


Abstract
The Ino4 protein belongs to the basic helix–loop–helix (bHLH) family of proteins. It is known to form a dimer with Ino2p, which regulates phospholipid biosynthetic genes. Mammalian bHLH proteins have been shown to form multiple dimer combinations. However, this flexibility in dimerization had not been documented for yeast bHLH proteins. Using the yeast two-hybrid assay and a biochemical assay we show that Ino4p dimerizes with the Pho4p, Rtg1p, Rtg3p and Sgc1p bHLH proteins. Screening a yeast cDNA library identified three additional proteins that interact with Ino4p: Bck2p, YLR422W and YNR064C. The interaction with Bck2p prompted us to examine if any of the Bck2p-associated functions affect expression of phospholipid biosynthetic genes. We found that hyperosmotic growth conditions altered the growth phase regulation of a phospholipid biosynthetic gene, CHO1. There are two recent reports of initial whole genome yeast two-hybrid interactions. Interestingly, one of these reports identified five proteins that interact with Ino4p: Ino2p, Hcs1p, Apl2p, YMR317W and YNL279W. Ino2p is the only protein in common with the data presented here. Our finding that Ino4p interacts with five bHLH proteins suggests that Ino4p is likely to be a central player in the coordination of multiple biological processes.
INTRODUCTION
A common mechanism used to coordinate the expression of multiple genes is regulation of transcription. For example, regulation of genes involved in proliferation and differentiation is governed in part by a set of basic helix–loop–helix (bHLH) proteins, Myc, Max, Mad and Mxi. The significance of this regulation is evident in the fact that abnormal regulation of these factors can result in malignancy (1). A hallmark of bHLH proteins in mammalian cells is their capacity to form multiple heterodimer combinations to regulate different pathways. Here we show that yeast bHLH proteins also have this capacity.
The role of the HLH domain is to create dimers by interactions between the amphipathic helices (2,3). Dimerization juxtaposes two regions, rich in basic residues, that create a DNA-binding interface which interacts with the consensus sequence, 5′-CANNTG-3′ (2,3). The ability of bHLH proteins to form multiple dimer combinations (4,5) allows them to recognize variations of the consensus sequence. However, there is dimerization specificity, since Myc and MyoD do not appear to form dimers. In the case of Max, dimerization is dependent on partner availability; Myc protein is transiently expressed in growth factor-stimulated cells and Mad protein is expressed during differentiation.
The INO2 and INO4 genes regulate expression of the phospholipid biosynthetic genes in response to inositol and choline (6). The Ino2p and Ino4p proteins share significant homology to the bHLH region of the Myc family (7,8). Neither Ino2p nor Ino4p can form homodimers but they do form a heterodimer which interacts with a conserved 10 bp element (5′-CATGTGAAAT-3′) called UASINO (9,10). The first six bases of the UASINO element contain the consensus bHLH binding site. Ino2p contains an activation domain while Ino4p is required for dimerization (10). INO2 gene expression is regulated ∼12-fold by inositol and choline in a manner similar to that of its genes (11). Unlike INO2, INO4 expression is only modestly regulated by inositol and choline at the post-transcriptional level (12).
We have previously suggested that Ino4p may form dimers with multiple bHLH proteins. This was based on the identification of INO4-dependent/INO2-independent elements in the promoters of the CTR1/HMN1 and INO1 genes (13,14). It has also been proposed that Ino4p interacts with an, as yet unknown, myristoylation-sensitive transcription factor (15). In addition to Ino2p and Ino4p, there are five well-studied bHLH proteins in yeast. The PHO4 gene was the first yeast gene shown to encode a bHLH protein (16). Binding of a Pho4p dimer (to the sequence 5′-CACGTG-3′) is required for the ∼1000-fold induction of gene expression in response to phosphate starvation. The CPF1 (CEP1/CBF1) gene encodes a protein that binds a centromeric sequence called CDEI (5′-RTCACRTG-3′) (17,18) to regulate chromosomal segregation. Cpf1p is also required for regulation of methionine biosynthetic genes. The RTG1 and RTG3 genes were identified as regulators of CIT2 gene expression (19,20). Expression of the nuclear encoded CIT2 gene is regulated in response to the functional state of the mitochondria (termed retrograde regulation) (19). In strains lacking mitochondrial DNA, CIT2 expression is induced 6- to 30-fold. The cis-acting sequence (UASr) required for induction of CIT2 expression forms an RTG1/RTG3-dependent complex but does not contain any sequences that resemble the consensus bHLH binding site (19,20). Rtg1p is responsible for recruiting the activation domain of Rtg3p to the UASr element, allowing for transcriptional activation of the target genes. The SGC1 gene is required for expression of glycolytic genes in yeast (21). It is not known whether Sgc1p binds a consensus bHLH binding site.
Recently, two published reports document initial results from whole genome two-hybrid arrays (22,23). One of the reports identified several Ino4p interactors using a high throughput screening procedure (22). Here we report that Ino4p interacts with five bHLH proteins (Ino2p, Pho4p, Rtg1p, Rtg3p and Sgc1p) and three non-bHLH proteins (Bck2p, YLR422W and YNR064C). The only overlap with the published report is Ino2p. We also show that hyperosmotic growth conditions alter growth phase regulation of the CHO1 phospholipid biosynthetic gene. This observation is consistent with the hypo-osmosensitive phenotype of a BCK2 null allele.
