

Abstract
Denitrification is a key metabolic process in the global nitrogen cycle and is performed by taxonomically diverse microorganisms. Despite the widespread importance of this metabolism, challenges remain in identifying denitrifying populations and predicting their metabolic end-products based on their genotype. Here, genome-resolved metagenomics was used to explore the denitrification genotype of Bacillota enriched in nitrate-amended high temperature incubations with confirmed N2O and N2 production. A set of 12 hidden Markov models (HMMs) was created to target the diversity of denitrification genes in members of the phylum Bacillota. Genomic potential for complete denitrification was found in five metagenome-assembled genomes from nitrate-amended enrichments, including two novel members of the Brevibacillaceae family. Genomes of complete denitrifiers encode N2O reductase gene clusters with clade II-type nosZ and often include multiple variants of the nitric oxide reductase gene. The HMM set applied to all genomes of Bacillota from the Genome Taxonomy Database identified 17 genera inferred to contain complete denitrifiers based on their gene content. Among complete denitrifiers it was common for three distinct nitric oxide reductases to be present (qNOR, bNOR, and sNOR) that may reflect the metabolic adaptability of Bacillota in environments with variable redox conditions.
Keywords: denitrification, metagenomics, endospores, Bacillota, nitrate
Introduction
Denitrification is a key metabolic process in the nitrogen cycle featuring sequential reduction of nitrate to nitrite and then gaseous metabolites (NO3− → NO2− → NO → N2O → N2). Different enzymes catalyze each of the four reduction reactions such that this modular metabolism can be performed by a single microorganism or a microbial consortium. When performed modularly, microorganisms can achieve complete denitrification by cross-feeding intermediates [1]. If denitrification is incomplete, this can give rise to the release of the greenhouse gas N2O to the atmosphere [2, 3]. Denitrification is prevalent in terrestrial and aquatic environments where oxic and anoxic conditions occur close to each other [4]. In soil environments, denitrification can contribute to losses of fixed nitrogen to the atmosphere reducing soil fertility and plant yield [5]. On the other hand, denitrification in wetlands mitigates the transport of nitrogen from land to lakes and coastal waters where excess nitrogen can cause eutrophication [6]. Nitrogen removal in wastewater and agricultural sectors via denitrification similarly represents a critical step that limits the release of excess to nitrogen into watersheds [7, 8]. Identifying microorganisms contributing towards denitrification in different environments is therefore of ecological and industrial importance.
Microorganisms known to perform denitrification are taxonomically diverse and span both bacterial and archaeal domains [9, 10]. The taxonomic diversity of denitrifiers means that this metabolism cannot be easily linked to phylogeny [11]. Instead, characterization of denitrifying populations relies on the presence of metabolic genes for each of the reduction steps. Nitrate reduction to nitrite (NO3− → NO2−) is catalyzed by the membrane-bound Nar enzyme or periplasmic Nap enzyme, both of which can be found in denitrifiers and nitrate-ammonifiers (NO3− → NH4+) [12, 13]. Thus, the presence of nitrite reductase (Nir) genes for nitrite reduction to nitric oxide (NO2− → NO), nitric oxide reductase (Nor) genes for nitric oxide reduction to nitrous oxide (NO → N2O), and the nitrous oxide reductase (Nos) gene for nitrous oxide reduction to dinitrogen (N2O → N2) differentiates the denitrification pathway from dissimilatory nitrate reduction to ammonium (DNRA).
Enzymes from the denitrification pathway exhibit broad taxonomic and sequence diversity. Nitrite reduction to nitric oxide is catalyzed by two structurally different enzymes, Cu-type NirK and cytochrome cd-1 type NirS, that have different evolutionary histories [14–16]. Nitric oxide reductases are members of the heme-copper oxidase (HCO) superfamily and are ancestral to terminal oxidases for aerobic respiration [17, 18]. Four Nor enzyme families have been biochemically characterized: cNOR [19], qNOR [20], bNOR (formerly CuANOR; [21]) and eNOR [22]. Cytochrome c-dependent (cNOR) and quinol-dependent (qNOR) Nor enzymes are related to C-family oxygen reductases whereas bNOR and the recently characterized eNOR are related to B-family oxygen reductases [18, 22]. Based on phylogenomic analysis and conserved proton channels, an additional three enzymes related to B-family oxygen reductases, sNOR, gNOR, and nNOR, have also been proposed [22].
In contrast to nitric oxide reduction, nitrous oxide reduction is catalyzed by just one enzyme, NosZ. This enzyme, however, forms two distinct groups known as clade I (typical) or clade II (atypical) characterized by different secretionary pathways (tat and sec, respectively) [10, 23]. Clade I is represented by well-studied denitrifying Proteobacteria (alpha-, beta-, and gamma-) whereas clade II is taxonomically diverse and encompasses at least 12 bacterial and archaeal phyla (subclades A-K) [10, 24].
The broad diversity and sequence divergence among denitrification enzymes gives rise to well-documented coverage limitations for PCR primers and probes [10, 11, 15, 25, 26]. This makes it challenging to accurately estimate the diversity and abundance of denitrification genes. Metagenomics circumvents primer bias limitations and is therefore advantageous for studying denitrification. Here, genome-resolved metagenomics was employed to explore the gene content of microorganisms enriched in the presence of nitrate in heated oil sands from outcrops in Alberta, Canada. Understanding thermophilic populations in oil sands and their nitrate-reducing metabolism is of interest for technologies that target in situ microbial activity [27]. Compared to conventional crude oil reservoir ecosystems, oil sands are not well characterized microbiologically but the presence of both mesophilic and thermophilic populations in riverbank outcrops and subsurface deposits has been reported [28, 29]. Our data show that dormant thermophilic endospore-forming (thermospore) populations with distinct denitrification genotypes are present in the oil sands microbiome.
Materials and Methods
Sample collection
Samples were collected from the Athabasca oil sands in Alberta, Canada, in June 2019. In this region, oil sands are present at various depths (up to 100 s of meters) and outcrops are naturally exposed along the riverbanks of the Athabasca River and its tributaries. Oil sands samples were collected from an outcrop at the Hangingstone River (56°42′38”N, 111°23′51″) in Fort McMurray. Samples were stored in a cold room at 4°C until incubations were established. Parallel samples were frozen for DNA extraction and analyzed to represent unincubated samples.
Nitrate-amended enrichments
Approximately 30 g oil sands inoculum containing ~14% bitumen, 4% water, and 82% sand was combined with 60 mL anoxic medium in 160 mL Wheaton glass serum bottles. Growth medium was based upon media previously used to isolate nitrate-reducing and fermentative thermophilic Bacillota (formerly Firmicutes) from hydrocarbon environments (Adkins et al., 1992; Salinas et al., 2004) and contained (L−1 distilled water): 0.2 g MgCl2•6H2O, 0.1 g KCl, 1 g NH4Cl, 0.1 g CaCl2•2H2O, 0.3 g K2HPO4, 0.3 g KH2PO4, 1 g NaCl, 0.2 g yeast extract. NaNO3 (2 g/L) was added as an electron acceptor to promote denitrification. Oil sands are heavily biodegraded such that microbial growth with bitumen as the sole carbon and electron source in these microcosms is negligible [30, 31]. Glucose (0.9 g/L) was therefore added to the enrichments as an easily biodegradable organic substrate. Cysteine hydrochloride (0.5 g/L), NaHCO3 (2.5 g/L), vitamins and trace minerals were added from sterile stock solutions. Anoxia was established with He to enable sub-samples of headspace gas to be analyzed for the presence of nitrogen compounds. Throughout the incubation period, sub-samples of the sand and water mixture (1.5 mL) were periodically removed from enrichments using a He-flushed syringe. Sub-samples were centrifuged (10 000 × g for 5 minutes) with supernatants filtered (0.2 μm) and frozen eventual for chemical analysis and pellets frozen for eventual DNA extraction.
