

Abstract
While a large number of chemically recyclable thermoplastics have been developed in recent years, technologically important thermoplastic elastomers (TPEs) that are not only bio-based and fully recyclable but also exhibit mechanical properties that can rival or even exceed those petroleum-based, non-recyclable polyolefin TPEs are critically lacking. The key challenge in developing chemically circular, bio-based, high-performance TPEs rests on the complexity of TPE’s block copolymer (BCP) structure involving block segments of different suitable monomers required to induce self-assembled morphologies responsible for performance as well as the control and monomer compatibility in their synthesis and the selectivity in their depolymerization. Here we demonstrate the utilization of bio-sourced δ-valerolactone (δVL) and its simple α-alkyl-substituted derivatives to produce all δVL-based polyester tri-BCP TPEs, which exhibit not only complete (closed-loop) chemical recyclability but also excellent toughness that is 2.5–3.8 times higher than commercial polyolefin-based TPEs. The visualized cylindrical morphology formed via crystallization-driven self-assembly in the new all δVL tri-BCP is postulated to contribute to the excellent TPE property.
Subject terms: Polymer synthesis, Sustainability, Polymers
While a large number of chemically recyclable thermoplastics have been developed in recent years, technologically bio-based and fully recyclable thermoplastic elastomers (TPEs) with excellent mechanical properties are lacking. Here the authors demonstrate an all δvalerolactone-based polyester tri-BCP TPEs, which exhibit not only closed-loop chemical recyclability but also exceptional toughness.
Introduction
Thermoplastic elastomers (TPEs) combining thermoplastics’ straightforward thermal processability with thermosets’ excellent elasticity have emerged as a more sustainable alternative to conventional thermosets or vulcanized natural rubber, neither of which can undergo chemical or mechanical recycling1,2. As a result, TPEs are applied in various fields including pressure-sensitive adhesives, elastomers, packaging, coatings, and fibers, as well as in additive manufacturing methods such as 3D printing3,4. The TPE market witnessed significant growth over the recent years and will continue to grow up to 5.55 million tonnes by 20265. To avoid the recurrence of plastic end-of-life problems, the design of TPEs with intrinsic chemical recyclability is needed as this strategy not only maintains the mechanical recycling of TPEs but also enables the chemical recycling of the TPEs at the end of their limited mechanical recycling lifetime to regenerate virgin-quality TPEs6–10. However, the significant challenge of achieving high selectivity and purity in the direct chemical recycling of TPEs into monomer still hinders the establishment of a closed-loop recycling pathway that reduces the reliance on continuous feedstock sourcing and eliminates the accumulation of TPE waste.
ABA-type triblock copolymers (tri-BCPs) made of aliphatic polyesters, consisting of hard end blocks A (a semicrystalline thermoplastic with a melting-transition temperature (Tm) > room temperature or an amorphous thermoplastic with a glass-transition temperature (Tg) >room temperature) and a soft midblock B (a soft, rubbery, and flexible amorphous polymer with a Tg < room temperature), show potential as eco-friendly substitutes for non-renewable or difficult-to-recycle/degrade TPEs because such polyesters are commonly biodegradable and can be conveniently obtained through effective ring-opening polymerization (ROP) of cyclic esters or lactones8,11–15. Nonetheless, three challenges remain in the context of developing high-performance polyester-based TPEs with closed-loop chemical circularity. One challenge has arisen in producing these tri-BCP polyesters with performance characteristics that can match or surpass those of current commodity TPEs such as robust polyolefin TPEs, which include tri-BCP polystyrene-b-polybutadiene-b-polystyrene (SBS) and tetra-BCP polystyrene-b-polyethylene-b-polybutadiene-b-polystyrene (SEBS)8,11–15. The second challenge is demonstration of complete chemical recyclability to cleanly recover monomers through selective depolymerization, with the ultimate objective of establishing a circular economy for polymers16,17. The third challenge is to provide an effective polymerization method to reconstruct a recovered monomer mixture, without further separation and purification, directly to new, recyclable TPEs with similar or even better properties relative to the original TPEs.
There are chemically recyclable TPEs that are able to chemically circulate a portion of their own composition, either hard or soft segments. For example, the poly(β-methyl-δ-valerolactones) chain segments in β-methyl-δ-valerolactones (βMeVL) based thermoplastic polyurethanes (TPUs) and cross-linked PU foams can degrade to recover βMeVL, while the isocyanic acid portion cannot degrade back to the original monomer18,19. In δ-methyl-δ-valerolactones (δMeVL)-based TPE poly(L-lactide (PLLA)-b-PδMeVL-b-PLLA), the PδMeVL chain segment can depolymerize to the original monomer δMeVL, but the PLLA segments can only be converted to ethyl lactate20. The PδMeVL chain segment in PδMeVL-based TPUs by chemolysis can depolymerize to δMeVL in an almost quantitative yield; however, recycling isocyanates proves to be difficult as they rapidly react with the remaining urethane bonds through urethane dissociation, resulting in the formation of allophanate linkages21.
Polyester-based BCP TPEs obtained through the ROP of lactones are appealing as the reversibility of ROP offers the possibility to convert all polyester blocks back into their respective lactone monomers. Discovering new intrinsically circular TPEs relies heavily on the design of monomers that produce both hard and soft segments. Recently, several lactone monomer families have been developed for the production of chemically recyclable polyesters. These include small (four-22,23, five-24–30 and six-31–45membered) lactones as well as large (seven-membered46–54 and macro55) lactones. Among these monomers, the six-membered lactones are especially attractive owing to the abundant availability of the parent bio-based δ-valerolactone (δVL). We design δVL-based chemically recyclable elastomers using the semicrystalline parent PVL (Tm ~ 60 °C) as the hard end block and the monosubstituted amorphous PVL as the soft midblock (Tg ~ –50 °C).
Additionally, the relatively high ring strain of δVL enables the efficient formation of poly(δ-valerolactone) (PVL) in high yields and molecular weight under mild conditions. Furthermore, the resulting semi-crystalline PVL can be chemically recycled back to δVL with nearly quantitative yield through catalytic thermolysis33. Addition of gem-dialkyl groups into the α-positions of δVL yielded disubstituted αR2VL that led to high molecular weight, semi-crystalline polyesters with high Tm (up to 140 °C) and chemical circularity34. On the other hand, the ROP of α,δ-disubstituted δVLs led to recyclable polyesters with only low molecular weight (Mn ~ 14000 g/mol)35,36. When a methyl group was introduced at the β- or δ-position of the δVL, the resulting βMeVL or δMeVL can be readily polymerized to high molecular weight, atactic, amorphous polymers PβMeVL37 or PδMeVL39. In the presence of a catalyst, both PβMeVL and PδMeVL can be depolymerized almost quantitatively to monomers. However, when directly pyrolyzed, a portion of the polymer forms a cross-linked residue upon pyrolysis (7% for PβMeVL and 5% for PδMeVL) as shown in Supplementary Figs. 1 and 2. A similar situation occurred for PδVL pyrolysis33. In addition, βMeVL and δMeVL exhibit only moderate equilibrium conversions (<85%) after polymerization with high monomer concentration ([M]0 = 4 M) (Supplementary Figs. 3 and 4) because of the low ceiling temperature (Tc) of 31 and 41 °C in 1.0 M, respectively (Supplementary Figs. 5 and 6). Thus, high-polymerizability lactones such as lactide (LA) (Tc = 284 °C, [LA]0 = 1.0 M) or ε-caprolactone (εCL, Tc = 2060 °C, [εCL]0 = 1.0 M) are required to form block polymers56.
