

Abstract
Rho-associated coiled-coil containing kinase (ROCK) plays an important role in inflammation. Herein, a series of compounds were designed and synthesized as ROCK inhibitors based on the structure-based drug design (SBDD) strategy and were evaluated for cytotoxicity, antioxidant activity and anti-inflammatory activity. Among them, compound DC24 was identified as the optimal hit in enzymatic screening with an IC50 value of 0.124 μM against ROCK2 and 50-fold selectivity over ROCK1. DC24 has a novel lipid amide scaffold with a bis(4-fluorophenyl)methyl substituent, and DC24 is the first ROCK2 inhibitor interacting with the hinge region of ROCK2 via the 1,2-dithiolan-3-yl motif, which has been confirmed by the binding model of DC24 with ROCK2. In a complete Freund's adjuvant (CFA) induced acute inflammation model, DC24 at a dose of 5 mg kg−1 exhibited an anti-inflammatory effect better than that of belumosudil. Furthermore, DC24 exhibits good safety in vivo.
Rho-associated coiled-coil containing kinase (ROCK) plays an important role in inflammation.
Introduction
Inflammation is a comprehensive array of physiological response to a foreign organism, including human pathogens, dust particles, and viruses. Inflammation is mainly divided into acute and chronic inflammation depending on various inflammatory processes and cellular mechanisms. Free radical production from different biological and environmental sources is due to an imbalance of natural antioxidants which further leads to various inflammatory associated diseases.1 If acute inflammation is not treated, whether due to prolonged exposure to stimuli outside or excessive autoimmune response, it can lead to chronic inflammation, during which tissue damage and fibrosis may occur.2 Rho-associated coiled-coil containing kinase (ROCK) belongs to the family of serine/threonine kinases and includes two isoforms: ROCK1 and ROCK2.3 They were first identified as downstream targets of the small GTP-binding protein, RhoA.4 All tissues express the ROCK1 transcript, although the brain and skeletal muscle express it less frequently. In contrast, the ROCK2 transcript is more prevalent in the brain, muscle, heart, lung, and placenta.5 ROCK is a key regulator of actin cytoskeletal dynamics, and also controls transcription factors that play critical roles in inflammation, including NF-κB, IRF4, and IRF7.6 In addition, ROCK participates in cell proliferation, and ROCK plays an important role in cell metabolism and survival.7–9 What's more, ROCKs play crucial roles in cytoskeletal rearrangements and gene expression.9 The inhibition of ROCK2 leads to the polarization of pathogenic macrophages while restoring beneficial macrophage balance, while inhibiting ROCK1 has the opposite effect. It should be noted that systemic inhibition of ROCK1 can lead to significant side effects, including decreased blood pressure and lymphocyte count. Therefore, developing selective ROCK2 inhibitors is crucial for effective treatment of inflammatory related diseases.10
Four ROCK inhibitors have been approved, including fasudil, ripasudil, netarsudil and belumosudil (Fig. 1), of which three are non-selective inhibitors and one is a ROCK2 selective inhibitor. Fasudil has been approved for the treatment of cerebral vasospasm and cerebral ischemic symptoms after subarachnoid hemorrhage surgery in China and Japan. Fasudil has also shown a protective effect on rheumatoid arthritis.11,12 However, several adverse effects have been observed since its approval, including hypotension, cerebral bleeding, and altered liver and kidney function, which is related to the inhibition of ROCK1.13 Ripasudil hydrochloride hydrate, developed by Kowa and the D Western Therapeutics Institute, is an ophthalmic solution for the treatment of several eye disorders. It was approved and launched in Japan for the treatment of glaucoma and ocular hypertension in 2014.14 Netarsudil is thought to lower intraocular pressure by increasing aqueous humour outflow through the trabecular meshwork, which was approved by the FDA in 2019.15 Belumosudil, a selective ROCK2 inhibitor developed by Kadmon Pharmaceuticals, was approved for the treatment of chronic graft-versus-host disease (cGVHD) and systemic sclerosis in the USA in 2021. This compound exhibits lower adverse effect on blood pressure due to selectivity to ROCK2 over ROCK1. Consequently, it is essential to discover novel ROCK2 inhibitors with sufficient effectiveness and safety.
Fig. 1. ROCK inhibitors launched on the market.

Despite this progress, the development of ROCK inhibitors for the treatment of rheumatoid arthritis is still in its early stage. In this work, fragments of diphenylmethyl and lipoic acid were introduced via hybridization and the structure-based drug design strategy, to investigate the potential of distinct chemical scaffolds as ROCK inhibitors. Subsequently, we found that the fragment of fasudil was not necessary, and hybridization on the lipoic acid fragment facilitated the discovery of lipoic amides, among which DC24 was the most potential ROCK2 inhibitor with acceptable selectivity against ROCK1.
Results and discussion
Molecular design
ROCK1 and ROCK2 harbor a high sequence homology within the kinase domain, causing great difficulty to the development of selective inhibitors.16 Over two decades, the structural diversity of ROCK inhibitors has been extensively explored, encompassing isoquinoline/isoquinolinone, indazole, pyridine, pyrimidine, pyrazole, aminofurazane, and boron derivatives.17,18 Studies have shown that the improved selectivity of compounds towards ROCK2 can be attributed to the hydrophobic groups.19 Additionally, the potential of a compound to bind to binding pockets of both subtypes may also be the reason for its lack of selectivity. The results of molecular docking indicate that fasudil can bind to the pockets of ROCK1 and ROCK2 due to its small volume, and there is partial overlap between the regions binding with fasudil and with co-crystal ligands (Fig. 2A and B).
Fig. 2. A) 3D diagram of fasudil (pink) and co-crystal ligand Y-27632 (yellow) docking in ROCK1 (PDB: 2ETR) B) 3D diagram of fasudil (pink) and co-crystal ligand (purple) docking in ROCK2 (PDB: 6ED6).

We attempted to increase the volume of fasudil derivatives by the hybridization of fragments with antioxidant potential, such as lipoic acid and cinnamic acid, which have the clinical benefit of anti-inflammation.20,21 In addition to fragments with antioxidant potential, we also introduced a diphenylmethyl fragment, a larger hydrophobic group, to provide DC21–DC24. Lipoic acid 5 is a key coenzyme of the pyruvate dehydrogenase complex in mitochondria, playing a crucial role in mitochondrial energy metabolism. Lipoic acid and its reduction product can both exert strong antioxidant effects.22 The cinnamic acid compounds are natural phenylpropanoid products extracted from tender branches of cinnamon, which have a wide range of biological activities, such as neuroprotective, antioxidant, anticoagulant, and cardiac protective effects (Fig. 3).23
Fig. 3. The discovery of DC24 with a lipoic amide scaffold.

The molecular docking results of target compounds are shown in Fig. 4. DC03, obtained by replacing homopiperazine with piperazine and hybridizing lipoic acid fragments, occupied more space in the binding pocket of ROCK2 compared to fasudil. And the occupied region and the hydrogen bonds of DC03 in the binding pocket of ROCK2 were similar to those of the co-crystal ligand. Interestingly, DC03 has two conformations when docked with ROCK2: one is where the isoquinoline moiety interacts with Met172 (conformation A, Fig. 4C); the other one is where a hydrogen bond is formed between Met172 and dithiopentane (conformation B, Fig. 4D), and the docking scores of the two conformations are equivalent.
Fig. 4. Docking results. A) 3D diagram of DC03 (conformation A (green), conformation B (orange), fasudil (pink) and co-crystal ligand (purple)) docking in ROCK2 (PDB: 6ED6). B) 2D diagram of the co-crystal ligand binding with ROCK2 (PDB: 6ED6). C) 2D diagram of DC03 (conformation A) binding with ROCK2 (PDB: 6ED6). D) 2D diagram of DC03 (conformation B) binding with ROCK2 (PDB: 6ED6).

Therefore, we speculate that dithiopentane can replace isoquinoline to interact with Met172, a key residue to inhibit ROCK2, which will lead to the discovery of a new skeleton. Employing dithiopentane to interact with Met172, compounds DC23–DC26 were designed by replacing the fasudil portion of DC03 with various motifs including diphenylmethyl and cinnamoyl groups to get more structural diversity. The fasudil fragment was replaced by the diphenylmethyl fragment with larger hydrophobic segments, providing compound DC24. The molecular docking results indicate that DC24 has a similar binding pattern to the co-crystal ligand in the ROCK2 protein, forming hydrogen bonds with Lys216 and Gly101 instead of Arg100 (Fig. 5), indicating that DC24 with a novel skeleton may have selectivity towards ROCK2.
Fig. 5. Docking results. A) 3D diagram of DC03 (conformation B, orange), DC24 (gray), and co-crystal ligand (purple) docking in ROCK2 (PDB: 6ED6). B) 2D diagram of DC24 binding with ROCK2 (PDB: 6ED6).

Chemistry
The synthetic route of fasudil 4 is depicted in Scheme 1. Isoquinoline-5-sulfonic acid 2 was obtained via sulfonation from isoquinoline, and then isoquinoline-5-sulfonyl chloride hydrochloride 3 was obtained by chlorination with thionyl chloride. Next, 3 is reacted with homopiperazine in the presence of sodium bicarbonate to give fasudil 4.
Scheme 1. Synthetic route to fasudil.

The synthesis of compounds DC01–DC03 is described in Scheme 2. 5-(1,2-Dithiolan-3-yl)pentan-1-ol 6 was obtained by reduction of lipoic acid 5 in borane tetrahydrofuran solution, then reacted with triphosgene to furnish 5-(1,2-dithiolan-3-yl)pentyl carbonochloridate 7, which then was reacted with fasudil to give DC01. Fasudil was taken as the starting material and protected by benzyloxyformyl to afford benzyl Cbz-protected fasudil 8, and then oxidized by m-chloroperoxybenzoic acid (m-CPBA) to furnish isoquinoline N-oxide compound 9. Protected hydroxy-fasudil 10 was obtained by reacting compound 6 in an aqueous solution of sodium acetate and benzoyl chloride. Hydroxy fasudil 11 was obtained by deprotection of compound 10 with trifluoroacetic acid. Compound DC02 was synthesized by coupling lipoic acid 5 and hydroxy fasudil 11 in the presence of EDCI, HOBt and Et3N in DCM. 5-(Piperazin-1-ylsulfonyl)isoquinoline 12 was obtained by the reaction of isoquinoline-5-sulfonyl chloride 3 with piperazine, then was reacted with lipoic acid 5 in the presence of EDCI, HOBt and Et3N in DCM to give amide compound DC03.
Scheme 2. Synthetic route of DC01–DC03.

The synthetic route for compounds DC04–DC18 is described in Schemes 3 and 4. According to the methods reported in the literature,24 a series of cinnamic acid derivatives were synthesized through esterification, hydrolysis, substitution, etc. By coupling fasudil and its derivatives with cinnamic acids or 2,2-dimethylbenzopyran derivatives in the presence of EDCI, HOBt and Et3N in DCM, compounds DC04–DC18 were obtained. The synthesis of 2,2-dimethylbenzopyran derivatives has been reported in our previous article.25,26
Scheme 3. Synthetic route of DC04–DC15.

Scheme 4. Synthetic route of DC16–DC18.

The synthetic route for compounds DC19–DC22 is described in Scheme 5. The alcohol intermediates 28a–b were obtained by reduction of benzophenone 27a and 4,4′-difluorobenzophenone 27b with sodium borohydride, and converted to compounds 29a–bvia reacting with thionyl chloride, which then reacted with fasudil to give compounds DC19 and DC20. Compounds DC21 and DC22 were obtained by reaction of intermediate 30 with diphenylmethylpiperazine 31 and 4,4′-difluorobenzylpiperazine 32, respectively.
Scheme 5. Synthetic routes of DC19–DC22.

The synthetic route for compounds DC23–DC26 is described in Scheme 6. The lipoic acid 8 was first converted to the corresponding acyl chloride and then reacted with diphenylmethyl piperazine 31 or 4, 4′-difluorophenylmethyl piperazine 32 to give compounds DC23 and DC24. In terms of compounds DC25 and DC26, the corresponding acid was used as the starting material, converted to the corresponding acyl chloride and then reacted with lipoic alcohol to give the target compound.
Scheme 6. Synthetic routes of DC23–DC26.

Structure–activity relationship
The inhibitory activities of DC01, the best among these compounds, against ROCKs were roughly equivalent to that of fasudil, but lacking selectivity. When the lipoic acid fragment was replaced by cinnamic acids, the inhibitory effect on ROCKs was significantly reduced (DC04–DC10). A nitrate ester moiety was introduced due to its NO releasing ability in order to improve the volume of target compounds and their activities against RA (DC11–DC15). However, the introduction of nitrate ester improved the activity of the compound against ROCK1, and the overall inhibition rate is about 50%. When the cinnamic acid fragment was replaced with the 2,2-dimethylbenzopyran fragment, the inhibitory activity further decreased, but the selectivity for ROCK2 increased (DC16–DC18). The introduction of the diphenylmethyl fragment also resulted in the decrease of activity against ROCKs, and the replacement of homopiperazine with piperazine caused the compound to lose its ability to inhibit ROCKs (DC19–DC22). The above results confirmed that the introduction of a larger substituent into the N atom of the fasudil homopiperazine group would affect its activity and selectivity. And the homopiperazine fragment of the fasudil motif is very important for the maintenance of activity. When replaced with the piperazine group, the inhibitory activity on ROCKs is greatly reduced (Table 1).
Table 1. The activities of DC series compounds on ROCKs.