Headspace gas measurement
Headspace gases (1 mL) were extracted from experimental incubations with a He-flushed syringe and immediately injected into two chain-connected sample loops on an Agilent 7890B gas chromatograph (GC). CO2 was first separated on a Hayesep N packing column (stainless steel tubing, 0.5 m length, 1/8″ OD, 2 mm ID, mesh size 80/100) followed by N2 separation on a MolSieve 5A packing column (UltiMetal tubing, 2.44 m length, 1/8″ OD, 2 mm ID, mesh size 60/80) with He carrier gas. Both CO2 and N2 were measured by thermal conductivity detector at 200°C. Through a second line, N2O was separated on a Hayseed Q packing column (stainless steel tubing, 6′ length, 1/8″ OD, 2.1 mm ID, mesh size 80/200) with Ar/CH4 5/95% carrier gas. N2O was measured by electron capture detector at 300°C. All columns were set in the same oven with a working temperature of 105°C.
Chemical analyses
Nitrate and nitrite were measured with a Dionex ICS-5000 reagent-free ion chromatography system equipped with an anion-exchange column (Dionex IonPac AS22; 4 × 250 mm). The eluent was 4.5 mM K2CO3/1.4 mM KHCO3, the flow rate was 1.3 mL/min, and the column temperature was 30°C. Organic acids (formate, acetate, propionate, lactate, butyrate, and succinate) were measured using UV (210 nm) on an HPLC RSLC Ultimate 3000 equipped with an Aminex HPX-87H, 7.8 × 300 mm analytical column. The isocratic eluent was 5 mM H2SO4, the flow rate was 0.6 mL/min, and the column oven was heated to 60°C. Glucose was measured by mixing samples with the Glucose (HK) Assay reagent (Sigma-Aldrich) following the manufacturer’s instructions and absorbance was measured on a spectrophotometer at 410 nm.
DNA extraction
DNA was extracted from frozen pellets (0.25 g) using the Qiagen DNeasy PowerLyzer PowerSoil kit according to the manufacturer’s protocol. DNA concentrations were measured using the dsDNA High Sensitivity assay kit on a Qubit 2.0 fluorometer. DNA yields from heated oil sands ranged between 329–7890 ng DNA g−1 oil sand. To represent unincubated samples, DNA was extracted from 8-10 g oil sands (i.e., samples that were frozen when original outcrop samples were collected) using the Qiagen DNeasy PowerMax Soil kit according to the manufacturer’s protocol. Triplicate DNA extractions from these oil sands yielded 172–228 ng DNA g−1.
16S rRNA gene amplicon sequencing
Amplicon sequencing of the 16S rRNA gene (V4-V5 region) was performed using the bacterial primer set 515F and 926R [32]. Triplicate PCR reactions were pooled then purified using the NucleoMag NGS clean-up and size select kit. Purified PCR products were indexed following Illumina’s 16S rRNA amplicon preparation instructions. Indexed amplicons were verified on an Agilent 2100 Bioanalyzer system and sequenced on a MiSeq benchtop sequencer (Illumina) using the v3 600-cycle (paired end) reagent kit. Primers were trimmed from sequence reads with Cutadapt v4.4 [33] and processed in DADA2 [34] following the recommended pipeline (https://benjjneb.github.io/dada2/tutorial.html). Taxonomy was assigned to amplicon sequence variants (ASVs) with “assignTaxonomy” in DADA2 using the Swedish Biodiversity Infrastructure Sativa curated 16S GTDB database from release R07-RS207 (https://doi.org/10.17044/scilifelab.14869077).
Metagenome sequencing, read processing, and binning
Metagenomic sequencing was performed on a NovaSeq 6000 (Illumina) with a S4 300 cycle flow cell. Libraries were prepared by shearing to an insert size of 200 bp using a Covaris instrument followed by library construction with the NEB Ultra II DNA library prep kit. Adaptors and low-quality reads were removed with Cutadapt v.1.18 [33] using the wrapper Trimgalore v0.6.7 [35]. Reads from each sample were assembled individually with Megahit v1.2.9 [36] using the “—meta-sensitive” option. Read and assembly statistics are provided in Table S1. Reads were cross-mapped to the assembled contigs with BBMap v38.95 [37] to generate coverage profiles for binning. Contigs from each assembly were then binned with MetaBAT2 [38] and CONCOCT [39] and refined with DAS Tool [40]. Bin completeness and contamination were calculated with CheckM2 [41] and bins with >50% completeness and <10% contamination were retained. Small subunit (SSU) rRNA gene sequences in redundant bins were identified with Metaxa2 v2.2.3 [42]. ASVs from amplicon sequencing were compared to SSU rRNA gene sequences in metagenome-assembled genomes (MAGs) with BLASTn 2.6.0+ [43]. Redundant bins were dereplicated with dRep [44] resulting in 17 non-redundant MAGs (Table S2). Relative abundance of non-redundant MAGs was determined with CoverM using default parameters for “coverm genome” [45].
Annotation and evaluation MAGs
MAGs were taxonomically classified with Genome Taxonomy Database Toolkit (GTDB-tk) v2.3.2 with reference data R214 [46]. Average amino acid identity (AAI) comparisons between MAGs without close relatives in GTDB (<70% AAI) were determined with AAI calculator [47]. Functional annotation with KEGG and EggNOG databases was performed with GhostKOALA [48] and eggnog-mapper v2.1.7 [49]. Optimal growth temperature was predicted from protein sequences with “tome predOGT” [50] (Table S2) and MAGs were checked for functional and regulatory genes involved in endospore formation (Table S3). MAGs of mesophilic non-endosporulating bacteria included Actinobacteriota (×1), Patescibacteria (×2), and Proteobacteria (×2). These MAGs binned from unheated oil sands inoculum and had low relative abundance in heated samples so were excluded from further analysis. The final non-redundant set of thermospore MAGs contained 12 high quality Bacillota genomes (Table S2).
Annotation of the Nos gene cluster
Genes identified as nosZ were checked for the presence of Sec/SPI signal peptides, characteristic of all clade II nosZ, with SignalP 6.0 [51]. Genes on the same contig as nosZ were then checked for transmembrane helices with DeepTMHMM [52] to identify NosB which contains 4 or 6 transmembrane helices and is typically situated adjacent to NosZ in clade II-type nosZ microorganisms [53]. A cytochrome c preceding nosZ was identified with eggNOG and was determined to be nosC [54]. Cellular localization of denitrification genes was predicted with PSORTb. [55]. Nos gene clusters were visualized with the gggenes extension [56] for gglot2 [57].
Hidden Markov models for denitrification
A denitrification gene set was compiled from both custom hidden Markov models (HMMs) and TIGRfams (Table S4). Six HMMs were created for nirK, nirS, qnor, bnor, snor, and nosB. Amino acid sequences from genes of interest were retrieved from studies biochemically characterizing and describing the enzymes [15, 20, 22, 58, 59]. Multiple sequence alignments of characterized genes and related amino acid sequences from MAGs in this study were created with Clustal-Omega v1.2.4 [60]. Each alignment was manually inspected for conserved active site residues essential for structure and function in AliView v1.28 [61]. HMMs were created from the inspected multiple sequence alignments with “hmmbuild” in HMMER 3.3.2 [62]. The HMMs were first tested on MAGs from this study using “hmmsearch” and the trusted cutoff (TC) values were iteratively adjusted to ensure only genes with conserved residues were captured. The resulting HMMs were used to retrieve denitrification gene sequences from Firmicutes genomes (Firmicutes and Firmicutes A-H, n = 13 543) downloaded from GTDB R07-RS207 [63] using “gtt-get-accessions-from-GTDB” in GToTree v1.6.34 [64]. Note that Firmicutes phyla were renamed Bacillota following the release of GTDB reference data R214. Denitrification gene sequences from GTDB and thermospore MAGs were dereplicated with “fastx_uniques” in USEARCH v11 [65]. Unique sequences were aligned with Clustal-Omega v1.2.4 [60], manually inspected, and included in a revised HMM. TIGRfams were used to identify the genes narGH, napA, nosZI (clade I), and nosZII (clade II) [66]. Following manual inspection, the TC for the napA HMM was amended to capture monomeric NapA found in Bacillota E genomes. The final HMM set is available at https://github.com/emma-bell/metabolism.
