

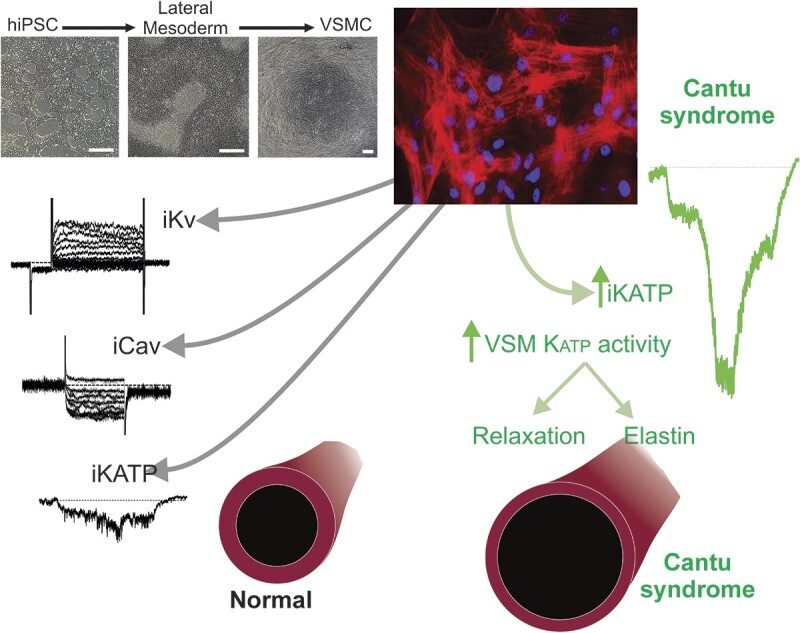
Abstract
Cantú syndrome (CS), a multisystem disease with a complex cardiovascular phenotype, is caused by gain-of-function (GoF) variants in the Kir6.1/SUR2 subunits of ATP-sensitive potassium (KATP) channels and is characterized by low systemic vascular resistance, as well as tortuous, dilated, vessels, and decreased pulse-wave velocity. Thus, CS vascular dysfunction is multifactorial, with both hypomyotonic and hyperelastic components. To dissect whether such complexities arise cell autonomously within vascular smooth muscle cells (VSMCs) or as secondary responses to the pathophysiological milieu, we assessed electrical properties and gene expression in human induced pluripotent stem cell-derived VSMCs (hiPSC-VSMCs), differentiated from control and CS patient-derived hiPSCs, and in native mouse control and CS VSMCs. Whole-cell voltage clamp of isolated aortic and mesenteric arterial VSMCs isolated from wild-type (WT) and Kir6.1[V65M] (CS) mice revealed no clear differences in voltage-gated K+ (Kv) or Ca2+ currents. Kv and Ca2+ currents were also not different between validated hiPSC-VSMCs differentiated from control and CS patient-derived hiPSCs. While pinacidil-sensitive KATP currents in control hiPSC-VSMCs were similar to those in WT mouse VSMCs, they were considerably larger in CS hiPSC-VSMCs. Under current-clamp conditions, CS hiPSC-VSMCs were also hyperpolarized, consistent with increased basal K conductance and providing an explanation for decreased tone and decreased vascular resistance in CS. Increased compliance was observed in isolated CS mouse aortae and was associated with increased elastin mRNA expression. This was consistent with higher levels of elastin mRNA in CS hiPSC-VSMCs and suggesting that the hyperelastic component of CS vasculopathy is a cell-autonomous consequence of vascular KATP GoF. The results show that hiPSC-VSMCs reiterate expression of the same major ion currents as primary VSMCs, validating the use of these cells to study vascular disease. Results in hiPSC-VSMCs derived from CS patient cells suggest that both the hypomyotonic and hyperelastic components of CS vasculopathy are cell-autonomous phenomena driven by KATP overactivity within VSMCs .
Keywords: KATP, KCNJ8, Kir6.1, ABCC9, SUR2, CRISPR, Cas9, cardiovascular system, whole-cell patch clamp, I Ca , Cantú syndrome, smooth muscle, VSMC, cardiac myocyte, blood pressure, vascular compliance, hiPSC, V m , LTC, Kv, BK, vascular tree, vascular tone, vasculopathy, cell-autonomous, RT-PCR, systemic vascular resistance
Graphical Abstract
Graphical Abstract.
Introduction
Cardiovascular events are often preceded by vascular abnormalities, and understanding fundamental vasculopathic mechanisms is critical to explaining disease progression. Interventional experimentation on bona fide vascular smooth muscle cells (VSMCs) can realistically only be carried out in non-human animal models, but the potential for species-dependent differences to obscure human relevance ultimately warrants replication in human cell-based systems. VSMCs derived from human induced pluripotent stem cells (hiPSCs) are therefore promising for the study of human-specific, as well as mutation-specific and cell-autonomous, mechanisms of cardiovascular disease and for pharmacotherapeutic testing.1,2
Notably, while the electrophysiology (EP) of cardiac myocytes derived from hiPSCs has been extensively studied,3 hiPSC-VSMC EP has remained unexplored, leaving a critical aspect of hiPSC-VSMC function unknown. VSMC excitability directly determines myocyte tone in resistance vessels and, in turn, systemic vascular resistance (SVR).4 Resistance vessel VSMCs are normally in a partially activated state that is dually responsive to myotonic stimulation or inhibition; small positive Vm deflections activate voltage-sensitive L-type Ca2+ (LTC) channels, while hyperpolarizing stimuli, such as activation of K+ channels, shift Vm away from the activation range of LTC channels, resulting in reduced excitability and vasodilation. Elastic vessels, such as the aorta, accommodate cardiac stroke volume during systole and passively recoil during diastole, delivering a nonpulsatile, laminar flow of blood to distal vessels. This passive function is conferred by the characteristic network of elastic fibers of large proximal vessels, which in turn is produced and deposited by elastogenic VSMCs. Increased elastin expression is a recognized consequence of potassium channel opener drugs in blood vessels,5–8 although the mechanism remains unexplained.
Vascular dysfunction underlies the complex cardiovascular phenotype of Cantú syndrome (CS), which encompasses cardiac hypertrophy and hypercontractility, patent ductus arteriosus, low systemic blood pressure, aortic root dilatation, and decreased pulse-wave velocity.9–11 CS is caused by gain-of-function (GoF) mutations in KCNJ8 and ABCC9, which encode the pore-forming Kir6.1 and regulatory SUR2B subunits of vascular KATP channels.9,12 Importantly, cardiac features of CS are driven by KATP GoF within the vasculature, mediated by elevated renin-angiotensin signaling and baroreceptor-mediated adrenergic signaling.10,13,14 CS vasculopathy is characterized by striking abnormalities in the large, elastic vessels, which exhibit gross morphological changes such as dilatation and tortuosity,15,16 as well as abnormal biomechanical properties, including excessive aortic compliance and decreased pulse wave velocity (PWV).10,12 Thus, CS vasculopathy is multifactorial with distinct hypomyotonic and hyperelastic components, which together drive broader cardiovascular consequences via elevated renin-angiotensin and adrenergic signaling.
The present study was undertaken to address multiple issues raised above, including (i) the current lack of electrophysiological characterization of hiPSC-derived VSMCs, and provide a first characterization of relevant vascular ionic currents; (ii) potential CS-dependent changes in other (ie, non-KATP) vascular currents; and (iii) the origin of hypomyotonic and hyperelastic components of CS vasculopathy, using native mouse VSMCs and hiPSC-VSMCs, and hence establish whether these phenomena arise as cell-autonomous consequences of VSMC KATP GoF or secondarily to non-VSMC KATP GoF.
