


Abstract
A novel approach for the amplification of cDNA ends is described. It requires only minimal amounts of material, a simple cDNA synthesis reaction and a single PCR reaction to amplify the desired 5′- or 3′-ends of a certain cDNA of interest. It combines the so called CapFinder approach with solid phase cDNA synthesis, thus almost eliminating background problems usually associated with 5′-RACE protocols. This approach could be used to generate complete 5′-ends of numerous cDNAs using only one cDNA synthesis reaction. In combination with LA PCR, several kilobases of unknown 5′-ends could be amplified. It is easy to perform, quick, inexpensive and reliable, which should enable it to replace most currently used 5′-RACE protocols.
INTRODUCTION
The extensive use of PCR approaches for the identification of novel genes requires the identification of those regions of the gene that are not enclosed by known sequences. These 3′- and 5′-regions could either be identified using conventional screening of cDNA libraries or using 5′- and 3′-RACE (rapid amplification of cDNA ends) approaches. 5′-RACE was made possible by tagging the 5′-end of a cDNA by means of different methods (1–3). Most approaches, such as homopolymeric tailing or ligation anchored tailing require a set of enzymatic reactions after completion of first strand cDNA synthesis (1). Each enzymatic step has the potential to introduce failures and to destroy the integrity of the cDNA. Recently, an alternative was introduced, the so called CapFinder approach. It depends on the ability of MMLV reverse transcriptase (RT) to add cytosine residues to the 3′-end of newly synthesised cDNAs upon reaching the 5′-end (cap region) of the mRNA. Usually 2–4 cytosine residues are added, depending on the reaction conditions (4). If an oligonucleotide with oligo(G) or oligo(rG) sequences at its 3′-most end is included in the incubation medium, its terminal 3–4 G residues could base pair with the 2–4 C residues of the newly synthesised cDNA, thus serving as a new template for the RT (template switch). The RT then switches the template and replicates the sequence of the CapFinder oligonucleotide, thus including the complementary CapFinder oligonucleotide sequence at the 3′-end of the newly synthesised cDNA (Fig. 1). This approach allows the amplification of whole cDNA libraries enriched in full-length clones. Enrichment of full-length clones is achieved because the MMLV RT adds C residues preferentially to the cDNA if complete (capped) mRNA serves as template. Variations in the composition of the RT buffer shifts the tendency of the RT to add more C residues to the cDNA end, which makes the entire process more effective (4). Amplification of the cDNA was successfully used for the generation of cDNA libraries and for the production of so-called virtual northern blots, a very useful alternative to mRNA northern blots (3). If these cDNAs are used for 5′-RACE experiments, however, background problems are obvious. Instead of distinct bands, a DNA smear appears if the 5′-RACE product is separated on an agarose gel. This has excluded this promising approach from the important field of 5′-RACE methods. High backgrounds mainly arise from contamination of the 5′-RACE reactions with components of the original CapFinder reaction. The most important contaminants are the CapFinder and oligo(dT) primers. Even residual amounts of these primers result in a high background, because both ideally fit to all cDNAs present in the reaction mixture. To circumvent this problem, strategies to remove all contaminants or to suppress unwanted PCR products are required. Matz and colleagues (5) introduced the PCR suppression effect in combination with CapFinder cDNA synthesis to achieve accurate 5′-RACE products. This refinement of the CapFinder approach seems to work, but, due to the relative complexity of the approach, it has gained insufficient interest.
Figure 1.


Schematic outline of the 5′-RACE approach. (A) cDNA synthesis. The biotin-labelled oligo(dT) primer is bound to avidin-coupled polystyrene-beads. Using these beads, mRNA is isolated and first strand cDNA synthesis starts directly on the beads (step 1). The intrinsic activity of the MMLV RT adds 2–4 C residues to the 3′-end of the newly synthesised cDNA. This region could base pair with the 3′-most part (containing G or rG residues) of the CapFinder A or B primer, which in turn functions as template for the RT. This includes a known sequence complementary to the CapFinder primer sequence at the 3′-end of the newly synthesised cDNA. If only minute amounts of material are available, the cDNA could be preamplified in a LA PCR reaction using 5′-Primer and 3′-Primer (steps 2 and 3). After the PCR reaction the biotinylated product is captured and washed and could now be used in a 5′-RACE–PCR reaction (step 4). (B) 5′-RACE reaction. For the 5′-RACE reaction either non-amplified CapFinder cDNA [see (A) step 1] or PCR amplified cDNA [see (A) step 4] could be used. The 5′-RACE reaction is performed with a gene-specific reverse primer (GSRP) and 5′-Primer, which represents the 5′-most 27 nt of the CapFinder primers. The PCR amplificate represents in both cases the 5′-region of the gene of interest surrounded by the 5′-Primer and gene-specific reverse primer (GSRP).