| |||||
|---|---|---|---|---|---|
| Compd. | R5 | R6 | n | ROCK1a | ROCK2a |
| DC01 | –H |

|
1 | 102.60 | 95.00 |
| DC02 | –OH |

|
1 | NAb | 59.38 |
| DC03 | –H |

|
0 | 80.67 | 49.17 |
| DC04 | –H |

|
1 | NAb | NAb |
| DC05 | –H |

|
1 | 0.74 | NAb |
| DC06 | –H |

|
1 | 37.17 | 10.47 |
| DC07 | –H |

|
1 | NAb | NAb |
| DC08 | –H |

|
1 | 53.28 | NAb |
| DC09 | –H |

|
1 | 69.39 | 29.95 |
| DC10 | –OH |

|
1 | NAb | 59.84 |
| DC11 | –H |

|
1 | 1.98 | NAb |
| DC12 | –H |

|
1 | 68.77 | 5.96 |
| DC13 | –H |

|
1 | 43.37 | 16.70 |
| DC14 | –H |

|
1 | 54.60 | 7.85 |
| DC15 | –H |

|
1 | 30.82 | 6.35 |
| DC16 | –H |

|
1 | 54.03 | 88.27 |
| DC17 | –H |

|
0 | NAb | 21.21 |
| DC18 | –H |

|
1 | NAb | 37.11 |
| DC19 | –H |

|
1 | 35.69 | 23.19 |
| DC20 | –H |

|
1 | 52.79 | 53.88 |
| DC21 | –H |

|
0 | NAb | NAb |
| DC22 | –H |

|
0 | NAb | NAb |
| Fasudil | –H | –H | 1 | 87.63 | 104.69 |
Inhibition rate % of compound at concentration of 1 μM.
NA: not affected.
As mentioned in the molecular design section, the results of molecular docking encourage the attempt to replace isoquinoline with dithiopentane. We combined the thiocaproic acid fragment with the larger volume diphenylmethylpiperazine fragment to obtain compounds DC23–DC24. The activity results are shown in Table 2. When the diphenylmethylpiperazine fragment is linked to lipoic acid, the compounds exhibit selective inhibitory towards ROCK2, and the inhibitory activity is significantly enhanced when F is introduced (DC23 and DC24). We identified the structure of lipoic acid as the dominant fragment and continued to combine it with other fragments. The inhibitory activities of the compounds were reduced when other fragments were used to replace the diphenylmethylpiperazine fragment (DC25 and DC26). (Table 2).
Table 2. The activities of DC series compounds on ROCKs.
| Compd. | Structure | ROCK1a | ROCK2a |
|---|---|---|---|
| DC23 |

|
NAb | 52.49 |
| DC24 |
|
33.21 | 102.25 |
| DC25 |

|
27.51 | 15.90 |
| DC26 |

|
39.90 | NAb |
| Fasudil | — | 87.63 | 104.69 |
Inhibition rate% of compound at concentration of 1 μM.
NA: not affected.
In summary, non-selective ROCK inhibitor DC01 and the ROCK2 inhibitor DC24 were discovered and their IC50 values are shown in Table 3. The IC50 value of compound DC01 for ROCK1 is 0.754 μM, while for ROCK2 it is 1.015 μM. The IC50 value of compound DC24 for ROCK1 is 6.354 μM, while for ROCK2 it is 0.124 μM. DC24 showed higher selectivity to ROCK2 compared to fasudil.
Table 3. The IC50 values and selectivities of DC01, DC24 and fasudil on ROCKs.
| Compd. | IC50 (μM) | Selectivity | |
|---|---|---|---|
| ROCK1 | ROCK2 | ||
| DC01 | 0.754 | 1.015 | — |
| DC24 | 6.354 | 0.124 | 51.24 |
| Fasudil | 0.358 | 0.158 | 2.27 |
Cytotoxicity of target compounds
The cytotoxicities of target compounds on BV2 cells were evaluated by the MTT method. The results show that 18 compounds have low toxicity, and their median toxic concentration (TC50) is more than 100 μM; DC25 has certain cytotoxicity, with TC50 less than 10 μM. Both DC07 and DC19 have cytotoxicity, with TC50 less than 50 μM (Table 4). The results indicate that the introduction of diphenylmethyl may increase the cytotoxicity of the compound, which is reduced when diphenylmethyl is replaced with 4,4′-difluorophenylmethyl.
Table 4. Cytotoxicities of target compounds.
| Compd. | TC50 (μM) | Compd. | TC50 (μM) | Compd. | TC50 (μM) |
|---|---|---|---|---|---|
| DC01 | >100 | DC10 | >20 | DC19 | >100 |
| DC02 | >100 | DC11 | >100 | DC20 | 62.0 |
| DC03 | >100 | DC12 | >100 | DC21 | >20 |
| DC04 | >100 | DC13 | >100 | DC22 | >20 |
| DC05 | >100 | DC14 | >100 | DC23 | 62.99 |
| DC06 | >100 | DC15 | >100 | DC24 | >100 |
| DC07 | 44.23 | DC16 | 64.41 | DC25 | 9.19 |
| DC08 | >100 | DC17 | >100 | DC26 | >100 |
| DC09 | >100 | DC18 | >100 |
Antioxidant activity analysis of target compounds
The increased production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) can cause oxidative damage to cells, leading to cell death, and cytokine activation, resulting in unfavorable effects on RA. The scavenging rate of the compounds against 1,1-diphenyl-2-trinitrophenylhydrazine free radical at 1 mM concentration was determined, and the findings are reported in Table 5. Compounds DC01, DC07, DC09 and DC24 exhibited good antioxidant activity.
Table 5. The free radical scavenging activities of target compounds.
| Compd. | Free radical scavenging ratea | Compd. | Free radical scavenging ratea | Compd. | Free radical scavenging ratea |
|---|---|---|---|---|---|
| DC01 | 91.39% | DC10 | 48.66% | DC19 | 22.39% |
| DC02 | 24.35% | DC11 | 23.86% | DC20 | 22.66% |
| DC03 | 51.22% | DC12 | 19.15% | DC21 | 26.46% |
| DC04 | 28.67% | DC13 | 20.71% | DC22 | 23.40% |
| DC05 | 22.69% | DC14 | 25.33% | DC23 | 50.45% |
| DC06 | 29.21% | DC15 | 24.34% | DC24 | 91.99% |
| DC07 | 82.40% | DC16 | 33.76% | DC25 | 28.48% |
| DC08 | 93.00% | DC17 | 25.44% | DC26 | 26.10% |
| DC09 | 90.86% | DC18 | 48.66% | Fasudil | 98.96% |
Free radical scavenging rate was determined by the DPPH method at 1 mM.
Study on inhibiting the release of inflammatory factors
After inflammation, the body can release various inflammatory cytokines, including TNF-α, IL-6 and IL-1β, etc. Therefore, we used enzyme-linked immunosorbent assay (ELISA) to detect the effect of compounds at different concentrations on LPS-stimulated BV2 cells to produce TNF-α and IL-6. The results are shown in Table 6. The inhibition rates of compounds at 1 mM were determined. The results showed that compounds DC01 and DC24 showed good anti-inflammatory activity, while DC09 had good antioxidant activity, but its anti-inflammatory activity was not good.
Table 6. Inhibitory activities of target compounds against cytokines in LPS-induced BV2 cells.
| Compd. | Inhibition ratio (%) | Compd. | Inhibition ratio (%) | ||
|---|---|---|---|---|---|
| TNF-α | IL-6 | TNF-α | IL-6 | ||
| DC01 | 53.88 | 86.72 | DC02 | NA | NA |
| DC03 | 9.02 | NA | DC04 | NA | NA |
| DC05 | 45.94 | 34.97 | DC06 | 49.49 | 26.33 |
| DC07 | 17.75 | 22.60 | DC08 | 0.08 | NA |
| DC09 | 34.47 | 0.07 | DC10 | 22.62 | 25.92 |
| DC11 | 45.77 | 2.78 | DC12 | 30.94 | 6.42 |
| DC13 | 57.55 | 36.30 | DC14 | 34.94 | 48.47 |
| DC15 | 53.80 | 56.06 | DC16 | 38.48 | NA |
| DC17 | 39.22 | NA | DC18 | 31.53 | NA |
| DC19 | NA | NA | DC20 | NA | NA |
| DC21 | 25.76 | NA | DC22 | NA | NA |
| DC23 | 41.02 | 49.63 | DC24 | 70.67 | 53.81 |
| DC25 | 51.27 | 27.83 | DC26 | NA | NA |
| Fasudil | 30.14 | 82.56 | |||
Based on the above experimental results, DC01 and DC24 have low intracellular toxicity and the best free radical scavenging and anti-inflammatory activity. Therefore, we selected the two compounds for further in vivo pharmacodynamic experiments.
Pharmacokinetic study
After intraperitoneal injection (ip) and intravenous (iv) administration at a single dose of 10 mg kg−1, blood levels were analyzed for 12 h and pharmacokinetic parameters are shown in Table 7. The maximum concentration of DC24 (9953.3 ng mL−1) was reached at 0.083 h after intraperitoneal injection; the area under the curve (AUC0–12h) was 1626.8 μg h L−1; DC24 had a plasma t1/2 of 0.7 h. The maximum concentration of DC24 (19 985 ng mL−1) was reached on 0.083 h after iv administration; the area under the curve (AUC0–12h) was 3354 μg h L−1. (Table 7). However, the low plasma exposure of DC01 after gavage and intraperitoneal injection suggests that this compound may not be suitable for oral use. DC24 has better pharmacokinetic properties than DC01.
Table 7. Pharmacokinetics of DC01 and DC24 in rats (−x ± s, n = 6).
| Compd. | DC24 | DC01 | |
|---|---|---|---|
| Parameters | ip | iv | iv |
| C max (ng mL−1) | 9953 ± 1640 | 19 985 ± 10 421 | 12 162 ± 3462 |
| AUC0–t (μg h L−1) | 1627 ± 165 | 3354 ± 1987 | 1943 ± 533 |
| AUC0−∞ (μg h L−1) | 1655 ± 174 | 3364 ± 2148 | 1948 ± 537 |
| T max (h) | 0.083 ± 0 | 0.083 ± 0 | 0.2 ± 0.1 |
| Vz/F (L kg−1) | 6.1 ± 0.1 | 0.7 ± 0.3 | 1.3 ± 0.6 |
| CLz/F (L h−1 kg−1) | 6.1 ± 0.6 | 2.9 ± 1.3 | 5.6 ± 1.9 |
| F% | 48.5% | ||
Compound DC01 is an amino ester prodrug of fasudil, so we further analyzed its metabolites in rat plasma. After intravenous administration (10 mg kg−1), compared with blank plasma samples, DC01 prototype drug (m/z 510.1550, [M + H]+) and two metabolites were detected in the plasma samples, with a mass charge ratio of 526.1496 (M1) and 542.1463 (M2) (Fig. S1†), respectively. Using precise molecular weight to identify metabolites, as shown in Fig. 6, DC01 is not metabolized as we hoped.
Fig. 6. Possible metabolic pathways of compound DC01.

Acute toxicity study
The effects of compounds DC01 and DC24 on the physiological status, body weight and organs of C57BL/6 mice were evaluated by single tail vein injection. The mice were divided into compound DC01 (50 mg kg−1), DC24 (50 mg kg−1) and Blank group. After administration, the mice were monitored for adverse effects or death, daily activities, mental state and weight.
After administration of DC24 and DC01 through tail vein, the mental state of mice in each group was normal within 14 days, without any adverse effects; there was no significant difference compared with the blank group. Body weight and organ index data are shown in Fig. 7. There is no significant difference in the body weight and organ index of mice between each administration group during the experiment.
Fig. 7. Long-term toxicity and safety of DC01 and DC24. (A) Body weight. (B) Organ index.

Acute inflammation induced by complete Freund's adjuvant
Based on the potential anti-inflammatory activity of DC24, we explored its effect on acute inflammation induced by complete Freund's adjuvant. Mice were randomly assigned in order to determine whether compound DC24 is effective in C57BL/6 mice with RA. The degree of swelling in mice was evaluated and it was found that DC24 had a very significant effect on eliminating foot swelling, and its anti-inflammatory effect was faster than that of belumosudil (Fig. 8).
Fig. 8. Evaluation of compound DC24 for the treatment of CFA-induced acute arthritis in a mouse model **p ≤ 0.01, ***p ≤ 0.001 and ****p ≤ 0.001 compounds versus CFA.

