

Summary
Brassica napus (rapeseed) is a recent allotetraploid plant and the second most important oilseed crop worldwide. The origin of B. napus and the genetic relationships with its diploid ancestor species remain largely unresolved. Here, chloroplast DNA (cpDNA) from 488 B. napus accessions of global origin, 139 B. rapa accessions and 49 B. oleracea accessions were populationally resequenced using Illumina Solexa sequencing technologies. The intraspecific cpDNA variants and their allelic frequencies were called genomewide and further validated via EcoTILLING analyses of the rpo region. The cpDNA of the current global B. napus population comprises more than 400 variants (SNPs and short InDels) and maintains one predominant haplotype (Bncp1). Whole‐genome resequencing of the cpDNA of Bncp1 haplotype eliminated its direct inheritance from any accession of the B. rapa or B. oleracea species. The distribution of the polymorphism information content (PIC) values for each variant demonstrated that B. napus has much lower cpDNA diversity than B. rapa; however, a vast majority of the wild and cultivated B. oleracea specimens appeared to share one same distinct cpDNA haplotype, in contrast to its wild C‐genome relatives. This finding suggests that the cpDNA of the three Brassica species is well differentiated. The predominant B. napus cpDNA haplotype may have originated from uninvestigated relatives or from interactions between cpDNA mutations and natural/artificial selection during speciation and evolution. These exhaustive data on variation in cpDNA would provide fundamental data for research on cpDNA and chloroplasts.
Keywords: Brassica napus, cpDNA variant, genetic diversity, haplotype, next‐genetation sequencing, maternal origin
Introduction
Rapeseed (Brassica napus L., AACC, 2n = 38) is a global economically important oil crop. It is the second most important oilseed crop in the world, and it is used in industry as lubricant and biodiesel (Hua et al., 2012). Brassica napus is a recent allotetraploid species, that is known to be derived from the spontaneous hybridization between the diploid species Brassica rapa (AA, 2n = 20) and Brassica oleracea (CC, 2n = 18; Nagaharu, 1935; Song and Osborn, 1992). B. napus has a short evolutionary (<10 000 years) and domestication (<500 years) history (Chalhoub et al., 2014; Prakash et al., 2011; Rana et al., 2004). One of the great achievements in regard to rapeseed has been the use of artificial breeding to increase its ecological adaptability and improve its nutritional quality, for example the reduction of the erucic acid and glucosinolate content of the seeds to make the rapeseed oil healthier and more nutrient‐rich (Li et al., 2014a); however, agronomic traits including seed yield, disease resistance and oil content are facing bottlenecks to further improvements. Identification and use of elite traits from diploid ancestor species (B. rapa and B. oleracea) and other relatives (e.g. B. juncea, AABB, 2n = 36) may be instrumental in overcoming these bottlenecks.
To date, the molecular mechanism underlying the origination and evolution of rapeseed remains largely ambiguous. It is a prerequisite to dissect the genetic relationships between B. napus and its diploid parental species, B. rapa and B. oleracea, which have longer evolutionary and domestication histories and are thought to possess greater genetic diversity than B. napus (Liu et al., 2014; Wang et al., 2011a); however, it is still difficult to resolve this issue via comparative analysis of the nuclear genomes, due to the tremendous cost of extensive resequencing of large number of rapeseed varieties. The cytoplasmic chloroplast genomes (cpDNA) are maternally inherited in most angiosperms (Li et al., 2013a; Mogensen, 1996). Additionally, cpDNA has a simplex genome structure that is only 100–200 kb in size, and it is inherited highly conservatively (Birky, 2001; Sato et al., 1999). Accordingly, cpDNA has been widely employed as reference DNA in phylogenetic studies and for the evaluation of population‐level genetic diversity. Therefore, cpDNA would be a robust and cost‐effective tool with which to examine the origination and evolution of B. napus.
