.1007/s10118-022-2847-5. [DOI] [Google Scholar]
- Peng H.; Li S.; Li G.; Dai S.; Ji M.; Liu Z.; Guo L. Rotation-restricted strategy to synthesize high molecular weight polyethylene using iminopyridyl nickel and palladium catalyst. Appl. Organomet. Chem. 2021, 35 (3), e6140 10.1002/aoc.6140. [DOI] [Google Scholar]
- D’Auria I.; Milione S.; Caruso T.; Balducci G.; Pellecchia C. Synthesis of hyperbranched low molecular weight polyethylene oils by an iminopyridine nickel(ii) catalyst. Polym. Chem. 2017, 8 (41), 6443–6454. 10.1039/C7PY01215B. [DOI] [Google Scholar]
- D’Auria I.; Saki Z.; Pellecchia C. Iminopyridine Ni(II) Catalysts Affording Oily Hyperbranched Ethylene Oligomers and/or Crystalline Polyethylenes Depending on the Reaction Conditions: Possible Role of In Situ Catalyst Structure Modifications. Macromolecules 2021, 1 (2), 121–129. 10.3390/macromol1020010. [DOI] [Google Scholar]
- Mahmood Q.; Li X.; Qin L.; Wang L.; Sun W.-H. Structural evolution of iminopyridine support for nickel/palladium catalysts in ethylene (oligo)polymerization. Dalton Trans. 2022, 51 (38), 14375–14407. 10.1039/D2DT02251F. [DOI] [PubMed] [Google Scholar]
- Laine T. V.; Piironen U.; Lappalainen K.; Klinga M.; Aitola E.; Leskelä M. Pyridinylimine-based nickel(II) and palladium(II) complexes: preparation, structural characterization and use as alkene polymerization catalysts. J. Organomet. Chem. 2000, 606 (2), 112–124. 10.1016/S0022-328X(00)00291-6. [DOI] [Google Scholar]
- Mogheiseh M.; Zohuri G. H.; Khoshsefat M. Synthesis of LDPE Using Pyridineimine-Based Nickel (II) Bromide Complexes: Effect of Catalyst Bulkiness on Thermal, Structural, and Morphological Properties. Macromol. React. Eng. 2018, 12 (5), 1800006 10.1002/mren.201800006. [DOI] [Google Scholar]
- Lovett D. M.; Thierer L. M.; Santos E. E. P.; Hardie R. L.; Dougherty W. G.; Piro N. A.; Kassel W. S.; Cromer B. M.; Coughlin E. B.; Zubris D. L. Structural analysis of imino- and amino-pyridine ligands for Ni(II):Precatalysts for the polymerization of ethylene. J. Organomet. Chem. 2018, 863, 44–53. 10.1016/j.jorganchem.2018.03.012. [DOI] [Google Scholar]
- Lindley B. M.; Wolczanski P. T.; Cundari T. R.; Lobkovsky E. B. First-Row Transition Metal and Lithium Pyridine-ene-amide Complexes Exhibiting N- and C-Isomers and Ligand-Based Activation of Benzylic C–H Bonds. Organometallics 2015, 34 (19), 4656–4668. 10.1021/acs.organomet.5b00385. [DOI] [Google Scholar]
- Li S.; Lu Z.; Fan W.; Dai S. Efficient incorporation of a polar comonomer for direct synthesis of hyperbranched polar functional ethylene oligomers. New J. Chem. 2021, 45 (8), 4024–4031. 10.1039/D0NJ05857B. [DOI] [Google Scholar]
- Li S.; Dai S. Highly efficient incorporation of polar comonomers in copolymerizations with ethylene using iminopyridyl palladium system. J. Catal. 2021, 393, 51–59. 10.1016/j.jcat.2020.11.015. [DOI] [Google Scholar]
- Falivene L.; Cao Z.; Petta A.; Serra L.; Poater A.; Oliva R.; Scarano V.; Cavallo L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11 (10), 872–879. 10.1038/s41557-019-0319-5. [DOI] [PubMed] [Google Scholar]
- Hayashi Y.; Rohde J. J.; Corey E. J. A Novel Chiral Super-Lewis Acidic Catalyst for Enantioselective Synthesis. J. Am. Chem. Soc. 1996, 118 (23), 5502–5503. 10.1021/ja960766s. [DOI] [Google Scholar]
- Chen Z.; Leatherman M. D.; Daugulis O.; Brookhart M. Nickel-Catalyzed Copolymerization of Ethylene and Vinyltrialkoxysilanes: Catalytic Production of Cross-Linkable Polyethylene and Elucidation of the Chain-Growth Mechanism. J. Am. Chem. Soc. 2017, 139 (44), 16013–16022. 10.1021/jacs.7b10281. [DOI] [PubMed] [Google Scholar]
- Normand A. T.; Richard P.; Balan C.; Daniliuc C. G.; Kehr G.; Erker G.; Le Gendre P. Synthetic Endeavors toward Titanium Based Frustrated Lewis Pairs with Controlled Electronic and Steric Properties. Organometallics 2015, 34 (10), 2000–2011. 10.1021/acs.organomet.5b00250. [DOI] [Google Scholar]