MATERIALS AND METHODS
Bacterial strains, yeast strains and growth conditions
Escherichia coli KC8 cells (hsdR, leuB600, trpC9830, pyrF::Tn5, hisB463, lacΔX74, strA, galU, galK) were used to shuttle the two-hybrid library plasmids out of yeast. KC8 cells were grown in M9 medium (24) supplemented with 10× Leu– dropout stock (230 mg/l lysine, 20 mg/l arginine, 20 mg/l methionine, 300 mg/l threonine, 20 mg/l tryptophan, 20 mg/l histidine, 20 mg/l adenine and 20 mg/l uracil), 40 mg/l proline, 1 mM thiamine HCl, 100 mg/l ampicillin. Escherichia coli BL21(DE3)pLysS cells (F–, ompT, hsdSB[rB–mB–], gal, dcm[DE3], pLysS[CmR]) (Novagen, Madison, WI) were grown in LB medium (24) containing 50 µl/ml ampicillin and 34 µl/ml chloramphenicol (to maintain the pLysS plasmid). All bacterial strains were grown at 37°C. Bacterial transformations were performed according to the recommendations of the manufacturer (Gibco BRL, Grand Island, NY).
The Saccharomyces cerevisiae strains used in this study were: BRS2004 (MATα, ade2-1, his3-11,15, leu2-3,112, can1-100, trp1-1, ura3-1, ino4Δ::LEU2) (12); BRS1189 (MATa, ade2-1, his3-11,15, leu2-3,112, can1-100, trp1-1, ura3-1::CHO1-lacZ::URA3) (25); Y190 (MATa, ura3-52, his3-Δ200, ade2-101, trp1-901, leu2-3,112, gal4Δ, gal80Δ, URA3::GAL-lacZ, cyhr2, LYS2::GAL-HIS3) (Clontech Laboratories, Palo Alto, CA). All yeast strains were grown at 30°C on YEPD plates or on a complete synthetic medium (26). Transformations with TRP1-, URA3- and LEU2-containing plasmids were plated on complete synthetic medium lacking the respective amino acid(s). Two-hybrid interactions were initially monitored on X-gal medium (27). INO4 complementation tests were performed on complete synthetic medium (28) either lacking or containing 75 µM inositol.
Plasmid constructions
Sequences encoding Ino4p and the other bHLH proteins were created lacking their respective AUG codons by PCR and ligated into pGEM®-T (Promega, Madison, WI). The primer pairs used to amplify each gene are listed by the gene name with 5′ (forward primer) and 3′ (reverse primer) designations (except for INO4-AD-5′ and INO4-AD-3′, which were used to amplify the INO4 sequences) (Table 1). The appropriate sequences were isolated from the pGEM-T derivatives with restriction enzymes as follows (shown as gene, restriction enzymes, fragment size): INO4, EcoRI and PstI, 462 bp; INO2, BamHI and PstI, 924 bp; CBF1, EcoRI and BglII, 1065 bp; RTG1, EcoRI and PstI, 543 bp; RTG3, EcoRI and PstI, 1470 bp; PHO4, EcoRI and PstI, 948 bp; SGC1, EcoRI and SalI, 885 bp. The INO4 sequences were ligated into pGBT9 (Clontech Laboratories) immediately downstream of the Gal4 DNA-binding domain (Gal4BD). The other bHLH sequences were cloned into pGAD424 (Clontech Laboratories) immediately downstream of the Gal4 activation domain (Gal4AD). The sequences encoded by the BCK2 gene and the YLR422W and YNR064C open reading frames (ORFs) were also cloned into pGAD424. These sequences were amplified by PCR using the appropriate primers (Table 1) and cloned into pGEM-T. The appropriate sequences were isolated from the pGEM-T derivatives with restriction enzymes as follows (shown as gene, restriction enzymes, fragment size): BCK2, SalI and PstI, 2565 bp; YLR422W, EcoRI and PstI, 2301 bp; YNR064C, EcoRI and PstI, 882 bp. In the case of YLR422W only the C-terminal region (representing ∼40% of the total protein) was cloned. All three were ligated into pGAD424 immediately downstream of the Gal4AD.
Table 1. Oligonucleotides used in this study.