Our initial attempts to produce all δVL-based tri-BCPs from copolymerization of βMeVL or δMeVL with a low Tc value (for favorable depolymerization selectivity) with δVL, which exhibits a moderate Tc of 243 °C ([δVL]0 = 1.0 M)33, failed to form ABA-type tri-BCPs (Supplementary Figs. 3–4 and 7–11). We reasoned that α-alkyl-substituted δ-valerolactones (αRVL) with potentially higher Tc values (due to higher ring-string energy) than β or δ substituted δVL derivatives, should be more thermodynamically compatible in copolymerization with the parent δVL. When a di-initiator is used, the first polymerization of αRVL builds a soft core-block (i.e., atactic, amorphous PαRVL with a low Tg); this is followed by polymerization of a second feed of δVL to construct the two semi-crystalline PVL outer hard blocks, thus furnishing ABA tri-BCPs, PVL-b-PαRVL-b-PVL (Scheme 1). However, answers to the following three open science questions are currently unknown: (1) The above hypothesis has not been tested. Can we design and synthesize a series of αRVL monomers to perform thermodynamic and kinetic (co)polymerization studies to confirm or dispute this hypothesis? (2) The chemical similarity and potential miscibility between the PVL and PαRVL block segments may hinder self-assembly to a microphase-separated morphology for manifesting robust TPE properties. Can we demonstrate crystallization-driven self-assembly of PVL-b-PαRVL-b-PVL into a discrete morphology responsible for potentially robust TPE properties that could rival incumbent polyolefin-based TPEs? (3) Chemical recycling of this tri-BCP will yield a mixture of two monomers, αRVL and δVL. Can we repolymerize these two recovered monomers, either further separated or used directly as a mixture, back to BCPs with similar properties to the virgin BCP, thereby demonstrating the closed-loop chemical circularity? Accordingly, this contribution documents the results of the experiments designed to answer the above three open science questions. Our efforts to address these fundamental questions have led to all δVL-based BCPs that not only possess complete chemical recyclability but also exhibit remarkable TPE mechanical performance, thanks to the self-assembled, crystallization-driven cylindrical morphology (Fig. 1).
Fig. 1. Synthesis and depolymerization of circular polyester BCPs based on α-alkyl-substituted δ-valerolactones.

αRVL (R = Me, Et, nPr, nBu, nPen, nHex).
Results and discussion
ROP of αRVL: thermodynamics, characteristics, and polymer properties
Monomers αRVL can be readily produced in a one-pot procedure from commercially accessible δVL, according to the preparation of αMeVL57. To investigate the polymerizability of αMeVL, equilibrium monomer concentrations [M]eq were determined at varying temperatures in its ROP, employing 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as a catalyst. A linear relationship was observed when plotting ln[αMeVL]eq versus 1/T in the van’t Hoff plot. The slope of the best-fit straight line was −1.44 and the intercept was 3.27 (Supplementary Fig. 12). Using these values, the standard-state thermodynamic parameters for the enthalpy change () and the entropy change () of polymerization were determined to be −12.0 kJ mol−1 and −27.2 J mol−1 K−1, respectively, based on the equation ln[M]eq = /RT − /R58 (Supplementary Fig. 12). The Tc was calculated to be 168 °C at [αMeVL]0 = 1.0 M, utilizing the equation Tc = /{+Rln[M]0}58. This result indicates that the polymerizability of αMeVL is indeed considerably higher than that of βMeVL (Tc = 31 °C at 1.0 M) and δMeVL (Tc = 41 °C at 1.0 M) although, as expected, lower than the parent δVL (Tc = 243 °C at [M]0 = 1.0 M)33. Substituting the Me group on the δVL ring with Et and nPr increased the Tc values to 211 and 216 °C (1.0 M), respectively, due to their lower entropy loss during polymerization (Supplementary Figs. 13 and 14). However, when the alkyl chain length was further increased to nBu, nPen, and nHex, Tc began to decrease to 192, 194, and 211 °C (1.0 M), respectively, and appeared to reach a plateau point (Fig. 2 and Supplementary Figs. 15–17).
Fig. 2. Dependence of Tc values on the singly substituted group and substitution pattern on the δVL ring.
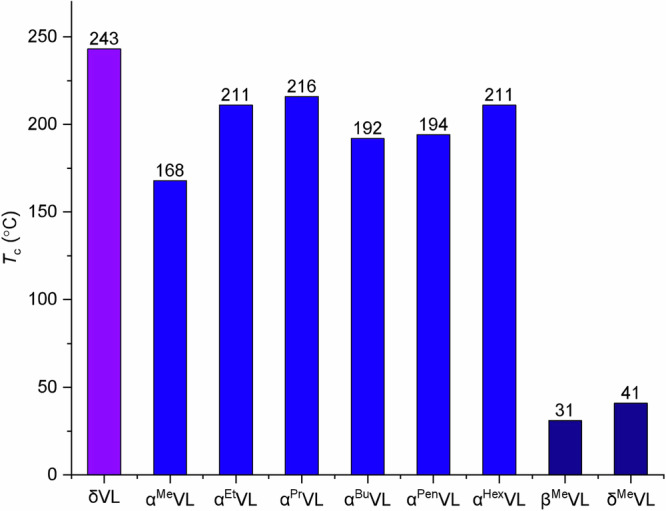
Tc values extrapolated to [M]eq = 1.0 M from the experimentally employed [M]0, the initial concentration used for the [M]eq measurements: αRVL compared with the parent δVL, βMeVL and δMeVL.
The aforementioned findings indicated that monomers with α-alkyl-substitutions have reduced thermodynamic polymerizability (or increased depolymerizability) compared to δVL. The main reason for the notable decrease in Tc after substitution is primarily due to the more pronounced decrease in the (negative) enthalpy change during polymerization, which surpasses the more positive change in entropy (Supplementary Fig. 18). The length of the R chain in the αRVL series impacts both and values, where the higher entropic penalty is balanced by the greater ring strain. Nevertheless, when examining the alterations in enthalpy and entropy weighted changes of the αRVL monomers (Supplementary Fig. 18), it becomes apparent that the variations in their Tc values are primarily attributed to the enthalpy factor. Notably, αMeVL exhibits the most favorable combination, resulting in the lowest Tc value in the series (Fig. 2). Hence, the resulting PαRVL should possess superior chemical recyclability compared to PVL, with PαMeVL exhibiting the greatest depolymerizability within the series based on the Tc value. Notably, when the methyl-substituent on the δVL ring is shifted to the β and δ position, the Tc value is much reduced to 31 and 41 °C (1.0 M), respectively (Supplementary Figs. 5 and 6). Overall, these thermodynamic data show αMeVL exhibits much higher polymerizability, or its ROP can achieve a much higher equilibrium monomer conversion, compared to monomers, αMeVL, βMeVL, and δMeVL when subjected to identical ROP conditions: catalyst TBD, initiator BnOH, [M]0: [TBD]0: [BnOH]0 = 200:1:1, [M]0 = 2.0 M. Comparatively, the ROP of αMeVL reached 89% conversion, however, the ROP of βMeVL and δMeVL only reached 56% and 69% conversion, respectively. Consistent with the Tc trend, the ROP of other α-alkyl- substituted monomers exhibit a higher polymerizability, achieving monomer conversions >90% (Supplementary Table 9, Supplementary Fig. 19).
Characteristics of the ROP of αRVL were investigated in more detail with commonly used metal-based catalysts for the ROP of lactones, including [La(OBn)3]x, [Y(OBn)3]x, [Zn(OBn)2]x, Zn[N(SiMe3)2][OBn] (isolated from alcoholysis of Zn[N(SiMe3)2]2 with BnOH), and Zn[N(SiMe3)2]2, the results of which were summarized in Supplementary Tables 10 and 11. Notably, the ROP of αMeVL by Zn[N(SiMe3)2][OBn] was shown to be living, as evidenced by the formation of PαMeVL, BnO–[αMeVL]n–H, with controlled Mn and a low Đ value of <1.11 (Supplementary Table 11, runs 3–7); pre-defined initiation chain end by 1H NMR spectrum (Supplementary Fig. 20); expected termination chain end by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) (Fig. 3A and Supplementary Fig. 21); and linear increase of PαMeVL Mn as a function of αMeVL conversion and [monomer]/[initiator] ratio while the Đ remained consistently narrow (1.07 − 1.11) across all conversions by size-exclusion chromatography (SEC) (Fig. 3B, C and Supplementary Fig. 22). The catalyst/initiator system [Zn(N(SiMe3)2)(OBn)] also exhibits control over the ROP of other αRVL monomers. For example, the ROP of 400 equivalents of αEtVL, αPrVL, αBuVL, αPenVL, and αHexVL resulted in the synthesis of PαEtVL with Mn = 35.5 kg mol−1 (Đ = 1.09), PαPrVL with Mn = 39.7 kg mol−1 (Đ = 1.12), PαBuVL with Mn = 42.2 kg mol−1 (Đ = 1.10), PαPenVL with Mn = 32.8 kg mol−1 (Đ = 1.08), and PαHexVL with Mn = 47.3 kg mol−1 (Đ = 1.07) (Supplementary Table 11, runs 8–12).
Fig. 3. Characterization of PαMeVL by MALDI-TOF MS analysis and living characteristics of the ROP of αMeVL.