Traditional cpDNA genotyping methods include Random Amplified Polymorphic DNA (RAPD), Restriction Fragment Length Polymorphism (RFLP) and chloroplast simple sequence repeats (cpSSRs; Provan et al., 2001; Van Den Berg et al., 2002). Genotyping cpDNA via sequence alignments of the selected hypervariable cpDNA regions (e.g. accD, rps16 and rbcL) is also a routine method (Dong et al., 2012). All the above‐mentioned methods are principally based on regional polymorphisms in the chloroplast genomes. The cpDNA haplotypes can be determined via the compositional derivations of the obtained individual variants, and the genetic diversity of the investigated population can be assessed accordingly. A set of cpSSR primers for Brassica genera has been developed and widely used in phylogenetic studies. Recently, the usefulness of these cpSSR primers was re‐assessed in evaluating the cytoplasmic diversity of a more diversified Brassica population (Zamani‐Nour et al., 2013). The maternal origin of B. napus has been systemically investigated by means of cpDNA diversities during the past decade. Based on the RFLP analyses of the cytoplasmic genomes, Song and Osborn (1992) showed that the cultivated forms of B. napus contain the same cpDNA as B. montana but have a unique mitochondrial genome (mtDNA) intermediate between those of B. montana and B. rapa. Using six cpSSR primer pairs, Allender and King (2010) determined that one predominant haplotype exists in 94 accessions of European B. napus and was also present in B. rapa at a low frequency; however, this major B. napus cpDNA haplotype rarely appeared in the investigated B. oleracea and other C‐genome species, for example B. montana, B. incana and B. villosa. Nonetheless, there are still significant controversies regarding the maternal origin and evolutionary mechanism underlying the origin of B. napus.
Recently, next‐generation sequencing technologies (e.g. 454, Solexa and the SOLID platform) have been successfully applied in cpDNA‐related research. With these high‐throughput technologies, a number of chloroplast genomes have recently been sequenced (Henry et al., 2014; Li et al., 2013b; Zhang et al., 2011). The cpDNA diversities have been systemically investigated and evaluated at the genome level in angiosperms (Heinze et al., 2014; Li et al., 2014b). A set of the most variable cpDNA loci (markers) has been identified and is thought to be highly suitable for evaluating plant phylogeny and DNA barcoding (Dong et al., 2012). Several phylogenetic studies have been substantially reinforced through the use of many cpDNA fragments (Fior et al., 2013) or by separate sequencing of the whole cpDNA genome (Nikiforova et al., 2013). The accuracy of the obtained phylogenetic relationship is substantially guaranteed; however, the experimental population is always limited to a small sample size due to the high cost of sufficient separate sequencing.
In this study, a novel high‐throughput pangenomic cpDNA sequencing method was developed and used to exhaustively identify global B. napus intraspecific cpDNA variants at the whole‐genome level. Based on the obtained vast data on variation in cpDNA, the genetic diversities of B. napus, B. rapa and B. oleracea and their genetic relationships were finely determined at the cpDNA level and the maternal origin of B. napus was further analysed.
Results
A novel high‐throughput pangenomic resequencing methodology facilitates identification of intraspecific Brassica cpDNA variants
The expeditious and economical identification of genomewide cpDNA variants in a large population remains difficult. To accomplish this aim, a method comprised of next‐generation sequencing technology and a deliberate sample‐multiplexing strategy was developed and used to identify intraspecific cpDNA variants in B. napus. A schematic diagram illustrating the experimental pipeline is shown in Figure 1. It has been suggested that cpDNA generally accounts for no more than 5% of the total leaf DNA in higher plants (Nock et al., 2011). In this study, only chloroplast DNA was sequenced. For chloroplast isolation, the step of Percoll gradient separation of the intact chloroplasts was omitted to increase the cpDNA yield and reduce the labour cost, whereas certain details of the protocol were modified to improve cpDNA purity. To further reduce the labour costs of chloroplast isolation, a sample‐mixing strategy was introduced. The newly developed leaves (with consistent sizes of approximately 40 cm2) were collected from the representative 8‐week‐old plants. Fresh leaves from each of the 24 accessions were weight‐equally pooled for chloroplast isolation. The obtained uniformly mixed chloroplasts were directly subjected to cpDNA isolation.
Figure 1.

A schematic diagram illustrating the experimental pipeline.
Five DNA libraries were eventually constructed from a total of 488 worldwide B. napus accessions, with one each for the 139 B. rapa and 49 wild B. oleracea accessions (Table S1), and finally sequenced using Illumina Solexa sequencing technologies. The amount of sequence data was set to 2 Gb (giga base pairs) for each samples. The obtained clean reads (100‐bp long) were directly assembled to the B. napus reference chloroplast genome (B. napus strain ZY036; GenBank: GQ861354.1; Hu et al., 2011) using the CLC Main Workbench DNA analysing platform (Version 6.0; CLC Bio, Aarhus, Denmark). Detailed information regarding the data amount and sequencing depth of each sample is given in Table 1. The mapped reads of each sample generally account for approximately 20% of their total reads, indicating that the cpDNA purity was improved up to 20% by our modification on chloroplast isolation. For the majority of the sequencing samples, the reference genome was almost completely covered with an average sequencing depth of 5000× (Figure 2), except for a few sites whose loss of coverage was likely caused by the disturbance of long (>10 bp) structural variations.