To exclude problems arising from contamination of the primer used for cDNA synthesis, we chose an alternative approach. In our hands, simple, physical removal of all primers is the best approach. To achieve this, we introduced a few, very simple modifications to the cDNA synthesis process. As reported earlier, we introduced a solid phase cDNA synthesis protocol (6) to handle the DNA.
MATERIALS AND METHODS
RNA (1–10 µg) was isolated from different parts of the locust (Schistocerca gregaria) brain (i.e. the optic lobes, thoracic ganglia or midbrain) or from trophozoites of the human protozoan parasites Entamoeba histolytica and Entamoeba dispar using standard protocols (Trizol Reagent; Life Technologies, Eggenstein, Germany). To isolate the mRNA, a biotinylated oligo(dT) primer [Fig. 2, oligo(dT) I] was bound to avidin-coated polystyrene beads according to the instructions of the manufacturer (Kisker, Mühlhausen, Germany); for each pmol primer, beads with a binding capacity of at least 5 pmol biotin were used. The mRNA is bound according to standard protocols (7) except that it is not eluted from the beads but washed an additional time in 1× cDNA synthesis buffer. For each 1 µg mRNA, 10 pmol oligo(dT) I primer bound to polystyrene beads were used. First strand cDNA synthesis was performed with the mRNA left on the beads using the bound oligo(dT) oligonucleotide as primer. The reaction conditions were as follows: 50 mM Tris–HCl pH 8.3 (45°C), 6 mM MgCl2, 2 mM MnCl2, 1 mM dNTPs, 0.01 M dithiothreitol, 0.2 mg/ml BSA, 10 pM CapFinder primer (CapFinder A or B), 200 U Superscript II (Life Technologies) in 20 µl total volume for 1 h at 45°C. After completion of the reaction the beads were centrifuged, the supernatant removed and the pellet, containing the cDNA bound to the beads, was resuspended in TE buffer. The beads were again centrifuged and the supernatant removed. This procedure was repeated twice, to obtain a washed cDNA preparation that is devoid of any residual primer not incorporated into the cDNA. This preparation could now be used for the 5′-RACE–PCR reaction or stored for several weeks at 4°C (Fig. 1A, step 1). Two alternative strategies could now be chosen. (i) The cDNA could be used for 5′- or 3′-RACE without additional treatment (Fig. 1B, step A). (ii) To enrich for full-length cDNAs, or if only small amounts of material are available, a preamplification with 5′-Primer and 3′-Primer is useful (Fig. 1). Both primers are devoid of the corresponding homopolymeric 3′-regions (Fig. 2), making them ideally suited for effective and specific PCR amplification (3). This amplified DNA has to be captured and bound via its biotin residues to avidin-coated beads to be suitable for subsequent 5′- or 3′-RACE amplification (Fig. 1A, step 4).
Figure 2.

Primers used for the 5′-RACE approach. Sequence of the primers used for the 5′-RACE protocols. Specific primers derived from sequenced genes are not included. mRNA is isolated and cDNA synthesis is primed using oligo(dT) primer I, which contains a biotin residue at its 5′-end. CapFinder primer A or B is included in the first strand synthesis medium to tag the first strand cDNA with a known sequence at its 5′-end. To preamplify the capped cDNA a LA PCR reaction with 5′-Primer and 3′-Primer could be performed; both primers are devoid of the 3′-homopolymeric regions. In the 5′- and 3′-RACE protocols the latter primers are used together with a gene-specific reverse primer (GSRP) and gene-specific sense primers, respectively.
Aliquots (∼1–2 µl of a 50 µl reaction mix) of this reaction are subjected to a PCR reaction using 5′-Primer and a gene-specific reverse primer (GSRP) derived from the cDNA under investigation (Fig. 1B, step B). 5′-RACE–PCR reactions were either performed under standard conditions (for small cDNAs of <1 kb) or with a LA PCR system (8) for longer cDNAs. Polystyrene beads are far better suited for this purpose compared with magnetic streptavidin beads, because they are smaller and stay without agitation in solution, which is a prerequisite for successful PCR reactions.