Visualization of denitrification genes in Bacillota
Bacillota genomes from GTDB with Nor and/or Nos genes identified in their genome (n = 433) were visualized in a phylogenomic tree with thermospore MAGs from this study. The tree was created with GToTree v1.6.34 [64] from a concatenated alignment of 119 single copy genes targeted by the Firmicutes HMM profile. Within GToTree, genes were first predicted with Prodigal v2.6.3 [67] and target genes were identified with HMMER3 v3.3.2 [62]. Target genes were individually aligned with muscle v5.1 [68] and trimmed with TrimAl v1.4.rev15 [69]. Concatenated sequence alignments were used to create a maximum-likelihood phylogenomic tree using the Jones-Taylor-Thornton substitution model in FastTree2 v2.1.11 [70]. An Actinobacteriota MAG from this study was used as outgroup to root the phylogenomic tree. The tree was transformed and annotated in Treeviewer v2.2.0 [71].
Results
Enrichment of thermophilic denitrifiers
Incubation of oil sands at 50°C with nitrate and glucose resulted in nitrogen compounds being sequentially reduced (Fig. 1A–C) coupled to the metabolism of glucose into organic acids and carbon dioxide (Fig. 1D–F). Gas production was variable between replicates, with ~88, 59, and 31% of added N-NO3− accounted for in the gas phase (i.e., combined N-N2O and N-N2; see Table S5) of the three incubations. This suggests ammonium was also produced by nitrate metabolism. Organic acid consumption was also variable. Consistent with these observations, 16S rRNA gene amplicon sequencing showed that distinct populations were enriched in different bottles (Fig. 1G–I). Amplicon sequence variants (ASVs) from the genera Brevibacillus, Neobacillus, Geobacillus, Paenibacillus, and JAGHKQ01 (family Bacillaceae G) were found in common across triplicate enrichments, whereas other taxonomic lineages were exclusive. For example, ASVs of the class Bacilli (ASVs 6 and 18) were enriched only in incubation 1 (Fig. 1G) whereas ASVs of the family Brevibacillaceae (ASVs 9, 33, 35) and genus Symbiobacterium (ASVs 4, 15, 24, 25, 27) were enriched only in incubation 2 (Fig. 2H). This experimental approach therefore showed potential to uncover a diverse range of thermospores, with different members of the oil sands microbial seed bank becoming enriched from within parallel inocula.
Figure 1.
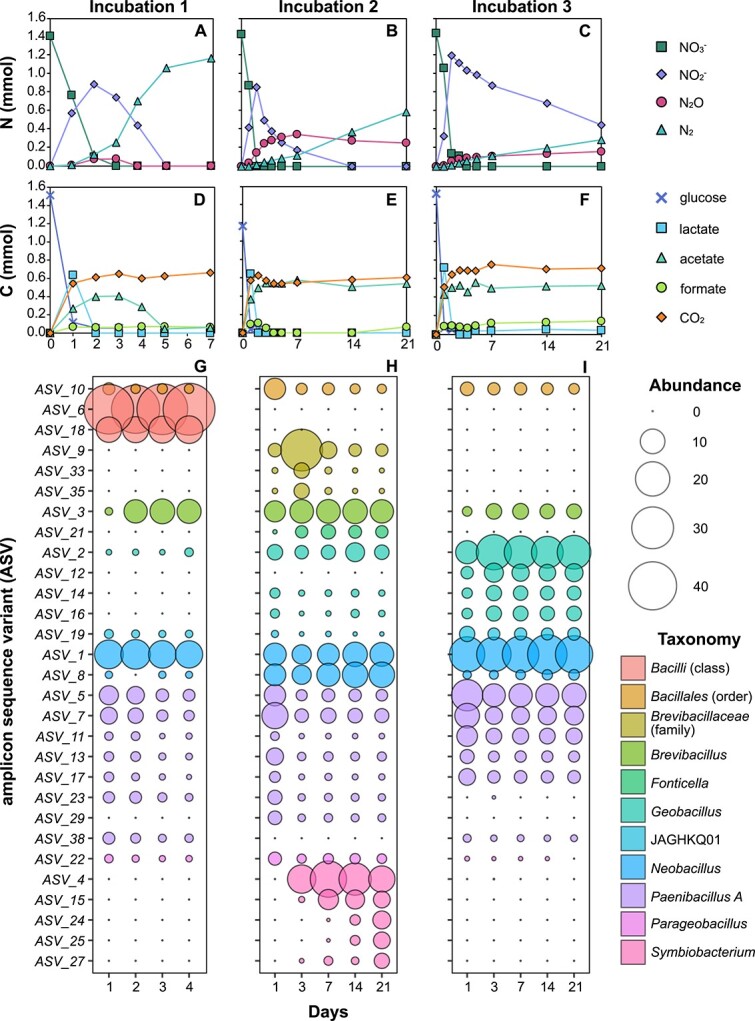
Thermospores enriched in heated nitrate-amended incubations. Nitrate reduction and production of nitrite, nitrous oxide and dinitrogen was monitored during incubation at 50°C (A, B, C) (nitric oxide was not measured). Glucose, organic acids, and CO2 measurements for each incubation are shown in corresponding panels underneath (D, E, F). All incubations were monitored over 21-days (Table S5) and measurements are shown for either 7-days (A) or 21-days (B, C) for clarity of nitrogen transformations. 16S rRNA gene amplicons were sequenced from multiple select time points (G, H, I) and Bacillota represented 94–98% read abundance in all cases. Only ASVs detected at >2% read abundance are included in the plots.
Figure 2.
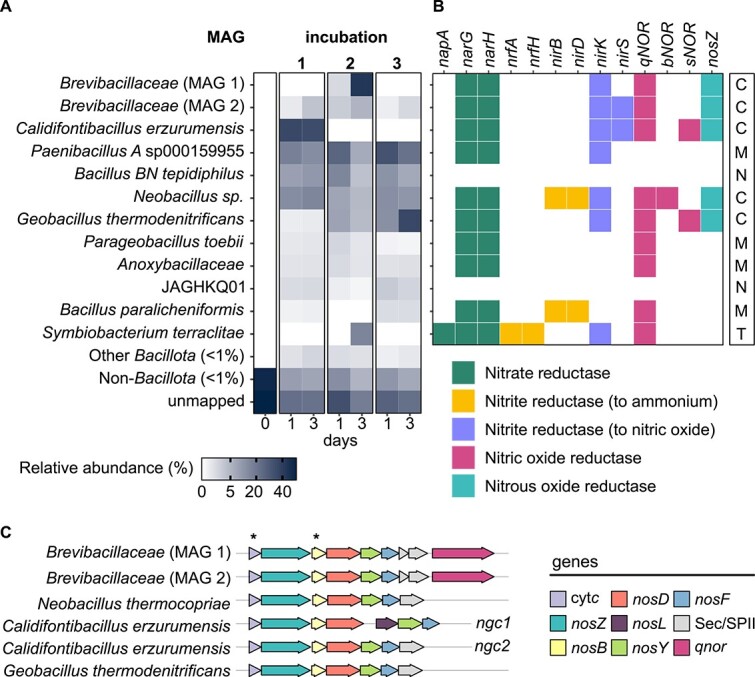
Denitrification genotypes of enriched thermospores. Relative abundance of thermospore MAGs in incubations with NO3− after one- and three-days incubation at 50°C (A). For comparison, the abundance of thermospores in the inoculum, prior to their enrichment, is shown as 0 days. Based on denitrification genes present (B), MAGs were classified as complete (C), truncated (T), modular (M) or non-denitrifying (N) with respect to their potential for denitrification (column at right). Complete denitrifiers encode clade II-type NosZ. The Nos gene cluster (ngc) of thermospore MAGs is shown (C) including two gene clusters in the single Calidifontibacillus erzurumensis MAG. Asterisks indicate conserved genes found in clade II.