Conclusion
In summary, a series of novel ROCK inhibitors were designed and synthesized based on combination principles and the structure-based drug design (SBDD) strategy. In order to increase the molecular volume of fasudil to increase its selectivity for two subtypes, long chain derivatives of lipoic acid and larger rigid hydrophobic groups are introduced into the structure of fasudil. Next, isoquinoline was replaced by dithiopentane to interact with Met172 leading to the discovery of the novel lipoic amide scaffold. After a systematical cellular and enzymatic-based SAR study, DC24 and DC01 were discovered as the optimal compounds which display potent inhibitory activity with IC50 of 0.124 μM and 1.015 μM against ROCK2, respectively. Notably, the introduction of fluorine atoms on diphenylmethyl increases the hydrogen bonding ability of the compound with the hydrophobic pocket of ROCK2, greatly enhancing the inhibitory activity of the compound against ROCK2. Meanwhile, the enzymatic assay showed that DC24 exhibits better selectivity than the lead compound fasudil (51.24 vs. 2.27). Furthermore, DC24 has low toxicity in both in vitro and in vivo experiments and exhibits strong ability to scavenge free radicals and inhibit inflammatory factors in cells. And DC24 has a significant inhibitory effect on mice feet swelling in vivo acute inflammation experiments. In a word, novel lipoic amide scaffold compound DC24 rather than fasudil turned out to be a promising ROCK2 inhibitor with broad perspective in inflammation relevant diseases.
Experimental section
Chemistry
Unless otherwise noted, all materials were obtained from commercially available sources and were used without further purification. Reaction progress was monitored by thin layer chromatography (TLC). Electrospray ionization mass spectrometry (ESI-MS) analyses were recorded on an Agilent 1100 Series MSD Trap SL (Santa Clara, CA, USA). 1H NMR and 13C NMR spectra were obtained using Bruker spectrometers (Bruker Bioscience, respectively, Billerica, MA, USA) with TMS as an internal standard. Melting points were determined with an X-4 apparatus and were uncorrected. Column chromatography was run on silica gel (200–300 mesh) from Qingdao Ocean Chemicals (Qingdao, Shandong, China).
Isoquinoline-5-sulfonic acid sulfate (2)
Isoquinoline 1 (50 g, 0.39 mol) was slowly added dropwise to 50% fuming sulfuric acid (124 g, containing 0.78 mol of SO3), with the temperature below 20 °C. After complete addition, the mixture was stirred at 60 °C for 40 h. Then it was cooled to room temperature and slowly poured into ice water (600 mL). A large amount of white solid was precipitated. It was filtered, washed with DCM, and dried to obtain 69.7 g of white solid with a yield of 59%. m.p. > 300 °C. 1H NMR (600 MHz, DMSO-d6): δ (ppm) 9.92 (s, 1H), 9.17 (d, J = 6.7 Hz, 1H), 8.74 (d, J = 6.7 Hz, 1H), 8.51 (d, J = 7.6 Hz, 2H), 7.99 (t, J = 7.7 Hz, 1H).
Isoquinoline-5-sulfonyl chloride hydrochloride (3)
A mixture of isoquinoline-5-sulfonic acid sulfate (2) (30.0 g, 0.1 mol), thionyl chloride (120 mL, 1.65 mol), and dry DMF (1 mL) was heated to reflux for 20 h. The mixture was then concentrated in vacuo and stirred with DCM (80 mL). The resulting mixture was filtered, washed with DCM, and dried, yielding 24.3 g of white powdered solid with a yield of 92%.
Isoquinoline-5-sulfonyl chloride
Isoquinoline-5-sulfonyl chloride hydrochloride (5 g) was poured into water (60 mL). The pH was adjusted to 6 with sodium bicarbonate. The aqueous phase was extracted with DCM (60 mL × 3), washed with water (20 mL × 2) and brine (20 mL), dehydrated over anhydrous Na2SO4, and then filtered. The filtrate was concentrated in vacuo, resulting in the formation of 3.6 g of pale yellow solid with a yield of 83.7%.
5-((1,4-Diazepan-1-yl)sulfonyl)isoquinoline (4, fasudil)
A solution of isoquinoline-5-sulfonyl chloride 3 (2.0 g, 8.78 mmol) in DCM (10 mL) was added dropwise to a solution of 1,4-diazepane (1.32 g, 13.2 mmol) and Et3N in dry DCM (10 mL) while maintaining the temperature below 5 °C. Then the mixture was stirred at room temperature for 30 min and poured into water (20 mL), the water phase was extracted with DCM (20 mL × 3), and the combined organic phase was washed with saturated sodium carbonate solution (20 mL × 2), water (20 mL × 2) and brine (20 mL), dried over anhydrous MgSO4, filtered, and concentrated in vacuo to give a residue. The residue was purified by silica gel column chromatography (DCM/MeOH = 20 : 1) to afford the pure final product (1.68 g) as a yellow solid, yield 66%. 1H NMR (600 MHz, DMSO-d6): δ (ppm) 9.35 (s, 1H), 8.69 (d, J = 6.1 Hz, 1H), 8.45 (d, J = 6.1 Hz, 1H), 8.35 (d, J = 7.3 Hz, 1H), 8.19 (d, J = 8.1 Hz, 1H),7.69 (t, J = 7.9 Hz, 1H), 3.50 (t, J = 5.3 Hz, 2H), 3.45 (t, J = 5.3 Hz, 2H), 2.98 (t, J = 5.3 Hz, 2H), 2.94 (t, J = 5.8 Hz, 2H), 2.05 (s, 1H), 1.86–1.82 (m, 2H). ESI MS: m/z 292.2 [M + H]+.
5-(1,2-Dithiolan-3-yl)pentan-1-ol (6)
To a solution of 5-(1,2-dithiolan-3-yl)pentanoic acid 5 (1.0 g, 4.85 mmol) in 20 mL tetrahydrofuran was added dropwise a solution of 1 M BH3–THF (10 mL, 9.69 mmol) at 0 °C and argon atmosphere. And the reaction mixture was stirred for 9 h and quenched by adding MeOH (10 mL), and the mixture was concentrated in vacuo. The residue was poured into water (20 mL), and the organic phase was extracted with EtOAc (20 mL × 2), water (20 mL × 3), and brine (10 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo to obtain 0.9 g oil. It was purified by silica gel column chromatography (EA/PE = 1/5) to give a yellow oil (0.60 g, 65%). ESI MS: m/z 175.1 [M − H2O + H]+, 215.1 [M + Na]+.
5-((4-((Benzyloxy)carbonyl)-1,4-diazepan-1-yl)sulfonyl) isoquinoline 2-oxide (9)
To a solution of benzyl 4-(isoquinolin-5-ylsulfonyl)-1,4-diazepane-1-carboxylate 8 (10.0 g, 23.5 mmol) in DCM (50 mL) was added dropwise a solution of m-chloroperoxybenzoic acid (6.1 g, 35.3 mmol) in DCM (40 mL) in an ice bath, then the reaction mixture was stirred for 6 h at room temperature. The saturated solution of sodium thiosulfate was added to the reaction solution until the starch potassium iodide test paper did not turn blue, the organic layer was separated, the water layer was extracted with DCM (30 mL × 2), the organic layers were combined, washed with 5% sodium hydroxide aqueous solution (30 mL × 2), washed with water (30 mL), and washed with brine (30 mL), and the organic phase was concentrated under vacuum to give 9.12 g oil, which was proceeded to the next step directly.
Benzyl 4-(isoquinolin-5-ylsulfonyl)-1,4-diazepane-1-carboxylate (8)
To a solution of fasudil 4 (20.0 g, 68.6 mmol), dried Et3N (21.0 g, 82.4 mmol), DMAP (0.80 g, 6.8 mmol) in DCM (200 mL) was added dropwise benzyl carbonochloridate (14.1 g, 82.4 mmol) in DCM (50 mL), and stirred at 0 °C for 2 h. The reaction solution was poured into water (100 mL) and the organic layer was separated. The aqueous layer was extracted with DCM (50 mL). The organic layers were combined and washed with water (100 mL × 2) and brine (20 mL) and dried with anhydrous MgSO4 and filtered. The filtrate was concentrated in vacuo to give 36.8 g of reddish brown oil. It was purified by silica gel column chromatography (EA/PE = 1/20 to 1 : 1) to give a colorless oil (25.4 g, 87%). 1H NMR (600 MHz, DMSO-d6): δ (ppm) 9.49 (s, 1H), 8.70 (t, J = 6.6 Hz, 1H), 8.46 (dd, J = 8.0, 4.0 Hz, 1H), 8.33 (t, J = 6.6 Hz, 2H), 7.83 (td, J = 15.4, 7.9, 3.2 Hz, 1H), 7.37–7.30 (m, 5H), 5.02 (s, 2H), 3.56–3.48 (m, 4H), 3.46–3.41 (m, 4H), 1.75–1.72 (m, 2H). ESI-MS: m/z 426.2 [M + H]+.
Benzyl 4-((1-hydroxyisoquinolin-5-yl)sulfonyl)-1,4-diazepane-1-carboxylate (10)
5-((4-((Benzyloxy)carbonyl)-1,4-diazepan-1-yl)sulfonyl)isoquinoline 2-oxide 9 (25.0 g, 56.6 mmol) was dissolved in DCM (80 mL). Then sodium acetate (13.9 g, 169.8 mmol) and TBAB (5.5 g, 17.0 mmol) aqueous solution (60 mL) were added. Benzoyl chloride (18.2 g, 113.3 mmol) in DCM (50 mL) was added dropwise in a water bath. After 12 h, the reaction was complete, and the organic layer was separated. The water layer was extracted with DCM (50 mL × 2), combined with organic layers, washed with water (50 mL × 2) and brine (50 mL), dried with anhydrous sodium and filtered. The filtrate was concentrated under vacuum to give a white solid (13.0 g, 52%). m.p. 189–191 °C. ESI-MS: m/z 440.1 [M − H]−.
5-(1,2-Dithiolan-3-yl) pentyl 4-(isoquinolin-5-ylsulfonyl)-1,4-diazepane-1-carboxylate (DC01)
Triphosgene (1.54 g, 5.20 mmol) was dissolved in dry DCM (10 mL) and cooled to below 0 °C in an ice bath. A solution of dry Et3N (1.1 g, 5.20 mmol) in 5 mL DCM was added dropwise, followed by dropwise addition of a solution of 5-(1,2-dithiolan-3-yl)pentan-1-ol (1.0 g, 5.20 mmol) in 10 mL DCM. The reaction mixture was stirred overnight at room temperature. The mixture was concentrated under vacuum and used directly for the next step.
To a solution of fasudil 4 (1.0 g, 3.47 mmol) and Et3N (1.1 g, 10.4 mmol) in 10 mL dry DCM was added dropwise a solution of 5-(1,2-dithiolan-3-yl)pentyl carbonochloridate 7 (1.33 g, 5.20 mmol) in DCM at −14 °C. The reaction mixture was stirred for 4 h. Then it was poured into water (20 mL), and the organic phase was extracted with DCM (20 mL × 4), water (20 mL × 2), and brine (20 mL × 2), dried over anhydrous Na2SO4, filtered and concentrated in vacuo to obtain 1.4 g brown oil. It was purified by silica gel column chromatography (EA/PE = 1/5 to 1 : 1) to give a yellow oil (0.46 g, 26%). 1H NMR (600 MHz, DMSO-d6): δ (ppm) 9.49 (s, 1H), 8.69 (d, J = 6.1 Hz, 1H), 8.46 (d, J = 7.8 Hz, 1 H), 8.33–8.31 (m, 2H), 7.85–7.82 (m, 1H), 3.91 (q, J = 11.9 Hz, 2H), 3.63–3.58 (m, 1H), 3.49–3.48 (m, 4H), 3.42–3.39 (m, 4H), 3.20–3.15 (m, 1H), 3.13–3.08 (m, 1H), 2.43–2.37 (m, 1H), 1.89–1.84 (m, 1H), 1.72–1.70 (m, 2H), 1.69–1.63 (m, 1H), 1.56–1.53 (m, 1H), 1.52–1.51 (m, 2H), 1.38–1.35 (m, 2H), 1.31–1.30 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 155.51, 155.44, 151.22, 135.48, 135.31, 134.90, 132.81, 129.15, 128.86, 120.10, 65.14, 63.09, 58.27, 56.65, 48.45, 47.86, 47.59, 47.47, 47.34, 45.90, 45.55, 38.55, 34.72, 29.35, 29.11, 28.80, 25.68. ESI MS: m/z 510.3 [M + H]+, 532.3 [M + Na]+.
Dry EtOAc (10 mL) was added to the yellow oily substance to dissolve, and 1.2 M hydrogen chloride solution of EtOAc was slowly added dropwise in an ice bath until no solids were precipitated. The solid was collected by filtration and washed with ether to obtain the yellow solid, which is the hydrochloride salt (DC01). The purity of compound DC01 was determined using a Shimadzu 2010A HPLC. The purity was 99.5% (area normalization method). The column was an Agilent ZORBA× 80A Extend-C18 (4.6× 150 mm, 5 μm), the mobile phase was MeCN/H2O = 55%/45%, the flow rate was 0.8 mL min−1, the column temperature was 25 °C, the detection wavelength was 210 nm, and the retention time was 9.830 min.
5-((1,4-Diazepan-1-yl)sulfonyl)isoquinolin-1-ol (11)