| Designation | Sequence |
|---|---|
| INO4-AD-5′ | 5′-GAATTCACGAACGATATTAAGGAG-3′ |
| INO4-AD-3′ | 5′-CTGCAGTCACTGACCACTCTGTCCATC-3′ |
| INO4-bHLH-5′ | 5′-GAATTCGATGGTCAAATACGTATA-3′ |
| INO4-bHLH-3′ | 5′-CTGCAGTCATTCCTTTCATACAACCA-3′ |
| INO4-INT-5′ | 5′-GAATTCAAGGGTGAACTGGCTAAT-3′ |
| INO4-INT-3′ | 5′-CTGCAGACTAAATCCCGAATGTTTCCATT-3′ |
| INO4-5′ | 5′-CTCGAGATGACGAACGATATT-3′ |
| INO4-3′ | 5′-CTCGAGTCACTGACCACTCTG-3′ |
| INO2-TH-5′ | 5′-GGATCCAACAAGCAACTGGGAAC-3′ |
| INO2-TH-3′ | 5′-CTGCAGTCAGCAATCATCCAGTAT-3′ |
| TH-DB-5′ | 5′-CTATTCGATGATGAAGATACCCCACCA-3′ |
| TH-DB-3′ | 5′-GAACTTGCGGGGTTTTTCAGTATCTACGAT-3′ |
| BCK2-5′ | 5′-GTCGACCGAAGAATAGTCACCACC-3′ |
| BCK2-3′ | 5′-CTGCAGTTAGTTGCTATTATCAAA-3′ |
| YLR422W-5′ | 5′-GAATTCCTTAGTTTATGCAAGGAT-3′ |
| YLR422W-3′ | 5′-CTGCAGTTATCCCGAATGTGAGTA-3′ |
| YNR064C-5′ | 5′-GAATTCTCAAATATCATTGCAAGA-3′ |
| YNR064C-3′ | 5′-CTGCAGTTAATTTTCTGCAAACAT-3′ |
| RTG1-5′ | 5′-GAATTCAGCAGCATTCCAGCTGGC-3′ |
| RTG1-5′g | 5′-ATGAGCAGCATTCCAGCTGGC-3′ |
| RTG1-3′ | 5′-CTGCAGTTAGCTACCATTACCGT-3′ |
| RTG3-5′ | 5′-GAATTCATGAACAATAACGAAAGT-3′ |
| RTG3-5′g | 5′-ATGATGAACAATAACGAAAGT-3′ |
| RTG3-3′ | 5′-CTGCAGCTACCCCGAACCAAATT-3′ |
| CBF1-5′ | 5′-GAATTCAACTCTCTGGCAAATAAT-3′ |
| CBF1-3′ | 5′-AGATCTTCAAGCCTCATGTGGAT-3′ |
| PHO4-5′ | 5′-GAATTCGGCCGTACAACTTCTGAG-3′ |
| PHO4-5′g | 5′-ATGGGCCGTACAACTTCTGAG-3′ |
| PHO4-3′ | 5′-CTGCAGTCACGTGCTCACGTTCT-3′ |
| SGC1-5′ | 5′-GAATTCAACTCTATTTTAGACAGA-3′ |
| SGC1-5′g | 5′-ATGAACTCTATTTTAGACAGA-3′ |
| SGC1-3′ | 5′-GTCGACTTATTTTTGGTCTTGTT-3′ |
Ino4p deletions fused to the Gal4BD in pGBT9 were created by PCR. Constructs lacking either the N-terminal 21 or 42 residues were amplified using primers INO4-INT-5′ and INO4-bHLH-5′ (Table 1), respectively. Constructs lacking either the C-terminal 16 or 50 residues were amplified using primers INO4-INT-3′ and INO4-bHLH-3′ (Table 1), respectively.
To create a 6×His-tagged Ino4p the INO4 coding sequences were amplified by PCR using primers INO4-5′ and INO4-3′. This created a PCR fragment containing the entire INO4 coding sequences flanked by XhoI sites. The 468 bp PCR product was inserted into pGEM-T and excised from the pGEM-T derivative by digestion with XhoI. The resulting fragment was inserted into a XhoI site in pET-15b to create pET-15b-INO4.
For the purpose of producing in vitro labeled Ino2p, Rtg1p, Rtg3p, Pho4p and Sgc1p, pGEM-T derivatives containing the entire coding sequences for these proteins were created. These plasmids differed from those described above in that they included the respective initiator codons and lacked any restriction sites flanking the initiator codon. The primer pairs used to amplify each gene are listed by the gene name with 5′g (forward primer) and 3′ (reverse primer) designations (Table 1).
Two-hybrid analyses
The yeast strain Y190 was transformed with the pGBT-INO4 and pGAD-bHLH protein vectors. Trp+/Leu+ transformants were replica plated onto Trp+/Leu+/X-gal plates and incubated overnight at 30°C. The plates were monitored daily, for ∼4 days, for the presence of blue colonies.
A S.cerevisiae cDNA library (Clontech Laboratories) was screened for proteins that interact with Ino4p. Yeast strain Y190 was transformed with 5 µg of the cDNA library and 5 µg pGBT9-INO42–151 (this is the entire INO4 coding sequence except the initiator AUG). Transformants were selected on Trp–/Leu–/His–/X-Gal plates (containing 15 mM 3-NH-1,2,4-triazole) at 30°C. Plates were monitored over a period of 4 days for the presence of blue colonies.
To isolate the library plasmid from the yeast transformants, cells were cultured in 1 ml of complete synthetic Leu– medium overnight to lose the pGBT9-INO42–151 plasmid. The next day the cells were pelleted, resuspended in 10 µl of lysis buffer (1.2 M sorbitol, 100 mM NaHPO4 and 5.0 U/µl lyticase) and incubated overnight at 37°C. Next, 10 µl of 20% SDS was added and the solution was mixed by vortexing. The solution was purified in a Chroma Spin+TE-400 column (Clontech Laboratories) according to the manufacturer’s recommendations. The purified nucleic acids were transformed into the leucine auxotrophic bacterial strain KC8. The yeast LEU2 gene can complement the leucine auxotrophy allowing for specific selection of the library plasmid. Restriction digests were done to ensure that the library domain was isolated and to estimate the approximate size of the insert. The plasmid inserts were sequenced using the primer TH-DB-5′ or TH-DB-3′ (Table 1). As a test for false positives the various Gal4AD fusions were retransformed into Y190 with either pVA3 (murine p53, amino acids 72–390) or pLAM5′ (human lamin C) (Clontech Laboratories).
β-Galactosidase assays
Assays were performed as previously described (29). Units of β-galactosidase activity were defined as OD420 nm/min/mg total protein.