A MALDI-TOF MS spectrum of PαMeVL produced with Zn[N(SiMe3)2][OBn] as catalyst ([αMeVL]/[Cat.] = 20/1). B Plot of Mn and Đ as a function of monomer conversion at [αMeVL]0/[Zn(N(SiMe3)2)(OBn)]0 = 400:1. Inset: Overlay of SEC curves at varied monomer conversion. C Plot of Mn and Đ as a function of [αMeVL]0/[Zn(N(SiMe3)2)(OBn)]0 ratio. Inset: Overlay of SEC curves at varied [αMeVL]0/[Zn(N(SiMe3)2)(OBn)]0 ratios.
As expected for these atactic polyesters, DSC analysis of the six PαRVL samples revealed amorphous materials with a low Tg ranging between −49 and −54 °C (Supplementary Figs. 23–29). This indicates the potential for utilizing PαRVL as the soft, flexible block in the creation of BCP-based TPE materials (vide infra). Thermal gravimetric analysis (TGA) of the six PαRVL polyesters revealed good thermal stability, with Td,5% (decomposition temperature at 5 wt% loss) varying between 233 and 258 °C (Supplementary Figs. 30–36). Thermolysis at elevated temperatures and chemolysis with a catalyst at milder temperatures were employed to investigate the chemical recyclability of these polyesters. By subjecting a sample of PαMeVL (Mn = 43.6 kg mol−1, Đ = 1.09) to heat in a sealed autoclave at 315 °C (which is the temperature at which the maximum rate of depolymerization occurs), for 2 h, the pure and colorless monomer αMeVL was obtained in a quantitative yield (Supplementary Fig. 37). Similarly, the thermal depolymerization of all the other polyester samples in the series at temperatures exceeding 300 °C for 2 h also resulted in recovery of the corresponding pure monomers in quantitative yield (Supplementary Figs. 38–42). Hence, the α-substitution in αRVL greatly enhances the depolymerizability of PαRVL, relative to the parent PVL, and avoids cross-linking of the PαRVL under thermolysis temperatures. Furthermore, to achieve the chemical recyclability of PαMeVL with a lower energy input, the depolymerization of PαMeVL took place at 90 °C using a commercial solid inorganic polyacid, phosphomolybdic acid (PMA)33, resulting in the near quantitative conversion to monomer (99% yield, Supplementary Fig. 37). PαMeVL with comparable Mn and Đ to the original PαMeVL (Mn = 43.3 kg mol−1, Đ = 1.11, Supplementary Fig. 43) was generated by repolymerizing the recovered αMeVL using Zn[N(SiMe3)2][OBn] in toluene at ambient temperature.
Thermal and mechanical properties of PVL-b-PαRVL-b-PVL
The sequential ROP of αMeVL and δVL using 1,4-benzenedimethanol as the di-initiator and TBD as the catalyst readily resulted in the preparation of five PVL-b-PαMeVL-b-PVL tri-BCPs with varying compositions and molecular weights (Supplementary Table 12). The well-defined structures of the synthesized PVL-b-PαMeVL-b-PVL tri-BCPs were characterized by 1H NMR and 13C NMR spectra (Supplementary Figs. 45–58) alongside SEC curves (Supplementary Figs. 59–65). The DOSY NMR spectrum of PVL-b-PαMeVL-b-PVL tri-BCP shows a single diffusion coefficient, indicating the successful formation of the block structure (Supplementary Fig. 66). The shifts of the unimodal SEC curves to higher molecular weight regions compared to telechelic PδVL precursors provided evidence for the expected increase in molecular weight (Supplementary Figs. 59–65). Supplementary Figs. 67–73 showed that all PVL-b-PαMeVL-b-PVL tri-BCP samples exhibit high thermal stability with Td,5% above 304 °C. The relatively low Td,5% of 252 °C observed for homopolymer PαMeVL is attributed to its lower Mn of 43.6 kg mol−1 than the BCP33,34. Despite the structural similarity between the blocks, microphase separation seems to take place, as evidenced by differential scanning calorimetry (DSC), which showed PVL-b-PαMeVL-b-PVL tri-BCPs exhibit separate Tg and Tm transitions for the amorphous, soft midblock and semi-crystalline hard endblock segments (Supplementary Figs. 74–80).
The resulting PVL-b-PαMeVL-b-PVL tri-BCPs were subsequently tested for mechanical performance by undergoing uniaxial tensile testing at a rate of 50 mm min−1 during extension. All PVL-b-PαMeVL-b-PVL tri-BCP exhibited TPE behavior. As anticipated, the composition and molecular weights of PVL-b-PαMeVL-b-PVL tri-BCPs significantly influenced their mechanical properties (Fig. 4, Supplementary Table 25). PVL-b-PαMeVL-b-PVL (Mn = 14.0–52.6–14.0 kg mol-1, FδVL (mol fractions of δVL) = 0.34, TPE-1) exhibited a good ductile behavior with a high elongation at break (εB) of 887 ± 6%, but it is a weak material with ultimate tensile strength (σB) = 5.9 ± 0.1 MPa (Fig. 4A and Supplementary Figs. 82, Table 13). With an increase of the PαMeVL midblock Mn to 61.4 kg mol-1, the σB of the tri-BCP PVL-b-PαMeVL-b-PVL (Mn = 15.3-61.4-15.3 kg/mol, FδVL = 0.33, TPE−2) rose to 12.1 ± 0.5 MPa (Fig. 4A and Supplementary Fig. 83, Table 14). Increasing the Mn of the PVL hard block to 21.0 kg mol-1 (Mn = 21.0-62.6-21.0 kg mol-1, FδVL = 0.41, TPE−3), the σB further increased to 14.9 ± 0.5 MPa, and the εB also increased to 1061 ± 11% (Fig. 4A and Supplementary Fig. 84, Table 15), corresponding to a high toughness (UT) of 68.4 ± 1.4 MJ m-3. By comparison, mechanically TPE-3 largely outperforms SEBS (YH-506T, σB = 11.3 ± 0.2 MPa, εB = 965 ± 6%, UT = 35.3 ± 1.1 MJ m-3, Supplementary Fig. 87, Table 18) and polyester-based BCP PLLA-b-PδCL-b-PLLA (σB = 13.4 ± 0.3 MPa, εB = 755 ± 33%)20. As FδVL of the PαMeVL midblock increased to 45% and 50%, the σB of tri-BCP PVL-b-PαMeVL-b-PVL (Mn = 27.5-67.1–27.5 kg mol-1, FδVL = 0.45, TPE−4) and PVL-b-PαMeVL-b-PVL (Mn = 36.0-72.0-36.0 kg mol-1, FδVL = 0.50, TPE−5) further increased to 22.2 ± 1.0 and 29.7 ± 1.9 MPa, while maintaining excellent flexibility (εB = 1059 ± 13% for TPE-4, Supplementary Fig. 85, Table 16; εB = 1041 ± 20% for TPE-5, Supplementary Fig. 86 and SupplementaryTable 17). TPE-4 (UT = 102 ± 4.6 MJ m-3) and TPE-5 (UT = 135 ± 8.2 MJ m-3) even outer-perform SBS (YH-791E, σB = 16.8 ± 1.6 MPa, εB = 752 ± 23%, UT = 55.2 ± 5.5 MJ m-3, Supplementary Fig. 88 and Supplementary Table 19). Remarkably, the tensile strength (29.7 ± 1.9 MPa) and elongation at break (εB = 1041 ± 20%) of TPE-5 were comparable with those of polyester-based BCP PLLA-b-PβMeVL-b-PLLA (σB = 28 ± 4 MPa, εB = 1720 ± 140%)37. To study the molecular weight influence on the mechanical performance of the TPE-5 having a fixed FδVL (FδVL ~ 0.50), the molecular weight was changed from 17.0 kg mol-1 to 34.5 kg mol-1, 69.0 kg mol-1, 120 kg mol-1, and 144 kg mol-1. The low molecular weight TPE-5 (17 kg mol-1 or 34.5 kg mol-1) is a viscous liquid (Supplementary Fig. 89). The PVL-b-PαMeVL-b-PVL (Mn = 69 kg mol-1, FδVL = 0.51) exhibited a good ductile behavior with a high εB of 626 ± 17%, but it is a weak material with an ultimate tensile strength of σB = 8.1 ± 0.3 MPa (Supplementary Fig. 90 and Supplementary Table 20). With an increase of the Mn to 120 kg mol-1, the σB of the tri-BCP PVL-b-PαMeVL-b-PVL (Mn = 120 kg mol-1, FδVL = 0.52) rose substantially to 27.9 ± 0.7 MPa, and the εB also largely increased to 984 ± 15% (Supplementary Fig. 91 and Supplementary Table 21). However, with a further increase of the Mn to 144 kg mol-1, the σB and εB of the PVL-b-PαMeVL-b-PVL (Mn = 144 kg mol-1, FδVL = 0.50) remain almost constant (σB = 29.7 ± 1.9 MPa, εB = 1059 ± 13%) (Supplementary Fig. 86, Table 17). To further assess the elastomeric performance, 10 reciprocating tensile cycles were applied to all tri-BCPs of PVL-b-PαMeVL-b-PVL at a rate of 50 mm min-1, subjecting them to a 100% strain. During the initial cycle 1, we observed plastic deformation and a decrease in tensile stress, which suggests an initial loss of energy due to the disentanglement of the PαMeVL midblock chains caused by stress, along with the rearrangement of microstructures59,60. The stress-strain response remained nearly the same throughout subsequent cycles 2–10, demonstrating a significant elastic recovery of 96.0 ± 0.4% for TPE-3 (Fig. 4B and Supplementary Figs. 92 and 93).