Table 1.
The properties of Brassica high‐throughput pangenomic sequencing
| Species | Sequencing samples | Sampling content | Data (Gb) | Mapped ratio (%) | Average depth |
|---|---|---|---|---|---|
| B. napus | 4‐1A | 96 | 4.25 | 20.39 | 5776× |
| B. napus | 4‐2B | 96 | 4.82 | 16.95 | 5456× |
| B. napus | 4‐3C | 96 | 6.65 | 19.50 | 8866× |
| B. napus | 4‐4D | 96 | 5 | 18.87 | 6283× |
| B. napus | 4‐5E | 104 | 5.29 | 16.75 | 5840× |
| B. rapa | BC139 | 139 | 3.39 | 33.33 | 7566× |
| B. oleracea | Bomix | 49 | 2.4 | 7.0 | 1099× |
Figure 2.

Sequencing depth of the samples 4‐1A and BC139. The horizontal axis indicates the reference genome (with a size of 152.86 kb), and the vertical axis indicates the genome coverage.
The parameters for cpDNA variant calling conducted by the CLC DNA analysing platform were optimized and further validated via visual inspection of the aligned sequences. Unreliable variants in regions (usually with high AT content) with low sequencing quality were manually removed. Finally, 538 variants, including single nucleotide polymorphisms (SNPs) and small insertions and deletions (InDels), were identified in the current B. napus and B. rapa populations. The allelic frequencies of each variant were calculated based on the numbers of allele‐specific reads. The variants with frequencies lower than 2% were omitted from analyses to avoid false‐positive variants caused by inevitable sequencing errors. Detailed information on all of these Brassica cpDNA variants is presented in Table S2. Up to 423 variants (294 SNPs and 129 InDels) were identified in the B. napus population, whereas 268 were identified for B. rapa. The two intraspecific populations shared 165 polymorphic loci with identical alleles, but most of these loci had opposite patterns of allelic frequencies.
EcoTILLING analyses validated the vast Brassica data on variation in cpDNA and identified a worldwide cpDNA haplotype in B. napus
The vast data on variation in cpDNA obtained above were experimentally validated, via EcoTILLING analysis followed by PCR and Sanger sequencing of the variants in the rpoC2‐C1‐B coding sequence (CDS) regions. Eight pairs of primers targeting PCR fragments covering 14 variants in the rpo region (B. napus reference genome position: 16 000–26 000 bp) were used in the EcoTILLING analyses of 256 B. napus accessions (Table S1, Entry No: 1‐256). A two‐dimensional sample‐multiplexing strategy was employed, and the frequency of each allele was estimated accordingly (Figure 3). The PCR products amplified from the corresponding individuals were Sanger sequenced to obtain their sequence information. Both the allelic frequencies and the sequence information coincided well with the corresponding data obtained above (Table 2), indicating that the data on variation in cpDNA obtained via the newly developed pangenomic sequencing method are reliable and relatively accurate.
Figure 3.

EcoTILLING analyses of the representative cpDNA variants. The 215F‐223R primer pair was used to examine two InDel variants located at bps 21 759 and 22 094 in the ZY036 reference genome. Black and grey arrows indicate the appearance of the two variants at 21 759 and 22 904 bp, respectively. The predicated sizes of the cleaved DNA fragments are labelled.
Table 2.
Validation of the cpDNA variants via EcoTILLING analyses
| Genome locus | Reference | Alleles | Allelic frequency | Frequencya from Tilling | Haplotypes | ||||
|---|---|---|---|---|---|---|---|---|---|
| Bncp1 | Bncp2 | Bncp3 | Bncp4 | Bncp5 | |||||
| 16953 | A | A/G | 85.2/14.8 | 18.3 | A | G | G | G | G |
| 17058 | T | T/C | 85.0/15.0 | 18.3 | T | C | C | C | C |
| 17370 | A | A/G | 85.5/14.5 | 14.8 | A | G | G | G | G |
| 19864 | T | T/– | 86.7/13.3 | 13.7 | T | – | – | – | – |
| 19983 | C | C/A | 86.3/13.7 | 13.7 | C | A | A | A | A |
| 21136 | T | T/C | 84.8/15.2 | 19.1 | T | C | C | C | C |
| 21759 | AGAAAAA | –/AGAAAAA | 64.9/35.1 | 30.4 | – | AGAAAAA | – | – | AGAAAAA |
| 22094 | T | –/T | 77.2/22.8 | 15.2 | – | T | T | T | T |
| 23619 | G | G/A | 83.9/16.1 | 19.9 | G | A | A | A | A |
| 24036 | T | T/C | 85.2/14.8 | 13.7 | T | C | C | C | C |
| 24705 | G | G/A | 84.5/15.5 | 16.2 | G | A | A | A | A |
| 24909 | A | A/C | 86.1/13.9 | 15.2 | A | C | C | C | C |
| 25149 | A | A/C | 84.4/15.6 | 11.3 | A | C | C | C | C |
| 25317 | T | T/G | 93.1/6.9 | 5.9 | T | G | G | T | T |
| 91.80% | 4.30% | 2.00% | 1.10% | 0.70% | |||||
| Haplotype frequency | |||||||||
Here, the frequencies derived from EcoTILLING analyses always indicate those of the infrequent alleles.