In vitro protein:protein interaction assay
Strain BL21(DE3)pLysS was transformed with pET-15b-INO4 and a culture (100 ml) was grown to OD600 = 0.6. Following a 3 h induction (with 1 mM IPTG) of 6×His-tagged Ino4p the cells were pelleted and frozen overnight at –80°C. The pLysS plasmid causes the cell to lyse upon thawing. The cell pellet was resuspended in 1 ml of lysis buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 10 mM imidazole) and pelleted by centrifugation. The cleared cell lysate was removed with a pipette and stored at –80°C.
Radioactively labeled bHLH proteins were generated using the plasmid described above in the TNT® Coupled Reticulocyte Lysate System. The reactions were carried out according to the specifications of the supplier (Promega) using SP6 RNA polymerase to transcribe the INO2, RTG3, SGC1 and PHO4 genes. T7 RNA polymerase was used to transcribe the RTG1 gene. Each 50 µl reaction was split into three aliquots: 5 µl were stored at –80°C; 22 µl were incubated with extracts from cells programmed with pET-15b-INO4; 22 µl were incubated with extracts from cells programmed with the parental plasmid, pET-15b.
Optimal conditions for binding to and elution from a nickel column (Ni–NTA) of 6×His-tagged Inop4 were determined empirically. The Ni–NTA columns (Qiagen, Valencia, CA) were equilibrated with 600 µl of lysis buffer and centrifuged for 2 min (700 g). The cleared lysate (600 µl) was incubated for 45 min with either 22 µl of the pET15b-INO4 extract or 22 µl of the pET-15b extract. Both reactions were added to the column and centrifuged for 2 min. The column was washed twice with 600 µl of wash buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 40 mM imidazole) and the bound proteins were eluted with 200 µl of elution buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 300 mM imidazole). The eluates from each of the reactions and 5 µl of the input were fractionated by SDS–PAGE and visualized using a phosphorimager (Molecular Dynamics, Sunnyvale, CA).
RESULTS
Ino4p can activate transcription
To gain a better understanding of the role of Ino4p, we sought to identify yeast proteins that interact with Ino4p using the two-hybrid system. The two-hybrid system uses a transcriptional reporter to assay for protein:protein interactions. Therefore, it was necessary to ensure that the Gal4–Ino4 fusion construct did not activate transcription of the reporter genes. Previous one-hybrid assays suggest that Ino4p does not contain an activation domain (10). However, we found that the Y190 yeast strain containing the pGBT9-INO42–151 construct did yield blue colonies after a 4 day incubation. Quantitation of β-galactosidase activity revealed that, unlike earlier reports, Ino4p can modestly activate transcription (Fig. 1A). However when strain Y190 was simultaneously transformed with pGBT9-INO42–151 and the pGAD424 vector this modest activity was eliminated (data not shown). This most likely results from a lower plasmid copy number in the double transformant, which brings the level of activation by pGBT9-INO42–151 below the level of sensitivity of the reporter.
Figure 1.

One-hybrid and complementation assays of Gal4BD–Ino4p deletions. (A) Yeast strain Y190 was transformed with the Gal4BD–Ino4p protein deletions. Yeast extracts prepared from transformants that were grown in Trp– synthetic medium were used to conduct β-galactosidase assays. The Ino4p protein deletions were also transformed into an ino4Δ strain and plated on Trp– synthetic medium. Cells were replica plated onto Trp– synthetic medium lacking inositol and scored for their ability to complement inositol auxotrophy. (B) Representative plate assay showing ino4Δ complementation by pGBT9-INO42–151 and pGBT9-INO422–135.
To map the transcriptional activation domain of Ino4p we created a set of Ino4p N- and C-terminal deletions fused to the Gal4BD. The various deletions were transformed into Y190 and lacZ expression was determined. Deleting 21 residues from the N-terminus and/or deleting 16 residues from the C-terminus eliminated expression of the lacZ reporter (Fig. 1A). Curiously, these deletions were still able to complement an ino4Δ strain (Fig. 1B), suggesting that the Gal4BD–Ino4p fusion was still capable of interacting with Ino2p. This also demonstrates that the ability to activate transcription is not required for complementation. Constructs deleting 42 residues from the N-terminus and/or 50 residues from the C-terminus were unable to complement an ino4Δ strain or activate transcription of the lacZ reporter (Fig. 1A).
Ino4p interacts with multiple yeast bHLH proteins
It has been established that in mammalian cells bHLH proteins can form homodimer and multiple pairwise heterodimer combinations. This prompted us to examine if yeast bHLH proteins also form multiple heterodimer combinations. In S.cerevisiae there are five other known bHLH proteins in addition to Ino4p and Ino2p (30). The coding sequence of INO4 was employed as the bait (in pGBT9) and the coding sequences of INO2 and the other five bHLH genes (PHO4, RTG1, RTG3, CBF1 and SGC1) were used as the prey (in pGAD424). These plasmids were simultaneously transformed into Y190. Trp+/Leu+ transformants were replica plated onto Trp+/Leu+/X-gal plates. The plates were monitored daily for 4 days for the blue colony phenotype and positive transformants were assayed for β-galactosidase activity using a liquid assay. By both assays, Ino4p and Ino2p were shown to interact, confirming previous biochemical and two-hybrid results (10,31; Fig. 2). These assays also demonstrated that the gene products of RTG1, RTG3, SGC1 and PHO4 all interact with Ino4p. Cpf1p was the only bHLH protein that did not interact with Ino4p (Fig. 2). However, the Gal4AD–Cbf1p fusion was able to complement the methionine auxotrophy of a cbf1Δ strain, indicating that the fusion protein was expressed (data not shown). The amount of β-galactosidase activity ranged from 15 (Sgc1p as the prey) to 104 U (Ino2p as the prey). However, these values do not reflect the strength of interaction, since some of the bHLH proteins have activation domains (Ino2p, Pho4p and Rtg3p).