Fig. 4. Mechanical properties of PVL-b-PαMeVL-b-PVL tri-BCPs.

A Stress–strain curves of PVL-b-PαMeVL-b-PVL tri-BCPs with various mole fractions of the hard blocks (FδVL = 0.34, 0.33, 0.41, 0.45, 0.50, for TPE-1, -2, -3, -4, -5, respectively) as compared with commercial SBS (YH-791E) and SEBS (YH-506T). B Cyclic tensile testing to 100% strain at a rate of 50 mm/min (10 cycles) for TPE-3 tri-BCP.
For other PVL-b-PαMeVL-b-PVL tri-BCPs, the plastic deformation and decline in tensile stress were also observed in the first cycle, after which the stress-strain behavior remained almost identical for cycles 2–10, also exhibiting a high elastic recovery of 92 ~ 96% (Supplementary Figs. 94–101). Figure 5 compares PVL-b-PαMeVL-b-PVL TPE-3 with PVL-b-PαEtVL-b-PVL TPE-6 (Mn = 21.7–52.7–21.7 kg mol-1), which is much less ductile (εB = 446 ± 38%) and weaker (σB = 5.2 ± 0.2 MPa) (Supplementary Fig. 115, Table 22), and PVL-b-PαPrVL-b-PVL TPE-7 (Mn = 23.5-59.0-23.5 kg mol-1), which further decreases ductility (εB = 312 ± 12%) and lowers the tensile strength (σB = 2.2 ± 0.1 MPa) (Supplementary Fig. 116 and Supplementary Table 23). The above TPE-3 was also examined by dynamic mechanical analysis (DMA) in a tension film mode. The thermomechanical spectrum of TPE-3 (Supplementary Fig. 117) shows that the maximum storage modulus (E’) in the glassy state is 2.06 GPa. After the glass-transition region with an alpha transition (Tα = -45 °C, defined by the peak maxima of tanδ), E’ dropped by about one order of magnitude and the material still maintained a high E’ in the rubbery plateau until reaching a flow temperature of about 50 °C, characteristic of a semi-crystalline material corresponding to its Tm (50 °C by DSC).
Fig. 5. Stress-strain curves of PVL-b-PαRVL-b-PVL.
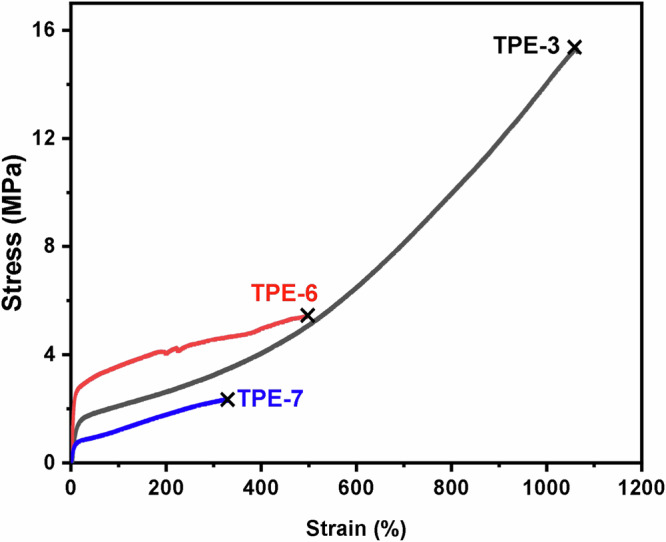
PVL-b-PαMeVL-b-PVL (Mn = 21.0–62.6–21.0 kg mol−1, FδVL = 0.41, TPE-3), PVL-b-PαEtVL-b-PVL (Mn = 21.7–52.7–21.7 kg mol-1, FδVL = 0.45, TPE-6) and PVL-b-PαPrVL-b-PVL (Mn = 23.5-59.0-23.5 kg mol-1, FδVL = 0.44, TPE-7) tri-BCPs at a strain rate of 50 mm/min, ambient temperature, fracture point indicated by ×.
Chemical circularity of PδVL-b-PαRVL-b-PδVL
Chemolysis was used to assess the chemical recyclability of the TPE materials under milder temperature conditions in the presence of a catalyst. Due to its low cost and demonstrated effectiveness in depolymerizing polyesters25,61 and nylon62, we employed ZnCl2 as a catalyst for the chemical recycling of TPEs to monomers. A straightforward vacuum distillation setup was utilized to perform the depolymerization process. Figure 6 shows that when TPE-3 was combined with 2.0 mol% ZnCl2 at 180 °C under reduced pressure (0.07 Torr), both pure δVL and αMeVL were obtained in essentially quantitative yield of 99%. To minimize energy consumption and prevent the evaporation of organic acids during the recycling procedure, PMA was also examined. Impressively, the depolymerization of 5.00 g of TPE-3 occurred at a relatively low temperature of 100 °C in 2 h, resulting in the recovery of 4.95 g of pure δVL and αMeVL monomers in 99% yield (Supplementary Fig. 118). Although the mixture can be separated by column chromatography (δVL 89% yield, αMeVL 83% yield), the loss of the monomer and additional energy cost are unavoidable. Likewise, catalyzed thermolysis of TPE-6 and TPE-7 at <120 °C with 2.0 mol% PMA for 2 h also afforded the corresponding pure monomer in 99% yield (Supplementary Figs. 119 and 120).
Fig. 6. Chemical recyclability of PVL-b-PαMeVL-b-PVL as shown by 1H NMR spectra (CDCl3).

Top, starting δVL. Second from the top, starting αMeVL. Third from top, clean recycled δVL and αMeVL were obtained using 2 mol% ZnCl2 after depolymerization at 180 °C for 3 h. Bottom, PVL-b-PαMeVL-b-PVL (Mn = 21.0–61.4−21.0 kg mol-1).
To reduce the recovery cost and avoid loss of the monomers in the separation and purification processes, direct conversion of the recovered δVL/αMeVL mixture to a BCP with similar properties to the original BCP was explored. The recovered monomer mixture ([δVL]/[αMeVL] = 41:59) was dried and mixed with an additional amount of δVL to achieve a [δVL]/[αMeVL] ratio of 92:8. The resulting mixture was then polymerized using TBD in toluene at room temperature to form telechelic OH-PVL-OH, and then αMeVL and δVL were added sequentially to form penta-BCP PVL-b-PαMeVL-b-PVL-b-PαMeVL-b-PVL (Mn = 22.2-59.6-39.7-59.6-22.2 kg mol-1, TPE-8). The well-defined structure of the obtained TPE-8 was characterized by 1H NMR and 13C NMR spectra (Fig. 7 and Supplementary Figs. 121) as well as by SEC (Supplementary Fig. 122). The DOSY NMR spectrum of TPE-8 showed only a single diffusion coefficient (Supplementary Fig. 123), further indicating the successful synthesis of the block structure. TGA showed that TPE-8 had the same, high thermal stability (Td,5% = 327 °C) as TPE-3 (Td,5% = 304 °C) (Supplementary Figs. 69 and 124). TPE-8 also exhibits separate Tg and Tm transitions for the midblock and endblock segments by DSC (Supplementary Fig. 125). Uniaxial tensile testing reveals that TPE-8 has good tensile strength, elongation at break and cyclic tensile results (σB = 17.8 ± 0.9 MPa, εB = 1077 ± 21%, UT = 65.7 ± 3.6 MJ m-3, Supplementary Fig. 126 and Table 24), which are the same quality of the virgin tri-BCP TPE-3 (Fig. 8).
Fig. 7. Stacked 13C NMR spectra (CDCl3) of tri-BCP and penta-BCP with those of homopolymers PαMeVL and PVL.

Top, PVL. Second from top, starting PαMeVL. Third from top, PVL-b-PαMeVL-b-PVL tri-BCP. Bottom, PVL-b-PαMeVL-b-PVL- b-PαMeVL-b-PVL penta-BCP.