Benzyl 4-((1-hydroxyisoquinolin-5-yl)sulfonyl)-1,4-diazepane-1-carboxylate 10 (1.67 g, 3.78 mmol) was dissolved in trifluoroacetic acid (10 mL) and raised to 65 °C for 12 h. The mixture was concentrated in vacuo to remove most of the trifluoroacetic acid. Then it was poured into water and the pH was adjusted to 8 with sodium bicarbonate. The white solid was precipitated, filtered and dried to obtain 1.12 g white solid with a yield of 97%. 1H NMR (600 MHz, DMSO-d6): δ (ppm) 11.71 (brs, 1H), 8.46 (d, J = 7.0 Hz, 1H), 8.18–8.13 (m, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.38 (d, J = 7.0 Hz, 1H), 7.10 (d, J = 7.4 Hz, 1H), 3.40–3.28 (m, 4H), 2.77–2.73 (m, 2H), 1.66 (s, 2H), 1.28–1.23 (m, 2H), 0.89–0.86 (m, 1H).
5-(1,2-Dithiolan-3-yl)-1-(4-((1-hydroxyisoquinolin-5-yl)sulfonyl)-1,4-diazepan-1-yl)pentan-1-one (DC02)
A mixture of 5-(1,2-dithiolan-3-yl)pentanoic acid 5 (0.74 g, 3.58 mmol), EDCI (0.47 g, 2.45 mmol), HOBt (0.33 g, 2.45 mmol), and Et3N (0.82 g, 8.15 mmol) in DCM (25 mL) was stirred for 30 min, then 5-((1,4-diazepan-1-yl)sulfonyl)isoquinolin-1-ol 11 (0.50 g, 1.63 mmol) was added and stirred for 12 h at 25 °C. The reaction mixture was poured into water (20 mL) and brine (20 mL), dried with anhydrous MgSO4, and filtered. The filtrate was concentrated in vacuo to obtain 0.50 g of light yellow solid, and then stirred with ether to give a white solid (0.30 g, 37%). m.p. 186–188 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 11.64 (s, 1H), 8.48 (d, J = 8.0 Hz, 1H), 8.17 (d, J = 7.6 Hz, 1H), 7.60 (td, J = 11.2, 7.8, 3.3 Hz, 1H), 7.39–7.34 (m, 1H), 7.05 (dd, J = 7.4, 3.4 Hz, 1H), 3.64–3.59 (m, 1H), 3.56–3.54 (m, 1H), 3.47–3.46 (m, 1H), 3.41–3.37 (m, 2H), 3.36–3.35 (m, 1H), 3.22–3.14 (m, 1H), 3.12–3.08 (m, 1H), 2.45–2.38 (m, 1H), 2.27–2.21 (m, 2H), 1.91–1.83 (m, 1H), 1.76–1.68 (m, 2H), 1.66–1.63 (m, 1H), 1.57–1.51 (m, 1H), 1.50–1.44 (m, 2H), 1.39–1.33 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.94, 171.89, 161.46, 135.11, 134.48, 134.29, 133.66, 133.40, 132.52, 132.01, 131.93, 128.41, 126.06, 101.28, 101.23, 56.67, 49.16, 48.83, 47.95, 47.79, 46.72, 46.56, 44.48, 38.57, 34.71, 32.37, 32.19, 29.72, 28.89, 28.34, 24.97, 24.87. HRMS (ESI) calcd for C22H29N3O4S3Na [M + Na]+: 518.1212, found: 518.1210.
5-(Piperazin-1-ylsulfonyl)isoquinoline (12)
A solution of isoquinoline-5-sulfonyl chloride 3 (2.0 g, 8.78 mmol) in DCM (10 mL) was added dropwise into a solution of piperazine (2.0 g, 8.78 mmol) and Et3N (4.4 g, 43.9 mmol) in dry DCM (15 mL) at 0 °C. The reaction mixture was stirred for 4 h, then poured into ice water (20 mL). The organic layer was separated and the water layer was extracted with DCM (20 mL × 3). The combined organic extracts were washed with water (20 mL × 2) and brine (20 mL). The organic layer was dried over anhydrous MgSO4 and then concentrated in vacuo. After concentration, 2.1 g white solid was obtained, which was directly used for the next step without purification.
5-(1,2-Dithiolan-3-yl)-1-(4-(isoquinolin-5-ylsulfonyl)piperazin-1-yl)pentan-1-one (DC03)
A mixture of 5-(1,2-dithiolan-3-yl)pentanoic acid 5 (0.41 g, 1.98 mmol), EDCI (0.53 g, 2.70 mmol), HOBt (0.36 g, 2.70 mmol), Et3N (0.90 g, 90.1 mmol), and dry DCM (10 mL) was stirred at room temperature for 1 h. Then intermediate 12 (0.50 g, 1.80 mmol) was added, and the stirring was continued at room temperature for 24 h. The reaction mixture was then poured into water (20 mL) and extracted with DCM (20 mL × 3). The combined organic layers were washed with water (20 mL × 2) and brine (20 mL), dried with anhydrous MgSO4, and filtered. The filtrate was concentrated in vacuo to obtain a yellow oil (0.7 g). It was purified by silica gel column chromatography to give a yellow solid (0.35 g, 38%), m.p. 154–155 °C. 1H NMR (600 MHz, DMSO-d6): δ (ppm) 9.51 (s, 1H), 8.70 (d, J = 6.2 Hz, 1H), 8.52 (d, J = 8.2 Hz, 1H), 8.45 (d, J = 6.1 Hz, 1H), 8.37 (dd, J = 7.4, 1.1 Hz, 1H), 7.89 (t, J = 7.9 Hz, 1H), 3.56–3.52 (m, 1H), 3.49–3.48 (m, 4H), 3.17–3.13 (m, 1H), 3.10–3.08 (m, 2H), 3.08–3.07 (m, 1H), 3.06–3.04 (m, 2H), 2.39–2.33 (m, 1H), 2.22 (t, J = 7.4 Hz, 2H), 1.84–1.78 (m, 1H), 1.63–1.57 (m, 1H), 1.51–1.46 (m, 1H), 1.45–1.38 (m, 2H), 1.31–1.25 (m, 2H). ESI MS: m/z 466.2 [M + H]+, 488.2 [M + Na]+.
(E)-3-(4-Acetoxy-3-methoxyphenyl)acrylic acid (14)
Sodium hydroxide (2.7 g, 66.9 mmol) and (E)-3-(4-hydroxy-3-methoxyphenyl) acrylic acid 13 (5.0 g, 25.7 mmol) were dissolved in 25 mL water, and acetic anhydride was added dropwise at 0 °C, then the reaction mixture was stirred for 12 h at room temperature. It was poured into water (20 mL), adjusted the pH to 3 with 6 N hydrochloric acid and filtered. The filter cake was recrystallized 15 times (m/v) with absolute ethanol to obtain 4.62 g of white needle-like solid with a yield of 76%. m.p. 197–200 °C.
Methyl (E)-3-(4-hydroxy-3-methoxyphenyl)acrylate (15)
(E)-3-(4-Hydroxy-3-methoxyphenyl)acrylic acid 13 (20.0 g, 0.10 mol), MeOH (250 mL) and concentrated sulfuric acid (10 mL) were added to a 500 mL single-neck flask. The mixture was heated to reflux and stirred for 4 h. Then it was cooled to room temperature, poured into a 500 mL beaker, pH was adjusted to 7 with sodium bicarbonate in an ice bath, and a large amount of dark green solid was precipitated. The solid was collected, washed with water, and dried to obtain a dark green solid (20.5 g, 96%). m.p. 62–64 °C.
General procedure A for the synthesis of compounds 16a–16e
A mixture of methyl (E)-3-(4-hydroxy-3-methoxyphenyl)acrylate 15 (19.2 mmol), potassium carbonate powder (19.2 mmol) and dibromoalkane (57.6 mmol) in DMF was stirred at 60 °C for 12 h. Then it was poured into water, extracted with DCM, washed with water and brine, dried with anhydrous MgSO4 and filtered. The filtrate was evaporated under vacuum and the crude was purified by silica gel column chromatography. According to this procedure, the following products have been obtained.
Methyl (E)-3-(4-(2-bromoethoxy)-3-methoxyphenyl)acrylate (16a)
According to general procedure A, compound 16a as a white solid (2.4 g, 40%) was obtained by taking ferulic acid methyl ester and 1,2-dibromoethane as starting materials.
Methyl (E)-3-(4-(3-bromopropoxy)-3-methoxyphenyl)acrylate (16b)
According to general procedure A, compound 16b as a white solid (2.8 g, 45%) was obtained by taking ferulic acid methyl ester and 1,3-dibromopropane as starting materials.
Methyl (E)-3-(4-(4-bromobutoxy)-3-methoxyphenyl)acrylate (16c)
According to general procedure A, compound 16c as a white solid (3.5 g, 53%) was obtained by taking ferulic acid methyl ester and 1,4-dibromobutane as starting materials.
Methyl (E)-3-(4-((5-bromopentyl)oxy)-3-methoxyphenyl)acrylate (16d)
According to general procedure A, compound 16d as a white solid (4.1 g, 59%) was obtained by taking ferulic acid methyl ester and 1,5-dibromopentane as starting materials.
Methyl (E)-3-(4-((6-bromohexyl)oxy)-3-methoxyphenyl)acrylate (16e)
According to general procedure A, compound 16e as a white solid (4.4 g, 62%) was obtained by taking ferulic acid methyl ester and 1,6-dibromohexane as starting materials. ESI MS m/z: 371.1 [M + H]+.
General procedure B for the synthesis of compounds 17a–17e
A mixture of methyl acrylate (2.70 mmol) and silver nitrate (4.05 mmol) in acetonitrile (50 mL) was stirred for 12 h at 60 °C away from light. The reaction mixture was cooled to room temperature and then filtered. The filtrate was concentrated under vacuum and then recrystallized with acetonitrile.
Methyl (E)-3-(3-methoxy-4-(2-(nitrooxy)ethoxy)phenyl) acrylate (17a)
According to general procedure B, compound 17a as a white solid (1.2 g, 55%) was obtained by taking compound 16a and silver nitrate as starting materials.
Methyl (E)-3-(3-methoxy-4-(3-(nitrooxy)propoxy)phenyl)acrylate (17b)
According to general procedure B, compound 17b as a white solid (1.6 g, 60%) was obtained by taking compound 16b and silver nitrate as starting materials.
Methyl (E)-3-(3-methoxy-4-(4-(nitrooxy)butoxy)phenyl) acrylate (17c)
According to general procedure B, compound 17c as a white solid (2.7 g, 80%) was obtained by taking compound 16c and silver nitrate as starting materials.
Methyl (E)-3-(3-methoxy-4-((5-(nitrooxy)pentyl)oxy)phenyl) acrylate (17d)
According to general procedure B, compound 17d as a white solid (3.2 g, 83%) was obtained by taking compound 16d and silver nitrate as starting materials.
Methyl (E)-3-(3-methoxy-4-((6-(nitrooxy)hexyl)oxy)phenyl) acrylate (17e)
According to general procedure B, compound 17e as a white solid (0.72 g, 76%) was obtained by taking compound 16e and silver nitrate as starting materials.
General procedure C for the synthesis of compounds 18a–18e
A mixture of methyl acrylate 18a–e (1.95 mmol), lithium hydroxide monohydrate (5.84 mmol), THF, MeOH and water was stirred for 24 h at room temperature. The mixture was filtered and most of the filtrate was evaporated under vacuum. Then the resulting residue was poured into water, pH was adjusted to 4 with 1 N hydrochloric acid in an ice bath and filtered. The filter cake was washed with water and dried.
(E)-3-(3-Methoxy-4-(2-(nitrooxy)ethoxy)phenyl)acrylic acid (18a)
According to general procedure C, compound 18a as a white solid (1.0 g, 88%) was obtained by taking compound 17a as a starting material.
(E)-3-(3-Methoxy-4-(3-(nitrooxy)propoxy)phenyl)acrylic acid (18b)
According to general procedure C, compound 18b as a white solid (1.4 g, 90%) was obtained by taking compound 17b as a starting material.
(E)-3-(3-Methoxy-4-(4-(nitrooxy)butoxy)phenyl)acrylic acid (18c)
According to general procedure C, compound 18c as a white solid (2.3 g, 90%) was obtained by taking compound 17c as a starting material.
(E)-3-(3-Methoxy-4-((5-(nitrooxy)pentyl)oxy)phenyl)acrylic acid (18d)
According to general procedure C, compound 18d as a white solid (2.8 g, 92%) was obtained by taking compound 17d as a starting material.
(E)-3-(3-Methoxy-4-((6-(nitrooxy)hexyl)oxy)phenyl)acrylic acid (18e)
According to general procedure C, compound 18e as a white solid (0.59 g, 86%) was obtained by taking compound 17e as a starting material.
(E)-3-(4-Acetoxy-3,5-dimethoxyphenyl)acrylic acid (22)
According to the synthetic process of compound 14, (E)-3-(4-acetoxy-3,5-dimethoxyphenyl)acrylic acid as a white solid (4.20 g, 70%) was obtained by taking (E)-3-(4-hydroxy-3,5-dimethoxyphenyl)acrylic acid 19 (5.0 g, 22.3 mmol) as a starting material.
(E)-4-(3-(4-(Isoquinolin-5-ylsulfonyl)-1,4-diazepan-1-yl)-3-oxoprop-1-en-1-yl)-2-methoxyphenyl acetate (DC04)
According to the synthetic process of compound DC03, DC04 as a white solid (2.40 g, 55%) was obtained by taking (E)-3-(4-acetoxy-3-methoxyphenyl)acrylic acid 14 (2.25 g, 9.52 mmol) as a starting material. m.p. 94–96 °C. 1H NMR (400 Hz, DMSO-d6): δ (ppm) 9.46 (s, 0.6H), 8.68 (t, J = 6.0 Hz, 1H), 8.45 (d, J = 8.2 Hz, 0.6H), 8.40 (d, J = 8.2 Hz, 0.4H), 8.34–8.29 (m, 2H), 7.85–7.79 (m, 1H), 7.48–7.41 (m, 2H), 7.31 (s, 0.4H), 7.29 (s, 0.6H), 7.13–7.05 (m, 2H), 3.87 (t, J = 5.4 Hz, 1H), 3.84 (s, 3H), 3.75 (t, J = 5.9 Hz, 1H), 3.69 (t, J = 5.0 Hz, 1H), 3.59 (t, J = 5.6 Hz, 1H), 3.57–3.52 (m, 2H), 3.45–3.42 (m, 2H), 2.26 (s, 3H), 1.81–1.77 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.93, 165.66, 165.61, 154.01, 153.97, 151.53, 145.44, 145.38, 141.79, 141.60, 140.81, 134.57, 134.52, 134.27, 134.23, 134.11, 132.93, 132.88, 132.83, 131.06, 129.31, 127.04, 126.99, 123.54, 121.35, 121.30, 118.71, 118.60, 117.43, 117.39, 112.70, 112.67, 56.51, 47.21, 30.36, 28.40, 20.88. HRMS (ESI) calcd for C26H27N3O6SNa [M + Na]+: 532.1513, found: 532.1523.