Figure 2.

Two-hybrid assay demonstrating interactions between Gal4BD–Ino4p and other yeast bHLH proteins. Y190 was transformed with pGBT9-INO42–151 and pGAD-bHLH. Extracts were prepared from these strains and assayed for β-galactosidase activity. Values represent the average of at least three separate trials. The location of the bHLH domain is shown (filled box).
A biochemical assay (Fig. 3) was used to confirm the interactions observed with the two-hybrid assay. To do this, each of the bHLH preys was labeled via an in vitro transcription–translation reaction. 6×His–Ino4p was produced in E.coli and incubated with each of the labeled preys. 6×His–Ino4p and associated proteins were purified through a nickel affinity column and the proteins fractionated by SDS–PAGE. As a negative control the labeled proteins were incubated with an extract from E.coli transformed with plasmid pET-15b (parental plasmid). This assay confirmed the interactions with Ino2p, Pho4p, Rtg3p and Sgc1p. The Rtg1p interaction was not confirmed because it was not produced by the in vitro labeling system.
Figure 3.
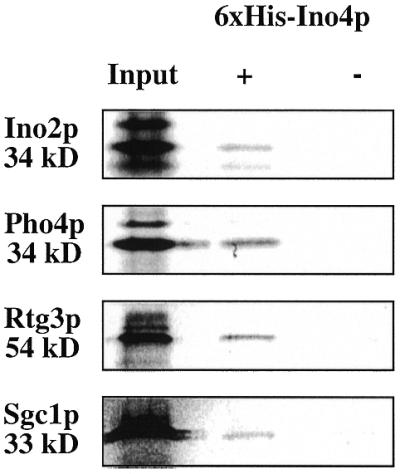
Biochemical assay demonstrating interactions between 6×His–Ino4p and radioactively labeled bHLH proteins. Each of the labeled bHLH proteins was incubated with extracts from E.coli programmed with either pET-15b-INO4 (+) or pET-15b (–). The input lanes contain 20% of the labeled bHLH proteins used in the reactions. The predicted molecular weights for each of the bHLH proteins are shown.
Ino4p interacts with YLR422W, YNR064C and Bck2p
We screened a yeast library to determine if there are other proteins that interact with Ino4p. The cDNA library and pGBT9-INO42–151 were simultaneously transformed into Y190. Approximately 33 000 transformants were plated onto Trp–/Leu–/His–/X-gal. The plates were monitored daily for the presence of blue colonies. After 4 days the Y190 strain transformed with pGBT9-INO42–151 by itself begins to turn blue. Therefore, any colony that turned blue after 4 days was considered to result from activation by plasmid pGBT9-INO42–151. The 32 Trp+/Leu+/His+/X-gal+ transformants that turned blue before day 4 were selected for further study.
A number of control transformations were performed to eliminate false positives. Initially, the isolated library plasmids were retransformed along with pGBT9-INO42–151 into Y190 to ensure that the original β-galactosidase activity was the result of the isolated library plasmid. Next, the library plasmid was transformed into Y190 alone, to ensure that the β-galactosidase activity was the result of an interaction between the library plasmid and pGBT9-INO42–151 and not autoactivation by the library clone. The library plasmids were transformed with three different control plasmids: pGBT9, Gal4BD; pLAM5′, human lamin C; pVA3, murine p53(72–390). These transformations are used to determine if the library plasmids encoded non-specific binding proteins or Gal4BD-specific binding proteins. In addition, the identity of the library inserts was determined by sequencing the prey plasmids and searching the Stanford yeast genome database (genome-www.stanford.edu/Saccharomyces ). Seven transformants were common false positives that have turned up in multiple two-hybrid screens and are confirmed as false positives by independent experiments (32). These false positives consist of, but are not limited to, mitochondrial proteins, splicing factors and cell cycle proteins. Seven transformants contained artifacts of library construction that consisted of either two genes fused together or sequences in the wrong orientation.
We selected for further study two ORFs of unknown function, YLR422W and YNR064C, and BCK2, a serine/threonine protein kinase in the cell wall integrity and osmoregulatory pathways. The library plasmids only contained small portions of YLR422W, YNR064C and BCK2, raising the possiblity that the interaction with the bait may be due to altered protein conformations present in the prey fusions. To eliminate this possibility, the entire coding sequences of YNR064C and BCK2 were cloned into pGEM-T and subsequently cloned into pGAD424. Because of the large size of YLR422W, only the C-terminal 767 amino acids were used. This region contains the sequences present in the original library isolate. These clones were transformed simultaneously into Y190 with pGBT9-INO42–151 and Trp+/Leu+ transformants were selected. The transformants were assayed for β-galactosidase activity. Similar to the original library clones, these new constructs also activated the lacZ reporter, suggesting that the encoded proteins are in fact interacting with Ino4p (Table 2).
Table 2. β-Galactosidase activity resulting from two-hybrid interactions between Ino4p and Bck2p, YLR422W and YNR064C.
| Gal4AD–ORF | β-Galactosidase activity (±SD)a |
|---|---|
| YLR422W (C-terminus) | 40.0 ± 1.40 |
| YNR064C | 46.8 ± 20.9 |
| BCK2 | 44.0 ± 3.31 |
| INO2 | 103.4 ± 10.7 |
aAssays were conducted in triplicate with extracts from yeast transformants containing the respective plasmids along with pGBT9-INO42–151.