Fig. 8. Comparisons of tensile properties of TPE−3 tri-BCP and TPE-8 penta-BCP.

A Overlay plots of stress/strain curves of TPE-3 (Mn = 21.0–61.4–21.0 kg mol-1) and TPE-8 (Mn = 22.2−59.6–39.7–59.6–22.2 kg mol−1) at 50 mm/min, ambient temperature, fracture point indicated by × (B) Plot of the elastic recovery of TPE−3 and TPE-8 determined from cyclic tensile tests. Error bars represent a standard deviation of 5 specimens.
Under the conditions of 120 °C and the presence of 2 mol% PMA, the depolymerization of 5.00 g of TPE-8 took 3 h and resulted in the recovery of 4.94 g of pure δVL and αMeVL monomers with 99% yield (Supplementary Fig. 127). Using the method described earlier to create TPE-8, the recovered δVL and αMeVL were again repolymerized with TBD in toluene to produce a new TPE of comparable molecular weight and structure to the original TPE-8 (Supplementary Figs. 128 and 129). Overall, these results demonstrate the chemical circularity of all δVL BCPs, through effective, direct utilization of the recovered BCP monomer mixture.
Morphological characteristics of PVL-b-PαMeVL-b-PVL
To better understand the thermal and mechanical material properties exhibited by tri-BCP PVL-b-PαRVL-b-PVL, we endeavored to analyze the surface morphology of PVL-b-PαMeVL-b-PVL. Atomic force microscopy (AFM) images of TPE-3 were obtained by analyzing spin-coated sheets of highly oriented pyrolytic graphite with a CHCl3 solution of TPE-3. Cylindrical nano-objects with an average length (L) of 96 ± 34 nm were directly observed in the thin film prepared from a dilute solution of TPE-3 (0.01 mg mL-1) (Fig. 9 and Supplementary Fig. 130). We posit that crystallization-driven self-assembly of this tri-BCP generated long cylindrical nano-objects, where the semicrystalline PVL cores are surrounded by the amorphous PαMeVL corona, by deposition of chains on growing crystal nuclei63,64. Imaging of the thin film prepared from a more concentrated solution (1 mg mL-1) revealed the same thin film morphology in more detail. Specifically, the packing of the PVL cylinders connected by the soft and amorphous PαMeVL matrix was observed along with consistent length (L = 115 ± 20 nm) and shape of the cylinder morphology (Fig. 9 and Supplementary Fig. 131). Considering the chemical similarity and miscibility between the PVL and PαMeVL blocks, the results suggested this self-assembled cylinder morphology is crystallization-driven, where the PVL crystallites are grown from homogeneous PVL-b-PαMeVL-b-PVL solutions or melts33,65. The morphology of TPE-3 was also characterized by small-angle X-ray scattering (SAXS) analysis of thin films under ambient conditions. A weak principal reflection was observed with the absence of a well-defined secondary peak, followed by a broad oscillation at higher q (Supplementary Fig. 132). Poor long-range ordering rendered the independent SAXS morphological assignment inconclusive. However, the broad reflection at low q indicated the presence of interparticle interference between cylinders. By using the cylinder length of 115 nm determined by AFM imaging, the form factor oscillation at high q showed good agreement with the simulated cylinder form factor model (Supplementary Fig. 133)66. This data supports the disorganized semicrystalline PVL cylinder dispersed in the rubbery matrix, as determined by AFM imaging. We reason that this crystallization-driven cylindrical morphology of the hard domain alleviates chain pull behavior upon extension leading to the observed high extensibility (TPE-3, εB = 1061 ± 11%) and resilience (TPE-3, 96.0 ± 0.4%)67,68.
Fig. 9. AFM images at different magnification of the surface of films.

The films prepared from both concentrated (1 mg/mL, top) and dilute (0.01 mg/mL, bottom) solutions of PVL-b-PαMeVL-b-PVL, revealing their crystallization-induced cylindrical morphology.
In summary, we have synthesized a range of simple, high-performance, new polyester TPEs derived from bio-based δVL and its α-alkyl-substituted δVL derivatives (αRVL). These α-alkyl-substituted αRVL lactones are polymerized to form amorphous, flexible, low-Tg polyesters that can be completely recycled back to the monomer through thermolysis. In comparison, PVL, PβMeVL, or PδMeVL produced a cross-linked residue after thermolysis. These α-alkyl-substituted lactones exhibit finely balanced (de)polymerization characteristics, allowing both the efficient generation of PVL-b-PαRVL-PVL tri-BCPs through controlled copolymerization of δVL with αRVL under ambient conditions and selective depolymerization of the tri-BCP to fully recover pure monomers (αRVL and δVL), under mild conditions (100 °C) in the presence of 2 mol% acid catalyst PMA. Importantly, the recovered monomer mixture containing δVL and αRVL can be directly repolymerized into a new recyclable TPE with the same properties as the original TPE without the need for further monomer separation and purification, thereby effectively completing the monomer-polymer-monomer circular cycle.
In addition to their complete chemical recyclability, these polyester TPEs also demonstrate high thermal stability (Td,5% > 300 °C) and varying degrees of mechanical properties, depending on the length of the side-chain alkyl group and the relative molar fraction of the hard and soft blocks. In particular, PVL-b-PαMeVL-b-PVL (FδVL = 0.50) exhibits remarkable mechanical properties, showcasing a high tensile strength of 29.7 ± 1.9 MPa, an impressive ductility of 1041 ± 20%, and a high toughness of 135 ± 8.2 MJ m-3, which significantly surpass the commercial SBS (σB = 16.8 ± 1.6 MPa, εB = 752 ± 23%, UT = 55.2 ± 5.5 MJ m-3) and SEBS (σB = 11.3 ± 0.2 MPa, εB = 965 ± 6%, UT = 35.3 ± 1.1 MJ m-3). We postulate the assigned crystallization-driven cylindrical morphology of PVL-b-PαMeVL-b-PVL contributes to such excellent TPE mechanical behavior.
In broader context, this study shows that the α-alkyl substitution in the simple bio-based six-membered lactone not only significantly improves the recyclability of the corresponding polyesters but also enhances the compatibility of αRVL block copolymerization with δVL. Compared to a number of known TPEs, fully recyclable, bio-based polyester TPEs that can rival or even exceed the properties of existing commercial TPE materials are still largely lacking. Continued efforts are required to develop new TPEs with even better performance, cost-effectiveness, and expedient chemical circularity.
Methods
Materials
The δ-valerolactone (δVL) was purchased from Energy Chemical Co., Ltd. Methyl iodide, CDCl3, diisopropylamine, and n-butyl lithium (2.5 M in hexanes) were purchased from Adamas-beta were used without further purification. CDCl3 was dried over CaH2, then degassed and stored over 4 Å molecular sieves. Benzyl alcohol (BnOH) obtained from Energy Chemical Reagent Co. was dried over CaH2 while stirring overnight and distilled before use. 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) and 1,4-Benzenedimethanol (BDM) obtained from Aladdin Reagent Co. were dried under vacuum at 50 °C for at least 48 h and placed in the glove box by sublimation. Toluene and tetrahydrofuran (THF) were distilled from sodium/benzophenone under nitrogen, degassed, and stored over fresh Na chips. CH2Cl2, monomers were dried over CaH2 while stirring overnight and distilled before use, and then degassed and stored over 4 Å molecular sieves. All monomers were dried over CaH2 overnight, vacuum distillated, and stored over activated 4 Å molecular sieves in the glovebox for further use.
Characterization methods
1H and 13C NMR
Spectra were recorded on a Bruker Avance II 400 MHz spectrometer, or a Varian Inova 400 MHz spectrometer. 1H and 13C NMR chemical shifts were referred to SiMe4 (TMS).
High-Resolution Mass Spectrometry (HRMS)
Analyses were performed on a Time-of-flight mass spectrometer (Agilent, G6224A) equipped with the Dual spray ESI/APCI ion source.
Size-Exclusion Chromatography (SEC)
Polymer number (Mn) and weight (Mw) average molecular weights, and dispersity index (Đ = Mw/Mn) were measured by SEC analyses carried out using Agilent 1260 GPC II instrument equipped with an autosampler and a refractive index detector. The system was equipped with two PLgel 5 μm mixed-C columns (Polymer Laboratories; linear range of molecular weight = 1000–3,000,000), which were eluted with dichloromethane (DCM) at 28 °C at a rate of 1.0 mL/min and calibrated using monodisperse polystyrene standards.
Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDITOF-MS)
MALDI-TOF-MS analyses were performed on a Bruker UltrafleXtreme. Polyester samples for MALDI analysis were prepared by preparing a solution of polyester, trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene] malononitrile (matrix), and sodium acetate (cationization agent) in CH2Cl2. The solution was spotted on a stainless steel MALDI target plate and allowed to air-dry. Sample solution was deposited on the MALDI target and allowed to dry completely to the measurement.
Differential Scanning Calorimetry (DSC)
Melting transition (Tm) and glass transition (Tg) temperatures were measured by differential scanning calorimetry (DSC) on an Auto Q20, TA Instrument. All Tm and Tg values were obtained from a DSC scan after the thermal history was removed from the first scan, unless noted otherwise. The DSC heating rate was 10 °C/min and the cooling rate was 10 °C/min unless indicated otherwise in the polymerization tables.
Thermogravimetric Analysis (TGA)
Decomposition temperatures (Td,5%, defined by the temperature of 5% weight loss) and maximum rate decomposition temperatures (Tmax) of the polymers were measured by thermal gravimetric analysis (TGA) on a Mettler-Toledo TGA/SDTA851. Polymer samples were heated from ambient temperatures to 500 °C at a heating rate of 10 °C/min. Values of Tmax were obtained from derivative (wt%/°C) vs. temperature (°C) plots, while Td were obtained from wt% vs. temperature (°C) plots.
Dynamic Mechanical Analysis (DMA)
Storage modulus (E’, MPa), loss modulus (E”, MPa), and tan δ (E”/E’) measurements for tri-BCPs were performed on a TA Q800 with an 18 N load cell in tension film mode on rectangular specimens (l = 29.4 mm, w = 4.1 mm, t = ~0.5 mm). Sample length between grips is 7.2 mm. Temperature-ramp frequency-sweep experiments were performed at 0.1 % oscillating strain at 1 Hz between -80 to 60 °C at a heating rate of 3 °C/min. Temperatures below ambient conditions were accessed via liquid-N2 GCA tank attachment. Post-run analysis was done on TA Universal analysis software.
Film preparation and mechanical property test
Polymer substrates are placed between non-stick Teflon sheets in a stainless-steel, rectangular (50 * 4 * 0.5 mm; length * width * thickness) mold and compressed on a hot stage at 7000 psi for roughly 20 min. All triblock copolymers were hot-pressed at a temperature of about 120 °C. After hot-pressing for 20 min, they were naturally cooled to room temperature, and placed at room temperature for 48 h before mechanical property testing.
Both uniaxial tensile tests and cyclic tensile tests are performed on an Instron 68TM-10 universal tensometer testing system. The uniaxial tensile test is carried out with the clamp clamped at 30 mm of the film at a tensile rate of 50 mm/min to sample failure. Reported measurements are obtained from the Instron readout in cooperation with Blue Hill Universal software (TA) normalizing force/displacement response to stress/strain, and reported curves were generated from an average of 3–5 individual specimens for reproducibility, with error margins included accordingly. Young’s modulus (E, MPa), ultimate strength (σB, MPa), yield strength (MPa), and elongation at break (εB, %) were obtained from software analysis.
The cyclic tensile tests were conducted to 100% strain at a tensile rate of 50 mm/min for 10 cycles. The elastic residual strain and recovery were reported as average values with standard deviations obtained from 3–5 specimens.
Atomic Force Microscopy (AFM)
AFM images were obtained under ambient conditions using a Bruker Bioscope Resolve AFM in Peak Force Tapping Scanasyst mode. Silicon cantilevers (SCANASYST-AIR, spring force constant: 0.12 N/m, frequency: 23 kHz) were used. The samples were prepared under ambient conditions by spin coating (2000 rpm for 30 s, then 4000 rpm for 10 s) freshly cleaved sheets (5 mm2) of highly ordered pyrolytic graphite (HOPG) with 10 µL of thoroughly dissolved polymer sample solutions (1 mg/mL and 0.01 mg/mL in chloroform).
Small-angle X-ray Scattering (SAXS)
SAXS was performed at the Stanford Synchrotron Radiation Lightsource (SSRL) beamline 1–5. The X-Ray beam energy was 15 keV (0.826 Å) with a slit size of 0.5 (horizontal) x 0.3 (vertical) mm2 for the beam-defining slits. The data was taken with a Pilatus 1 M detector comprising 981 × 1043 pixels with a pixel size of 0.172 mm × 0.172 mm in transmission geometry. The sample-to-detector distance was 2848 mm downstream of the sample. Silver behenate (AgBe) was used as the standard to calibrate the sample-to-detector distance. The two-dimensional scattering patterns were azimuthally integrated to afford one-dimensional profiles presented as scattering vector (q) versus scattering intensity, where the magnitude of scattering vector was calculated with q = (4π/λ) sin(θ/2). The bulk polymer sample was prepared by annealing at 140 °C in the vacuumed oven for 1 day and subsequent cooling at RT. Data was fitted to cylinder models using SasView v5.06 small-angle scattering software package with 115 nm long cylinders and a 4 nm radius69.
Supplementary information
Acknowledgements
The work done at Dalian University of Technology was supported by the National Natural Science Foundation of China (No. 22371030), Liaoning Provincial Science and Technology Program Joint Program (2023JH2/101700291), Fundamental Research Funds for the Central Universities (DUT24ZD112) and CNPC Innovation Found (2020D-5007-0406). The work done at Colorado State University was supported by the U.S. National Science Foundation (NSF-2305058). We thank S. Hesse and C. Tassone of SLAC for collecting raw scattering data for TPE-3.
Author contributions
K.M., T.-Q.X. and E.Y.-X.C. conceived the idea and designed the experiments. K.M., H.-Y.A., J. N., L. T. R., and Y.-L.Z. carried out experiments and analyzed and processed the data. All authors co-wrote the manuscript and participated in data analyses and discussions.
Peer review
Peer review information
Nature Communications thanks Junpeng Wang and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Data availability
All of the data necessary to support the conclusions of this paper are provided in the manuscript and its Supplementary information File. All data are available from the corresponding author upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-52229-1.
References
- 1.Bonart, R. Thermoplastic elastomers. Polymer20, 1389–1403 (1979). 10.1016/0032-3861(79)90280-5 [DOI] [Google Scholar]
- 2.Spontak, R. J. & Patel, N. P. Thermoplastic elastomers: fundamentals and applications. Curr. Opin. Colloid Interface Sci.5, 333–340 (2000). 10.1016/S1359-0294(00)00070-4 [DOI] [Google Scholar]
- 3.Steube, M., Johann, T., Barent, R. D., Mueller, A. H. E. & Frey, H. Rational design of tapered multiblock copolymers for thermoplastic elastomers. Frey. Prog. Polym. Sci.124, 101488 (2022). 10.1016/j.progpolymsci.2021.101488 [DOI] [Google Scholar]
- 4.Haque, F. M. et al. Defining the macromolecules of tomorrow through synergistic sustainable polymer research. Chem. Rev.122, 6322–6373 (2022). 10.1021/acs.chemrev.1c00173 [DOI] [PubMed] [Google Scholar]
- 5.Smithers. The Future of Thermoplastic Elastomers To 2026.www.smithers.com/services/market-reports/materials/the-future-of-thermoplastic-elastomers-to-2024 (2024).