(E)-4-(3-(4-(Isoquinolin-5-ylsulfonyl)-1,4-diazepan-1-yl)-3-oxoprop-1-en-1-yl)-2,6-dimethoxyphenyl acetate (DC05)
According to the synthetic process of compound DC03, DC05 as a white solid (2.10 g, 46%) was obtained by taking acetylsinapic acid (2.28 g, 9.52 mmol) as a starting material. m.p. 128–129 °C. 1H NMR (400 Hz, CDCl3): δ (ppm) 9.36 (s, 0.7H), 9.33 (s, 0.3H), 8.70 (d, J = 5.6 Hz, 1H), 8.43–8.38 (m, 1H), 8.36–8.29 (m, 1H), 8.24–8.18 (m, 1H), 7.73–7.68 (m, 1H), 7.62 (d, J = 15.2 Hz, 0.7H), 7.56 (d, J = 15.4 Hz, 0.3H), 6.72 (s, 1H), 6.69–6.59 (m, 2H), 3.85 (s, 6H), 3.83–3.77 (m, 3H), 3.58–3.43 (m, 4H), 2.34 (s, 3H), 2.10–2.03 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 168.52, 165.69, 165.62, 153.98, 152.38, 145.44, 145.39, 142.31, 142.14, 134.24, 134.21, 133.92, 133.88, 132.96, 132.86, 131.06, 129.54, 129.31, 127.05, 126.99, 118.75, 118.69, 117.43, 11 739, 105.57, 56.70, 30.41, 28.39, 30.61. HRMS (ESI) calcd for C27H30N3O7S [M + H]+: 540.1799, found: 540.1805.
(E)-3-(4-Acetoxyphenyl)acrylic acid (23)
According to the synthetic process of compound 14, (E)-3-(4-acetoxyphenyl)acrylic acid as a white solid (5.0 g, 80%) was obtained by taking (E)-3-(4-hydroxyphenyl)acrylic acid 20 (5.0 g, 30.5 mmol) as a starting material.
(E)-4-(3-(4-(Isoquinolin-5-ylsulfonyl)-1,4-diazepan-1-yl)-3-oxoprop-1-en-1-yl)phenyl acetate (DC06)
According to the synthetic process of compound DC03, DC06 as a white solid (2.4 g, 58%) was obtained by taking compound 23 (1.77 g, 9.52 mmol) as a starting material. m.p. 119–120 °C. 1H NMR (400 MHz, CDCl3): δ (ppm) 9.34 (t, J = 8.9 Hz, 1H), 8.69 (d, J = 6.1 Hz, 1H), 8.42–8.37 (m, 1H), 8.35–8.30 (m, 1H), 8.23–8.16 (m, 1H), 7.72–7.68 (m, 1H), 7.64–7.60 (m, 1H), 7.56–7.47 (m, 2H), 7.19–7.16 (m, 0.3H), 7.12–7.10 (m, 1.7H), 6.73 (d, J = 15.2 Hz, 0.6H), 6.65 (d, J = 15.4 Hz, 0.3H), 3.87–3.76 (m, 4H), 2.35 (s, 0.4H), 2.31 (s, 2.6H), 2.10–2.04 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 169.56, 166.03, 159.52, 153.97, 145.43, 145.37, 142.56, 142.37, 134.21, 132.87, 131.06, 130.37, 129.31, 127.04, 122.65, 117.42, 116.04, 114.67, 114.51, 56.51, 49.60, 48.30, 47.50, 47.08, 30.27, 21.34. HRMS (ESI) calcd for C25H26N3O5S [M + H]+: 480.1588, found: 480.1592.
(E)-3-(3,4-Diacetoxyphenyl)acrylic acid (24)
According to the synthetic process of compound 14, (E)-3-(3,4-diacetoxyphenyl)acrylic acid as a white solid (4.7 g, 65%) was obtained by taking (E)-3-(3,4-dihydroxyphenyl)acrylic acid 21 (5.0 g, 27.8 mmol) as a starting material.
(E)-4-(3-(4-(Isoquinolin-5-ylsulfonyl)-1,4-diazepan-1-yl)-3-oxoprop-1-en-1-yl)-1,2-phenylene diacetate (DC07)
According to the synthetic process of compound DC05, DC07 as a white solid (2.1 g, 45%) was obtained by taking compound 24 (2.27 g, 9.52 mmol) as a starting material. 1H NMR (600 Hz, CDCl3): δ (ppm) 9.36 (s, 0.7H), 9.33 (s, 0.3H), 8.70 (d, J = 6.1 Hz, 1H), 8.45 (d, J = 6.1 Hz, 0.7H), 8.42 (d, J = 6.0 Hz, 0.3H), 8.35 (d, J = 7.7 Hz, 0.3H), 8.33 (d, J = 7.4 Hz, 0.7H), 8.22 (d, J = 8.2 Hz, 0.7H), 8.19 (d, J = 8.2 Hz, 0.3H), 7.62 (d, J = 15.3 Hz, 0.7H), 7.53 (d, J = 15.2 Hz, 0.3H), 7.37 (dd, J = 8.4, 1.9 Hz, 0.7H), 7.33–7.32 (m, 1H), 7.28 (d, J = 1.6 Hz, 0.3H), 7.22–7.19 (m, 1H), 6.69 (d, J = 15.3 Hz, 0.7H), 6.59 (d, J = 15.3 Hz, 0.3H), 3.85 (t, J = 5.6 Hz, 1H), 3.81–3.76 (m, 3H), 3.53–3.49 (m, 2H), 3.46–3.43 (m, 2H), 2.31 (s, 3H), 2.30 (s, 3H), 2.07–2.02 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.50, 166.03, 165.99, 153.85, 147.96, 147.91, 145.90, 145.14, 142.97, 142.80, 134.30, 134.26, 132.99, 132.89, 131.13, 129.30, 127.13, 127.06, 121.30, 117.53, 116.07, 115.31, 114.56, 114.36, 48.32, 46.63, 45.26, 30.23, 28.54, 21.52. HRMS (ESI) calcd for C27H27N3O7SNa [M + Na]+: 560.1452, found: 560.1476.
(E)-3-(4-Hydroxy-3-methoxyphenyl)-1-(4-(isoquinolin-5-ylsulfonyl)-1,4-diazepan-1-yl)prop-2-en-1-one (DC08)
Compound DC07 (1.3 g, 2.55 mmol), methanol (20 ml), water (100 ml), and sodium hydroxide (1 g, 25.5 mmol) were added to a 250 ml single mouth bottle and stirred at room temperature for 8 h. After the reaction solution was cooled to room temperature, the pH was slowly adjusted to 6 with 1 N hydrochloric acid in an ice bath, and a white solid was precipitated. 1.4 g of white solid was obtained by suction filtration. Acetone was added to recrystallize the obtained white solid (0.5 g, 42%). m.p. 187–188 °C. 1H NMR (400 Hz, CDCl3): δ (ppm) 9.36 (s, 0.7H), 9.33 (s, 0.3H), 8.69 (d, J = 5.8 Hz, 1H), 8.46–8.41 (m, 1H), 8.37–8.30 (m, 1H), 8.25–8.18 (m, 1H), 7.74–7.70 (m, 1H), 7.63–7.56 (m, 1H), 7.09 (d, J = 8.1 Hz, 0.7H), 7.05 (d, J = 8.0 Hz, 0.3H), 6.96–6.90 (m, 2H), 6.60 (d, J = 15.4 Hz, 0.7H), 6.53 (d, J = 15.4 Hz, 0.3H), 5.92 (brs, 1H), 3.93 (s, 3H), 3.86–3.76 (m, 4H), 3.59–3.55 (m, 1H), 3.51–3.40 (m, 3H), 2.10–2.03 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 166.08, 166.01, 153.91, 149.04, 148.28, 145.28, 145.24, 143.00, 134.24, 132.95, 132.87, 131.10, 129.30, 127.08, 127.04, 122.88, 117.49, 117.45, 115.99, 114.82, 114.70, 112.14, 112.10, 56.33, 48.23, 47.48, 47.03, 30.33, 28.50. HRMS (ESI) calcd for C24H25N3O5SNa [M + Na]+: 490.1407, found: 490.1430.
(E)-3-(4-Hydroxy-3,5-dimethoxyphenyl)-1-(4-(isoquinolin-5-ylsulfonyl)-1,4-diazepan-1-yl)prop-2-en-1-one (DC09)
According to the synthetic process of compound DC08, DC09 as a white solid (0.48 g, 52%) was obtained by taking compound DC05 (2.27 g, 9.52 mmol) as a starting material. m.p. 100–101 °C. 1H NMR (400 Hz, CDCl3): δ (ppm) 9.42 (s, 1H), 8.73 (s, 1H), 8.50 (s, 1H), 8.38–8.35 (m, 1H), 8.25–8.21 (m, 1H), 7.76–7.72 (m, 1H), 7.62–7.55 (m, 1H), 6.73 (s, 1.4H), 6.72 (s, 0.6H), 6.60 (d, J = 15.2 Hz, 0.7H), 6.53 (d, J = 15.2 Hz, 0.7H), 5.78 (brs, 1H), 3.93 (s, 6H), 3.87–3.78 (m, 4H), 3.58–3.42 (m, 4H), 2.07–2.04 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 161.64, 155.38, 155.26, 137.38, 137.32, 136.73, 134.50, 134.39, 134.06, 132.85, 128.86, 128.83, 128.24, 127.86, 127.84, 127.58, 126.70, 101.34, 66.74, 66.67, 62.19, 48.48, 48.19, 45.94, 29.31, 29.01. HRMS (ESI) calcd for C25H27N3O6SNa [M + Na]+: 520.1513, found: 520.1530.
(E)-4-(3-(4-((1-hydroxyisoquinolin-5-yl)sulfonyl)-1,4-diazepan-1-yl)-3-oxoprop-1-en-1-yl)-2-methoxyphenyl acetate (DC10)
A mixture of (E)-3-(4-hydroxy-3,5-dimethoxyphenyl)acrylic acid 19 (0.81 g, 3.42 mmol), EDCI (0.47 g, 2.45 mmol), HOBt (0.33 g, 2.45 mmol), and Et3N (0.82 g, 8.15 mmol) in DCM (25 mL) was stirred for 30 min at room temperature. 1-Hydroxyfasudil (0.50 g, 1.63 mmol) was added and stirred for 12 h, then it was poured into water, extracted with DCM (20 mL × 2), water (20 mL × 2) and brine (20 mL), dried with anhydrous MgSO4, and filtered. The filtrate was concentrated in vacuo to obtain 0.94 g of yellow oil. It was recrystallized to give a white solid (0.28 g, 33%). m.p. 230–232 °C. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 11.62 (d, J = 5.0 Hz, 1H), 8.48–8.43 (m, 1H), 8.19–8.16 (m, 1H), 7.62–7.56 (m, 1H), 7.49 (d, J = 9.6 Hz, 1H), 7.46–7.43 (m, 1H), 7.37–7.29 (m, 1H), 7.11 (t, J = 9.0 Hz, 2H), 7.05 (t, J = 7.7 Hz, 1H), 3.87–3.83 (m, 1H), 3.76 (t, J = 6.1 Hz, 1H), 3.68 (t, J = 4.8 Hz, 1H), 3.59 (t, J = 5.6 Hz, 1H), 3.50 (q, J = 11.2 Hz, 2H), 3.40 (q, J = 7.2 Hz, 2H), 2.26 (s, 3H), 1.82–1.77 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.19, 154.34, 154.09, 145.50, 142.20, 135.02, 134.75, 132.07, 131.59, 131.33, 130.02, 129.25, 128.32, 127.07, 126.38, 121.77, 121.40, 117.52, 116.56, 115.62, 77.22, 28.27. ESI-MS: 548.7 [M + Na]+.
General procedure D for synthesis of compounds DC11–DC15
A mixture of acid 18a–e (1.66 mmol), EDCI (0.48 g, 2.49 mmol), HOBt (2.49 mmol), and Et3N (8.28 mmol) in dry DCM was stirred for 30 min at room temperature. Fasudil (1.66 mmol) was added and stirred for 8 h, then it was poured into water, extracted with DCM, washed with saturated sodium carbonate solution, water and brine, dried with anhydrous MgSO4, and filtered. The filtrate was concentrated in vacuo. The crude was purified by silica gel column chromatography.
(E)-2-(4-(3-(4-(Isoquinolin-5-ylsulfonyl)-1,4-diazepan-1-yl)-3-oxoprop-1-en-1-yl)-2-methoxyphenoxy)ethyl nitrate (DC11)
According to general procedure D, compound DC11 as a white solid (0.39 g, 42%) was obtained by taking compound 18a as a starting material. 1H NMR (600 Hz, CDCl3): δ (ppm) 9.34 (s, 0.7H), 9.32 (s, 0.3H), 8.69 (d, J = 5.7 Hz, 1H), 8.40 (d, J = 6.1 Hz, 0.7H), 8.38 (d, J = 6.5 Hz, 0.3H), 8.34 (d, J = 7.4 Hz, 0.3H), 8.20 (d, J = 8.2 Hz, 0.7H), 8.18 (d, J = 8.2 Hz, 0.3H), 7.71–7.67 (m, 1H), 7.62 (d, J = 15.2 Hz, 0.7H), 7.58 (d, J = 15.2 Hz, 0.3H), 7.09–7.05 (m, 1H), 7.01 (brs, 0.7H), 6.99 (brs, 0.3H), 6.89–6.86 (m, 1H), 6.64 (d, J = 15.2 Hz, 0.7H), 6.58 (d, J = 15.1 Hz, 0.3H), 4.84 (t, J = 4.6 Hz, 2H), 4.33–4.29 (m, 2H), 3.89 (s, 3H), 3.86–3.76 (m, 4H), 3.56–3.40 (m, 4H), 2.08–2.04 (m, 2H). HRMS (ESI) calcd for C26H28N4O8SNa [M + Na]+: 579.1520, found: 579.1531.
(E)-3-(4-(3-(4-(isoquinolin-5-ylsulfonyl)-1,4-diazepan-1-yl)-3-oxoprop-1-en-1-yl)-2-methoxyphenoxy)propyl nitrate (DC12)
According to general procedure D, compound DC12 as a white solid (0.43 g, 45%) was obtained by taking compound 18b as a starting material. m.p. 143–144 °C. 1H NMR (600 Hz, CDCl3): δ (ppm) 9.34 (s, 0.7H), 9.32 (s, 0.3H), 8.69 (d, J = 5.8 Hz, 1H), 8.41–8.38 (m, 1H), 8.34 (d, J = 7.4 Hz, 0.3H), 8.31 (d, J = 6.9 Hz, 1H), 8.20–8.17 (m, 1H), 7.70–7.67 (m, 1H), 7.63–7.57 (m, 1H), 7.08 (d, J = 8.5 Hz, 0.7H), 7.06 (d, J = 7.9 Hz, 1H), 7.00 (s, 0.7H), 6.98 (s, 0.3H), 6.86–6.85 (m, 1H), 6.63 (d, J = 15.4 Hz, 0.7H), 6.57 (d, J = 14.8 Hz, 0.3H), 4.70 (t, J = 5.8 Hz, 2H), 4.14 (t, J = 5.8 Hz, 2H), 3.89 (s, 1H), 3.86–3.76 (m, 4H), 3.57–3.40 (m, 4H), 2.28–2.24 (m, 2H), 2.09–2.03 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.93, 153.96, 149.72, 149.65, 145.41, 145.35, 142.54, 142.37, 134.22, 132.92, 131.06, 129.31, 128.84, 127.04, 122.59, 117.44, 115.99, 113.63, 111.69, 71.37, 65.31, 56.31, 32.21, 30.36, 28.46, 26.73. HRMS (ESI) calcd for C27H30N4O8SNa [M + Na]+: 593.1677, found: 593.1693.
(E)-4-(4-(3-(4-(Isoquinolin-5-ylsulfonyl)-1,4-diazepan-1-yl)-3-oxoprop-1-en-1-yl)-2-methoxyphenoxy)butyl nitrate (DC13)
According to general procedure D, compound DC13 as a white solid (0.47 g, 48%) was obtained by taking compound 18c as a starting material. m.p. 125–126 °C. 1H NMR (600 Hz, CDCl3): δ (ppm) 9.34 (s, 0.7H), 9.32 (s, 0.3H), 8.69 (d, J = 5.9 Hz, 1H), 8.40 (d, J = 6.1 Hz, 0.7H), 8.38 (d, J = 6.4 Hz, 0.3H), 8.34 (d, J = 7.3 Hz, 0.3H), 8.31 (d, J = 7.3 Hz, 0.7H), 8.20 (d, J = 8.2 Hz, 0.7H), 8.18 (d, J = 7.9 Hz, 0.3H), 7.71–7.67 (m, 1H), 7.62 (d, J = 15.3 Hz, 0.7H), 7.58 (d, J = 15.2 Hz, 0.3H), 7.09–7.05 (m, 1H), 7.00 (brs, 0.7H), 6.98 (brs, 0.3H), 6.85–6.83 (m, 1H), 6.63 (d, J = 15.2 Hz, 0.7H), 6.57 (d, J = 15.5 Hz, 0.3H), 4.57 (t, J = 5.5 Hz, 2H), 4.08 (t, J = 5.5 Hz, 2H), 3.88 (s, 3H), 3.86–3.76 (m, 4H), 3.56–3.40 (m, 4H), 2.08–2.04 (m, 4H), 1.97–1.96 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 165.91, 165.84, 153.95, 149.70, 149.64, 145.40, 145.35, 144.52, 142.52, 142.35, 134.25, 134.21, 132.90, 132.82, 131.05, 129.30, 128.88, 128.38, 127.03, 126.98, 122.58, 117.41, 116.10, 115.98, 113.62, 111.68, 71.37, 65.30, 56.31, 32.21, 30.36, 28.46, 26.73. HRMS (ESI) calcd for C28H32N4O8SNa [M + Na]+: 607.1833, found: 607.1847.