CHO1 expression is regulated by growth phase
The above results indicate that Ino4p interacts with several yeast proteins. This suggested the possibility that these interactions might affect regulation of the phospholipid biosynthetic genes under previously uncharacterized conditions. We investigated the effect of several conditions on regulation of the phospholipid biosynthetic genes using several lacZ reporter fusions. We found that hyperosmotic conditions affected phospholipid biosynthetic gene expression. This is consistent with the interaction with BCK2, since bck2Δ mutants are hypo-osmosensitive.
In addition to being regulated by inositol and choline, the phospholipid biosynthetic genes are also regulated by growth phase (33). Confirming previous results, we found that CHO1–lacZ expression was regulated by growth phase (Fig. 4A). More specifically, CHO1–lacZ expression levels in I–C– medium (derepressing) start out low at the beginning of the lag phase and increase throughout the log phase (Fig. 4B). β-Galactosidase levels peak at the beginning of stationary phase and decrease precipitously until it reaches the initial expression levels seen in early lag phase. The expression pattern is similar in I+C+ medium, however, because expression levels are repressed in this medium, the overall expression levels are lower than those observed in I–C– medium (Fig. 4A).
Figure 4.
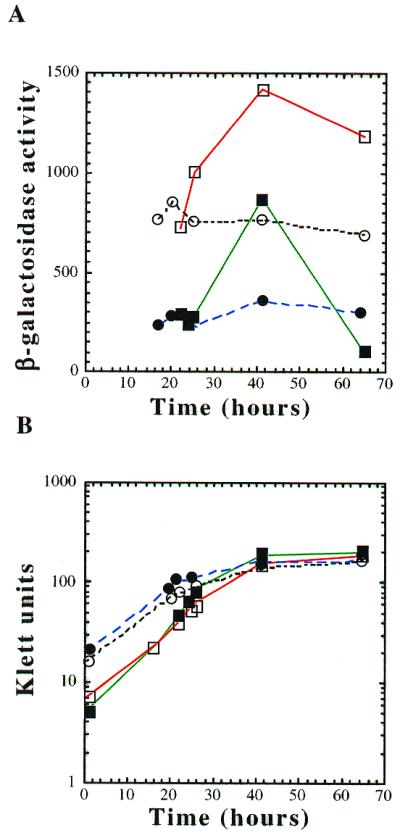
CHO1–lacZ expression in response to hyperosmotic stress and growth phase. (A) Cultures of strain BRS1189 were grown in synthetic medium: lacking inositol and choline (open squares); containing 75 µM inositol and 1 mM choline (solid squares); containing 0.7 M NaCl (open circles); containing inositol, choline and NaCl (solid circles). Aliquots were taken at various times throughout the growth phase. (B) Klett readings were taken for each aliquot.
Expression of the CHO1–lacZ gene was altered in response to external solute concentration. In the presence of 0.7 M NaCl CHO1–lacZ expression begins at approximately the same levels as cultures grown in the absence of solutes (Fig. 4A). However, in the presence of 0.7 M NaCl CHO1–lacZ gene expression stays at this initial level throughout the entire growth phase. Because CHO1–lacZ gene expression is not completely repressed by inositol and choline, we were also able to look at both growth phase regulation and the osmotic response under I+C+ conditions. In the presence of 0.7 M NaCl CHO1–lacZ gene expression levels were again equal to those seen in cultures grown without solutes (Fig. 4A). This initial expression level was maintained with no obvious growth phase regulation. Therefore, the addition of solutes separates growth phase regulation from the inositol and choline response.
DISCUSSION
In contrast to the previously reported one-hybrid results (33), we observed a low level of activation from the Gal4p–Ino4p fusion protein (Fig. 1A). Short deletions from either the N- or C-terminus of Ino4p eliminated the activation function (Fig. 1A). However, these deletions were still able to activate transcription of the INO1 gene. One possible explanation for the loss of activation is that regions at the N- and C-termini may include an activation domain. Consistent with this possibility, the sequences that were deleted included short stretches of acidic residues. However, the question remains, is this activation activity physiologically relevant? To address this question, it will be necessary to determine if the activation function is required for expression of the INO4 target genes.
In mammalian cells bHLH proteins have been shown to form multiple heterodimers in order to regulate a diverse set of genes (4,5). Our results demonstrate that Rtg1p, Rtg3p, Pho4p and Sgc1p all interact with Ino4p (Figs 2 and 3). Mutant alleles of the yeast bHLH genes are not inositol auxotrophs, suggesting that these proteins do not play a direct role in phospholipid biosynthesis (B.P.Ashburner and J.M.Lopes, unpublished data; 21). These observations suggest that Ino4p is at the center of a network designed to coordinate multiple cellular functions. RTG1 and RTG3 are required for communication between the mitochondria and nucleus (19,34) which allows the cell to adjust to changes in mitochondrial activity and biogenesis. Increased mitochondrial activity may signal increased biosynthesis of mitochondria and therefore, an increased need for phospholipids. Sgc1p is a regulator of glycolytic gene expression. This interaction would suggest that energy production might be coordinated with phospholipid biosynthesis. An increase in the glycolytic flow may increase growth rates that might require an increase in phospholipid biosynthesis.