Reconstruction of thermospore MAGs
Amplicon sequencing profiles showed the microbial communities to be consistent over time (Fig. 1G-I), which led to the selection of two time points from each incubation to more comprehensively survey the diversity of thermospores with metagenomic sequencing. In addition to sub-samples taken from the 50°C incubations after 1 and 3 days, metagenomic sequencing was performed on the inoculum (i.e., unheated oil sands). Twelve high quality non-redundant MAGs from Bacillota phyla (i.e., Bacillota ×11; Bacillota E × 1) were recovered from heated incubations (Fig. 2A). 16S rRNA gene sequences were present in MAGs of Geobacillus thermodenitrificans and Brevibacillaceae (MAG 2), which correspond to ASV_2 (99% identity) and ASV_3 (100% identity), respectively (Fig. 1G-I). Reads from the Bacillota MAGs were not detected in the unheated oil sands metagenome (Fig. 2A) which is consistent with dormant thermospore populations only germinating upon heating. Genomic potential for endospore formation and germination in Bacillota MAGs was also confirmed by the presence of functional and regulatory genes conserved within endospore-forming taxa (Table S3).
Identifying genes for denitrification
KEGG orthologs (KO) did not capture the diversity of denitrification enzymes present in Bacillota. With KEGG, the Nor qNOR was annotated as subunit B of cNOR (norB, K04561) and Nors bNOR and sNOR are annotated as the related but functionally distinct cytochrome c oxidase (coxA, K02274). Nitrous oxide reductase is identified with KEGG (nosZ, K00376) but clade I and clade II enzymes are not differentiated. Furthermore, the only gene annotated in addition to nosZ from the Nos gene cluster was the accessory protein nosD (K07218), found in both clade I and clade II nosZ microorganisms [72]. A set of HMMs was therefore created to capture denitrification potential in genomes of Bacillota. The HMM set includes distinct HMMs for Nors and differentiates between clade I and clade II NosZ. An HMM for the membrane lipoprotein nosB is also included. NosB is essential for N2O respiration in clade II nosZ microorganisms but is commonly absent in clade I microorganisms [24, 53].
Using the HMM set, MAGs were designated as complete-, truncated-, modular-, or non-denitrifiers based on genes present (Fig. 2B). A MAG was designated complete if genes for each step of the denitrification pathway are present. A MAG was called truncated if the genome lacked only nosZ, suggesting the end product of nitrate metabolism is N2O rather than N2. A MAG was considered modular if it lacked nosZ in addition to any other genes from the denitrification pathway, suggesting it can only participate in certain reductive steps. Finally, a MAG was called non-denitrifying if it contains no genes for reductive N metabolism.
Genomic potential for complete denitrification in thermospores
Potential for complete denitrification was found in five thermospore MAGs (Fig. 2B). Each of the five MAGs contain a membrane-associated nosZ with Sec-type signal peptide characteristic of clade II enzymes [23] and all enzymes in the pathway were predicted to be located in the cytoplasmic membrane, as expected for gram-positive denitrifiers [73]. Complete denitrifiers are taxonomically classified as Calidifontibacillus erzurumensis, G. thermodenitrificans, Neobacillus sp., and novel members of the family Brevibacillaceae (×2). The two Brevibacillaceae MAGs shared just 71.4% AAI with each other and comparison to members of this family in GTDB revealed no close relatives (>70% AAI). The greatest AAI was shared with an uncharacterized thermophilic soil bacterium, Brevibacillaceae species CFH-S0501 sp011059135, at 68.4 and 70.4% AAI, respectively.
Three Nor genes (qnor, bnor, and snor) were present in thermospore MAGs. The genes bnor and snor were found in genomes of complete denitrifiers in addition to qnor (Fig. 2B). Two complete denitrifiers, Calidifontibacillus erzurumensis and Brevibacillaeace (MAG 2), contained two nitric oxide-producing Nirs with both a nirS and a nirK gene. In addition to nitric oxide-producing nirK, Neobacillus sp. also contained an ammonium-producing Nir (nirBD) that can support both assimilation and dissimilation [74].
Clade II denitrifiers have a Nos gene cluster that differs to clade I denitrifiers, featuring some conserved genes that are absent in clade I genomes [24, 72]. Assessment of the Nos gene cluster (Fig. 2C) showed a cytochrome c preceding nosZ in all five thermospore genomes, as found in other clade II microoorganisms [54]. A gene with four transmembrane helices characteristic of nosB was adjacent to the putative ABC transporter complex nosD, -Y, -F in all of those genomes. Calidifontibacillus erzumensis had two Nos gene clusters, one of which had the copper chaperone nosL but this gene was absent from the genomes of other thermospores. The Nos gene cluster of Brevibacillaceae also differed from the other thermospores in that it contained a Nor (qNOR) immediately adjacent to the Nos gene cluster, whereas this gene was found elsewhere in the genome in the other three thermospore genomes.
Truncated or modular denitrification potential in thermospores
Genes from the denitrification pathway were detected in five nosZ-lacking thermospore MAGs. These thermospores were designated truncated or modular in their metabolic potential for denitrification (Fig. 2B). Symbiobacterium terraclitae was the only MAG designated as truncated and was the only MAG to contain both nar and nap nitrate reductases (Fig. 2B). The nap nitrate reductase in S. terraclitae is monomeric (napA) and is distinct from the heterodimeric napAB commonly found in Gram-negative bacteria [75]. Ammonium-producing nitrite reductase (nrfAH) was present in this MAG, suggesting S. terraclitae can also perform DNRA. Production of NH4+ by S. terraclitae could account for the proportion of added N-NO3− that is unaccounted for in the gas phase of incubations featuring germination and enrichment of this thermospore (Fig. 1B and Table S5).
Ammonium-producing Nir (nirBD) was also present in Bacillus paralichenformis (Fig. 2B), which was present in low relative abundance in incubations 1 and 3 (Fig. 2A). The B. paralichenformis MAG also contained Nor (qnor). In isolates of this species qnor is reported to play a role in detoxification of nitrite during DNRA resulting in the concomitant production of nonstoichiometric N2O [74, 76]. DNRA metabolism by B. paralichenformis could therefore contribute to both NH4+ and N2O production. Other MAGs designated modular have in common a respiratory nitrate reductase (membrane-bound nar), quinol-dependent nitric oxide reduction (qnor) and/or Cu-type nitrite reductase (nirK).
Non-denitrifying thermospores
Two thermospore MAGs from the denitrifying enrichments contain no genes for respiratory nitrate metabolism. Bacillus BN tepidiphilus reached >10% relative abundance within one day of incubation and JAGHKQ01 (family Bacillaceae G) maintained a comparatively lower abundance (<2.5%) in all enrichments (Fig. 2A). Both of these genomes encode potential for glucose metabolism (mixed acid fermentation, sugar transport) indicating that they became enriched by fermentative growth. Populations that ferment sugars likely provided substrates to nitrate-reducing populations in the form of fermentation products such as lactate, acetate and formate that were observed to increase in the early hours of 50°C incubations (Fig. 1D–F).
Denitrification genotypes of Bacillota
Representative genomes from Bacillota phyla (Bacillota and Bacillota A-H) were retrieved from GTDB and screened for nitric oxide (cNOR, qNOR, bNOR, sNOR) and Nos (NosZI, NosZII). Nitric oxide and/or Nos genes were present in ~10% of Bacillota genomes (n = 392/4216), ~5% of Bacillota C genomes (n = 20/395), ~4% of Bacillota B genomes (n = 12/323), and ~ 1.5% of Bacillota E genomes (n = 5/65). Just four genomes from Bacillota A (n = 8243) and Bacillota G (n = 131) contained either gene and all genomes from the phyla Bacillota D, F, and H (170 genomes) lacked both.
Genomes from Bacillota phyla with nitric oxide and/or Nos (n = 433) were screened with the full denitrification HMM set and included in a phylogenomic tree with MAGs from this study (Fig. 3). Genomic potential for complete denitrification is constrained to the phylum Bacillota and was only found within members of the class Bacilli. Seventeen genera contain complete denitrifiers (Fig. 3; 41 GTDB MAGs +5 MAGs from this study). This includes 11 genomes that encode bNOR and/or sNOR but lack qNOR (or cNOR) and would have been considered incomplete denitrifiers using KO annotations only.