Methods
Study Approval
Animal studies were performed in compliance with the standards for the care and use of animal subjects defined in the NIH Guide for the Care and Use of Laboratory Animals17 and were reviewed and approved by the Washington University Institutional Animal Care and Use Committee. Human studies were approved by the Washington University Human Studies Committee and carried out with the full consent of participating patients.
Mouse Lines
Generation of knock-in mice carrying human GoF mutations in the ABCC9 or KCNJ8 genes using CRISPR/Cas9-mediated genome engineering technology was described previously.10 Briefly, guide RNA (gRNA) target sequences were predicted using the MIT CRISPR design tool (http://crispr.mit.edu). abcc9 gRNA (5′ CATTGCCACGAAGCTGGCGG 3′) or kcnj8 gRNA (5′ ACGCCACTTCAGGTCTACCA 3′) was cloned into BbsI-digested plasmid pX330 (Addgene # 42230). T7 sgRNA template and T7 Cas9 template were prepared by polymerase chain reaction (PCR) amplification and gel purification, and injected into zygotes from superovulated B6CBA F1/J female mice mated with B6CBA F1/J male mice. After injection, zygotes were incubated at 5.5% CO2 at 37°C for 2 h, and surviving embryos were transferred to recipient mice by oviduct transfer. We identified 12 positive founder animals carrying CS mutation mouse SUR2[A476V] (equivalent to human SUR2[A478V]) and 2 positive founder animals carrying the CS mutation Kir6.1[V65M]. All founders were viable and fertile. Successful mutation of founder (F0) mice was verified by Sanger sequencing of genomic DNA, and mutant mice were subsequently crossed with C57BL/6J mice to generate heterozygous F1 Kir6.1wt/V65M and SUR2wt/A478V lines. PCR was used to generate amplicons of KCNJ8 and ABCC9 spanning >1 kb either side of the introduced mutation, from gDNA isolated from mouse tails, and resultant PCR products were sequenced to confirm the absence of unintended additional mutations. After verification, 1 F1 animal from 1 line of each genotype was selected and further crossed against C57BL/6J to generate F2 Hets, which were intercrossed to generate subsequent generation Het and Homo as well as wild-type (WT) littermates that were used in experiments.
Human iPSC Generation and Differentiation to VSMCs
CS patient-derived hiPSCs heterozygous for the R1154Q and R1154W variants were generated, respectively, from peripheral blood mononuclear cells and renal epithelial cells (RECs), which were generously provided by CS patients who had been genetically confirmed to carry these variants. Human R1154W patient RECs were reprogrammed to hiPSCs by the WUSM Genome Engineering and iPSC Core (GEiC) using Sendai virus-based reprogramming vectors. After 4 unsuccessful attempts to reprogram human R1154Q patient RECs, peripheral blood mononuclear cells were provided by the patient, and were successfully reprogrammed by the GEiC using the Sendai virus-based reprogramming cocktail. hiPSCs were maintained on a 4-d passaging cycle. Two subclonal hiPSC lines were generated for each patient sample, and DNA sequencing analysis confirmed the expected gene variant in each line. Two control lines, C2a (gift of Dr. Gordana Vunjak-Novakovic, Columbia University) and AN1.1 (gift of Amber Neilson), were used as controls. hiPSCs were differentiated to hiPSC-VSMCs by adapting the chemically defined protocol published by Patsch et al.,1 which involves initially “priming” hiPSCs into lateral plate mesodermal progenitors through Wnt activation and hBMP4 treatment, with subsequent induction to VSMC fate through co-treatment with PDGF-BB and activin A. The original protocol was followed exactly, except that differentiation was carried out in 12-well plates, rather than in T175 flasks. Cryopreserved human aortic smooth muscle cells were obtained from Lonza Pharma and Biotech (Catalog # CC-2571). Cells were plated on gelatin-coated coverslips and grown in Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) for 3-5 d before analysis. Immunohistochemical (IHC) staining for smooth muscle actin was carried out using Anti-Alpha-Smooth Muscle-Cy3 monoclonal antibody C6198 (Sigma-Aldrich).
RNA Analysis and Immunocytochemistry in Native VSMCs and hiPSC-VSMCs
For analysis of elastin transcript levels in aortae and hiPSC-VSMCs, RNA was isolated using Trizol following the manufacturer’s protocol (Life Technologies, Grand Island, NY, USA). For iPSCs, first-strand cDNA was synthesized using SuperScript III First-Strand Synthesis System (Thermo Fisher). For aortic samples, 1 μg of total RNA was reverse transcribed using a high-Capacity RNA-to-cDNA Kit, per the manufacturer’s protocol (Life Technologies, Grand Island, NY, USA). Quantitative real-time PCR was carried out using 100 ng of cDNA template, TaqMan Fast Universal PCR Master Mix, and TaqMan assays (primers/probes) purchased from Life Technologies (Grand Island, NY, USA). Reactions were run in duplicate on the QuantStudio-3 real-time PCR system (Applied Biosystems) using the FastPCR parameters (95°C for 20 s, followed by 40 cycles of 95°C for 1 s, and 60°C for 20 s). Experimental gene expression was normalized to that of Gapdh or β2-microglobulin (B2m) transcript levels, as indicated in the figures. TaqMan assays used in this study are Mm00514670_m1 (Eln), Mm99999915_g1 (Gapdh), and Mm00437762_m1 (B2m).
For immunocytochemistry, cells were fixed with 4% PFA for 5 h, permeabilized with 100% ice-cold methanol for 10 min, blocked with 1% BSA, 22.52 mg/mL glycine in PBS with 0.1% Tween 20 (PBST) for 30 min, and incubated in primary antibody diluted in 1% BSA/PBST for 1 h. After washing with PBS, slides were incubated in secondary antibody with Hoechst 34580 (0.5 µg/mL; #63493; Sigma) in 1% BSA/PBST for 1 h. After mounting with Fluoromount™ Aqueous Mounting Medium (F4680; Sigma), images were obtained using a 20× objective with a W1 Spinning Disk confocal microscope, a Fusion camera, and the Nikon Eclipse Ti2-E base. Images were then deconvolved using Nikon NIS-Elements software. Primary antibodies used for immunocytochemistry were mouse anti-PDGFRβ (1:100; ab69506; Abcam), mouse anti-Smoothelin (1:100; sc-376902, AF647 conjugated; Santa Cruz), rabbit anti-MYH11 (1:100; ab82541; Abcam), rabbit anti-α-SMA (1:100; D4K9N, XP; Cell Signaling Technology), and rabbit anti-TAGLN (1:100; ab14106; Abcam). Secondary antibodies used were acquired from Invitrogen and used at a 1:750 dilution: Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 (A-11032), Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (A-11034), and Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 633 (A-21071).
Patch Clamp EP
Mice were anesthetized with 2.5% avertin (10 mL/kg, intraperitoneal Sigma-Aldrich), and the descending aorta or mesenteric arteries were rapidly dissected and placed in ice-cold physiological saline solution (PSS) containing (in m m) NaCl 134, KCl 6, CaCl2 2, MgCl2 1, HEPES 10, and glucose 10, with pH adjusted to 7.4 with NaOH. Smooth muscle cells were enzymatically dissociated in dissociation solution containing (in m m) NaCl 55, sodium glutamate 80, KCl 5.6, MgCl2 2, HEPES 10, and glucose 10, pH 7.3 with NaOH, then placed into dissociation solution containing papain 12.5 µg/mL, dithioerythreitol 1 mg/mL, and BSA 1 mg/mL for 25 min (at 37°C), before transfer to dissociation solution containing collagenase (type H:F = 1:2) 1 mg/mL, and BSA 1 mg/mL for 5 min (at 37°C). Cells were dispersed by gentle trituration using a Pasteur pipette, plated onto glass coverslips on ice, and allowed to adhere for >1 h before transfer to the recording chamber.