The two-hybrid assay established an interaction with Pho4p (Figs 2 and 3). Previous studies demonstrated that PHO5, a gene directly regulated by Pho4p, contains a putative UASINO element (5′-CATGTGCGAT-3′) and is responsive to inositol in an INO2-dependent manner (35). This suggests that Ino2p/Ino4p heterodimers may bind to the PHO5 promoter to regulate its expression in an inositol-dependent manner. Ino2p/Ino4p binding to UASINO-like elements as in the PHO5 promoter is one mechanism for cross-regulation of pathways. Another mechanism is the formation of different heterodimers, like Pho4p/Ino4p. However, it is unlikely that these heterodimers regulate PHO5, since its regulation is INO2-dependent. Using microarrays, we have recently discovered that Ino4p represses expression of the PHO13 and PHO84 genes (K.A.Robinson and J.M.Lopes, unpublished data). This repression may be the result of an Ino4p:Pho4p interaction.
One of the next challenges will be to identify the binding sites for the Ino4p:bHLH dimer combinations. The crystal structure of Max and Pho4p allows one to make some predictions as to how the yeast bHLH dimers might interact with their cognate binding sites (3,36). Pho4p homodimers bind the DNA sequence 5′-CACGTG-3′, with each monomer forming three contacts with each half-site. The complementary base at the first position (G residue) is bound by H5 of the basic region (3′-GTGCAC-5′), while the CA residues are bound by E9 (5′-CACGTG-3′) and the complementary base at the third position (G residue) is bound by R13 (3′-GTGCAC-5′) (Fig. 5A). Rtg1p:Rtg3p binding to the UASr is difficult to predict because Rtg1p contains a truncated basic region and the UASr (5′-GGTCAC-3′) is not a consensus bHLH binding site (19). However, Rtg3p does contain a complete basic region which likely interacts with the distal half-site (5′-GGTCAC-3′) (Fig. 5B). The distal half-site in the UASr resembles the proximal bHLH half-site. Thus, Rtg3p may be facing in the opposite direction from the other bHLH proteins. It is difficult to predict how Rtg1p binds to the UASr element because of the absence of certain conserved amino acids in the basic region. The UASINO element 5′-CATGTGAAAT-3′ is not a palindrome, which complicates predicting the orientation of the Ino2p/Ino4p interaction with the bHLH binding site. The distal half-site probably forms the same contacts described above with either Ino2p or Ino4p (Fig. 5C). However, the proximal half-site has an A instead of a G residue at the complementary base of the third position. Therefore, R13 of Ino2p or Ino4p might interact with this A residue. The orientation of Ino2p/Ino4p on the UASINO element may be directional, since the binding site is not a palindrome. These heterodimer:DNA interactions allow one to infer possible interactions for the new heterodimer combinations. For example, Pho4p:Ino4p may bind to the following elements: 5′-CACGTG-3′, 5′-CATGTG-3′ or 5′-CACATG-3′. However, the actual binding sites will have to be determined experimentally.
Figure 5.

Consensus E-box and conserved amino acid contacts for various yeast bHLH proteins. (A) Binding of Pho4p homodimer (36) and putative binding of Cpf1p homodimers to the consensus DNA sequence. (B) Putative binding of Rtg1p(b) and Rtg3p(a) heterodimers to the UASr element. (C) Putative interaction between the UASINO element and conserved amino acids of Ino2p and Ino4p. It is currently unknown which protein binds to each half-site. Arg13* of either Ino2p or Ino4p may interact with the internal A residue of the UASINO element.
A two-hybrid screen of a yeast cDNA library demonstrated that the products of two ORFs and the gene BCK2 interact with Ino4p (Table 2). One ORF, YNR064C, encodes a 33 kDa protein with homology to the family of serine esterases. There is no additional information about the other ORF, YLR422W. BKC2 encodes a serine/threonine kinase that plays a role in the PCK pathway that is responsible for cell integrity (reviewed in 37). As the yeast cell grows and divides, as well as during mating and sporulation, the cell wall must be continuously remodeled and expanded. These are also conditions under which the phospholipid membrane must be remodeled and expanded. Therefore, it is not unreasonable to suggest that these two pathways would be coordinately regulated. The BCK2 pathway also helps maintain proper osmolyte concentrations, since bck2Δ strains are hypo-osmosensitive. Consistent with this phenotype, we discovered that the concentration of salt in the medium affected the expression of some of the phospholipid biosynthetic genes. Growth phase regulation of the CHO1–lacZ (Fig. 4) and INO1–lacZ reporter genes (data not shown) was eliminated or altered in the presence of 0.7 M NaCl.
The interaction of Ino4p with the other bHLH proteins demonstrates the potential to regulate a vast set of genes with a few regulatory genes. This information places a premium on determining what genes are regulated by these new heterodimers. Most importantly, the finding that yeast bHLH proteins also form multiple dimer combinations and the wealth of knowledge on the roles of the individual proteins make yeast a model organism for the study of cross-pathway regulation by bHLH proteins.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Kyle Gardenour, Mohan R. Kaadige, Mary Elizabeth Gardocki and Lisa Lepeak for comments on the manuscript. We also thank Xuan Kou for constructing some of the plasmids used in this study. This work was supported by a grant (RPG-97-002-01) from the American Cancer Society to J.M.L.