Figure 3.
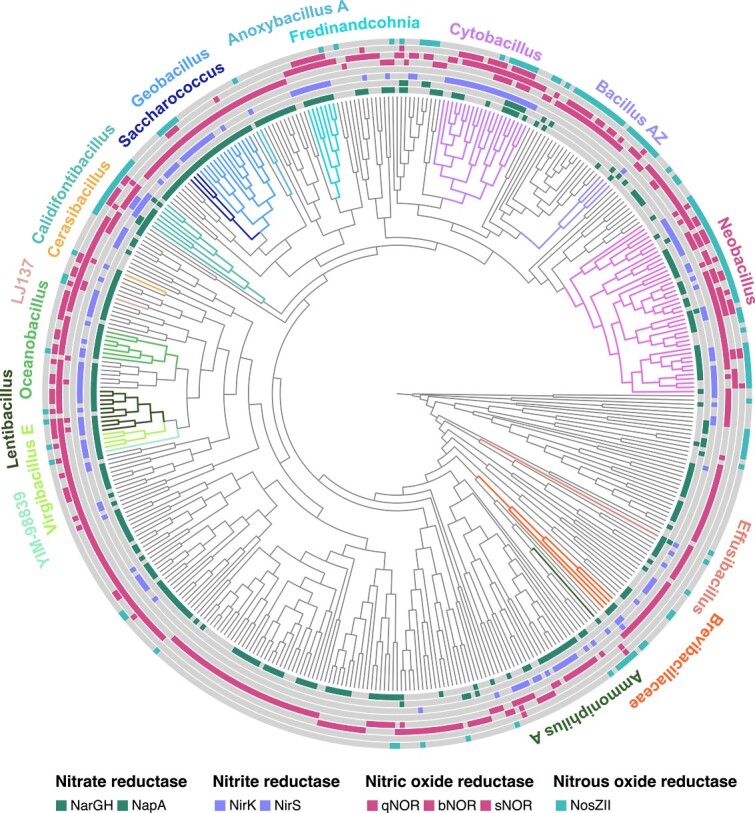
Bacillota genera with genomic potential for complete denitrification. A phylogenomic tree was constructed with Bacillota MAGs from this study and 433 genomes from GTDB that contain nitric oxide reductase and/or Nos. Colored wedges at the tips of branches indicate gene presence (filled) or absence (grey). Genes shown from the innermost circle to the outer: narGH, napA, nirK, nirS, qnor, bnor, snor, clade II-type nosZ. The phylogenomic tree was constructed using a concatenated alignment of 119 single copy genes conserved within Bacillota and is presented as a cladogram. Within the phylum Bacillota, 17 genera have potential for complete denitrification (colored clades with bold text).
Among denitrification genotypes, the absence of one or more of the reduction steps is common. A further 74 MAGs were categorized as truncated denitrifiers i.e., missing only Nos, 73 were categorized as nitric oxide reducers i.e., contain Nor only, and 15 were categorized as non-denitrifying nitrous oxide reducers i.e., contain Nos only (Fig. 3 and Table S6). The remaining genomes were categorized as modular i.e., they contain Nor and/or Nos in addition to one or more denitrification pathway genes. Many genomes from Bacillota phyla encode qNOR (172/433) or bNOR (123/433) whereas sNOR was rarely found without the presence of another Nor gene (2/433). Cytochrome c-dependent Nor (cNOR) was only encoded in genomes within the family Desulfitobacteriaceae from the phylum Bacillota B (6/433). Desulfitobacteriaceae also contain clade II-type nosZ but lack genes for nitrite reduction. Occurrences of three NOR genes in the same genome (qNOR, bNOR, and sNOR) was observed in 26 genomes, most of which belong to Neobacillus and other genera containing complete denitrifiers (Fig. 3).
Discussion
Targeted enrichment with nitrate resulted in the germination and activity of denitrifying thermospore populations. This approach uncovered multiple lineages of denitrifiers, including novel members of the family Brevibacillaceae. Different thermospore taxa responded in parallel incubations suggesting there are heterogeneous populations of dormant thermospores in Athabasca oil sands outcrops, which is consistent with similar observations of oil sands microbiomes generally [29, 77]. It is well documented that thermospores comprise part of the microbial seedbank in cold sedimentary [78, 79] and soil environments [80, 81]. Germination can be activated by sublethal heat shock and addition of nutrients [82] with enrichment of different thermospore populations being influenced by both temperature and available substrates [78, 83, 84]. When conditions change favorably, dormant populations germinate and become active members of the microbial community. This premise underpins strategies for engineered microbial activity in situ with the objective of pressure generation and maintenance via microbial biogas such as N2 [27]. Our results demonstrate the feasibility for denitrifying thermospores to be stimulated in oil sands.
Biogas production was variable between replicates and can be attributed to the enrichment of distinct thermospore populations in different incubations. Genomic analysis showed potential for both denitrification and DNRA in different individual genomes as well as within the same genome, a feature that is not uncommon among Bacillus spp. [54, 85]. Co-occurrence of both pathways in a single genome highlights the challenge associated with predicting metabolic end-products based on gene content and the importance of environmental factors for determining metabolic activity [86, 87]. Metabolic end-products can also be influenced by the accumulation of intermediate metabolites. The protonated form of nitrite (HNO2) is toxic at high concentration [88] and could have influenced the germination and enrichment of thermospores detected in this study. Certain Bacillus spp. capable of DNRA have a high tolerance to nitrite, but detoxification results in a greater production of N2O [74, 76]. Nitrite has also been shown to inhibit N2O reduction during denitrification [89] resulting in reduced production of N2.
Denitrifying thermospores enriched from oil sands have clade II-type nosZ genes (subclade H) for catalyzing reduction of N2O to N2. Clade II nosZ-bearing microorganisms are numerically significant in the environment [90–93], though they are often considered to be non-denitrifiers lacking genes needed for the stepwise reduction of nitrate to dinitrogen [24, 94]. While this is true for certain lineages within the diverse clade II-type NosZ, analysis of MAGs from N2-producing enrichments in this study, as well as Bacillota from GTDB, shows that multiple genera within the class Bacilli contain a full complement of denitrification genes. This is consistent with multiple isolated representatives from this class that have been experimentally shown to perform complete denitrification [25]. Examples include G. thermodenitrificans isolated from a deep oil reservoir [95] and Calidifontibacillus azotoformans (formerly Bacillus azotoformans) isolated from soil [73].
Genomic potential for complete denitrification was present in 17 genera within the class Bacilli. Within these genera, it was common for microorganisms to possess multiple Nors (qNOR, bNOR, and sNOR). This highlights that NOR enzymes are not mutually exclusive, though the different conditions under which they are not expressed in Bacillota are not clear. It has been suggested that bNOR in B. azotoformans can be used for aerobic NO reduction in microoxic environments [54]. The recently characterized enzyme eNOR, that descends from the same family of oxygen reductases as bNOR, also reduces nitric oxide under microoxic conditions [22, 96]. Members of the Bacillota are often identified as contributors to denitrification in environments with variable redox conditions, including agricultural soil, deep vadose zone soil, and rice paddy soil [8, 97, 98] and can be present in the soil microbiome generally [99]. Having multiple Nors could provide Bacillota with metabolic versatility in environments like soils, where combined oxic and anoxic conditions are commonly found [4].
So-called functional redundancy has also been found with other enzymes within the denitrification pathway. B. azotoformans contains five Nos gene clusters, three of which include a nosZ gene (Heylen 2012). In addition, a recent survey of nitrite reductases (NirK and NirS) in isolates and MAGs showed that possessing both enzymes is more common than previously appreciated and potentially allows microorganisms bearing both enzymes to denitrify across a wider range of environments [100]. Functionally redundant enzymes within a genome may also reflect an ability of the microorganisms to adapt to changing environmental conditions. This would be a beneficial trait for members of the Bacillota as endospore-formers undergo periods of dormancy and respond rapidly through germination to changes in their environment.