Conventional whole-cell voltage-clamped currents were recorded using an Axopatch 200B amplifier and Digidata 1200 (Molecular Devices). Recordings were sampled at 3 kHz and filtered at 1 kHz. Voltage-gated K currents were measured in a high [Na+] bath solution containing (in m m) NaCl 134, KCl 5.4, CaCl2 100 μm, MgCl2 1, HEPES 10, and glucose 10, with pH adjusted to 7.4 with NaOH. The pipette solution contained (in m m) KCl 140, MgCl2 1, HEPES 10, glucose 10, EGTA 10, and ATP 5, with pH adjusted to 7.2 with KOH. Voltage-gated LTC currents were measured in high-choline bath solution containing (in m m) choline chloride 124, BaCl2 20, MgCl2 1, HEPES 10, and glucose 5, with pH adjusted to 7.4 with NaOH. The pipette solution contained (in m m) CsCl 130, MgCl2 2, HEPES 10, glucose 10, EGTA 10, NaATP 3.5, and free Ca <1 n m, with pH adjusted to 7.3 with KOH. To assess KATP conductances, currents were initially measured at a holding potential of −70 mV in high Na+ bath solution containing (in m m) NaCl 136, KCl 6, CaCl2 2, MgCl2 1, HEPES 10, and glucose 10, with pH adjusted to 7.4 with NaOH, before switching to a high K+ bath solution (KCl 140, CaCl2 2, MgCl2 1, HEPES 10, and glucose 10, with pH adjusted to 7.4 with KOH) in the absence and presence of pinacidil and glibenclamide, as indicated. The pipette solution contained (in m m) potassium aspartate 110, KCl 30, NaCl 10, MgCl2 1, HEPES 10, CaCl2 0.5, K2HPO4 4, and EGTA 5, with pH adjusted to 7.2 with KOH.
Arterial Compliance
After mice were euthanized under isoflurane anesthesia, as above the aortae of 3-wk-old mice were excised and placed in a physiological saline solution (PSS) containing (m m) 130 NaCl, 4.7 KCl, 1.18 MgSO4-, 1.17 KH2PO4, 14.8 NaHCO3, 5.5 dextrose, and 0.026 EDTA (pH 7.4). The vessels were then cleaned from surrounding fat, mounted on a pressure arteriograph (Danish Myo Technology), and maintained in PSS at 37°C. Vessels were visualized with an inverted microscope connected to a charge-coupled device camera and a computerized system, which allows continuous recording of vessel diameter. Intravascular pressure was increased from 0 to 175 mmHg by 25 mmHg increments, and the vessel outer diameter was recorded at each step (12 s per step). The average of three measurements at each pressure was reported.
Data Analysis
Unless otherwise noted, data are presented as mean ± SEM. The Real Statistics add-in package was used to run statistical analysis in Microsoft Excel. All data were tested for statistical significance using Mann-Whitney U test or Kruskal-Wallis test with post hoc Dunn’s test.
Results
Non-KATP Ionic Currents that Govern Vascular Excitability Are Not Altered in CS Vasculopathy
VSMC excitability depends on the concerted activity of multiple ion conductances, in particular voltage-gated K+ (Kv) currents and LTC currents. To provide a benchmark of these conductances in native WT VSMCs, whole-cell voltage clamp was first used to assess functional activity of Kv channels in acutely dissociated WT mouse aortic VSMCs (Figure 1). The voltage protocol was designed to inclusively survey the entire ensemble of VSMC Kv channels, with high (5 m m) ATP included in the internal solution to exclude KATP currents. Very small, linear, K+ conductance was detected below ∼−25 mV, but additional conductance with apparently instantaneous and time-dependent components were increasingly activated at more positive voltages (Figure 1A). Additionally, more positive voltage steps revealed observable single-channel activity, consistent with large-conductance calcium-activated potassium (BK) channels (Figure 1A). We carried out the same survey in aortic VSMCs acutely dissociated from mice carrying the Kir6.1[V65M] (V65M) mutation, a CS-associated variant that causes marked KATP GoF and severe CS vasculopathy.10,18 Both cell capacitance (15.5 ± 1.9 pF, n = 26 and 16.3 ± 1.8 pF, n = 20 in WT and V65M, respectively) and measured currents were very similar in V65M and WT VSMCs (Figure 1), and summarized I-V relationships show that overall current amplitudes (measured during the final 100 ms of the voltage pulse) were indistinguishable between WT and V65M VSMCs (Figure 1B). To separate Kv and BK currents, and to assess Kv current kinetics, a monoexponential function representing “idealized” Kv current activation was fitted directly to each recording at the 3 most positive voltage steps (ie, +25, +35, and + 45 mV), as shown in Figure 1C. This allowed separation of instantaneous Kv amplitudes, time-dependent Kv amplitudes, and assessment of Kv time constant (Tau). BK channel activity (NPo) was separately estimated for each record at +25, +35, and +45 mV by subtracting the idealized Kv current from each raw recording. Instantaneous and time-dependent Kv amplitudes, as well as Kv kinetics, and BK activity, were all statistically unaltered in V65M VSMCs (Figure 1C).
Figure 1.

Voltage-gated K and Ca currents in mouse CS and WT VSMCs. (A) Representative whole-cell voltage-clamp recordings show Kv currents from acutely isolated aortic VSMCs from a WT (left) and a V65M (right) mouse (scale bars = 50 pA, 100 ms). Voltage was stepped from holding potential of −65 mV to prepulse at −95 mV and then stepped more positive in 10 mV increments. Hashed box demarcates the portion depicted in the representative patch-clamp traces. (B) Peak I-V relationships (measured during the final 100 ms of the voltage pulse) show mean ± SEM for n = 9 cells from 4 mice (WT) and 5 cells from 3 mice (Vm) cells in each case. Reversal potential of −75 to −85 mV indicates that currents are carried by K+ ions. (C) Representative traces following step to +45 mV from WT (left) and V65M (right) VSMCs, fit with monoexponential functions to the minimal currents as a function of time, assumed to reflect Kv currents without the contaminating fluctuating, presumably BK current. From such traces, Kv amplitudes and kinetics, as well as BK currents, were calculated, as shown to the right (n as above). Statistical significance was determined by Mann-Whitney U test (α = 0.05, no significant differences detected). (D) Representative whole-cell voltage-clamp recordings of LTC currents were obtained from acutely isolated mesenteric VSMCs from WT (above) and V65M (below) mice (scale bars = 10 pA, 75 ms). Voltage was ramped from −70 to −45 mV over 400 ms to inactivate any high-voltage-activated currents and then stepped to voltages between −30 and +40 mV in 10 mV steps. Hashed box demarcates the portion depicted in the representative patch-clamp traces. Recordings were obtained in the presence of 1 μm Bay K8644 (left), and LTC conductance was confirmed by >95% inhibition with 10 μm nifedipine (right). (E) (Top) Peak I-V relationships on Bay K8644 are summarized, with (bottom) peak current at 0 mV for individual traces. Error bars show mean + SEM (n = 8 cells from 4 WT mice and 6 cells from 4 V65M mice in each case). Statistical significance was determined by Mann-Whitney U test (α = 0.05, no significant differences detected).