Based on the EcoTILLING analyses of the 14 rpo variants, a total of 5 cpDNA haplotypes were identified in the 256 investigated B. napus accessions. PCR products were amplified from the accessions representing the corresponding haplotypes and then were Sanger sequenced. The sequencing results verified the existence of the above 5 haplotypes, one of which was found to be predominant, with a population‐level frequency of 91.8%; this haplotype is referred to as Bncp1 (Table 2).
Genomewide determination of the predominant cpDNA haplotype for Brasssica napus
To obtain genomewide sequence information for the Bncp1 haplotype, four B. napus accessions (Fengyou 1, P7, Zheyou 18 and Zhongshuang 11) classified into that group were individually resequenced by high‐throughput methods. Separate assembly of the reads derived from these accessions to the ZY036 reference genome yielded 27 completely identical variants (Table S2). Of these variants, up to 12 were found to lack polymorphisms in the current B. napus and B. rapa populations. The obtained sequence data shared by the 4 B. napus accessions was referred to as the Bncp1 cpDNA sequence. The systemic examination of allelic frequencies at the 423 global B. napus polymorphic cpDNA loci showed that Bncp1 cpDNA maintained the predominant alleles at nearly all these loci, indicating that Bncp1 is the predominant B. napus cpDNA haplotype worldwide.
Two B. rapa accessions, Tianmendaye Rape and Qingyuanbendi Rape, were also individually sequenced. They were found to possess only a few distinct variants differing from the B. rapa reference genome (B. rapa subsp. Pekinensis; GenBank: DQ231548.1) (Table S2). Both the B. napus and B. rapa reference genomes kept most of the predominant alleles identified in their respective populations, indicating that the ZY036 (B. napus) and Pekinensis (B. rapa) haplotypes are appropriate species‐specific cpDNA representatives of B. napus and B. rapa, respectively.
Interspecific comparison of the cpDNA variants between B. napus and B. rapa
Brassica napus may carry either B. rapa or B. oleracea cpDNA depending on the direction of hybridization at the time of its origination. The cytoplasmic diversity of this species was certainly enriched by the postspeciation introgression of other cpDNA variants from genetically close species and by the artificial resynthesization of novel B. napus individuals via the interspecific crossing of B. oleracea and B. rapa accessions. The distribution of the predominant B. napus alleles in the B. rapa population was also investigated. Of the predominant alleles at all the 423 B. napus cpDNA polymorphic loci, those at 116 loci existed at low frequencies in the B. rapa population, whereas those at 216 loci never appeared in it; only those at 82 loci were found at high frequencies (Figure 4a). These results suggest that the pattern of variation in B. napus cpDNA is distinct from that observed in B. rapa. A large proportion (116/423) of the predominant B. napus alleles may originate from a few rare B. rapa accessions of the cpDNA haplotype with the same alleles. Notably, up to 50% (216/423) of the predominant alleles that are absent in B. rapa seem to be specific to B. napus.
Figure 4.

Patterns of variation in Brassica cpDNA. (a) A pie chart illustrating the distribution of 423 predominant B. napus alleles in B. rapa. These alleles (with population‐level frequencies of >60% in B. napus) were grouped into infrequent, absent and predominant types, according to the frequency of their appearance in B. rapa. The total numbers of each grouped alleles were labelled on their respective regions. (b) A pie chart illustrating the genomic distribution information of the 538 Brassica cpDNA variants. IGR, intergenic region; CDS, coding sequence. Noncoding RNA includes transfer RNA (tRNA) and ribosomal RNA (rRNA). The total numbers of each grouped variants were labelled on their respective regions.