Genome-resolved metagenomics is a useful approach for studying denitrification as it provides the gene content of populations and circumvents challenges with PCR-based marker gene approaches. However, we found that certain denitrification genes were missed, or pathways appeared incomplete, using standard annotation databases that are biased towards clade I denitrifiers. For example, the denitrification reference pathway in KEGG includes nitrate reductase composed of subunits napAH and Nor composed of subunits norBC. However, nitrate reductases in Symbiobacterium (Bacillota E) are monomeric [75] and lack the napH subunit. Similarly, qNOR Nors are fused and lack the norC subunit [18]. This can result in modules or pathways appearing incomplete. To date bNOR has only been found in Bacillota and was originally isolated from B. azotoformans [21, 73]. Despite being biochemically characterized bNOR genes were not identified with commonly used gene annotation databases KEGG, eggNOG, or TIGRfam. Finally, while putative NOR enzyme families that have been recently proposed [22, 96] are not expected to be represented in curated annotation databases, their absence nevertheless highlights that interpretation of community gene content is limited by the breadth of gene databases. Considering the complete diversity of NOR enzymes reveals a greater diversity of microorganisms capable of denitrification. This is an important consideration for studies attempting to quantify capacity for denitrification or N2O emissions based on gene content in both natural environments and engineered systems to ensure that metabolic potential is not underestimated.
Supplementary Material
Contributor Information
Emma Bell, Department of Biological Sciences, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada.
Jianwei Chen, Department of Biological Sciences, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada.
William D L Richardson, Department of Biological Sciences, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada.
Milovan Fustic, Department of Biological Sciences, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada; Department of Geology, Nazarbayev University, 53 Kabanbay Batyr Ave, Astana 010000, Kazakhstan.
Casey R J Hubert, Department of Biological Sciences, University of Calgary, 2500 University Drive NW, Calgary, Alberta T2N 1N4, Canada.
Conflicts of interest
CRJH and MF have patented oil sands emissions reduction technology that depends on biogas production by thermophilic bacteria, related to the presented research. EB, JC and WDLR declare no conflict of interest.
Funding
This work was funded by a Campus Alberta Innovates Program chair awarded to CRJH.
Data availability
DNA sequencing data (16S rRNA gene amplicon, metagenome, and metagenome-assembled genomes) are available at the NCBI Sequence Read Archive under BioProject ID PRJNA1110647.
References
- 1. Gowda K, Ping D, Mani Met al. Genomic structure predicts metabolite dynamics in microbial communities. Cell 2022;185:530–546.e25. 10.1016/j.cell.2021.12.036 [DOI] [PubMed] [Google Scholar]
- 2. Tian H, Xu R, Canadell JGet al. A comprehensive quantification of global nitrous oxide sources and sinks. Nature 2020;586:248–56. 10.1038/s41586-020-2780-0 [DOI] [PubMed] [Google Scholar]
- 3. Harris E, Yu L, Wang Y-Pet al. Warming and redistribution of nitrogen inputs drive an increase in terrestrial nitrous oxide emission factor. Nat Commun 2022;13:4310. 10.1038/s41467-022-32001-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Christensen S, Rousk K. Global N2O emissions from our planet: which fluxes are affected by man, and can we reduce these. iScience 2024;27:109042. 10.1016/j.isci.2024.109042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bowles TM, Atallah SS, Campbell EEet al. Addressing agricultural nitrogen losses in a changing climate. Nat Sustain 2018;1:399–408. 10.1038/s41893-018-0106-0 [DOI] [Google Scholar]
- 6. Nilsson JE, Weisner SEB, Liess A. Wetland nitrogen removal from agricultural runoff in a changing climate. Sci Total Environ 2023;892:164336. 10.1016/j.scitotenv.2023.164336 [DOI] [PubMed] [Google Scholar]
- 7. Conthe M, Lycus P, Arntzen MØet al. Denitrification as an N2O sink. Water Res 2019;151:381–7. 10.1016/j.watres.2018.11.087 [DOI] [PubMed] [Google Scholar]
- 8. Zhang L, Zhao H, Qin Set al. Genome-resolved metagenomics and denitrifying strain isolation reveal new insights into microbial denitrification in the deep Vadose zone. Environ Sci Technol 2024;58:2323–34. 10.1021/acs.est.3c06466 [DOI] [PubMed] [Google Scholar]
- 9. Philippot L. Denitrifying genes in bacterial and archaeal genomes. Biochim Biophys Acta BBA - Gene Struct Expr 2002;1577:355–76. 10.1016/S0167-4781(02)00420-7 [DOI] [PubMed] [Google Scholar]
- 10. Chee-Sanford JC, Connor L, Krichels Aet al. Hierarchical detection of diverse clade II (atypical) nosZ genes using new primer sets for classical- and multiplex PCR array applications. J Microbiol Methods 2020;172:105908. 10.1016/j.mimet.2020.105908 [DOI] [PubMed] [Google Scholar]
- 11. Heylen K, Gevers D, Vanparys Bet al. The incidence of nirS and nirK and their genetic heterogeneity in cultivated denitrifiers. Environ Microbiol 2006;8:2012–21. 10.1111/j.1462-2920.2006.01081.x [DOI] [PubMed] [Google Scholar]
- 12. Verbaendert I, De Vos P, Boon Net al. Denitrification in gram-positive bacteria: an underexplored trait. Biochem Soc Trans 2011;39:254–8. 10.1042/BST0390254 [DOI] [PubMed] [Google Scholar]
- 13. Saghaï A, Pold G, Jones CMet al. Phyloecology of nitrate ammonifiers and their importance relative to denitrifiers in global terrestrial biomes. Nat Commun 2023;14:8249. 10.1038/s41467-023-44022-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wei W, Isobe K, Nishizawa Tet al. Higher diversity and abundance of denitrifying microorganisms in environments than considered previously. ISME J 2015;9:1954–65. 10.1038/ismej.2015.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Decleyre H, Heylen K, Tytgat Bet al. Highly diverse nirK genes comprise two major clades that harbour ammonium-producing denitrifiers. BMC Genomics 2016;17:527. 10.1186/s12864-016-2812-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun H, Jiang S. A review on nirS-type and nirK-type denitrifiers via a scientometric approach coupled with case studies. Environ Sci Process Impacts 2022;24:221–32. 10.1039/D1EM00518A [DOI] [PubMed] [Google Scholar]
- 17. Saraste M, Castresana J. Cytochrome oxidase evolved by tinkering with denitrification enzymes. FEBS Lett 1994;341:1–4. 10.1016/0014-5793(94)80228-9 [DOI] [PubMed] [Google Scholar]
- 18. Hemp J, Gennis RB. Diversity of the Heme–copper superfamily in archaea: Insights from Genomicsand structural Modeling. In: Schäfer G., Penefsky H.S. (eds.), Bioenergetics: Energy Conservation and Conversion, Springer. Berlin, Heidelberg: Berlin Heidelberg, 2008, 1–31. [Google Scholar]