Next, whole-cell voltage clamp was used to assess functional activity of voltage-sensitive LTC channels. Ba2+ was used as a charge carrier, and included in the bath was BayK8644 (1 μm), a potentiator of vascular LTC channels. K+ currents were minimized by inclusion of Cs+ (130 m m), as well as 3.5 m m ATP to inhibit KATP currents, in the internal solution. LTC currents were undetectable in acutely isolated WT mouse aortic VSMCs (n = 8 recordings from 3 animals). However, characteristic, presumably LTC currents that were fully inhibited by nifedipine (10 μm) were present in VSMCs acutely dissociated from first- through fourth-order mesenteric arteries, which are resistance vessels (Figure 1D). Again, LTC currents recorded from mesenteric VSMCs acutely dissociated from V65M mice were essentially identical to WT (Figure 1E).
Together, the above experiments indicate that, despite marked changes in VSMC KATP currents in Cantú mice,10 there are no obvious accompanying compensatory or downstream changes in other vascular K+ or LTC currents.
Differentiation of Human iPSCs to VSMCs
To examine the EP of hiPSC-VSMCs, two genetically unrelated control human iPSC lines (C2a and AN-1.1 from individuals with no known disease-associated variants) were first differentiated to VSMCs. Human induced pluripotent stem cell-VSMCs were differentiated using the protocol developed by Patsch et al.,1 which allows rapid and efficient differentiation to VSMCs that resemble native VSMCs, based on expression of key marker genes, global transcriptomic and metabolomic signatures, and key functional characteristics including contractility and extracellular fibronectin deposition mediated by TGF-β signaling.1 This protocol is completely chemically defined and, in our hands, was reproducible in all technically successful (C2a n = 7, AN-1.1 n = 3) differentiations, where cells survived the lateral mesoderm induction and VSMC differentiation steps, under the published conditions. In each case, hiPSCs began as relatively small, round, gray cells with dark nuclei (Figure 2A). For 4 d, the cells were induced to become lateral mesodermal progenitors, which appeared slightly enlarged but morphologically similar to hiPSCs. At this stage, the cells form a confluent monolayer, and patches of dead cells begin to accrue near the end of the 4-d lateral mesoderm induction. Finally, the cells were committed to a VSMC fate via exposure to PDGF-β and Activin A. Once differentiated and cultured on a collagen-coated surface, the hiPSC-VSMCs possess a tapered, spindle-shaped morphology, and tend to align with one another, forming networks of multicellular whorls (Figure 2B).
Figure 2.

Differentiation of hiPSCs to VSMCs. (A) Images of hiPSCs (left) prior to differentiation, (center) after initial differentiation to lateral mesodermal progenitors, and (right) after full differentiation to hiPSC-VSMCs cultured on a collagen-coated surface (scale bars = 200 μm). (B) Composite fluorescent image of hiPSC-VSMCs with DAPI nuclear stain (blue) and Cy3-conjugated alpha-SMA stain (red), showing a dense network of discrete, co-aligned filaments expressed by hiPSC-VSMCs (scale bar = 50 μm). (C) Composite fluorescent images of undifferentiated iPSCs, hiPSC-VSMCs, and human aortic VSMCs, stained with DAPI nuclear stain (blue) versus antibodies for human αSMA (green), Smoothelin (magenta), Myh11 (green), Pdgfrβ (magenta), and Tagln (green) (scale bar = 50 μm in all panels).
We verified that the differentiation was effective by fixing undifferentiated hiPSCs, hiPSC-VSMCs, and cultured human aortic VSMCs and staining for multiple VSMC markers: alpha-SMA (smooth muscle actin), Smoothelin, Myh11, Pdgfrβ, and Tagln. While the ultimate maturity of the cells as VSMCs cannot be determined, both the morphology and the VSMC protein expression pattern of human iPSC-VSMCs are strikingly similar to those of native human aortic VSMCs, with alpha-SMA filaments expressed as organized, co-aligned networks (Figure 2~).
hiPSC-VSMCs Express Very Similar Ion Channels to Those in Native Mouse Arterial VSMCs
To assess the vascular ensemble of Kv currents functionally expressed in hiPSC-VSMCs, recordings were obtained from these cells using the same protocols and solutions as above in mouse VSMCs (Figure 3). Cell capacitance 18.2 ± 1.8 pF, n = 25) was similar to that measured in native mouse VSMCs above. Consistently, we observed Kv currents that were very similar to those measured in native VSMCs, with nearly identical current amplitudes and time-dependence (Figure 3A). BK currents were also evident in some recordings, although less consistently than in mouse VSMCs, and were not characterized. We also detected nifedipine-sensitive LTC currents with essentially identical time dependence and current density to those in native mesenteric VSMCs (Figure 3B).
Figure 3.

Electrophysiological characterization of hiPSC-VSMCs. (A) (Left) Representative whole-cell voltage-clamp recordings show Kv currents from a hiPSC-VSMC (scale bars = 50 pA, 100 ms). Voltage-clamp protocol was as in Figure 1. (right) Peak I-V relationship (measured during the final 100 ms of the voltage pulse) shows mean ± SEM, for n = 9 cells, as well as the WT mouse I-V from Figure 1. (B) (Top) Representative whole-cell voltage-clamp recordings of LTC currents were obtained from a hiPSC-VSMC (scale bars = 10 pA, 75 ms). Voltage-clamp protocol as in Figure 2. Recordings were obtained in the presence of 1 μm Bay K8644 (left), and LTC conductance was confirmed by >95% inhibition with 10 μm nifedipine (right). (Bottom) Peak I-V relationships in Bay K8644 are summarized (together with data from WT mouse VSMCs from Figure 2), with peak current at 0 mV displayed right for individual traces. Error bars show mean + SEM (n = 8 WT and 6 V65M cells in each case). (C) (Left) Representative whole-cell voltage-clamp recordings of KATP channel conductance from control C2a hiPSC-VSMC using an intracellular pipette solution containing no nucleotides. Cells were voltage clamped at −70 mV. (Right) Summary of mean currents in basal conditions, in 100 μm pinacidil, and in 100 μm pinacidil plus 10 μm glibeclamide (mean + SEM, n = 5 cells). Statistical significance was determined by pairwise t-test with Bonferroni correction for multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001.
These experiments provide a first survey of hiPSC-VSMC EP. While more extensive pharmacological dissection would be needed to confirm individual components, the similarity to native VSMC currents provides confidence that the essential EP of the latter matches that of the former, necessary for future studies using these hiPSC-VSMCs to understand vascular function in general, and particularly for assessing vascular myocyte excitability, contractility, and response to electrically active agents.
Pinacidil-sensitive KATP Channels Are Present in hiPSC-VSMCs
Pinacidil-sensitive KATP channels, formed by Kir6.1 and SUR2B subunits, are key determinants of VSMC membrane voltage and excitability.19 In previous studies, we characterized vascular KATP activity and pharmacology in mouse aortic VSMCs with an intracellular pipette solution containing no ATP.10 Using identical experimental conditions, we obtained whole-cell voltage-clamp recordings of basal K+ conductance in hiPSC-VSMCs (Figure 4C). K+ conductance increased substantially in the presence of a supra-maximal concentration20 of the SUR2-selective KATP channel activator pinacidil (100 μm), and current decreased following the addition of the KATP inhibitor glibenclamide (10 μm) in the continued presence of pinacidil (Figure 3C), reflecting activation of similar KATP currents to those recorded in WT mouse VSMCs.10 Pinacidil responsivity indicates that hiPSC-VSMC KATP channels comprise the same vascular-type SUR2 isoform that is expressed in native VSMCs. Reduction of pinacidil-activated currents to essentially basal levels by glibenclamide further confirms that these currents are conducted by KATP, with glibenclamide efficacy closely resembling that previously measured in native VSMCs.10 Given these observations, hiPSC-VSMCs constitute a promising approach for the study of molecular and cellular consequences of CS variants.