The population‐level genetic diversities were evaluated using the variant‐dependent polymorphism information content (PIC) values (Nagy et al., 2012). The PICs were called based on the numbers of alleles and the related frequency information for each polymorphic locus. The results (Figure 5) showed that the PIC values of 400 B. napus cpDNA variants tended to be densely distributed, with 58% clustered within a range between 0.2 and 0.3; however, the distribution for B. rapa was scattered and even, with 26% exceeding 0.3. These findings indicate that the genetic diversity of B. napus cpDNA is distinctly lower than that of B. rapa cpDNA.
Figure 5.
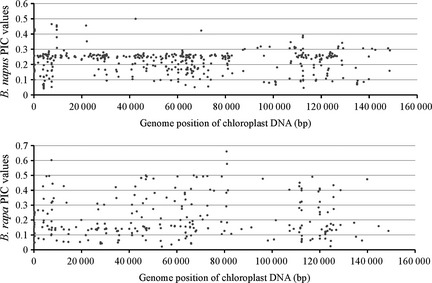
Genomic distribution of PIC values of the B. napus and B. rapa cpDNA polymorphic loci. The PIC values were calculated as , where n is the number of alleles and p i is the frequency of each allele at the corresponding polymorphic locus in the corresponding population. Each data point therefore indicates the PIC value for a single locus. The site with alleles at equal frequencies has the highest PIC value; the maximum PIC score for sites containing two alleles is 0.5 and that for sites containing three alleles is 0.67.
The cpDNA diversity is low in Brassica oleracea rather than in its wild relatives
To examine the genetic relationships between B. napus and B. oleracea, 49 wild B. oleracea (Table S1) accessions were also pangenomically analysed. The majority of these materials were collected from the Eastern Mediterranean (Spain), which is recognized as the centre of origin for B. napus. The reads were directly assembled to the B. napus and B. rapa chloroplast reference genomes. The cpDNA of the wild B. oleracea population lacked genetic diversity and appeared to consist of a single cpDNA haplotype (Bocp1) that is distinct from the B. napus and B. rapa reference sequences. Over 400 variants were identified by referencing to the ZY036 cpDNA reference sequence (Table S2). These results indicate that the cpDNA of wild B. oleracea is distinct from that of B. rapa and B. napus.
Five cpDNA regions were also amplified from 15 diversified cultivated B. oleracea accessions (Table S1). The Sanger sequencing results showed that all of the investigated cultivated B. oleracea accessions carry the same variations in the 5 cpDNA regions as observed in wild B. oleracea, suggesting that the cultivated and wild B. oleracea share a single cpDNA haplotype. The cpDNA diversity in 5 accessions of the B. oleracea wild relatives (B. cretica, B. incana, B. insularis and B. villosa) was also investigated via Sanger sequencing of their rpo regions. The cpDNA variants from the B. oleracea wild relatives showed that the four B. oleracea wild relatives kept distinct haplotypes that differed from the major haplotype Bocp1 in B. oleracea. These results indicated a marked contrast in cpDNA diversity between B. oleracea and its wild relatives.
Natural mutation patterns in Brassica cpDNA
The patterns of cpDNA variations in the Brassica populations discussed above were systematically identified. The cpDNA variants appeared to be distributed in a sites‐dependent manner in both the B. napus and B. rapa populations. Approximately three‐fourths (409/538) of the variants are located in the intergenic regions (IGR) (Figure 4b). The IRG‐enriched genome regions, for example the majority of the tRNA regions were identified as hypervariable, whereas the coding regions, for example the atpA‐rpoC1 and psbD‐psaA regions, were identified to be relatively stable. This pattern resembled the intergeneric pattern of cpDNA variations identified in angiosperms (Dong et al., 2012). These findings highlight that noncoding hypervariable cpDNA regions, which evolved rapidly, are the most effective molecular markers for phylogenetic studies and DNA barcoding, especially for studies at the intraspecific level.
In contrast, the 52‐kb inverted repeated regions (IR) contained only 27 pairs of symmetrically distributed variants, with a distribution density four times lower than the average genomewide density (538 variations in 152 kb), indicating that the IR regions are conserved cpDNA elements. There are over 100 cpDNA variants in the coding sequence (CDS) regions, 53 variants of which are nonsynonymous mutations (Table S2). Ycf1, ycf2, ccsA, rpoB, rpoC2, matK, ndhF, psaA, psaB, accD and ndhI were identified as hypervariable protein‐coding genes, and ycf1 with a total of 30 nonsynonymous variations appeared to be the most hypervariable. These results indicate that cpDNA also has a rapid intraspecific evolutionary rate, even in a recent species B. napus.