REFERENCES
- 1.Haluska F.G., Tsujimoto,Y. and Croce,C.M. (1987) Annu. Rev. Genet., 21, 321–345. [DOI] [PubMed] [Google Scholar]
- 2.Ma P.C., Weintraub,H. and Pabo,C.O. (1994) Cell, 77, 451–459. [DOI] [PubMed] [Google Scholar]
- 3.Ferré-D’Amaré A.R., Prendergast,G.C., Ziff,E.B. and Burley,S.K. (1993) Nature, 363, 38–45. [DOI] [PubMed] [Google Scholar]
- 4.Ryan K.M. and Birnie,G.D. (1996) Biochem. J., 314, 713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Massari M.E. and Murre,C. (2000) Mol. Cell. Biol., 20, 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greenberg M.L. and Lopes,J.M. (1996) Microbiol. Rev., 60, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoshizaki D.K., Hill,J.E. and Henry,S.A. (1990) J. Biol. Chem., 265, 4736–4745. [PubMed] [Google Scholar]
- 8.Nikoloff D.M., McGraw,P. and Henry,S.A. (1992) Nucleic Acids Res., 20, 3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bachhawat N., Ouyang,Q. and Henry,S.A. (1995) J. Biol. Chem., 270, 25087–25095. [DOI] [PubMed] [Google Scholar]
- 10.Schwank S., Ebbert,R., Rautenstrauss,K., Schweizer,E. and Schüller,H.-J. (1995) Nucleic Acids Res., 23, 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashburner B.P. and Lopes,J.M. (1995) Mol. Cell. Biol., 15, 1709–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson K.A. and Lopes,J.M. (2000) J. Bacteriol., 182, 2746–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Z. and Brendel,M. (1993) Mol. Gen. Genet., 241, 680–684. [DOI] [PubMed] [Google Scholar]
- 14.Swift S. and McGraw,P. (1995) Nucleic Acids Res., 23, 1426–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cok S.J., Martin,C.G. and Gordon,J.I. (1998) Nucleic Acids Res., 23, 1426–1433. [Google Scholar]
- 16.Berben G., Legrain,M., Gilliquet,V. and Hilger,F. (1990) Yeast, 6, 451–454. [DOI] [PubMed] [Google Scholar]
- 17.Cai M. and Davis,R.W. (1990) Cell, 61, 437–446. [DOI] [PubMed] [Google Scholar]
- 18.Mellor J., Jiang,W., Funk,M., Rathjen,J., Barnes,C.A., Hinz,T., Hegemann,J.H. and Philippsen,P. (1990) EMBO J., 9, 4017–4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao X. and Butow,R.A. (1993) Cell, 72, 61–71. [DOI] [PubMed] [Google Scholar]
- 20.Jia Y., Rothermel,B., Thornton,J. and Butow,R.A. (1997) Mol. Cell. Biol., 17, 1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishi K., Seo Park,C., Pepper,A.E., Eichinger,G., Inis,M.A. and Holland,M.J. (1995) Mol. Cell. Biol., 15, 2646–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uetz P., Giot,L., Cagney,G., Mansfield,T.A., Judson,R.S., Knight,J.R., Lockshon,D., Narayan,V., Srinivasan,M., Pochart,P., Qureshi-Emili,A., Li,Y., Godwin,B., Conover,D., Kalbfleisch,T., Vijayadamodar,G., Yang,M., Johnston,M., Fields,S. and Rothberg,J.M. (2000) Nature, 403, 623–627. [DOI] [PubMed] [Google Scholar]
- 23.Ito T., Tashiro,K., Muta,S., Ozawa,R., Chiba,T., Nishizawa,M., Yamamoto,K., Kuhara,S. and Sakaki,Y. (2000) Proc. Natl Acad. Sci. USA, 97, 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 25.Bailis A.M., Lopes,J.M., Kohlwein,S.D. and Henry,S.A. (1992) Nucleic Acids Res., 20, 1411–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherman F. (1991) Methods Enzymol., 94, 1–20. [Google Scholar]
- 27.Hudak K.A., Lopes,J.M. and Henry,S.A. (1994) Genetics, 136, 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelly B.L. and Greenberg,M.L. (1990) Biochim. Biophys. Acta, 1046, 144–150. [DOI] [PubMed] [Google Scholar]
- 29.Lopes J.M., Hirsch,J.P., Chorgo,P.A., Schulze,K.L. and Henry,S.A. (1991) Nucleic Acids Res., 19, 1687–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson K.A. and Lopes,J.M. (2000) Nucleic Acids Res., 28, 1499–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ambroziak J. and Henry,S.A. (1994) J. Biol. Chem., 269, 15344–15349. [PubMed] [Google Scholar]
- 32.Hengen P.N. (1997) Trends Biochem. Sci., 22, 33–34. [DOI] [PubMed] [Google Scholar]
- 33.Lamping E., Lückl,J., Paltauf,F., Henry,S.A. and Kohlwein,S. (1995) Genetics, 137, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rothermel B.A., Thornton,J.L. and Butow,R.A. (1997) J. Biol. Chem., 272, 19801–19807. [DOI] [PubMed] [Google Scholar]
- 35.Paltauf F., Kohlwein,S.D. and Henry,S.A. (1992) In Jones,E.W., Pringle,J. and Broach,J.R. (eds), The Molecular Biology of the Yeast Saccharomyces cerevisiae: Gene Expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. pp. 415–499.
- 36.Shimizu T., Tuomoto,A., Ihara,K., Shimizu,M., Kyohoku,Y., Ogawa,N., Oshima,Y. and Hakoshima,T. (1997) EMBO J., 16, 4689–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hohmann S. (1997) In Hohmann,S. and Willem,M. (eds), Yeast Stress Responses. R.G. Landes, Austin, TX, pp. 101–145.