Figure 4.

Electrophysiological characterization of CS hiPSC-VSMCs. (A) Composite fluorescent image of sparsely plated control C2a, R1154Q, and R1154W hiPSC-VSMCs with DAPI nuclear stain (blue) and Cy3-conjugated alpha-SMA stain (red) (scale bar = 200 μm). (B) Representative whole-cell voltage-clamp recordings from control hiPSC-VSMCs (left), R1154Q hiPSC-VSMCs (middle), and R1154W hiPSC-VSMCs (right) using an intracellular pipette solution containing 100 μm MgATP and 500 μm MgADP. Cells were voltage-clamped at −70 mV. (Bottom) Summary of mean currents in basal conditions, in 100 μm pinacidil, and in 100 μm pinacidil plus 10 μm glibeclamide (mean + SEM, n = 5 cells in each case), as well as fraction of current inhibited by glibenclamide (right). (C) (Left) Representative whole-cell voltage-clamp recordings show Kv currents from control, R1154Q, and R1154W hiPSC-VSMCs (scale bars = 50 pA, 100 ms), voltage-clamp protocol as in Figure 1. (Bottom) I-V relationships show mean ± SEM for n = 9 (control), 5 (R1154Q), and 5 (R1154W) cells in each case. (D) cDNA PCR product from control and R1154Q hiPSCs reveals only a single band corresponding to a 642 bp fragment from full-length SUR2 cDNA (predicted site indicated by upper red arrowhead) and no band corresponding to the predicted 549 bp from exon 28-excluded cDNA (predicted site indicated by lower red arrowhead) (representative gel from 3 different differentiations of R1154Q patient hiPSC-derived VSMCs). For hiPSC-VSMCs, two control samples and two R1154Q samples are shown, generated from two separate differentiations. Statistical significance was determined by Mann-Whitney U test (α = 0.05). *P < 0.05, **P < 0.01, ***P < 0.001.
Increased Basal KATP Activity, and Decreased Sensitivity to Glibenclamide, in CS Patient-Derived hiPSC-VSMCs
We next developed human iPSC-derived vascular myocyte models for CS using two patient-derived iPSC lines. These were generated using Sendai virus-based reprogramming vectors on PBMCs and RECs obtained from CS patients carrying the R1154Q (R1154Q) and R1154W (R1154W) mutations.12 Two subclonal hiPSC lines were produced for each mutation, and DNA sequencing analysis confirmed the expected mutation in each. Expression of human pluripotency-associated genes and a normal karyotype were confirmed for all hiPSCs prior to subsequent experiments, and CS hiPSC-VSMCs were generated as described above. Differentiation was highly efficient for both R1154Q and R1154W hiPSC-VSMCs in 5 differentiations for each genotype, and differentiated cells were morphologically indistinguishable from control hiPSC-VSMCs, with alpha-SMA consistently expressed in control, R1154Q, and R1154W hiPSC-VSMCs (>99% positive for each cell line, Figure 4A).
Whole-cell voltage clamp was used to measure KATP currents in R1154Q and R1154W hiPSC-VSMCs, as well as control C2a hiPSC-VSMCs (Figure 4B). Experimental conditions were similar to those described above, but the pipette solution contained 100 μm MgATP and 500 μm MgADP (see the “Methods” section) to determine channel behavior under nucleotide regulation, relevant to KATP activity in vivo. Cell capacitance (21.4 ± 1.9 pF, n = 21) was similar to that measured in control hiPSC-VSMCs and native mouse VSMCs above. As shown in Figure 4B, basal K+ conductances were elevated in each of the CS patient-derived hiPSC-VSMCs compared to control cells (>4-fold in R1154Q cells and > 2-fold in R1154W cells, note logarithmic scale). Additional pinacidil-activated current was ∼2.5× basal in WT (Figure 4B) and ∼5.5× in R1154Q hiPSC-VSMCs (Figure 4B), but only ∼1.9× in R1154W hiPSC-VSMCs (Figure 4B).
In control hiPSC-VSMCs, glibenclamide inhibited almost 100% of the pinacidil-activated current, but glibenclamide action was reduced in mutant cells, inhibiting only ∼80% and ∼55% of pinacidil-activated current in R1154Q and R1154W, respectively (Figure 4B). Increased basal currents in the CS mutant cells are consistent with elevated basal KATP activity due to the known molecular consequences of the mutations.21,22 However, since glibenclamide fails to reduce these currents to basal levels, we cannot rule out the possibility that increased basal K+ currents are non-KATP-mediated. To test this possibility, whole-cell voltage clamp was used, as described above, to characterize the vascular ensemble of functional Kv currents with high (5 m m) ATP in the pipette (Figure 4C). I-V relationships revealed no significant basal K+ conductances. Voltage-activated Kv currents were nonsignificantly reduced in R1154Q and R1154W hiPSC-VSMCs compared to control, confirming that elevated basal currents in Figure 4B are indeed KATP mediated, and further suggesting that KATP GoF in CS vasculopathy does not lead to secondary, cell-autonomous changes in vascular Kv currents.
Cell-autonomous Consequences of Genetic KATP Overactivity Underlie Both Hypomyotonic and Hyperelastic Components of CS Vasculopathy
We previously generated a third CS mouse model in which the SUR2[R1154Q] was knocked into the endogenous locus.23 In contrast to mice carrying the SUR2[A478V] or Kir6.1[V65M] CS-associated variants, SUR2-dependent KATP currents and the resultant CS phenotype were both minimal in R1154Q mice. This was associated with alternate splicing leading to excision of exon 28, 3’ of the introduced variant, and to non-functional SUR2.23 If occurring in human patients with this variant, such splice excision could significantly ameliorate the CS features resulting from the molecular GoF itself. However, RT-PCR analysis of cDNA amplified from RNA isolated from control and R1154Q hiPSC-VSMCs (Figure 4D) revealed only a single transcript band, with no evidence of exon 28 exclusion, in agreement with prior observations from primary human tissue samples.23 This indicates that exon 28 exclusion is only seen in the R1154Q mouse and that a purely GoF phenotype will be expected in human R1154Q cells. Consistent with this, there was a striking increase in KATP activity in R1154Q hiPSC-VSMC patch-clamp recordings (Figure 4B).
To understand how KATP GoF impacts VSMC excitability under physiological conditions, whole-cell current-clamp (I = 0) was used to measure membrane voltage (Vm) immediately (<2 s) upon break-in. In the two genetically unrelated control C2a and AN-1.1 hiPSC-VSMC lines, we observed identical mean Vm values (−44 mV), consistent with reports of native VSMC resting Vm, which lies in the range of −35 to −45 mV.24–26 Consistent with enhanced basal KATP conductance (Figure 4B), R1154Q and R1154W hiPSC-VSMCs were both hyperpolarized by 10-15 mV relative to both controls (Figure 5A).
Figure 5.

Consequences of CS mutations on human iPSC-VSMC excitability and elastin expression. (A) (Top) Representative whole cell current-clamp (at zero current) recordings from two control hiPSC-VSMC lines (left; C2a, AN-1.1) and two CS variant hiPSC-VSMC lines (right; R1154Q, R1154W) using an intracellular pipette solution absent of nucleotides. (Bottom) Individual and mean (± SEM, n = 6-16 cells) initial Vm following break-in from experiments as above. (B) Vessel compliance in pressurized aortae of p21 WT, heterozygous and homozygous Kir6.1[V65M] mice (left), and WT, heterozygous and homozygous SUR2[A478V] (right) mice. Data show mean + SEM, n = 4-5 animals in each case. (C) Elastin mRNA expression (normalized to GAPDH expression) in aortae from WT and V65M or A478V CS mice (left 2 panels), and from C2a control or R1154W and R1154Q hiPSC-VSMCs, normalized to β2 microglobulin (right panel). Data are from separate differentiations in each case. Statistical significance was determined by Mann-Whitney U test (α = 0.05). *P < 0.05, **P < 0.01, ***P < 0.001.