- 6.Geyer, R., Jambeck, J. R. & Law, K. L. Production, use, and fate of all plastics ever made. Sci. Adv.3, e1700782 (2017). 10.1126/sciadv.1700782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borrelle, S. B. et al. Predicted growth in plastic waste exceeds efforts to mitigate plastic pollution. Science369, 1515–1518 (2020). 10.1126/science.aba3656 [DOI] [PubMed] [Google Scholar]
- 8.Coates, G. W. & Getzler, Y. D. Y. L. Chemical recycling to monomer for an ideal, circular polymer economy. Nat. Rev. Mater.5, 501–516 (2020). 10.1038/s41578-020-0190-4 [DOI] [Google Scholar]
- 9.Sathe, D. et al. Olefin metathesis-based chemically recyclable polymers enabled by fused-ring monomers. Nat. Chem.13, 743–750 (2021). 10.1038/s41557-021-00748-5 [DOI] [PubMed] [Google Scholar]
- 10.Xie, X., Huo, Z., Jang, E. & Tong, R. Recent advances in enantioselective ring-opening polymerization and copolymerization. Commun. Chem.6, 202 (2023). 10.1038/s42004-023-01007-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jehanno, C. et al. Critical advances and future opportunities in upcycling commodity polymers. Nature603, 803–814 (2022). 10.1038/s41586-021-04350-0 [DOI] [PubMed] [Google Scholar]
- 12.Hong, M. & Chen, E. Y.-X. Chemical recycling to monomer for an ideal, circular polymer economy. Trends Chem.1, 148–151 (2019). 10.1016/j.trechm.2019.03.004 [DOI] [Google Scholar]
- 13.Meys, R. et al. Achieving net-zero greenhouse gas emission plastics by a circular carbon economy. Science374, 71–76 (2021). 10.1126/science.abg9853 [DOI] [PubMed] [Google Scholar]
- 14.Li, X.-L., Ma, K., Xu, F. & Xu, T.-Q. Advances in the synthesis of chemically recyclable polymers. Chem. Asian J.18, e202201167 (2023). 10.1002/asia.202201167 [DOI] [PubMed] [Google Scholar]
- 15.Zhao, W. et al. One-pot synthesis of supertough, sustainable polyester thermoplastic elastomers using block-like, gradient copolymer as soft midblock. CCS Chem.4, 1263–1272 (2022). 10.31635/ccschem.021.202100897 [DOI] [Google Scholar]
- 16.Schneiderman, D. K. & Hillmyer, M. A. 50th anniversary perspective: there is a great future in sustainable polymers. Macromolecules50, 3733–3749 (2017). 10.1021/acs.macromol.7b00293 [DOI] [Google Scholar]
- 17.Worch, J. C. & Dove, A. P. 100th Anniversary of macromolecular science viewpoint: toward catalytic chemical recycling of waste (and future) plastics. ACS Macro Lett.9, 1494–1506 (2020). 10.1021/acsmacrolett.0c00582 [DOI] [PubMed] [Google Scholar]
- 18.Brutman, J. P., De Hoe, G. X., Schneiderman, D. K., Le, T. N. & Hillmyer, M. A. Renewable, degradable, and chemically recyclable cross-linked elastomers. Ind. Eng. Chem. Res.55, 11097–11106 (2016). 10.1021/acs.iecr.6b02931 [DOI] [Google Scholar]
- 19.Schneiderman, D. K. et al. Chemically recyclable biobased polyurethanes. ACS Macro Lett.5, 515–518 (2016). 10.1021/acsmacrolett.6b00193 [DOI] [PubMed] [Google Scholar]
- 20.Li, C. et al. Rapid and controlled polymerization of bio‐sourced δ‐caprolactone toward fully recyclable polyesters and thermoplastic elastomers. Angew. Chem. Int. Ed.16, e202201407 (2022). [DOI] [PubMed] [Google Scholar]
- 21.Yan, Q., Li, C. J., Yan, T., Shen, Y. & Li, Z.-B. Chemically recyclable thermoplastic polyurethane elastomers via a cascade ring-opening and step-growth polymerization strategy from bio-renewable δ-caprolactone. Macromolecules55, 3860–3868 (2022). 10.1021/acs.macromol.2c00439 [DOI] [Google Scholar]
- 22.Zhou, L. et al. Chemically circular, mechanically tough, and melt-processable polyhydroxyalkanoates. Science380, 64–69 (2023). 10.1126/science.adg4520 [DOI] [PubMed] [Google Scholar]
- 23.Yuan, J. et al. 4-Hydroxyproline-derived sustainable polythioesters: controlled ring-opening polymerization, complete recyclability, and facile functionalization. J. Am. Chem. Soc.141, 4928–4935 (2019). 10.1021/jacs.9b00031 [DOI] [PubMed] [Google Scholar]
- 24.Hong, M. & Chen, E. Y.-X. Completely recyclable biopolymers with linear and cyclic topologies via ring-opening polymerization of γ-butyrolactone. Nat. Chem.8, 42–49 (2016). 10.1038/nchem.2391 [DOI] [PubMed] [Google Scholar]
- 25.Zhu, J.-B., Watson, E. M., Tang, J. & Chen, E. Y.-X. A synthetic polymer system with repeatable chemical recyclability. Science360, 398–403 (2018). 10.1126/science.aar5498 [DOI] [PubMed] [Google Scholar]
- 26.Zhu, J.-B. & Chen, E. Y.-X. Living coordination polymerization of a six‐five bicyclic lactone to produce completely recyclable polyester. Angew. Chem. Int. Ed.57, 12558–12562 (2018). 10.1002/anie.201808003 [DOI] [PubMed] [Google Scholar]
- 27.Shi, C., Clarke, R. W., McGraw, M. L. & Chen, E. Y.-X. Closing the “one monomer–two polymers–one monomer” loop via orthogonal (de) polymerization of a lactone/olefin hybrid. J. Am. Chem. Soc.144, 2264–2275 (2022). 10.1021/jacs.1c12278 [DOI] [PubMed] [Google Scholar]
- 28.Xiong, W. et al. Geminal dimethyl substitution enables controlled polymerization of penicillamine-derived β-thiolactones and reversed depolymerization. Chem6, 1831–1843 (2020). 10.1016/j.chempr.2020.06.003 [DOI] [Google Scholar]
- 29.Yuan, P., Sun, Y., Xu, X., Luo, Y. & Hong, M. Towards high-performance sustainable polymers via isomerization-driven irreversible ring-opening polymerization of five-membered thionolactones. Nat. Chem.14, 294–303 (2022). 10.1038/s41557-021-00817-9 [DOI] [PubMed] [Google Scholar]
- 30.Xia, Y., Yuan, P., Zhang, Y., Sun, Y. & Hong, M. Converting non-strained γ-valerolactone and derivatives into sustainable polythioesters via isomerization-driven cationic ring-opening polymerization of thionolactone intermediate. Angew. Chem. Int. Ed.62, e202217812 (2023). 10.1002/anie.202217812 [DOI] [PubMed] [Google Scholar]
- 31.Xu, T.-Q., Yu, Z.-Q. & Zhang, X.-M. Recyclable vinyl‐functionalized polyesters via chemoselective organopolymerization of bifunctional α‐methylene‐δ‐valerolactone. Macromol. Chem. Phys.220, 1900150 (2019). 10.1002/macp.201900150 [DOI] [Google Scholar]
- 32.Li, J., Liu, F., Liu, Y., Shen, Y. & Li, Z.-B. Functionalizable and chemically recyclable thermoplastics from chemoselective ring-opening polymerization of bio-renewable bifunctional α-methylene-δ-valerolactone. Angew. Chem. Int. Ed.61, e202207105 (2022). 10.1002/anie.202207105 [DOI] [PubMed] [Google Scholar]
- 33.Li, X.-L. et al. Dual recycling of depolymerization catalyst and biodegradable polyester that markedly outperforms polyolefins. Angew. Chem. Int. Ed.62, e20230379 (2023). [DOI] [PubMed] [Google Scholar]
- 34.Li, X. L., Clarke, R. W., Jiang, J. Y., Xu, T. Q. & Chen, E. Y.-X. A circular polyester platform based on simple gem-disubstituted valerolactones. Nat. Chem.15, 278–285 (2023). 10.1038/s41557-022-01077-x [DOI] [PubMed] [Google Scholar]
- 35.Rapagnani, R. M., Dunscomb, R. J., Fresh, A. A. & Tonks, I. A. Tunable and recyclable polyesters from CO2 and butadiene. Nat. Chem.14, 877–883 (2022). 10.1038/s41557-022-00969-2 [DOI] [PubMed] [Google Scholar]
- 36.Lou, Y., Xu, L., Gan, N., Sun, Y. & Lin, B.-L. Chemically recyclable polyesters from CO2, H2, and 1, 3-butadiene. Innovation3, 100216 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiong, M., Schneiderman, D. K., Batesa, F. S., Hillmyer, M. A. & Zhang, K. Scalable production of mechanically tunable block polymers from sugar. PNAS111, 8357–8362 (2014). 10.1073/pnas.1404596111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martello, M. T., Schneiderman, D. K. & Hillmyer, M. A. Synthesis and melt processing of sustainable poly(ε-decalactone)-block-poly(lactide) multiblock thermoplastic elastomers. ACS Sustain. Chem. Eng.2, 2519–2526 (2014). 10.1021/sc500412a [DOI] [Google Scholar]