(E)-5-(4-(3-(4-(Isoquinolin-5-ylsulfonyl)-1,4-diazepan-1-yl)-3-oxoprop-1-en-1-yl)-2-methoxyphenoxy)pentyl nitrate (DC14)
According to general procedure D, compound DC14 as a white solid (0.45 g, 45%) was obtained by taking compound 18d as a starting material. m.p. 138–139 °C. 1H NMR (400 Hz, CDCl3): δ (ppm) 9.36 (s, 0.7H), 9.34 (s, 0.3H), 8.69 (d, J = 6.0 Hz, 1H), 8.45–8.42 (m, 1H), 8.36–8.32 (m, 1H), 8.23–8.18 (t, J = 8.2 Hz, 1H), 7.73–7.69 (m, 1H), 7.64–7.59 (m, 1H), 7.09–7.04 (m, 1H), 7.00–6.98 (m, 1H), 6.84 (d, J = 8.3 Hz, 1H), 6.62 (d, J = 15.3 Hz, 0.7H), 6.56 (d, J = 15.2 Hz, 0.3H), 4.48 (t, J = 6.5 Hz, 2H), 4.05 (t, J = 6.4 Hz, 2H), 3.89 (s, 3H), 3.86–3.78 (m, 4H), 3.56–3.40 (m, 4H), 2.08–2.04 (m, 2H), 1.93–1.79 (m, 4H), 1.65–1.58 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 165.96, 165.90, 153.95, 150.15, 149.54, 145.39, 145.34, 142.65, 142.47, 134.27, 134.22, 132.92, 132.83, 131.07, 129.31, 128.41, 128.36, 127.05, 127.00, 122.71, 117.45, 117.40, 115.83, 115.70, 113.15, 111.56, 111.52, 74.24, 68.43, 56.27, 48.16, 46.99, 30.36, 28.62, 28.47, 26.25, 22.34. HRMS (ESI) calcd for C29H34N4O8SNa [M + Na]+: 620.1990, found: 620.2010.
(E)-6-(4-(3-(4-(Isoquinolin-5-ylsulfonyl)-1,4-diazepan-1-yl)-3-oxoprop-1-en-1-yl)-2-methoxyphenoxy)hexyl nitrate (DC15)
According to general procedure D, compound DC15 as a white solid (0.41 g, 40%) was obtained by taking compound 18e as a starting material. m.p. 112–113 °C. 1H NMR (600 Hz, CDCl3): δ (ppm) 9.35 (s, 0.7H), 9.32 (s, 0.3H), 8.69 (d, J = 5.7 Hz, 1H), 8.42 (d, J = 6.0 Hz, 0.7H), 8.40 (d, J = 5.9 Hz, 0.3H), 8.35 (d, J = 7.2 Hz, 0.3H), 8.32 (d, J = 7.3 Hz, 0.7H), 8.20 (d, J = 8.2 Hz, 0.7H), 8.18 (d, J = 8.2 Hz, 0.3H), 7.71–7.68 (m, 1H), 7.63 (d, J = 15.2 Hz, 0.7H), 7.59 (d, J = 15.3 Hz, 0.3H), 7.09 (d, J = 8.1 Hz, 0.7H), 7.05 (d, J = 8.2 Hz, 0.3H), 7.00 (s, 0.7H), 6.97 (s, 0.3H), 6.86–6.83(m, 1H), 6.63 (d, J = 15.2 Hz, 0.7H), 6.56 (d, J = 15.2 Hz, 0.3H), 4.46 (t, J = 6.6 Hz, 2H), 4.04 (t, J = 6.5 Hz, 2H), 3.89 (s, 3H), 3.86–3.78 (m, 4H), 3.56–3.40 (m, 4H), 2.09–2.08 (m, 2H), 1.89–1.84 (m, 2H), 1.79–1.74 (m, 2H), 1.59–1.47 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 165.97, 165.91, 153.93, 150.20, 149.54, 145.36, 145.31, 142.67, 142.50, 134.28, 134.20, 132.91, 132.82, 131.08, 129.82, 129.31, 128.38, 128.32, 127.03, 126.97, 122.72, 117.46, 117.42, 115.79, 115.70, 115.67, 113.08, 111.55, 111.52, 74.26, 68.51, 56.25, 28.92, 26.43, 25.55, 25.29. ESI HRMS m/z: [M + Na]+ cacld for C30H36N4O8SNa: 635.2146, found: 635.2168.
(E)-3-(2,2-Dimethyl-2H-chromen-6-yl)-1-(4-(isoquinolin-5-ylsulfonyl)-1,4-diazepan-1-yl)prop-2-en-1-one (DC16)
A mixture of (E)-3-(2,2-dimethyl-2H-chromen-6-yl)acrylic acid 25, EDCI (0.34 g, 1.78 mmol), HOBt (0.24 g, 1.78 mmol), Et3N (0.60 g, 5.92 mmol) and dry DCM (10 mL) was stirred at room temperature for 30 min. Then fasudil 4 (0.43 g, 1.18 mmol) was added, and stirred for 24 h. The reaction mixture was then poured into water (15 mL) and extracted with DCM (20 mL × 3). The combined organic layers were washed with saturated sodium carbonate solution (20 mL × 2), water (20 mL × 2) and brine (20 mL), dried with anhydrous MgSO4, and filtered. The filtrate was concentrated in vacuo to obtain a yellow oil (1.0 g). It was purified by silica gel column chromatography to give a white solid (0.36 g, 55%), m.p. 152–153 °C 1H NMR (600 MHz, DMSO-d6): δ (ppm) 9.45 (d, J = 6.4 Hz, 1H), 8.68 (t, J = 6.1 Hz, 1H), 8.45–8.41 (m, 1H), 8.33–8.30 (m, 2H), 7.84–7.80 (m, 2H), 7.45 (t, J = 6.4 Hz, 2H), 7.37 (t, J = 15.5 Hz, 1H), 6.94 (q, J = 2.3, 8.1 Hz, 1H), 6.42 (d, J = 9.8 Hz, 1H), 5.81 (d, J = 9.8 Hz, 1H),3.84 (t, J = 5.4 Hz, 1H), 3.73 (t, J = 6.0 Hz, 1H), 3.68 (t, J = 5.0 Hz, 1H), 3.59–3.51 (m, 3H), 3.43 (q, J = 5.9 Hz, 2H), 1.82–1.75 (m, 2H), 1.39 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 165.88, 165.81, 153.97, 145.42, 145.37, 142.10, 141.90, 134.27, 134.21, 132.88, 132.83, 132.04, 131.06, 130.11, 130.05, 129.30, 128.44, 128.37, 127.04, 126.99, 126.48, 121.85, 121.45, 117.43, 117.40, 116.60, 115.85, 115.68, 77.22, 49.57, 48.66, 48.22, 47.50, 47.19, 47.05, 46.66, 45.33, 30.32, 28.50, 28.28. HRMS (ESI) calcd for C28H29N3O4SNa [M + Na]+: 526.1771, found: 526.1785.
(E)-3-(2,2-Dimethyl-2H-chromen-6-yl)-1-(4-(isoquinolin-5-ylsulfonyl)piperazin-1-yl)prop-2-en-1-one (DC17)
A mixture of (E)-3-(2,2-dimethyl-2H-chromen-6-yl)acrylic acid 25, EDCI (0.53 g, 2.70 mmol), HOBt (0.36 g, 2.70 mmol), Et3N (0.60 g, 5.92 mmol) and dry DCM (10 mL) was stirred at room temperature for 30 min. Then 12 (0.50 g, 1.80 mmol) was added, and stirred for 24 h. The reaction mixture was then poured into water (15 mL) and extracted with DCM (20 mL × 3). The combined organic layers were washed with saturated sodium carbonate solution (20 mL × 2), water (20 mL × 2) and brine (20 mL), dried with anhydrous MgSO4, and filtered. The filtrate was concentrated in vacuo to obtain a yellow oil (1.0 g). It was purified by silica gel column chromatography to give a white solid (0.4 g, 45%), m.p. 196–197 °C. 1H NMR (600 MHz, DMSO-d6): δ (ppm) 9.50 (s, 1H), 8.71 (d, J = 6.1 Hz, 1H), 8.51 (d, J = 8.2 Hz, 1H), 8.47 (d, J = 6.2 Hz, 1H), 8.37 (d, J = 7.4, 0.9 Hz, 1H), 7.89 (t, J = 7.9 Hz, 1H), 7.40–7.39 (m, 2H), 7.31 (d, J = 15.3 Hz, 1H), 6.97 (d, J = 15.3 Hz, 1H), 6.72 (d, J = 8.9 Hz, 1H), 6.37 (d, J = 9.9 Hz, 1H), 5.79 (d, J = 4.9 Hz, 1H), 3.75 (brs, 2H), 3.61 (brs, 2H), 3,12 (brs, 4H), 1.37 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 165.19, 154.34, 154.09, 145.50, 142.20, 135.02, 134.75, 132.07, 131.59, 131.33, 130.02, 129.25, 128.32, 127.07, 126.38, 121.77, 121.40, 117.52, 116.56, 115.62, 77.22, 28.27. HRMS (ESI) calcd for C27H27N3O4SNa [M + Na]+: 512.1614, found: 512.1626.
(2,2-Dimethyl-2H-chromen-6-yl)(4-(isoquinolin-5-ylsulfonyl)-1,4-diazepan-1-yl)methanone (DC18)
According to the synthetic process of compound DC17, compound DC18 as a yellow oil (0.47 g, 50%) was obtained by taking fasudil 4 (0.52 g, 1.78 mmol) and 2,2-dimethyl-2H-chromene-6-carboxylic acid 26 (0.40 g, 1.96 mmol) as starting materials. 1H NMR (600 MHz, DMSO-d6): δ (ppm) 9.45 (d, J = 6.4 Hz, 1H), 8.68 (t, J = 6.1 Hz, 1H), 8.45–8.41 (m, 1H), 8.33–8.30 (m, 2H), 7.84–7.80 (m, 2H), 7.45 (t, J = 6.4 Hz, 2H), 7.37 (t, J = 15.5 Hz, 1H), 6.94 (q, J = 8.1 Hz, 1H), 6.42 (d, J = 9.8 Hz, 1H), 5.81 (d, J = 9.8 Hz, 1H),3.84 (t, J = 5.4 Hz, 1H), 3.73 (t, J = 6.0 Hz, 1H), 3.68 (t, J = 5.0 Hz, 1H), 3.59–3.51 (m, 3H), 3.43 (q, J = 11.5 Hz, 2H), 1.82–1.75 (m, 2H), 1.39 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 165.20, 154.34, 154.09, 145.50, 142.20, 135.02, 134.75, 132.07, 131.59, 131.33, 130.02, 129.25, 128.32, 127.07, 126.38, 121.77, 121.40, 117.52, 116.57, 115.62, 77.22, 28.27. HRMS (ESI) calcd for C26H27N3O4SNa [M + Na]+: 500.1614, found: 500.1628.
(Chloromethylene)dibenzene (29a)
To a solution of benzophenone 27a (5.0 g, 27.4 mmol) in anhydrous MeOH (10 mL) was added NaBH4 (1.56 g, 41.2 mmol) at 0 °C. The resulting solution was stirred for 30 min, quenched by adding 1 N hydrochloric acid and the mixture was concentrated in vacuo. The residue was poured into water (20 mL), the organic phase was extracted with EtOAc (20 mL × 2), water (20 mL × 2), and brine (10 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo to obtain 5 g colorless transparent liquid, and acicular crystals were precipitated after long-term storage with a yield of 99%.
To a solution of diphenylmethanol 28a (5.0 g, 27.1 mmol) and 2 drops of dried pyridine in dry DCM (20 mL) was added dropwise a solution of thionyl chloride (3 mL, 40.7 mmol) in DCM (5 mL) at 0 °C. The reaction mixture was stirred for 1 h, then poured into ice water (20 mL). The organic layer was separated and the water layer was extracted with DCM (20 mL × 2). The combined organic extracts were washed with saturated sodium carbonate solution (20 mL × 2), water (20 mL × 2) and brine (20 mL). The organic layer was dried over anhydrous MgSO4 and then concentrated in vacuo. After concentration, 5.2 g colorless liquid was obtained with a yield of 95%.
4,4′-(Chloromethylene)bis(fluorobenzene) (29b)
NaBH4 (1.28 g, 33.9 mmol) was added in batches into a solution of bis(4-fluorophenyl)methanone 27b (5.0 g, 22.9 mmol) in anhydrous MeOH (10 mL) at 0 °C. The reaction was stirred for 30 min, quenched by adding 1 N hydrochloric acid, and the mixture was concentrated in vacuo. The residue was poured into water (20 mL), the organic phase was extracted with EtOAc (20 mL × 2), water (20 mL × 2), and brine (10 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo to obtain 5 g colorless transparent liquid with a yield of 99%.
To a solution of bis(4-fluorophenyl)methanol 28b (5.0 g, 22.7 mmol) and 3 drops of dried pyridine in dry DCM (20 mL) was added dropwise a solution of thionyl chloride (2.5 mL, 34.0 mmol) in DCM (10 mL) at 0 °C, while maintaining the temperature below 5 °C. The reaction mixture was stirred for 1 h, then poured into ice water (20 mL). The organic layer was separated and the water layer was extracted with DCM (20 mL × 2). The combined organic extracts were washed with a saturated solution of Na2CO3 (20 mL × 2), water (20 mL × 2) and brine (20 mL). The organic layer was dried over anhydrous MgSO4 and then concentrated in vacuo. After concentration, 5.3 g colorless liquid was obtained with a yield of 98%.
5-((4-Benzhydryl-1,4-diazepan-1-yl)sulfonyl)isoquinoline (DC19)
Fasudil 4 (0.40 g, 1.37 mmol) was combined with (chloromethylene)dibenzene 29a (0.30 g, 1.51 mmol), K2CO3 (0.57 g, 4.12 mmol) and CH3CN (10 mL). The suspension was heated to reflux for 16 h then cooled to room temperature, and concentrated in vacuo. The residue was poured into ice water (20 mL), and the organic phase was extracted with DCM (20 mL × 2), water (20 mL × 2), and brine (20 mL), dried over anhydrous MgSO4, filtered and concentrated in vacuo to obtain 0.74 g oil. It was purified by silica gel column chromatography (EA/PE = 1/30 to 1/10) to give a white solid (0.12 g, 21%). m.p. 160–162 °C. 1H NMR (600 MHz, CDCl3): δ (ppm) 9.36 (s, 1H), 8.69 (d, J = 6.1 Hz, 1H), 8.47 (d, J = 6.1 Hz,1H), 8.36 (d, J = 7.3 Hz, 1H), 8.19 (d, J = 8.1 Hz, 1H), 7.69 (t, J = 7.7 Hz, 1H), 7.32–7.30 (m, 4H), 7.25–7.22 (m, 4H), 7.17–7.15 (m, 2H), 4.59 (s, 1H), 3.57 (t, J = 5.8 Hz, 2H), 3.40 (t, J = 4.8 Hz, 2H), 2.70 (t, J = 5.0 Hz,2H), 2.65 (t, J = 5.6 Hz, 2H), 1.80–1.76 (m, 2H). ESI MS: m/z 458.3 [M + H]+.
5-((4-(Bis(4-fluorophenyl)methyl)-1,4-diazepan-1-yl)sulfonyl) isoquinoline (DC20)
According to the synthetic process of compound DC19, compound DC20 as a white solid (0.70 g, 41%) was obtained by taking 4,4′-(chloromethylene) bis(fluorobenzene) 29b (0.82 g, 3.43 mmol) and potassium carbonate powder (1.42 g, 10.30 mmol) as starting materials. Compound DC20, m.p. 129–130 °C. 1H NMR (600 MHz, DMSO-d6): δ (ppm) 9.52 (s, 1H), 8.72 (d, J = 6.1 Hz, 1H), 8.48 (d, J = 8.2 Hz,1H), 8.39 (d, J = 6.1 Hz, 1H), 8.35 (dd, J = 1.0 Hz, 7.4 Hz, 1H), 7.86 (t, J = 7.9 Hz, 1H), 7.31–7.29 (m, 4H), 7.10–7.07 (m, 4H), 4.74 (s, 1H), 3.53 (t, J = 5.9 Hz, 2H), 3.38–3.36 (m, 2H), 2.60–2.58 (m, 2H), 2.52–2.50 (m, 2H), 1.67–1.63 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 162.70, 160.28, 154.04, 145.39, 139.53, 139.49, 134.53, 134.15, 133.14, 131.10, 129.71, 129.63, 129.36, 127.07, 117.64, 115.84, 115.63, 71.21, 53.70, 52.30, 48.55, 46.55, 28.95. ESI MS: m/z 494.3 [M + H]+.