Finally, tortuous, dilated blood vessels with decreased PWV,15,16,27 as well as joint hyperelasticity12,28 and tracheomalacia,12 are major features of CS. Potentially, these might all be related to excess elastin production, which has been reported to occur in blood vessels and in cultured VSMCs treated with the KATP activators minoxidil5–8 and pinacidil.29 As shown in Figure 5B, compliance was increased in aortae from homozygous A478V and both hetero- and homozygous V65M mice at P21, the effect being proportional to the severity of the molecular consequences (from WT < Het < Hom in each case). In each case, similar trends in increased elastin mRNA expression in isolated vessels, were measured at 3 mo of age (Figure 5C). We further measured mRNA expression from control (C2a), R1154Q, and R1154W hiPSC-VSMCs. Elastin gene expression was barely detectable in control C2a samples and was higher in both of the CS lines, significantly so in R1154W (Figure 5C). Taken together, these data are consistent with the increased elastin expression seen in CS being a direct, cell-autonomous, response to the increased VSMC KATP activity.
Discussion
The Lack of Electrical Compensation for KATP GoF in CS
Blood pressure depends on vascular tone, increasing or decreasing in response to membrane depolarization or hyperpolarization from a potential (Vm) that is typically close to the activation threshold for LTC currents.24–26 In this voltage range, subtle changes in the activity of KATP or other channels, whether genetic or in response to neurohumoral signaling, will be expected to change vascular excitability, unless compensated by changes in another current. The finding that VSMCs from CS mice are basally hyperpolarized23 (as too are GI SMCs30) is consistent with our finding of ~enhanced basal KATP conductance in CS that is uncompensated by changes in other conductances. This is consistent with previous studies in cardiac muscle, which showed no compensation for KATP subunit knockout by other KATP subunits nor changes in Kv currents in response to KATP GoF in other KATP channel subunits.31,32 More broadly, genetic knockout of background Kir2.1 channels in vascular or cardiac myocytes is also not compensated for by changes in Kv or LTC currents.33,34
Therefore, while we did not utilize pharmacological dissection to determine specific conductances and cannot completely rule out changes in all vascular ion channels, no changes in the major Kv and LTC currents that govern vascular excitability appear to contribute to, or compensate for, CS vasculopathy. While different vascular beds can have quite different properties, lack of compensation by other currents in mesenteric and aortic cells helps to explain why, despite the homeostatic imperative, manipulation of VSMC KATP conductance leads to marked changes in blood pressure in experimental animals, from a mean BP of ∼115 mmHg in Kir6.1−/− animals to ∼85 mmHg in smooth muscle-direct KATP GoF transgenic animals.35 Interestingly, the results contrast with the finding that cardiac LTC currents are actually increased with cardiac KATP GoF,32,36 which is a potentially compensatory consequence in response to enhanced adrenergic signaling.36
Human iPSC-derived VSMCs Recapitulate Native VSMC EP
Human VSMCs are not readily accessible, and the EP of human VSMCs has received relatively little attention.37–39 Human iPSC-derived VSMCs offer a potentially profitable model in which to explore human VSMC biology in general, including EP. We used an established protocol consisting of fully chemically defined conditions to differentiate hiPSCs into alpha-SMA-positive hiPSC-VSMCs via lateral mesodermal lineage, and then used whole-cell patch-clamp EP to characterize ion currents in identical conditions to those used above for native mouse VSMCs. The Kv current ensemble in human iPSC-VSMCs closely resembles that in native mouse VSMCs, confirming that hiPSC-VSMCs recapitulate key components of native vascular EP. Moreover, the non-inactivating time-dependent delayed rectifier K+ currents, as well as noisy, non-inactivating currents that we ascribe to Ca2+-activated maxi-K (BK) channels, are quite similar to those originally reported in human mesenteric arterial myocytes.39 In addition, we detected LTC currents at similar levels, and with identical voltage dependence, to those in mouse mesenteric myocytes, and again quite similar to those reported in human mesenteric arterial myocytes.40
Cantú Syndrome Patient-derived hiPSC-VSMCs Exhibit Elevated KATP Activity, With Decreased Glibenclamide Sensitivity
We also found that KATP channels are expressed in hiPSC-VSMCs at similar levels to those in murine VSMCs.10 Pharmacological sensitivity to the SUR2-selective drug pinacidil shows that these KATP channels comprise the appropriate “vascular-type” architecture. In CS patient-derived hiPSC-VSMCs, KATP conductances were much higher than control under basal conditions in R1154W myocytes, and even more so in the R1154Q myocytes. This is consistent with the effect of both R1154W and particularly R1154Q mutations to enhance Mg-nucleotide activation of recombinant KATP channels.22 Both mutations also increased the pinacidil-activated current—an effect that was much more prominent in R1154Q cells. Since pinacidil acts to stabilize Mg-nucleotide-activated channels, this is also consistent with the much greater MgATP-activating effect in R1154Q versus R1154W channels.22 Finally, the relative inhibitory effect of glibenclamide was lower for R1154Q, and more so for R1154W hiPSC-VSMCs, again consistent with findings from recombinant mutant channels.22 These data suggest that individuals with the two most common CS variants12 may receive limited therapeutic effect from glibenclamide or other sulfonylurea drugs and may instead benefit from drugs that act through a separate binding site and/or distinct allosteric mechanism.
Distinct Components of CS Vasculopathy Can Arise From Cell-autonomous Effects in VSMCs
Our recent studies have made it increasingly clear that multiple pathological features of CS arise primarily from KATP GoF in smooth muscle.10,13,41 At least one component involves decreased SVR,10,13,41 which is a reflection of vascular tone and is in turn determined by VSMC membrane potential. Importantly, control hiPSC-VSMCs from two genetically unrelated origins exhibited essentially the same membrane potential, similar to prior reports in native VSMCs,24–26 and somewhat depolarized relative to that of nonvascular myocytes, near the activation range for LTC channels. Consistent with KATP currents being major determinants of vascular membrane potential and hence excitability,42 the resting membrane potential was markedly more negative in both CS lines, providing direct evidence for increased basal KATP in VSMCs themselves, driving cellular hyperpolarization, reducing excitability, and promoting vasodilation and lowered SVR.9,12,13,30
R1154Q and R1154W are the most common CS-associated variants in SUR212 and cause very marked molecular GoF.21,22 Unexpectedly, the phenotype of a CS mouse in which SUR2[R1154Q] was knocked in23 was minimal compared to mice carrying the CS-associated SUR2[A478V] or Kir6.1[V65M], due to alternate splicing leading to excision of exon 28, which led to nonfunctional channels. Such splice excision could significantly ameliorate the CS features resulting from the mutation itself if it happened in humans. However, the lack of any detectable exon 28 exclusion in hiPSC-VSMCs, or in CS patient biopsies,23 suggests that this is not occurring in humans.