RESULTS AND DISCUSSION
We used this approach to identify 5′-regions of genes isolated in a differential display PCR (DD PCR) (9) screen using different parts of the insect brain (10). In addition, 5′-ends of genes from the protozoan parasites E.histolytica and E.dispar were identified. Among the most problematical tasks in a DD PCR screen is identification of the amplicon 5′-ends. DD PCR makes only small fragments in the most 3′-region of the cDNA accessible, which is a major drawback of this approach. To overcome this, the 5′-RACE has to combine effective amplification with a high reproducibility and a low hands-on time. This is realised in our new DD PCR approach.
Both alternative modifications outlined in this study, the direct use of first strand cDNA reactions and the use of PCR preamplified reactions, gave almost identical results. Currently, these methods are in use in our laboratories. One project where the approach is successfully in use is a large DD PCR screen looking for transcripts present in only a single area of the insect brain. We isolated numerous DNAs, each representing a transcript exclusively present in one defined area of the insect brain. To obtain access to the full-length cDNAs we routinely employed our 5′-RACE approach and obtained 5′-regions ranging from a few hundred to several kilobases in length (data not shown). To demonstrate the usefulness of our approach we used known genes of the protozoan parasites E.histolytica and E.dispar. To complete the respective 5′-regions, GSRPs, designed from known regions of the corresponding genes, were used, together with 5′-Primer and the corresponding solid phase CapFinder cDNA. As seen in Figure 3, all reactions gave the anticipated result, yielding a single amplificate of the desired length. The identity of the amplified DNA was revealed by sequencing. In all cases the complete 5′-regions were isolated, including the start codon and a short 5′-untranslated region. The chosen genes are all single copy genes, with some of them transcribed at rather low levels.
Figure 3.
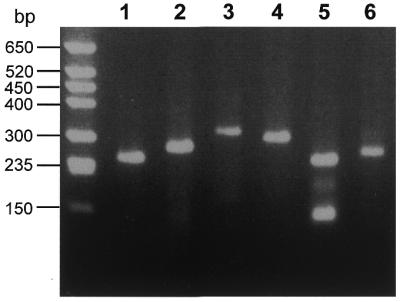
5′-RACE experiments with different genes of the protozoan parasite E.histolytica. Using the 5′-RACE protocol outlined in Figure 1, a number of different 5′-regions of identified genes of the human protozoan parasite E.histolytica were detected. Gene-specific reverse primers (GSRP) were designed from the known sequences of the respective genes and used, together with the 5′-Primer and CapFinder cDNA to amplify the 5′-regions. In lanes 1 and 2 two different 5′-RACE products corresponding to the 5′-ends of cysteine protease cDNAs, in lanes 3 and 4 those of two thioredoxin cDNAs and in lanes 5 and 6 those of two protein disulphide isomerase cDNAs are separated. The smaller band in lane 5 represents a PCR artefact, as it appears even if only the GSRP is present in the PCR reaction.
If we perform these experiments with raw, uncoupled first strand cDNA or amplified cDNA material we obtain a smear of DNA rather than a single DNA population. This holds true even if a second PCR with a nested gene-specific primer is used. The reactions were performed with small aliquots (∼1 µl) of a total reaction (either cDNA or PCR preamplified cDNA) resuspended in a volume of 50 µl. As cDNA synthesis is primed with an oligo(dT) primer rather than with a gene-specific primer, this reaction could be used for the amplification of numerous different 5′-regions.