5-((4-Benzhydrylpiperazin-1-yl)sulfonyl)isoquinoline (DC21)
1-Benzhydrylpiperazine 31 (0.50 g, 1.98 mmol) and potassium carbonate powder (1.40 g, 9.90 mmol) were added into 5 mL DMF and cooled to 5 °C. Isoquinoline-5-sulfonyl chloride 30 (0.55 g, 2.01 mmol) was added into the reaction mixture and stirred for 12 h. The mixture was poured into water, leading to the precipitation of a solid. The precipitate was filtered, washed with water and dried to give 0.72 g crude product. It was recrystallized from MeOH, yielding white solid DC21 (0.40 g, 45%) m.p. 78–79 °C. 1H NMR (600 MHz, DMSO-d6): δ (ppm) 9.53 (s, 1H), 8.68 (d, J = 6.1 Hz, 1H), 8.54 (d, J = 8.2 Hz, 1H), 8.45 (d, J = 6.1 Hz, 1H), 8.34 (d, J = 7.3 Hz, 1H), 7.90 (t, J = 7.7 Hz, 1H), 7.32 (d, J = 7.4 Hz, 4H), 7.23 (t, J = 7.5 Hz, 4H), 7.14 (t, J = 7.3 Hz, 2H), 4.28 (s, 1H), 3.10 (s, 1H), 2.32 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 154.08, 145.44, 142.75, 134.91, 134.85, 131.65, 131.31, 129.21, 129.04, 127.95, 127.43, 127.09, 117.67, 74.69, 51.07, 46.19. ESI MS: m/z 444.2 [M + H]+.
5-((4-(Bis(4-fluorophenyl)methyl)piperazin-1-yl)sulfonyl) isoquinoline (DC22)
According to the synthetic process of compound DC22, compound DC22 as a white solid (0.23 g, 34%) was obtained by taking 1-(bis(4-fluorophenyl)methyl) piperazine 32 (0.40 g, 1.39 mmol) and potassium carbonate powder (0.96 g, 6.96 mmol) as starting materials. Compound DC22, m.p. 87–90 °C. 1H NMR (600 MHz, DMSO-d6): δ (ppm) 9.52 (s, 1H), 8.69 (d, J = 6.1 Hz, 1H), 8.54 (d, J = 8.2 Hz, 1H), 8.45 (d, J = 6.1 Hz, 1H), 8.35 (d, J = 6.8 Hz, 1H), 7.90 (t, J = 7.8 Hz, 1H), 7.34 (d, J = 8.0 Hz, 4H), 7.06 (t, J = 8.7 Hz, 4H), 4.37 (s, 1H), 3.09 (s, 1H), 2.30 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.76, 160.35, 154.08, 145.45, 138.65, 138.63, 134.93, 134.86, 131.29, 129.84, 129.76, 127.09, 117.65, 115.92, 115.71, 72.60, 50.86, 46.16. ESI MS: m/z 480.2 [M + H]+.
1-(4-Benzhydrylpiperazin-1-yl)-5-(1,2-dithiolan-3-yl)pentan-1-one (DC23)
To a solution of 5-(1,2-dithiolan-3-yl)pentanoic acid 5 (1.0 g, 4.85 mmol) and dried pyridine (2 drops) in dried methylene chloride (10 mL) was added dropwise a solution of thionyl chloride (0.86 g, 7.26 mmol) in DCM (5 mL) at 0 °C and stirred for 2 h. The remaining thionyl chloride was concentrated under reduced pressure and dissolved in dry DCM (5 mL).
To a solution of 1-benzhydrylpiperazine 31 (1.02 g, 4.04 mmol), Et3N (2.0 g, 20.2 mmol), and DMAP (0.05 g, 0.4 mmol) in dry DCM (10 mL) was added dropwise a solution of the above-mentioned 5-(1,2-dithiolan-3-yl)pentanoyl chloride (1.1 g, 4.85 mmol) in DCM (5 mL) at 0 °C and stirred for 4 h. Then the mixture was poured into water (20 mL), and extracted with DCM (20 mL × 3). The organic layers were combined, washed with saturated sodium carbonate solution (20 mL × 2), water (20 mL × 2), and brine (20 mL), dried with anhydrous MgSO4 and filtered. The filtrate was evaporated under vacuum. The crude was purified by silica gel column chromatography to obtain a yellow oil (0.98 g, 55%). 1H NMR (600 MHz, CDCl3): δ (ppm) 7.41 (d, J = 7.5 Hz, 4H), 7.28 (d, J = 7.4 Hz, 4H), 7.19 (t, J = 7.3 Hz, 2H), 4.23 (s, 1H), 3.71 (q, J = 7.0 Hz, 1H), 3.61 (brs, 2H), 3.59–3.54 (m, 1H), 3.45 (brs, 2H), 3.19–3.15 (m, 1H), 3.12–3.08 (m, 1H), 2.47–2.42 (m, 1 H), 2.37–2.36 (m, 4H), 2.29 (t, J = 7.3 Hz, 2H), 1.93–1.87 (m, 1H), 1.73–1.60 (m, 5H), 1.50–1.44 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.22, 135.97, 129.84, 129.42, 128.93, 74.81, 56.64, 51.80, 51.46, 41.56, 38.60, 37.82, 34.64, 32.20, 28.89, 24.79. ESI MS: m/z 441.4 [M + H]+.
Dry EtOAc (10 mL) was added to the above yellow oil to dissolve, and 1.2 M hydrogen chloride solution of EtOAc was slowly added dropwise in an ice bath until no solids were precipitated. The mixture was stirred for a while to become sticky, and concentrated under reduced pressure to dryness. Next, 20 mL of ether was added and stirred to solidify the mixture. The solid was collected by filtration and washed with ether to obtain a yellow solid, which is the hydrochloride salt. m.p. 115–118 °C.
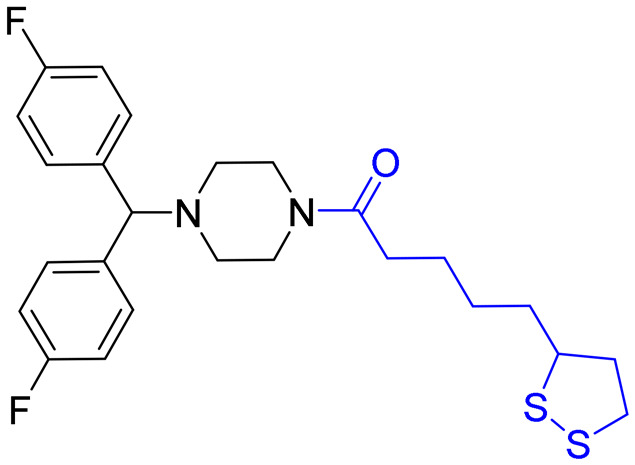
1-(4-(Bis(4-fluorophenyl)methyl)piperazin-1-yl)-5-(1,2-dithiolan-3-yl)pentan-1-one (DC24)
According to the synthetic process of compound DC23, compound DC24 as a yellow oil (0.87 g, 45%) was obtained by taking 1-(bis(4-fluorophenyl)methyl) piperazine 32 (1.16 g, 4.04 mmol) as a starting material. 1.2 M hydrogen chloride solution of EtOAc was added dropwise into the oil to obtain the corresponding hydrochloride. 1H NMR (600 MHz, CDCl3): δ (ppm) 7.34 (t, J = 6.4 Hz, 4H), 6.98 (t, J = 8.3 Hz, 4H), 4.23 (s, 1H), 4.12 (q, J = 7.1 Hz, 1H), 3.61 (brs, 2H), 3.58–3.55 (m, 1H), 3.45 (brs, 2H), 3.20–3.16 (m, 1H), 3.13–3.09 (m, 1H), 2.48–2.43 (m, 1 H), 2.34 (brs, 4H), 2.29 (t, J = 7.4 Hz, 2H), 1.93–1.88 (m, 1H), 1.71–1.62 (m, 5H), 1.48–1.44 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 171.22, 135.96, 129.83, 129.41, 128.93, 74.82, 56.64, 51.80, 51.46, 41.56, 38.60, 37.82, 34.64, 32.20, 28.89, 24.79. ESI MS: m/z 477.2 [M + H]+. The purity of compound DC24 was determined using a Shimadzu 2010A HPLC. The purity was 95.3% (area normalization method). The column was an Agilent ZORBA× 80A Extend-C18 (4.6 × 150 mm, 5 μm), the mobile phase was MeCN/H2O = 75%/25%, the flow rate was 0.8 mL min−1, the column temperature was 25 °C, the detection wavelength was 210 nm, and the retention time was 6.877 min.
5-(1,2-Dithiolan-3-yl)pentyl 2-acetoxybenzoate (DC25)
To a solution of 2-acetoxybenzoic acid 33 (1.0 g, 5.54 mmol) and DMF (4 drops) in dry DCM (10 mL) was added dropwise a solution of oxalyl chloride (1.4 g, 11.1 mmol) in DCM (5 mL) at 0 °C. After dropping, it was stirred for 2 h at 25 °C. The reaction mixture was concentrated under reduced pressure and dissolved in dry DCM (10 mL).
To a solution of lipoic alcohol 6 (0.85 g, 4.43 mmol), Et3N (2.80 g, 27.8 mmol) and dried DCM (20 mL) was added dropwise a solution of 2-(chlorocarbonyl)phenyl acetate in DCM (10 mL) at 0 °C and stirred for 5 h. Then the mixture was poured into water (20 mL), and extracted with DCM (20 mL × 2). The organic layers were combined, washed with saturated sodium carbonate solution (20 mL × 2), water (20 mL × 2), and brine (20 mL), dried with anhydrous MgSO4 and filtered. The filtrate was evaporated under vacuum to obtain a yellow oil (1.1 g). The crude was purified by silica gel column chromatography to obtain the yellow oil (0.3 g, 19%). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.01 (dd, J = 7.8, 1.7 Hz, 1H), 7.55 (td, J = 9.4, 7.8, 1.7 Hz, 1H), 7.31 (td, J = 9.0, 7.8, 1.7 Hz, 1H), 7.10 (dd, J = 8.1, 0.84 Hz, 1H), 4.27 (t, J = 6.7 Hz, 2H), 3.61–3.54 (m, 1H), 3.21–3.08 (m, 2H), 2.50–2.42 (m, 1H), 2.35 (s, 3H), 1.95–1.87 (m, 1H), 1.79–1.67 (m, 4H), 1.50–1.45 (m, 4H). 13C NMR (101 MHz, Chloroform-d) δ 169.66, 169.59, 164.51, 164.43, 150.73, 133.82, 131.72, 126.03, 123.84, 123.43, 119.15, 65.08, 65.03, 56.51, 40.27, 38.49, 34.84, 28.97, 28.63, 28.54, 25.90, 25.84, 21.09, 1.03. HRMS (ESI) calcd for C17H22O4S2Na [M + Na]+: 377.0852, found: 377.0880.
(5-(1,2-Dithiolan-3-yl)pentyl (E)-3-(4-acetoxy-3-methoxyphenyl)acrylate (DC26)
According to the synthetic process of compound DC25, compound DC26 as a yellow oil (0.25 g, 22%) was obtained by taking (E)-3-(4-acetoxy-3-methoxyphenyl) acrylic acid 14 (0.8 g, 3.39 mmol) as a starting material. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.63 (d, J = 15.9 Hz, 1H), 7.13–7.11 (m, 2H), 7.05 (d, J = 7.9 Hz, 1H), 6.38 (d, J = 15.9 Hz, 1H), 4.21 (t, J = 6.6 Hz, 2H), 3.87 (s, 3H), 3.62–3.55 (m, 1H), 3.21–3.09 (m, 1H), 2.51–2.43 (m, 1H), 2.32 (s, 3H), 1.96–1.88 (m, 1H), 1.76–1.68 (m, 4H), 1.55–1.43 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 169.29, 167.18, 163.50, 149.82, 148.42, 145.43, 126.07, 123.63, 115.97, 114.99, 111.65, 107.78, 64.10, 56.66, 38.56, 34.74, 28.83, 25.77, 22.84, 19.29, 18.64. HRMS (ESI) calcd for C20H26O5S2Na [M + Na]+: 433.1114, found: 433.1118.
Biological assays
Kinase Inhibition
Kinase inhibition data were obtained externally at Reactions Biology Corporation (Malvern, PA, USA) in a radiometric assay. For IC50 determination on ROCK1 and ROCK2, the competing ATP concentration was 1 μM (initial screen) or 200 μM (secondary assay for the most active compounds).
The formal enzyme concentration was 1 nM in both cases.
Kinase selectivity data were collected externally at Reactions Biology Corporation (Malvern, PA, USA) in a radiometric assay, against a panel of 335 kinases. The competing ATP concentration was 10 μM.
Further details regarding the assay technology can be obtained from Anastassiadis et al.27
Toxicity study on BV2 cells
Cells were cultured and subcultured with complete culture medium (DMEM + 10% FBS), until the fusion degree was about 90%, and were used in the experiment. The cells were digested with trypsin, centrifuged at 800 rpm for 5 min, the supernatant was discarded, resuspended with fresh medium (DMEM + 10% FBS), counted, inoculated with a density of 3000 cells per well to a 96-well cell culture plate, and cultured overnight in a 5% CO2 incubator at 37 °C. 20 μL sample was taken into line A of the V-hole plate, marked, 12 μL DMSO was diluted to the G-hole 3 times by the continuous dilution method, and each dilution was shaken evenly. The test compounds were taken 5 μL and added to 120 μL medium (25 times dilution), while a DMSO control hole was also created. The cells were cultured overnight, the medium was removed, 195 μL DMEM and 5 μL diluted medium containing the corresponding concentration of compounds were added respectively, and then the culture plate was placed in a 5% CO2 incubator for overnight cultivation at 37 °C. The stock solution was removed, afterward 100 μL fresh serum-free DMEM medium containing MTT (0.5 mg mL−1) was added to each hole, and culturing was continued. The stock solution was removed after 4 hours, and 100 μL DMSO was added to each hole, keeping away from light shock for 10 min, then the light absorption value at 552/690 nm was read with multi-function readout.
Study on antioxidant activity
The selected sample powder was dissolved into 10 mM solution with DMSO, and then mixed into different concentrations of solution according to the volume of sample and ethanol = 1 : 9 (DMSO content is 10%). DPPH mother liquor: 0.0079 g of DPPH powder was weighed and dissolved in 20 mL of anhydrous ethanol to make the mother liquor with a concentration of 1 mM, which was stored in the dark at 4 °C for standby, then diluted to 0.2 mM with anhydrous ethanol. Quercetin solution: 5 mg of quercetin was weighed and dissolved in 1 mL of DMSO, 100 μL was added to 900 μL DMSO, diluted to 0.5 mg mL−1, and sub-packed into small tubes, each tube = 40 μL. When used, 360 μL anhydrous ethanol was added (DMSO content is 10%). Solvent control group: DMSO was mixed with anhydrous ethanol in a volume ratio of 1 : 9 (DMSO content was 10%), and was prepared immediately.
2) Operation steps