Discussion
Exhaustive identification of cpDNA variants at the population level using a novel pangenomic sequencing methodology
The successful identification of hundreds of cpDNA variants in current Brassica populations verified that our newly developed highly multiplex sequencing methodology is feasible for expeditious identification of the intraspecific variants in a large population, which is highly effective and economical. Isolation of intact chloroplasts requires a large quantity of plant materials, with high costs in labour and money. Considering that cpDNA is generally maintained at high levels in developing leaves and is apt to attach membranes even in broken chloroplasts (Rowan et al., 2009; Sato et al., 1998), chloroplast isolation was optimized for quick isolation of cpDNA with high purity. The cpDNA purity was increased at least fivefold compared to the leaf total DNA (from <3% to 30%; Nock et al., 2011; Table 1). The uniform mixing of leaf samples collected from each individual accession prior to chloroplast isolation can greatly amplify the sample size, reaching up to hundreds of accessions in a single population. The amount of sequence data should be principally set according to the sample size of the sequencing population to guarantee coverage of low‐frequency cpDNA variants. Natural cpDNA variants generally appear in the form of SNPs and short InDels in low‐taxonomic populations, especially in intraspecific populations (Hu et al., 2011; Wang et al., 2013); therefore, sequence reads up to 100 bp long generated by the next‐generation sequencing platform, enabled the direct calling of the cpDNA variants and their allelic frequencies. The above details eventually facilitated the successful development of novel pangenomic high‐throughput cpDNA sequencing methodology.
Conventional cpDNA‐based phylogenetic studies primarily depend on polymorphisms in hypervariable cpDNA regions. The conclusions derived from these studies may diverge from their true relationships due to the fluctuations in the evolutionary rates among cpDNA regions or species (Dong et al., 2012). One of the most important low‐taxonomic studies for crops is to dissect the molecular mechanisms regarding its origination, domestication and artificial breeding selection, with the purpose of exploring and utilizing elite alleles from their wild relatives or ancestor species. These studies primarily focus on intraspecific populations but are generally limited by the lack of sufficient effective molecular markers. The pangenomic sequencing strategy developed here would greatly aid in obtaining information on the overall genetic diversity in large collections. Then, sufficient markers with high intraspecific resolution can be exploited based on the hypervariable regions, accelerating the accurate determination of the intraspecific genetic relationships.
The genetic diversity among B. napus, B. rapa and B. oleracea
The PIC values of B. napus cpDNA variants were densely distributed at a range of 0.2–0.3. In addition, one rpo region‐derived haplotype (Bncp1) was identified as predominant via EcoTILLING analyses, with a frequency of 91.8% in B. napus. A total of three haplotypes were detected in 94 European B. napus accessions via cpSSR genotyping analyses (Allender and King, 2010). The populational frequency for each haplotype was 79.8%, 17% and 3.2%. The coexistence of one predominant and several rare haplotypes in the B. napus populations caused their lower PIC values and simplex distribution patterns. These results demonstrated that B. napus had lower genetic diversity at the cpDNA level. To some extent, these data also reflect low genetic diversity at the nuclear‐DNA level. In contrast, from the PIC values, B. rapa appeared to have much higher genetic diversity than B. napus. This result was also suggested by the cpSSR analysis of European B. rapa (Allender and King, 2010). In addition, the nucleotide sequence and allelic frequencies of intraspecific cpDNA variants have been systemically identified. Therefore, the genetic diversities of B. napus and B. rapa have been finely determined at the cpDNA level. The low genetic diversity of B. napus may be the result of its recent speciation and strong artificial selection during its evolution.
Interestingly, cpDNA of the investigated cultivated and wild B. oleracea was found to consist of only one haplotype, in strong contrast to its wild relatives (Allender and King, 2010; current study). The lack of cpDNA diversity in B. oleracea cpDNA was also revealed via cpSSR genotyping of 106 cultivated and wild B. oleracea accessions (Allender and King, 2010; Allender et al., 2007), the vast majority of which (102/106) shared a single haplotype (C:01). All the haplotypes identified in the B. oleracea population and in its wild relatives are distinct from the B. napus Bncp1 haplotype. These results clearly exclude the introgression of the Bncp1 haplotype from any of the investigated B. oleracea populations into B. napus. Compared to B. rapa and B. napus, B. oleracea speciated earlier (Liu et al., 2014) and have diverged morphologically for multiple agricultural purposes (Wang et al., 2011b). B. oleracea has high genetic diversity at the nuclear‐DNA level, especially in wild groups (Liu et al., 2014; Mei et al., 2011; Wu et al., 2014). Thus, there is a stark contrast between the low cpDNA diversity and high nuclear diversity in B. oleracea, suggesting that its cpDNA likely evolves much slower than the nuclear DNA.