- 19. Hino T, Matsumoto Y, Nagano Set al. Structural basis of biological N2O generation by bacterial nitric oxide reductase. Science 2010;330:1666–70. 10.1126/science.1195591 [DOI] [PubMed] [Google Scholar]
- 20. Matsumoto Y, Tosha T, Pisliakov AVet al. Crystal structure of quinol-dependent nitric oxide reductase from Geobacillus stearothermophilus. Nat Struct Mol Biol 2012;19:238–45. 10.1038/nsmb.2213 [DOI] [PubMed] [Google Scholar]
- 21. Al-Attar S, de Vries S. An electrogenic nitric oxide reductase. FEBS Lett 2015;589:2050–7. 10.1016/j.febslet.2015.06.033 [DOI] [PubMed] [Google Scholar]
- 22. Murali R, Pace LA, Sanford RAet al. Diversity and evolution of nitric oxide reduction in bacteria and archaea. Proc Natl Acad Sci 2024;121:e2316422121. 10.1073/pnas.2316422121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sanford RA, Wagner DD, Wu Qet al. Unexpected nondenitrifier nitrous oxide reductase gene diversity and abundance in soils. Proc Natl Acad Sci 2012;109:19709. 10.1073/pnas.1211238109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hallin S, Philippot L, Löffler FEet al. Genomics and ecology of novel N2O-reducing microorganisms. Trends Microbiol 2018;26:43–55. 10.1016/j.tim.2017.07.003 [DOI] [PubMed] [Google Scholar]
- 25. Verbaendert I, Hoefman S, Boeckx Pet al. Primers for overlooked nirK, qnorB, and nosZ genes of thermophilic gram-positive denitrifiers. FEMS Microbiol Ecol 2014;89:162–80. 10.1111/1574-6941.12346 [DOI] [PubMed] [Google Scholar]
- 26. Ma Y, Zilles JL, Kent AD. An evaluation of primers for detecting denitrifiers via their functional genes. Environ Microbiol 2019;21:1196–210. 10.1111/1462-2920.14555 [DOI] [PubMed] [Google Scholar]
- 27. Hubert CRJ, Fustic M. Microbially Influenced Steam Assisted Gravity Drainage (MISAGD). Ottawa: Ontario, 2017. [Google Scholar]
- 28. Wong ML, An D, Caffrey SMet al. Roles of thermophiles and fungi in bitumen degradation in mostly cold oil sands outcrops. Appl Environ Microbiol 2015;81:6825–38. 10.1128/AEM.02221-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ridley CM, Voordouw G. Aerobic microbial taxa dominate deep subsurface cores from the Alberta oil sands. FEMS Microbiol Ecol 2018;94:fiy073. 10.1093/femsec/fiy073 [DOI] [PubMed] [Google Scholar]
- 30. de Rezende JR, Oldenburg TBP, Korin Tet al. Anaerobic microbial communities and their potential for bioenergy production in heavily biodegraded petroleum reservoirs. Environ Microbiol 2020;22:3049–65. 10.1111/1462-2920.14995 [DOI] [PubMed] [Google Scholar]
- 31. Pannekens M, Voskuhl L, Mohammadian Set al. Microbial degradation rates of natural bitumen. Environ Sci Technol 2021;55:8700–8. 10.1021/acs.est.1c00596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Caporaso JG, Lauber CL, Walters WAet al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci 2011;108:4516–22. 10.1073/pnas.1000080107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 2011;17:10. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- 34. Callahan BJ, McMurdie PJ, Rosen MJet al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 2016;13:581–3. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krueger F, James F, Ewels Pet al. TrimGalore: v0.6.10. 2023. Zenodo [Google Scholar]
- 36. Li D, Liu CM, Luo Ret al. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015;31:1674–6. 10.1093/bioinformatics/btv033 [DOI] [PubMed] [Google Scholar]
- 37. Bushnell B, Rood J, Singer E. BBTools software package. PLoS One . https://sourceforge.net/projects/bbmap/. [Google Scholar]
- 38. Kang DD, Li F, Kirton Eet al. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019;7:e7359–9. 10.7717/peerj.7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alneberg J, Bjarnason BS, De Bruijn Iet al. Binning metagenomic contigs by coverage and composition. Nat Methods 2014;11:1144–6. 10.1038/nmeth.3103 [DOI] [PubMed] [Google Scholar]
- 40. Sieber CMKK, Probst AJ, Sharrar Aet al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol 2018;3:836–43. 10.1038/s41564-018-0171-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chklovski A, Parks DH, Woodcroft BJet al. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat Methods 2023;20:1203–12. 10.1038/s41592-023-01940-w [DOI] [PubMed] [Google Scholar]
- 42. Bengtsson-Palme J, Hartmann M, Eriksson KMet al. metaxa2: improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data. Mol Ecol Resour 2015;15:1403–14. 10.1111/1755-0998.12399 [DOI] [PubMed] [Google Scholar]
- 43. Camacho C, Coulouris G, Avagyan Vet al. BLAST+: architecture and applications. BMC Bioinformatics 2009;10:421. 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Olm MR, Brown CT, Brooks Bet al. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J 2017;11:2864–8. 10.1038/ismej.2017.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aroney STN, Newell RJP, Nissen Jet al. CoverM: Read Coverage Calculator for Metagenomics. Zenodo, 2024, 10.5281/zenodo.10531253 [DOI] [Google Scholar]
- 46. Chaumeil P-A, Mussig AJ, Hugenholtz Pet al. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 2022;38:5315–6. 10.1093/bioinformatics/btac672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rodriguez-R LM, Konstantinidis KT. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr 2016;4:e1900v1. [Google Scholar]
- 48. Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol 2016;428:726–31. 10.1016/j.jmb.2015.11.006 [DOI] [PubMed] [Google Scholar]
- 49. Huerta-Cepas J, Szklarczyk D, Heller Det al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 2019;47:D309–14. 10.1093/nar/gky1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li G, Rabe KS, Nielsen Jet al. Machine learning applied to predicting microorganism growth temperatures and enzyme catalytic optima. ACS Synth Biol 2019;8:1411–20. 10.1021/acssynbio.9b00099 [DOI] [PubMed] [Google Scholar]
- 51. Teufel F, Almagro Armenteros JJ, Johansen ARet al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat Biotechnol 2022;40:1023–5. 10.1038/s41587-021-01156-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hallgren J, Tsirigos KD, Pedersen MDet al. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. 2022; bioRxiv. , 2022.04.08.487609.
- 53. Hein S, Witt S, Simon J. Clade II nitrous oxide respiration of Wolinella succinogenes depends on the NosG, -C1, -C2, -H electron transport module, NosB and a Rieske/cytochrome bc complex. Environ Microbiol 2017;19:4913–25. 10.1111/1462-2920.13935 [DOI] [PubMed] [Google Scholar]
- 54. Heylen K, Keltjens J. Redundancy and modularity in membrane-associated dissimilatory nitrate reduction in bacillus. Front Microbiol 2012;3:371. 10.3389/fmicb.2012.00371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu NY, Wagner JR, Laird MRet al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinforma Oxf Engl 2010;26:1608–15. 10.1093/bioinformatics/btq249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wilkins D. Gggenes: Draw Gene Arrow Maps in ‘ggplot2’, 2023.
- 57. Wickham H. ggplot2: Elegant Graphics for Data Analysis, Springer-Verlag New York, 2016.