To further evaluate the abilities of this approach, we amplified larger 5′-regions of already identified genes of the human protozoan parasites E.histolytica and E.dispar, which share ∼95% sequence homology. These genes, the cysteine proteinases 1 and 112, are transcribed at relatively low abundance, which is especially true for the second, larger cysteine protease (EhCP112). Alternative 5′-RACE approaches, such as homopolymeric tailing (1), failed to give the desired amplificate. The 5′-RACE reactions were performed with non-amplified first strand cDNA, a gene-specific reverse primer (GSRP; Fig. 1B) and 5′-Primer. These PCRs, which were performed under standard conditions, revealed single DNA bands in the cDNA populations of both species. All of them were of the expected length (∼800 bp for EhCP1 and 1400 bp for EhCP112; Fig. 4). The identities of the amplified DNA populations were verified by sequencing. In addition, the amplified 5′-regions contained the complete 5′-regions including the start codon, which is problematical for amoebae as they usually have very short 5′-untranslated regions. This leads to a loss of the first coding sequences in conventional cDNA libraries made from material of these organisms. Simple and reliable 5′-RACE approaches are therefore very useful to obtain full-length sequences of all cloned genes of these and comparable organisms. Loss of the first few bases of a cDNA during the cDNA library production process is a common problem found in most cDNA libraries. To complete these sequences, very simple and reliable 5′-RACE protocols are required. In addition to its high performance and high efficiency, a single cDNA synthesis is sufficient for numerous 5′-RACE–PCR reactions, reducing hands-on time and use of material to a minimum. The amplification of 1.4 kb transcripts without any specific PCR precautions (without the use of LA PCR systems) demonstrates the usefulness of our novel approach. We were able to obtain even larger parts of unknown 5′-regions using LA PCR reactions (7). Amplifications between 2 and 3 kb were possible in some cases (data not shown). This should significantly facilitate the cloning of larger genes.
Figure 4.
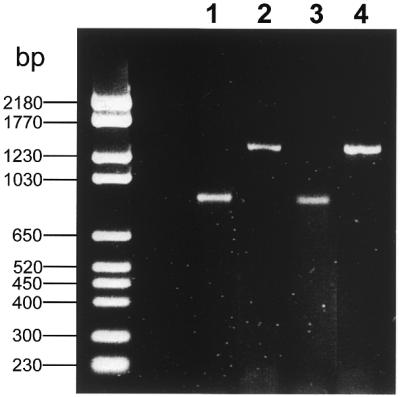
5′-RACE experiments with cysteine proteases of the protozoan parasite E.histolytica. 5′-Regions of the cysteine proteases CP 1 (lanes 1 and 3) and CP 112 (lanes 2 and 4) were amplified from cDNA derived from the protozoan parasites E.histolytica (lanes 1 and 2) and its close relative E.dispar (lanes 3 and 4). Gene-specific reverse primers (GSRP) were chosen that could be anticipated to amplify ∼900 (CP 1, lanes 1 and 3) or 1400 bp (CP 112, lanes 2 and 4). The 5′-RACE amplifications revealed single bands, whose identity was verified by sequencing.
Our 5′-RACE approach combines numerous advantages compared with other protocols currently in use. It is very effective and yields, in almost every case, the correct amplificate. Usually it is of the desired length, without any contaminating DNA populations. The preference for complete 5′-regions might result from the phenomenon that effective addition of C residues to the cDNA occurs only if complete (capped) mRNA served as the template.
The present paper describes a 5′-RACE method that combines simplicity with extraordinary performance. The hands-on time and the chance to introduce experimental errors are reduced to a minimum and optimal results could be obtained in most experiments. The simple solid phase approach opens the potential of the CapFinder approach, which could complement numerous protocols in molecular biology.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank M. Gewecke for support. This work was funded by grants from the Deutsche Forschungsgemeinschaft to T.R. (DFG Ro 1241) and to I.B. (DFG IB 1744).
REFERENCES
- 1.Schaefer B.C. (1995) Anal. Biochem., 227, 255–273. [DOI] [PubMed] [Google Scholar]
- 2.Fromont-Racine M., Bertrand,E., Pictet,R. and Grange,T. (1993) Nucleic Acids Res., 21, 1683–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franz O., Bruchhaus,I. and Roeder,T. (1999) Nucleic Acids Res., 27, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmidt W.M. and Mueller,M.W. (1999) Nucleic Acids Res., 27, e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matz M., Shagin,D., Bogdanova,E., Britanova,O., Lukyanov,S., Diatchenko,L. and Chenchik,A. (1999) Nucleic Acids Res., 27, 1558–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roeder T. (1998) Nucleic Acids Res., 26, 3451–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barnes W.M. (1994) Proc. Natl Acad. Sci. USA, 91, 2216–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dynal (1995) Biomagnetic Techniques in Molecular Biology,Technical Handbook, 2nd Edn. Dynal A. S., Oslo, Norway.
- 9.Liang P. and Pardee,A.B. (1992) Science, 257, 967–971. [DOI] [PubMed] [Google Scholar]
- 10.Franz O., Roeder,T. and Gewecke,M. (1998) J. Comp. Physiol., 182A, 627–633. [Google Scholar]