The possible origins of the predominant rapeseed cpDNA haplotype
It is interesting that a vast majority of the current worldwide B. napus tend to carry one predominant cpDNA haplotype (Bncp1). The genomewide sequence for Bncp1 cpDNA has been determined here. Studies of genetic diversity in B. rapa demonstrated that the classical Old World (Europe and west Asia–north Africa) was the centre of origin, whereas East Asia and the Asian ancient trade routes were the diversity centres for this species (Guo et al., 2014). Thus, the Chinese and European B. rapa are thought to have high genetic diversity; however, approximately half of the 423 predominant (Bncp1 haplotype) B. napus alleles have never been detected in the investigated Chinese B. rapa. Besides, the Bncp1 haplotype was not present in a foreign B. rapa population (38 accessions) whose haplotypes were identified via PCR and Sanger sequencing of the rpo regions (Table S2). Phylogenetic analyses of these Brassica cpDNA haplotypes demonstrated that they clustered into three species‐specific groups, and Bncp1 haplotype was distinct from others (Figure 6). The predominant B. napus haplotype has been found in a few wild and weedy accessions by investigating a large A‐genome B. rapa population (83 accessions) (Allender and King, 2010); however, these data were not verified genomewide and may have resulted from the cpDNA introgression from B. napus into B. rapa due to their wild co‐cultivation.
Figure 6.

Phylogenetic relationships of Brassica cpDNA haplotypes based on the rpo variants. The subclades representing evolutionary linkage are marked. Numbers at nodes indicate the bootstrap values (%) calculated from 1000 trials, the values lower than 50% were hidden. The length of branches indicates the evolutionary divergence according to the scale bar (relative units) at the bottom.
Therefore, the origin of Bncp1 haplotype remains a mystery. It may be that their genuine diploid parents have disappeared or were never successfully collected by researchers, or they may exist at a low frequency under the detection limit of currently available methods. Multiple breeding approaches may also lead to the introduction of cytoplasmic genomes from relatives into B. napus, which would complicate the origination of its cytoplasmic genomes. There is also a major possibility that the parental cytoplasmic genomes can genetically recombine during fertilization to generate a novel cpDNA genotype, as was reported in in vitro cell fusion (Yamagishi and Bhat, 2014). Therefore, the predominant B. napus haplotype may also arise from currently unknown cpDNA mutations and then be maintained by natural (or artificial) selection during speciation and evolution. This hypothesis will require rigorous testing via investigations of the resynthesized or manually mutagenized B. napus over successive generations.
Potential applications of these cpDNA variants
It is surprising that the essential genes ycf1, ycf2, ccsA, rpoB, rpoC2, rpoC1 and accD (De Santis‐MacIossek et al., 1999; Drescher et al., 2000; Kode et al., 2005; Xie and Merchant, 1996) were identified as the most variable coding genes containing nonsynonymous mutations. YCF1 was recently identified as an essential component of protein translocon at the chloroplast inner envelope membrane (Kikuchi et al., 2013). Its six predicted transmembrane segments and two moderately hydrophobic stretches are conserved and are likely the functional domains, and the remaining portions are highly divergent. A protein alignment showed that all of the B. napus ycf1 variants are located in the highly divergent portions, suggesting their nonmutational functionality, which would partially explain why ycf1 has a rapid evolutionary rate (Hernández‐León et al., 2013).
Chloroplast genes encode products that are used in the assembly of photosynthetic machinery, the regulation of plastid gene expression, energy metabolism and the synthesis of essential metabolites (Qiao et al., 2011; Sato et al., 1999); however, to date, the functions and their detailed mechanisms for many chloroplast genes (e.g. ycf1, ycf2, accD and certain rps genes) remain unresolved (Pfalz et al., 2006; Qiao et al., 2013). A number of noncoding organelle DNA regions have high transcription rates and may participate in the regulation of chloroplast functions (Hodgkinson et al., 2014; Zhelyazkova et al., 2012). Specific plants with related functional cpDNA variations are ideal subjects for an investigation of cpDNA function. Therefore, the exhaustive data on variation in cpDNA obtained in this study would be helpful in obtaining natural plant accessions with specific cpDNA variants and promoting research on plastid genes.