CS vasculopathy includes not only the hypomyotonic component considered above, but also a hyperelastic component, characterized by tortuous vessels with increased compliance and decreased PWV, as well as aortic root dilation, associated with aortic insufficiency, and aortic aneurysms.15,16,27,43,44 These latter phenomena are not trivially explained by any recognized effect of KATP activation, but are consistent with multiple previous studies that have shown increased elastin expression in cultured cells and blood vessels exposed to pharmacological KATP activators, including minoxidil and pinacidil.5–8,29 VSMCs in culture behave differently from that in a vessel, and do not generate layers of elastin, such that our experiments cannot formally exclude additional systemic consequences in vivo, but detection of enhanced elastin mRNA expression in CS variant hiPSC-VSMCs implicates at least a cell-autonomous effect of KATP GoF. The connection between KATP activity and elastin gene expression remains unexplained and will require further study, but, like the hypomyotonic component, it is potentially linked to effects of membrane potential on cellular calcium levels.
Conclusions
Our results represent a first characterization of hiPSC-VSMC EP and demonstrate that hiPSC-VSMCs recapitulate the typical electrical currents seen in native VSMCs, an important validation for the use of such cells for the study of human vascular pathologies in general. While we cannot be certain of the ultimate maturity of the hiPSC-VSMCs, and hence how well they model proliferative versus contractile myocytes, our data also shed light on cell-autonomous nature of both the hypomyotonic and hyperelastic components of CS vasculopathy. Our findings validate a CS patient-derived human cell model that is suitable both for detailed study of the earliest mechanisms of CS vascular pathologies and for identification of new pharmacotherapeutic candidates.
Novelty and Significance
CS, which is caused by GoF in vascular KATP channels, is characterized by large-vessel dilatation with decreased pulse-wave velocity, as well as low systemic vascular resistance, reflecting hyperelastic and hypomyotonic components. First, we have shown that the functional expression of voltage-gated K+ and Ca2+ currents is essentially the same in mouse VSMCs from WT and CS mice, revealing lack of any compensatory electrical response. We then derived VSMCs from hiPSC-VSMCs and made a first characterization of their EP, showing that functional expression of voltage-gated K+ and Ca2+ currents in control hiPSC-VSMCs is very similar to that in mouse arterial VSMCs. Both basal and pinacidil-activated KATP currents were considerably larger in CS hiPSC-VSMCs. Consistent with lack of cell-autonomous modulation of other currents, this resulted in membrane hyperpolarization, explaining the hypomyotonic basis of CS vasculopathy. Consistent with the hyperelastic component of CS, we also observed increased compliance and dilatation in isolated CS mouse aortas, which was associated with increased elastin mRNA expression (homozygous > heterozygous). We also found increased elastin mRNA in 2 genetically unrelated lines of CS hiPSC-VSMCs. These results show that increased VSMC elastin expression is also a cell-autonomous consequence of KATP channel activation.
Acknowledgement
We are very grateful to Kristina Hinman and Robert Mecham (Department of Cell Biology and Physiology) for help with compliance measurements.
Contributor Information
Alex Hanson, Center for the Investigation of Membrane Excitability Diseases, Washington University School of Medicine, St. Louis, MO 63110, USA; Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, MO 63110, USA.
Conor McClenaghan, Center for the Investigation of Membrane Excitability Diseases, Washington University School of Medicine, St. Louis, MO 63110, USA; Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, MO 63110, USA.
Kuo-Chan Weng, Department of Biomedical Engineering, Washington University in Saint Louis, St. Louis, MO 63110, USA.
Sarah Colijn, Center for the Investigation of Membrane Excitability Diseases, Washington University School of Medicine, St. Louis, MO 63110, USA; Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, MO 63110, USA.
Amber N Stratman, Center for the Investigation of Membrane Excitability Diseases, Washington University School of Medicine, St. Louis, MO 63110, USA; Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, MO 63110, USA.
Carmen M Halabi, Department of Pediatrics, Washington University in St. Louis, St. Louis, MO 63130, USA.
Dorothy K Grange, Department of Pediatrics, Washington University in St. Louis, St. Louis, MO 63130, USA.
Jonathan R Silva, Department of Biomedical Engineering, Washington University in Saint Louis, St. Louis, MO 63110, USA.
Colin G Nichols, Center for the Investigation of Membrane Excitability Diseases, Washington University School of Medicine, St. Louis, MO 63110, USA; Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, MO 63110, USA.
Author Contributions
A.H., C.M., and C.G.N. originally conceived the study; D.K.G. was responsible for clinical contact with patients; A.H., K.-C.W., and J.R.S. developed the iPSC-VSMCs; A.H. carried out all electrophysiology; A.H., S.C., and A.N.S. carried out histology; A.H. and C.G.N. carried out analysis; C.M.H. carried out compliance measurements; A.H., C.M.H. carried out expression analysis; A.H. and C.G.N. wrote the paper, which was edited by all authors.
Funding
This work was supported by NIH grants R35 HL171542 to C.G.N. and R21 HD103347 to C.G.N. and D.K.G., and by Pilot and Feasibility Grant [CIMED-18-04] to C.H. A.H. was supported by NIH grant T32 HL125241. C.M. was supported by NIH grant R00 HL150277. C.M.H. was supported by NIH grant K08 HL135400.
Conflict of Interest Statement
C.G.N. holds the position of Executive Editor for Function and is blinded from reviewing or making decisions for the manuscript.
Data Availability
The data underlying this article will be shared on reasonable request to the corresponding author.
References
- 1. Patsch C, Challet-Meylan L, Thoma EC et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat Cell Biol. 2015;17(8):994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maguire EM, Xiao Q, Xu Q. Differentiation and application of induced pluripotent stem cell-derived vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2017;37(11):2026–2037. [DOI] [PubMed] [Google Scholar]
- 3. Wu JC, Garg P, Yoshida Y et al. Towards precision medicine with human iPSCs for cardiac channelopathies. Circ Res. 2019;125(6):653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cole WC, Gordon GR, Braun AP. Cellular and ionic mechanisms of arterial vasomotion. Adv Exp Med Biol. 2019;1124:297–312. [DOI] [PubMed] [Google Scholar]
- 5. Hayashi A, Suzuki T, Wachi H et al. Minoxidil stimulates elastin expression in aortic smooth muscle cells. Arch Biochem Biophys. 1994;315(1):137–141. [DOI] [PubMed] [Google Scholar]
- 6. Tajima S, Hayashi A, Suzuki T, Nishikawa T. Stimulation of elastin expression by minoxidil in chick skin fibroblasts. Arch Dermatol Res. 1995;287(5):494–497. [DOI] [PubMed] [Google Scholar]
- 7. Knutsen RH, Beeman SC, Broekelmann TJ et al. Minoxidil improves vascular compliance, restores cerebral blood flow, and alters extracellular matrix gene expression in a model of chronic vascular stiffness. Am J Physiol Heart Circ Physiol. 2018;315(1):H18–H32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fhayli W, Boyer M, Ghandour Z et al. Chronic administration of minoxidil protects elastic fibers and stimulates their neosynthesis with improvement of the aorta mechanics in mice. Cell Signal. 2019;62:109333. [DOI] [PubMed] [Google Scholar]