- Punzalan E.; Froese R. D.; Zimmerman P. M. Revealing the Interactions between the Flexible Polymer and Counteranion during Propagation and Termination for Olefin Polymerization with the Ti(IV) Constrained Geometry Catalyst. Organometallics 2023, 42 (22), 3236–3248. 10.1021/acs.organomet.3c00365. [DOI] [Google Scholar]
- Macchioni A. Ion Pairing in Transition-Metal Organometallic Chemistry. Chem. Rev. 2005, 105 (6), 2039–2074. 10.1021/cr0300439. [DOI] [PubMed] [Google Scholar]
- Bochmann M.; Cannon R. D.; Song F. Kinetic and mechanism of alkene polymerization. Kinet. Catal. 2006, 47 (2), 160–169. 10.1134/S0023158406020029. [DOI] [Google Scholar]
- Lassahn P.-G.; Lozan V.; Wu B.; Weller A. S.; Janiak C. Dihalogeno(diphosphane)metal(ii) complexes (metal = Co, Ni, Pd) as pre-catalysts for the vinyl/addition polymerization of norbornene – elucidation of the activation process with B(C6F5)3/AlEt3 or Ag[closo-1-CB11H12] and evidence for the in situ formation of “naked” Pd2+ as a highly active species. Dalton Trans. 2003, 23, 4437–4450. 10.1039/B302937A. [DOI] [Google Scholar]
- Zhang R.; Gao R.; Gou Q.; Lai J.; Li X. Recent Advances in the Copolymerization of Ethylene with Polar Comonomers by Nickel Catalysts. Polymers 2022, 14 (18), 3809. 10.3390/polym14183809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y.; Li S.; Wang H.; Dai S. Synthesis of Branched Polyethylene and Ethylene-MA Copolymers Using Unsymmetrical Iminopyridyl Nickel and Palladium Complexes. Organometallics 2021, 40 (17), 3033–3041. 10.1021/acs.organomet.1c00388. [DOI] [Google Scholar]
- Ge Y.; Lu Z.; Dai S. Flexible Axial Shielding Strategy for the Synthesis of High-Molecular-Weight Polyethylene and Polar Functionalized Polyethylene with Pyridine-Imine Ni(II) and Pd(II) Complexes. Organometallics 2022, 41 (15), 2042–2049. 10.1021/acs.organomet.2c00207. [DOI] [Google Scholar]
- Guan Z.; Cotts P. M.; McCord E. F.; McLain S. J. Chain Walking: A New Strategy to Control Polymer Topology. Science 1999, 283 (5410), 2059–2062. 10.1126/science.283.5410.2059. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Nadres E. T.; Brookhart M.; Daugulis O. Synthesis of Highly Branched Polyethylene Using “Sandwich” (8-p-Tolyl naphthyl α-diimine)nickel(II) Catalysts. Organometallics 2013, 32 (18), 5136–5143. 10.1021/om400704h. [DOI] [Google Scholar]
- Fall W. S.; Baschnagel J.; Lhost O.; Meyer H. Role of Short Chain Branching in Crystalline Model Polyethylenes. Macromolecules 2022, 55 (19), 8438–8450. 10.1021/acs.macromol.2c00938. [DOI] [Google Scholar]
- Galland G. B.; de Souza R. F.; Mauler R. S.; Nunes F. F. 13C NMR Determination of the Composition of Linear Low-Density Polyethylene Obtained with [η3-Methallyl-nickel-diimine]PF6 Complex. Macromolecules 1999, 32 (5), 1620–1625. 10.1021/ma981669h. [DOI] [Google Scholar]
- O’Connor K. S.; Lamb J. R.; Vaidya T.; Keresztes I.; Klimovica K.; LaPointe A. M.; Daugulis O.; Coates G. W. Understanding the Insertion Pathways and Chain Walking Mechanisms of α-Diimine Nickel Catalysts for α-Olefin Polymerization: A 13C NMR Spectroscopic Investigation. Macromolecules 2017, 50 (18), 7010–7027. 10.1021/acs.macromol.7b01150. [DOI] [Google Scholar]
- Pei L.; Liu F.; Liao H.; Gao J.; Zhong L.; Gao H.; Wu Q. Synthesis of Polyethylenes with Controlled Branching with α-Diimine Nickel Catalysts and Revisiting Formation of Long-Chain Branching. ACS Catal. 2018, 8 (2), 1104–1113. 10.1021/acscatal.7b03282. [DOI] [Google Scholar]
- Jiang S.; Zheng Y.; Oleynik I. V.; Yu Z.; Solan G. A.; Oleynik I. I.; Liu M.; Ma Y.; Liang T.; Sun W.-H. N N-Bis(2,4-Dibenzhydryl-6-cycloalkylphenyl)butane-2,3-diimine-Nickel Complexes as Tunable and Effective Catalysts for High-Molecular-Weight PE Elastomers. Molecules 2023, 28 (12), 4852. 10.3390/molecules28124852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.