Sample test group (A0): 50 μL DPPH solution + 50 μL absorbance value of sample solution;
Sample color control group (A1): 50 μL anhydrous ethanol + 50 μL absorbance value of sample solution;
Solvent control group (A2): 50 μL DPPH solution + 50 μL absorbance value of absolute ethanol of 10% DMSO; at the same time, three compound wells were set and incubated in a 25 °C incubator for 30 min, and then the absorbance value at 517 nm wavelength was measured.
Test of inhibiting the release of inflammatory factors from BV2 cells
The experiment was divided into solvent control group (C), model group (M) and sample group. The cells were digested with 0.25% pancreatin–EDTA, and the cell density was adjusted to 1 × 105 mL−1 in DMEM medium containing 10% FBS, evenly inoculated to a 24-hole plate, 400 μL per hole cell suspension, containing 10 000 cells, was incubated in a 5% CO2 incubator for 24 hours at 37 °C. 1 μL was added to 999 μL DMEM to the test sample, mixed before using; DMSO was diluted 1000 times with DMEM for group C and group M. After incubation for 24 hours, the 24-well plate was taken out, the supernatant was sucked out, and 495 μL serum-free DMEM containing 0.1% DMSO was added into the solvent control group and model group, 495 μL diluted sample was added to the sample group, and the 24-well plate was put into the incubator for 1 h after dosing.
In vivo safety
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Shenyang Pharmaceutical University and approved by the Animal Ethics Committee of Shenyang Pharmaceutical University. Mice used in this study were C57BL/6 (half females and half males, 6–8 weeks old, body weight 16–18 g) and purchased from Henan Scribes Biotechnology Co., Ltd. with license number SCXK (Henan) 2020–0005. The animals were raised in the Experimental Animal Center of Nanjing Research Institute of Jiangsu Kangyuan Pharmaceutical Co., Ltd. The animals were housed under sterile conditions at a constant temperature of 22–24 °C under daily cycles of light/darkness (12 h). After three days of adaptive feeding, the mice were randomly divided into vehicle group, DC01 50 mg kg−1 group, and DC24 50 mg kg−1 group (n = 9–10). The vehicle was DMSO : Tween 80 : normal saline = 5 : 5 : 90, and the volume of administration was 10 mL kg−1. Animals were weighed before each administration to adjust the required volume. 1, 3 and 6 h after administration, clinical symptoms and death/near death situation were observed (including general conditions, behavioral activities, gait and posture, eyes, nose and mouth, gastrointestinal function, skin coat, urogenital tract). After that, clinical symptoms were monitored for 14 days. The animals' weight was monitored for 14 days. Statistical analysis for the body weight was performed using GraphPad Prism 6 software.
Pharmacokinetic analysis
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Shenyang Pharmaceutical University and approved by the Animal Ethics Committee of Shenyang Pharmaceutical University. SD rats were used in this study (12 females, 12 males, and 8 weeks old), and purchased from Qinglongshan Animal Breeding Farm, with animal production license number 20180004030364. The animals were raised in the Experimental Animal Center of Nanjing Research Institute of Jiangsu Kangyuan Pharmaceutical Co., Ltd. The SD rats were raised under sterile conditions (18–22 °C, humidity 35–65%, and ventilation) under daily cycles of light/darkness with a 12 h interval. They went through a period of 3 days of acclimatization before the experimental procedure.
24 SD rats were assigned to 4 groups randomly (6 females and 6 males for DC01, 6 females and 6 males for DC24). Compounds formulated in DMSO : Tween 80 : normal saline = 5 : 5 : 90 were administered via injection at the indicated doses. SD rats were weighed to calculate the required volume before each administration. Blood samples were obtained at indicated time points (0, 0.033, 0.083, 0.17, 0.33, 0.5, 0.75, 1, 2, 4, 12 h). The blood sample was taken from the orbit vein at indicated time points, collected on an Eppendorf containing of anticoagulant EDTA-K2, and centrifuged (4 °C) at 1100 rpm for 5 min. After that, each plasma sample (50 mL) was mixed with tadalafil (50 mL) in MeCN (20 ng mL−1) and MeCN (300 mL), and the resulting mixture was further centrifuged at 13 000 rpm for 10 min. Then, the supernatant (50 mL) was reconstituted with MeCN (200 mL), and the resulting concentration of compounds was respectively analyzed by using a TSQ Altis Triple Quadrupole LC/MS instrument (Thermo Scientific). All the pharmacokinetic parameters were obtained using Phoenix WinNonlin software.
Evaluation of the efficacy of compound DC24 in the acute inflammation model
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Shenyang Pharmaceutical University and approved by the Animal Ethics Committee of Shenyang Pharmaceutical University. 32 C57BL/6 mice (female, body weight 20–22 g) were randomly divided into four groups, namely, CFA, CFA + Belumosudil groups, DC24 low-dose group (5 mg kg−1), and DC24 high-dose group (10 mg kg−1), with 8 animals in each group; all other groups of animals were injected with Freund's complete adjuvant (25 μL) into the right sole of the foot; different drugs were prepared into corresponding concentrations before administration. After 12 h of modeling, each group of animals was given different test substances at a rate of 20 mL kg−1 by intraperitoneal injection. The normal control group and model control group were given equal volumes of pure water once a day. The thickness of the right foot at 100% and 0.5, 1, 2, 4, 6, 8, 10 and 12 hours after administration was measured.
Author contributions
Conceptualization and supervision: Guoliang Chen, Xuefei Bao and Wei Xiao; methodology: Ruolin Cao, Fangyu Du, Pengcheng Cai, Minggang Qi, Wei Xiao; writing – original draft: Ruolin Cao, Zhiqiang Liu; writing – review & editing: Xuefei Bao, Guoliang Chen.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was funded by the National Natural Science Foundation of China (No. 82273793), Key Research Project of Department of Education of Liaoning Province (LJKZZ20220108). the China Postdoctoral Science Foundation (No. 2019M661132), and the Foundation of Liaoning Provincial Department of Education (No. LJKQZ20222351).
Electronic supplementary information (ESI) available: Supplemental figures and tables; NMR and MS spectra. See DOI: https://doi.org/10.1039/d4md00438h
Data availability
The data supporting this article have been included as part of the ESI.†
Notes and references
- Arulselvan P. Fard M. T. Tan W. S. Gothai S. Fakurazi S. Norhaizan M. E. Kumar S. S. Oxid. Med. Cell. Longevity. 2016;2016:5276130. doi: 10.1155/2016/5276130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germolec D. R. Shipkowski K. A. Frawley R. P. Evans E. Methods Mol. Biol. 2018;1803:57–79. doi: 10.1007/978-1-4939-8549-4_5. [DOI] [PubMed] [Google Scholar]
- Nakagawa O. Fujisawa K. Ishizaki T. Saito Y. Nakao K. Narumiya S. FEBS Lett. 1996;392:189–193. doi: 10.1016/0014-5793(96)00811-3. [DOI] [PubMed] [Google Scholar]
- Matsui T. Amano M. Yamamoto T. Chihara K. Nakafuku M. Ito M. Nakano T. Okawa K. Iwamatsu A. Kaibuchi K. EMBO J. 1996;15:2208–2216. doi: 10.1002/j.1460-2075.1996.tb00574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung T. Chen X. Q. Manser E. Lim L. Mol. Cell. Biol. 1996;16:5313–5327. doi: 10.1128/MCB.16.10.5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T. Nishimura D. Wu R. C. Amano M. Iso T. Kedes L. Nishida H. Kaibuchi K. Hamamori Y. J. Biol. Chem. 2006;281:15320–15329. doi: 10.1074/jbc.M510954200. [DOI] [PubMed] [Google Scholar]
- Amano M. Nakayama M. Kaibuchi K. Cytoskeleton. 2010;67:545–554. doi: 10.1002/cm.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield A. V. Bernard O. Crit. Rev. Biochem. Mol. Biol. 2013;48:301–316. doi: 10.3109/10409238.2013.786671. [DOI] [PubMed] [Google Scholar]
- Julian L. Olson M. F. Small GTPases. 2014;5:e29846. doi: 10.4161/sgtp.29846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi S. Nakao S. Chun K. H. Fiorina P. Sun D. Arita R. Zhao M. Kim E. Schueller O. Campbell S. Taher M. Melhorn M. I. Schering A. Gatti F. Tezza S. Xie F. Vergani A. Yoshida S. Ishikawa K. Yamaguchi M. Sasaki F. Schmidt-Ullrich R. Hata Y. Enaida H. Yuzawa M. Yokomizo T. Kim Y. B. Sweetnam P. Ishibashi T. Hafezi-Moghadam A. Cell Rep. 2015;10:1173–1186. doi: 10.1016/j.celrep.2015.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S.-L. Cipolla M. J. J. Cereb. Blood Flow Metab. 2017;37:3262–3270. doi: 10.1177/0271678X17718665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H. Yoshio T. Kaneko H. Yamanaka H. Arthritis Rheum. 2010;62:82–92. doi: 10.1002/art.25063. [DOI] [PubMed] [Google Scholar]
- Shimokawa H. Takeshita A. Arterioscler., Thromb., Vasc. Biol. 2005;25:1767–1775. doi: 10.1161/01.ATV.0000176193.83629.c8. [DOI] [PubMed] [Google Scholar]
- Garnock-Jones K. P. Drugs. 2014;74:2211–2215. doi: 10.1007/s40265-014-0333-2. [DOI] [PubMed] [Google Scholar]
- Hoy S. M. Drugs. 2018;78:389–396. doi: 10.1007/s40265-018-0877-7. [DOI] [PubMed] [Google Scholar]
- Boland S. Bourin A. Alen J. Geraets J. Schroeders P. Castermans K. Kindt N. Boumans N. Panitti L. Fransen S. Vanormelingen J. Stassen J. M. Leysen D. Defert O. J. Med. Chem. 2015;58:4309–4324. doi: 10.1021/acs.jmedchem.5b00308. [DOI] [PubMed] [Google Scholar]
- Xie Y. Yue L. Shi Y. Su X. Gan C. Liu H. Xue T. Ye T. J. Med. Chem. 2023;66:4342–4360. doi: 10.1021/acs.jmedchem.2c01753. [DOI] [PubMed] [Google Scholar]
- Feng Y. LoGrasso P. V. Defert O. Li R. J. Med. Chem. 2016;59:2269–2300. doi: 10.1021/acs.jmedchem.5b00683. [DOI] [PubMed] [Google Scholar]
- Huang Y. Mao C. R. Lou Y. Zhan S. Chen Z. Ding W. Ma Z. J. Med. Chem. 2023;66:15205–15229. doi: 10.1021/acs.jmedchem.3c01297. [DOI] [PubMed] [Google Scholar]
- van Vugt R. M. Rijken P. J. Rietveld A. G. van Vugt A. C. Dijkmans B. A. Clin. Rheumatol. 2008;27:771–775. doi: 10.1007/s10067-008-0848-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaswal S. Mehta H. C. Sood A. K. Kaur J. Clin. Chim. Acta. 2003;338:123–129. doi: 10.1016/j.cccn.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Packer L. Cadenas E. J. Clin. Biochem. Nutr. 2011;48:26–32. doi: 10.3164/jcbn.11-005FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunia-Krzyżak A. Słoczyńska K. Popiół J. Koczurkiewicz P. Marona H. Pękala E. Int. J. Cosmet. Sci. 2018;40:356–366. doi: 10.1111/ics.12471. [DOI] [PubMed] [Google Scholar]
- Ruwizhi N. Aderibigbe B. A. Int. J. Mol. Sci. 2020;21(16):5712. doi: 10.3390/ijms21165712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du F. Zhou Q. Fu X. Shi Y. Chen Y. Fang W. Yang J. Chen G. RSC Adv. 2019;9:2498–2508. doi: 10.1039/C8RA10424G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R. Du F. Cui Y. Qi M. Zhuang J. Wang J. Zhang M. Zhang X. Liu Z. Zou L. Xiao W. Chen G. Bioorg. Chem. 2024;143:107064. doi: 10.1016/j.bioorg.2023.107064. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T. Deacon S. W. Devarajan K. Ma H. Peterson J. R. Nat. Biotechnol. 2011;29(11):1039. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting this article have been included as part of the ESI.†