- 9. Nichols CG, Singh GK, Grange DK. KATP channels and cardiovascular disease: suddenly a syndrome. Circ Res. 2013;112(7):1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang Y, McClenaghan C, Harter TM et al. Cardiovascular consequences of KATP overactivity in Cantú syndrome. JCI Insight. 2018;3(15):e121153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singh GK, McClenaghan C, Aggarwal M et al. A unique high-output cardiac hypertrophy phenotype arising from low systemic vascular resistance in Cantú syndrome. J Am Heart Assoc. 2022;11(24):e027363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grange DK, Roessler HI, McClenaghan C et al. Cantú syndrome: findings from 74 patients in the International Cantú Syndrome Registry. Am J Med Genet C Semin Med Genet. 2019;181(4):658–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McClenaghan C, Huang Y, Matkovich SJ et al. The mechanism of high-output cardiac hypertrophy arising from potassium channel gain-of-function in Cantú syndrome. Function (Oxf). 2020;1(1):zqaa004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McClenaghan C, Huang Y, Yan Z et al. Glibenclamide reverses cardiovascular abnormalities of Cantú syndrome driven by KATP channel overactivity. J Clin Invest. 2020;130(3):1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leon Guerrero CR, Pathak S, Grange DK et al. Neurologic and neuroimaging manifestations of Cantú syndrome: a case series. Neurology. 2016;87(3):270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kisilevsky E, Kohly RP, Margolin EA. Dilated and tortuous retinal vessels as a sign of Cantú syndrome. Ophthalmic Genet. 2019;40(5):453–454. [DOI] [PubMed] [Google Scholar]
- 17. Jahangir A, Terzic A. K(ATP) channel therapeutics at the bedside. J Mol Cell Cardiol. 2005;39(1):99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cooper PE, McClenaghan C, Chen X, Stary-Weinzinger A, Nichols CG. Conserved functional consequences of disease-associated mutations in the slide helix of Kir6.1 and Kir6.2 subunits of the ATP-sensitive potassium channel. J Biol Chem. 2017;292(42):17387–17398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flagg TP, Enkvetchakul D, Koster JC, Nichols CG. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev. 2010;90(3):799–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Quayle JM, Bonev AD, Brayden JE, Nelson MT. Pharmacology of ATP-sensitive K+ currents in smooth muscle cells from rabbit mesenteric artery. Am J Physiol. 1995;269(5):C1112–C1118. [DOI] [PubMed] [Google Scholar]
- 21. Harakalova M, van Harssel JJ, Terhal PA et al. Dominant missense mutations in ABCC9 cause Cantú syndrome. Nat Genet. 2012;44(7):793–796. [DOI] [PubMed] [Google Scholar]
- 22. McClenaghan C, Hanson A, Sala-Rabanal M et al. Cantú syndrome-associated SUR2 (ABCC9) mutations in distinct structural domains result in KATP channel gain-of-function by differential mechanisms. J Biol Chem. 2018;293(6):2041–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang H, Hanson A, de Almeida TS et al. Complex consequences of Cantú syndrome SUR2 variant R1154Q in genetically modified mice. JCI Insight. 2021;6(5):e145934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Welsh DG, Jackson WF, Segal SS. Oxygen induces electromechanical coupling in arteriolar smooth muscle cells: a role for L-type Ca2+ channels. Am J Physiol. 1998;274(6):H2018–2024. [DOI] [PubMed] [Google Scholar]
- 25. Welsh DG, Nelson MT, Eckman DM, Brayden JE. Swelling-activated cation channels mediate depolarization of rat cerebrovascular smooth muscle by hyposmolarity and intravascular pressure. J Physiol. 2000;527 Pt 1(1):139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Loutzenhiser R, Chilton L, Trottier G. Membrane potential measurements in renal afferent and efferent arterioles: actions of angiotensin II. Am J Physiol. 1997;273(2 Pt 2):F307–314. [DOI] [PubMed] [Google Scholar]
- 27. Brownstein CA, Towne MC, Luquette LJ et al. Mutation of KCNJ8 in a patient with Cantú syndrome with unique vascular abnormalities—support for the role of K(ATP) channels in this condition. Eur J Med Genet. 2013;56(12):678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Czeschik JC, Voigt C, Goecke TO et al. Wide clinical variability in conditions with coarse facial features and hypertrichosis caused by mutations in ABCC9. Am J Med Genet A. 2013;161A(2):295–300. [DOI] [PubMed] [Google Scholar]
- 29. Slove S, Lannoy M, Behmoaras J et al. Potassium channel openers increase aortic elastic fiber formation and reverse the genetically determined elastin deficit in the BN rat. Hypertension. 2013;62(4):794–801. [DOI] [PubMed] [Google Scholar]
- 30. York NW, Parker H, Xie Z et al. Kir6.1- and SUR2-dependent KATP over-activity disrupts intestinal motility in murine models of Cantú syndrome. JCI Insight. 2020;5(23):e141443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Koster JC, Knopp A, Flagg TP et al. Tolerance for ATP-insensitive K(ATP) channels in transgenic mice. Circ Res. 2001;89(11):1022–1029. [DOI] [PubMed] [Google Scholar]
- 32. Flagg TP, Charpentier F, Manning-Fox J et al. Remodeling of excitation-contraction coupling in transgenic mice expressing ATP-insensitive sarcolemmal KATP channels. Am J Physiol Heart Circ Physiol. 2004;286(4):H1361–H1369. [DOI] [PubMed] [Google Scholar]
- 33. Zaritsky JJ, Eckman DM, Wellman GC, Nelson MT, Schwarz TL. Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K(+) current in K(+)-mediated vasodilation. Circ Res. 2000;87(2):160–166. [DOI] [PubMed] [Google Scholar]
- 34. Zaritsky JJ, Redell JB, Tempel BL, Schwarz TL. The consequences of disrupting cardiac inwardly rectifying K+ current (IK1) as revealed by the targeted deletion of the murine Kir2.1 and Kir2.2 genes. J Physiol. 2001;533(3):697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li A, Knutsen RH, Zhang H et al. Hypotension due to Kir6.1 gain-of-function in vascular smooth muscle. J Am Heart Assoc. 2013;2(4):e000365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Levin MD, Singh GK, Zhang HX et al. KATP channel gain-of-function leads to increased myocardial L-type Ca2+ current and contractility in Cantú syndrome. Proc Nat Acad Sci USA. 2016;113(24):6773–6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Akbarali HI, Wyse DG, Giles WR. Ionic currents in single cells from human cystic artery. Circ Res. 1992;70(3):536–545. [DOI] [PubMed] [Google Scholar]
- 38. Hu ZY, Lin PT, Liu J, Liao DQ. Remifentanil induces L-type Ca2+ channel inhibition in human mesenteric arterial smooth muscle cells. Can J Anaesth. 2008;55(4):238–244. [DOI] [PubMed] [Google Scholar]
- 39. Smirnov SV, Aaronson PI. Ca(2+)-activated and voltage-gated K+ currents in smooth muscle cells isolated from human mesenteric arteries. J Physiol. 1992;457(1):431–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li PY, Zeng XR, Cheng J, Wen J, Inoue I, Yang Y. Rhynchophylline-induced vasodilation in human mesenteric artery is mainly due to blockage of L-type calcium channels in vascular smooth muscle cells. Naunyn Schmiedebergs Arch Pharmacol. 2013;386(11):973–982. [DOI] [PubMed] [Google Scholar]
- 41. McClenaghan C, Huang Y, Yan Z et al. Glibenclamide reverses cardiovascular abnormalities of Cantú syndrome driven by KATP channel overactivity. J Clin Invest. 2020;130(3):1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. 1997;77(4):1165–1232. [DOI] [PubMed] [Google Scholar]
- 43. Hiraki Y, Miyatake S, Hayashidani M et al. Aortic aneurysm and craniosynostosis in a family with Cantú syndrome. Am J Med Genet A. 2014;164A(1):231–236. [DOI] [PubMed] [Google Scholar]
- 44. Parrott A, Lombardo R, Brown N, Tretter JT, Riley L, Weaver KN. Cantú syndrome: a longitudinal review of vascular findings in three individuals. Am J Med Genet A. 2020;182(5):1243–1248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.