- 58. Rinaldo S, Giardina G, Castiglione Net al. The catalytic mechanism of Pseudomonas aeruginosa cd1 nitrite reductase. Biochem Soc Trans 2011;39:195–200. 10.1042/BST0390195 [DOI] [PubMed] [Google Scholar]
- 59. Gomaa F, Utter DR, Powers Cet al. Multiple integrated metabolic strategies allow foraminiferan protists to thrive in anoxic marine sediments. Sci Adv 2021;7:eabf1586. 10.1126/sciadv.abf1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sievers F, Wilm A, Dineen Det al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol Syst Biol 2011;7:539. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Larsson A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014;30:3276–8. 10.1093/bioinformatics/btu531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol 2011;7:e1002195–5. 10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Parks DH, Chuvochina M, Rinke Cet al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res 2022;50:D785–94. 10.1093/nar/gkab776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee MD. GToTree: a user-friendly workflow for phylogenomics. Bioinformatics 2019;35:4162–4. 10.1093/bioinformatics/btz188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010;26:2460–1. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 66. Selengut JD, Haft DH, Davidsen Tet al. TIGRFAMs and genome properties: tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res 2007;35:D260–4. 10.1093/nar/gkl1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hyatt D, Chen G-L, Locascio PFet al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010;11:119. 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Edgar RC. Muscle5: high-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nat Commun 2022;13:6968. 10.1038/s41467-022-34630-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009;25:1972–3. 10.1093/bioinformatics/btp348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Price MN, Dehal PS, Arkin AP. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One 2010;5:e9490. 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bianchini G, Sánchez-Baracaldo P. TreeViewer: flexible, modular software to visualise and manipulate phylogenetic trees. Ecol Evol 2024;14:e10873. 10.1002/ece3.10873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hein S, Simon J. Chapter four - bacterial nitrous oxide respiration: Electron transport chains and copper transfer reactions. In: Poole R.K. (ed.), Advances in Microbial Physiology. Academic Press, London, United Kingdom, 2019, 137–75. [DOI] [PubMed] [Google Scholar]
- 73. Suharti, de Vries S. Membrane-bound denitrification in the gram-positive bacterium bacillus azotoformans. Biochem Soc Trans 2005;33:130–3. 10.1042/BST0330130 [DOI] [PubMed] [Google Scholar]
- 74. Sun Y, De Vos P, Heylen K. Nitrous oxide emission by the non- denitrifying, nitrate ammonifier bacillus licheniformis. BMC Genomics 2016;17:68. 10.1186/s12864-016-2382-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jepson BJN, Marietou A, Mohan Set al. Evolution of the soluble nitrate reductase: defining the monomeric periplasmic nitrate reductase subgroup. Biochem Soc Trans 2006;34:122–6. 10.1042/BST0340122 [DOI] [PubMed] [Google Scholar]
- 76. Sun Y, De Vos P, Willems A. Influence of nitrate and nitrite concentration on N2O production via dissimilatory nitrate/nitrite reduction to ammonium in bacillus paralicheniformis LMG 6934. MicrobiologyOpen 2018;7:e00592. 10.1002/mbo3.592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. An D, Caffrey SM, Soh J, et al. Metagenomics of Hydrocarbon Resource Environments Indicates Aerobic Taxa and Genes to be Unexpectedly Common. 2013, Metagenomics of hydrocarbon resource environments indicates aerobic taxa and genes to be unexpectedly common, Environ Sci Technol47:10708–17. 10.1021/es4020184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hubert C, Loy A, Nickel Met al. A constant flux of diverse thermophilic bacteria into the cold Arctic seabed. Science 2009;325:1541–4. 10.1126/science.1174012 [DOI] [PubMed] [Google Scholar]
- 79. Müller AL, de Rezende JR, Hubert CRJet al. Endospores of thermophilic bacteria as tracers of microbial dispersal by ocean currents. ISME J 2014;8:1153–65. 10.1038/ismej.2013.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Marchant R, Banat IM, Rahman TJet al. The frequency and characteristics of highly thermophilic bacteria in cool soil environments. Environ Microbiol 2002;4:595–602. 10.1046/j.1462-2920.2002.00344.x [DOI] [PubMed] [Google Scholar]
- 81. Marchant R, Franzetti A, Pavlostathis SGet al. Thermophilic bacteria in cool temperate soils: are they metabolically active or continually added by global atmospheric transport? Appl Microbiol Biotechnol 2008;78:841–52. 10.1007/s00253-008-1372-y [DOI] [PubMed] [Google Scholar]
- 82. Setlow P. Germination of spores of bacillus species: what we know and do not know. J Bacteriol 2014;196:1297–305. 10.1128/JB.01455-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bell E, Sherry A, Pilloni Get al. Sediment cooling triggers germination and sulfate reduction by heat-resistant thermophilic spore-forming bacteria. Environ Microbiol 2020;22:456–65. 10.1111/1462-2920.14866 [DOI] [PubMed] [Google Scholar]
- 84. Bell E, Rattray JE, Sloan Ket al. Hyperthermophilic endospores germinate and metabolize organic carbon in sediments heated to 80°C. Environ Microbiol 2022;24:5534–45. 10.1111/1462-2920.16167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sun Y, De Vos P, Willems A. Nitrogen assimilation in denitrifier bacillus azotoformans LMG 9581T. Antonie Van Leeuwenhoek 2017;110:1613–26. 10.1007/s10482-017-0911-x [DOI] [PubMed] [Google Scholar]
- 86. van den Berg EM, Boleij M, Kuenen JGet al. DNRA and denitrification coexist over a broad range of acetate/N-NO3− ratios, in a Chemostat enrichment culture. Front Microbiol 2016;7:7. 10.3389/fmicb.2016.01842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. van den Berg EM, Elisário MP, Kuenen JGet al. Fermentative bacteria influence the competition between Denitrifiers and DNRA bacteria. Front Microbiol 2017;8:8. 10.3389/fmicb.2017.01684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Almeida JS, Júlio SM, Reis MAMet al. Nitrite inhibition of denitrification by Pseudomonas fluorescens. Biotechnol Bioeng 1995;46:194–201. 10.1002/bit.260460303 [DOI] [PubMed] [Google Scholar]
- 89. Zhou Y, Pijuan M, Zeng RJet al. Free nitrous acid inhibition on nitrous oxide reduction by a denitrifying-enhanced biological phosphorus removal sludge. Environ Sci Technol 2008;42:8260–5. 10.1021/es800650j [DOI] [PubMed] [Google Scholar]
- 90. Jones CM, Graf DR, Bru Det al. The unaccounted yet abundant nitrous oxide-reducing microbial community: a potential nitrous oxide sink. ISME J 2013;7:417–26. 10.1038/ismej.2012.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Coyotzi S, Doxey AC, Clark IDet al. Agricultural soil denitrifiers possess extensive nitrite reductase gene diversity. Environ Microbiol 2017;19:1189–208. 10.1111/1462-2920.13643 [DOI] [PubMed] [Google Scholar]
- 92. Mosley OE, Gios E, Close Met al. Nitrogen cycling and microbial cooperation in the terrestrial subsurface. ISME J 2022;16:2561–73. 10.1038/s41396-022-01300-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tang W, Jayakumar A, Sun Xet al. Nitrous oxide consumption in oxygenated and anoxic estuarine waters. Geophys Res Lett 2022;49:e2022GL100657. 10.1029/2022GL100657 [DOI] [Google Scholar]
- 94. Conthe M, Wittorf L, Kuenen JGet al. Life on N2O: deciphering the ecophysiology of N2O respiring bacterial communities in a continuous culture. ISME J 2018;12:1142–53. 10.1038/s41396-018-0063-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Feng L, Wang W, Cheng Jet al. Genome and proteome of long-chain alkane degrading Geobacillus thermodenitrificans NG80-2 isolated from a deep-subsurface oil reservoir. Proc Natl Acad Sci USA 2007;104:5602–7. 10.1073/pnas.0609650104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Murali R, Hemp J, Gennis RB. Evolution of quinol oxidation within the heme-copper oxidoreductase superfamily. Biochim Biophys Acta BBA - Bioenerg 2022;1863:148907. 10.1016/j.bbabio.2022.148907 [DOI] [PubMed] [Google Scholar]
- 97. Satoshi I, Michihiro Y, Mami Ket al. Microbial populations responsive to denitrification-inducing conditions in Rice Paddy soil, as revealed by comparative 16S rRNA gene analysis. Appl Environ Microbiol 2009;75:7070–8. 10.1128/AEM.01481-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Anderson CR, Peterson ME, Frampton RAet al. Rapid increases in soil pH solubilise organic matter, dramatically increase denitrification potential and strongly stimulate microorganisms from the Firmicutes phylum. PeerJ 2018;6:e6090. 10.7717/peerj.6090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Felske A, Akkermans ADL, De Vos WM. In situ detection of an uncultured predominant bacillus in Dutch grassland soils. Appl Environ Microbiol 1998;64:4588–90. 10.1128/AEM.64.11.4588-4590.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Pold G, Bonilla-Rosso G, Saghaï Aet al. Phylogenetics and environmental distribution of nitric oxide forming nitrite reductases reveals their distinct functional and ecological roles. ISME Commun 2024;4:ycae020. 10.1093/ismeco/ycae020 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA sequencing data (16S rRNA gene amplicon, metagenome, and metagenome-assembled genomes) are available at the NCBI Sequence Read Archive under BioProject ID PRJNA1110647.