- 39.Li, C. et al. Rapid and controlled polymerization of bio-sourced δ-caprolactone toward fully recyclable polyesters and thermoplastic elastomers. Angew. Chem. Int. Ed.61, e202201407 (2022). 10.1002/anie.202201407 [DOI] [PubMed] [Google Scholar]
- 40.Fahnhorst, G. W. & Hoye, T. R. A carbomethoxylated polyvalerolactone from malic acid: synthesis and divergent chemical recycling. ACS Macro Lett.7, 143–147 (2018). 10.1021/acsmacrolett.7b00889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruckmoser, J., Remke, S. & Rieger, B. Ring-opening polymerization of a bicyclic lactone: polyesters derived from norcamphor with complete chemical recyclability. ACS Macro Lett.11, 1162–1166 (2022). 10.1021/acsmacrolett.2c00445 [DOI] [PubMed] [Google Scholar]
- 42.Wang, Y., Li, M., Chen, J., Tao, Y. & Wang, X. O‐to‐S substitution enables dovetailing conflicting cyclizability, polymerizability, and recyclability: dithiolactone vs dilactone. Angew. Chem. Int. Ed.60, 22547–22553 (2021). 10.1002/anie.202109767 [DOI] [PubMed] [Google Scholar]
- 43.Zhou, T., Guo, Y. T., Du, F. S. & Li, Z. C. Ring-opening polymerization of 2-oxabicyclo [2.2.2] octan-3-one and the influence of stereochemistry on the thermal properties of the polyesters. Chin. J. Polym. Sci.40, 1173–1182 (2022). 10.1007/s10118-022-2725-1 [DOI] [Google Scholar]
- 44.Zhu, Y., Li, M., Wang, Y., Wang, X. & Tao, Y. Performance‐advantaged stereoregular recyclable plastics enabled by aluminum‐catalytic ring‐opening polymerization of dithiolactone. Angew. Chem. Int. Ed.62, e202302898 (2023). 10.1002/anie.202302898 [DOI] [PubMed] [Google Scholar]
- 45.Wang, Y., Zhu, Y., Lv, W., Wang, X. & Tao, Y. Tough while recyclable plastics enabled by monothiodilactone monomers. J. Am. Chem. Soc.145, 1877–1885 (2023). 10.1021/jacs.2c11502 [DOI] [PubMed] [Google Scholar]
- 46.MacDonald, J. P. & Shaver, M. P. An aromatic/aliphatic polyester prepared via ring-opening polymerisation and its remarkably selective and cyclable depolymerisation to monomer. Polym. Chem.7, 553–559 (2016). 10.1039/C5PY01606A [DOI] [Google Scholar]
- 47.Lizundia, E., Makwana, V. A., Larrañaga, A., Vilas, J. L. & Shaver, M. P. Thermal, structural and degradation properties of an aromatic–aliphatic polyester built through ring-opening polymerization. Polym. Chem.8, 3530–3538 (2017). 10.1039/C7PY00695K [DOI] [Google Scholar]
- 48.Li, L.-G., Wang, Q.-Y., Zheng, Q.-Y., Du, F.-S. & Li, Z.-C. Tough and thermally recyclable semiaromatic polyesters by ring-opening polymerization of benzo-thia-caprolactones. Macromolecules54, 6745–6752 (2021). 10.1021/acs.macromol.1c00497 [DOI] [Google Scholar]
- 49.Tu, Y.-M. et al. Biobased high-performance aromatic–aliphatic polyesters with complete recyclability. J. Am. Chem. Soc.143, 20591–20597 (2021). 10.1021/jacs.1c10162 [DOI] [PubMed] [Google Scholar]
- 50.Tu, Y.-M., Gong, F.-L., Wu, Y.-C., Cai, Z. & Zhu, J.-B. Insights into substitution strategy towards thermodynamic and property regulation of chemically recyclable polymers. Nat. Comm.14, 3198 (2023). 10.1038/s41467-023-38916-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fan, H.-Z. et al. Advancing the development of recyclable aromatic polyesters by functionalization and stereocomplexation. Angew. Chem. Int. Ed.61, e202117639 (2022). 10.1002/anie.202117639 [DOI] [PubMed] [Google Scholar]
- 52.Li, Z., Zhao, D., Shen, Y. & Li, Z.-B. Ring-opening polymerization of enantiopure bicyclic ether-ester monomers toward closed-loop recyclable and crystalline stereoregular polyesters via chemical upcycling of bioplastic. Angew. Chem. Int. Ed.62, e202302101 (2023). 10.1002/anie.202302101 [DOI] [PubMed] [Google Scholar]
- 53.Su, J. et al. Closed-loop chemical recycling of poly(ε-caprolactone) by tuning reaction parameters. Polym. Chem.13, 5897–7904 (2022). 10.1039/D2PY00953F [DOI] [Google Scholar]
- 54.Cao, Q. et al. Torsional strain enabled ring‐opening polymerization towards axially chiral semiaromatic polyesters with chemical recyclability. Angew. Chem. Int. Ed.136, e202400196 (2024). 10.1002/ange.202400196 [DOI] [PubMed] [Google Scholar]
- 55.Hu, Z. et al. Catalytically controlled ring-opening polymerization of 2-oxo-15-crown-5 for degradable and recyclable peg-like polyesters. ACS Macro Lett.11, 792–798 (2022). 10.1021/acsmacrolett.2c00210 [DOI] [PubMed] [Google Scholar]
- 56.Save, M., Schappacher, M. & Soum, A. Controlled ring‐opening polymerization of lactones and lactides initiated by lanthanum isopropoxide, 1. General aspects and kinetics. Macromol. Chem. Phys.203, 889–899 (2002). [DOI] [Google Scholar]
- 57.Yang, X. H. et al. Iridium-catalyzed asymmetric hydrogenation of racemic α-substituted lactones to chiral diols. Chem. Sci.8, 1811–1814 (2017). 10.1039/C6SC04609F [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Du, A. & Kowalski, A. Handbook of Ring-Opening Polymerization. (eds. Dubois, P., Coulembier, O. & Raquez, J.-M.) 425 (Wiley-VCH, 2009).
- 59.Gregory, G. L. et al. Triblock polyester thermoplastic elastomers with semi-aromatic polymer end blocks by ring-opening copolymerization. Chem. Sci.11, 6567–6581 (2020). 10.1039/D0SC00463D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martello, M. T. & Hillmyer, M. A. Polylactide–poly (6-methyl-ε-caprolactone)–polylactide thermoplastic elastomers. Macromolecules44, 8537–8545 (2011). 10.1021/ma201063t [DOI] [Google Scholar]
- 61.Gallin, C. F., Lee, W.-W. & Byers, J. A. A simple, selective, and general catalyst for ring closing depolymerization of polyesters and polycarbonates for chemical recycling. Angew. Chem. Int. Ed.135, e202303762 (2023). 10.1002/ange.202303762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cywar, R. M. et al. Redesigned hybrid nylons with optical clarity and chemical recyclability. J. Am. Chem. Soc.144, 5366–5376 (2022). 10.1021/jacs.1c12611 [DOI] [PubMed] [Google Scholar]
- 63.Yu, W., Foster, J. C., Dove, A. P. & O’Reilly, R. K. Length control of biodegradable fiber-like micelles via tuning solubility: a self-seeding crystallization-driven self-assembly of poly(ε-caprolactone)-containing triblock copolymers. Macromolecules53, 1514–1521 (2020). 10.1021/acs.macromol.9b02613 [DOI] [Google Scholar]
- 64.Foster, J. C., Varlas, S., Couturaud, B., Coe, Z. & O’Reilly, R. K. Getting into shape: reflections on a new generation of cylindrical nanostructures’ self-assembly using polymer building blocks. J. Am. Chem. Soc.141, 2742–2753 (2019). 10.1021/jacs.8b08648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koo, C. M. et al. Microstructure and mechanical properties of semicrystalline-rubbery-semicrystalline triblock copolymers. Macromolecules38, 6090–6098 (2005). 10.1021/ma0501794 [DOI] [Google Scholar]
- 66.Pedersen, J. Analysis of small-angle scattering data from colloids and polymer solutions: modeling and least-squares fitting. Adv. Colloid Interface Sci. 70, 171–210 (1997).
- 67.Watts, A., Kurokawa, N. & Hillmyer, M. A. Strong, resilient, and sustainable aliphatic polyester thermoplastic elastomers. Biomacromolecules18, 1845–1854 (2017). 10.1021/acs.biomac.7b00283 [DOI] [PubMed] [Google Scholar]
- 68.Burns, A. B. & Register, R. A. Thermoplastic elastomers via combined crystallization and vitrification from homogeneous melts. Macromolecules49, 269–279 (2016). 10.1021/acs.macromol.5b02546 [DOI] [Google Scholar]
- 69.Doucet, M. et al. SasView Version 4.2.2.https://www.sasview.org/2019-05-20-release-4.2.2/ (2019).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the data necessary to support the conclusions of this paper are provided in the manuscript and its Supplementary information File. All data are available from the corresponding author upon request.