Experimental procedures
Plant materials
The B. napus and B. rapa samples were conserved in the National Mi‐term GenBank for Oil Crops of China. B. napus were globally collected and included various ecological types with abundant genetic diversity. This population was previously used in our genomewide association studies (Li et al., 2014a; Wang et al., 2014). The B. rapa sampling population contained primarily landraces that were collected throughout China. All of these materials were cultivated in experimental fields in Wuhan (N 30.52, E 114.31) from October 2012 to May 2013. The wild B. oleracea were collected from the rocky Atlantic coasts of Spain (Bay of Biscay) and have been perennially cultivated in screenhouses in Wuhan since 2010. The cultivated B. oleracea and its wild relatives were cultivated in a greenhouse. The accession names, geographic origins and other related information for all these plant materials are given in Table S1.
Chloroplast and DNA isolation
The isolation of leaf chloroplasts was performed according to Fitzpatrick and Keegstra (2001), with minor modifications to reduce contamination by non‐cpDNA. Leaf tissues were homogenized in chloroplast isolation buffer (CIB, 0.33 m sorbitol, 50 mm Tricine, 2 mm EDTA, 2 mm MgCl2, 1 mm DTT, 0.1% BSA, pH 7.8). The volume of CIB was increased to 15 volumes of fresh weight of the corresponding leaves (e.g. 15 mL of CIB for 1 g of fresh leaves). An additional centrifugation step (240 g , 3 min) was performed to remove the unwanted whole plant cells and cell debris after filtration through a nylon mesh. The chloroplast pellets were washed twice with CIB (without BSA) prior to DNA extraction. Chloroplast DNA was extracted using the cetyltrimethylammonium bromide (CTAB) methods described by Lutz et al. (2011). The accession‐specific DNA used for the EcoTILLING analyses was directly extracted from the juvenile leaves of representative plants using the above‐mentioned CTAB method.
High‐throughput sequencing
Approximately 1 μg of multiplexed cpDNA was randomly ultrasonically sheared. Fragments with sizes ranging from 300 to 400 bp were recovered, adaptor ligated (TruSeq DNA sample preparation kit; Illumina, San Diego, CA) and PCR‐amplified to construct the DNA libraries (TruSeq PE Cluster Kit; Illumina). The DNA libraries were then subjected to an Illumina HiSeq 2000 sequencing system for sequencing at both single ends. The obtained raw data were preprocessed to remove low‐quality reads and adaptor sequences according to the Illumina fixed criteria.
Default parameters were set for the assembly of obtained sequence reads to the reference genomes. The ‘no‐specific matches’ parameter was set to ‘Random’ to avoid any asymmetric assembly caused by plastome inverted repeated regions (IR). The average sequencing depth was calculated through the total nucleotide content of the mapped reads divided by the size of the reference genome (152.86 kb). The cpDNA variants were called using the following optimized parameter settings: neighbourhood radius: 5; maximum gap and mismatch count: 2; minimum neighbourhood quality: 20; minimum central quality: 20; minimum coverage: 200; minimum variant frequency: 1.0%; maximum expected alleles: 4; requirement for both the forward and reverse reads; and with a filter for 454/Ion homopolymer indels.
Tilling analysis, Sanger sequencing and phylogenetic analyses
EcoTILLING analyses were performed as described in the previous study (Zeng et al., 2012). An eightfold two‐dimensional pooling strategy was introduced to increase the sample throughput, as in Till et al. (2006). Equal volumes of the leaf total DNA (200 μg/μL) from eight individuals were pooled as one sample. A mutation results in the appearance of appropriate bands in two separate gel lanes. Therefore, the unique individuals harbouring this mutation can be identified by its coordinates. First‐round PCR was performed using gene‐specific primers with M13F and M13R tags at each end. The obtained PCR products were then diluted 50 times and used as templates for second‐round PCR using the M13F‐labelled (IRDye 700) and M13R‐labelled (IRDye 800) primers. Final PCR products were denatured by heating and then gradually annealed to form heteroduplexes. Any mismatches (SNPs and InDels) were cleaved using the endonuclease CEL I and detected in the 700 and 800 Dye channel on a Li‐COR 4300 DNA analyzer. A mutation was identified by judging whether the summed size of the bands in the two dye channels was equal to the size of the corresponding PCR product. PCR products amplified from the corresponding individual DNA samples were Sanger sequenced using a 3730 DNA analyzer (Life Technologies). All the primers used here are given in Table S3. Evolutionary analyses were conducted in MEGA5 (Tamura et al., 2011) using the neighbour‐joining method.
Conflict of interest
There is no conflict of interest for us to declare.
Supporting information
Table S1 List of plant materials for B. napus, B. rapa and B. oleracea.
Table S2 Total cpDNA variants in the current B. napus, B. rapa and B. oleracea populations. The information regarding genome locus, allelic frequency and related changes in coding amino acids for each variant is given.
Table S3 Primers used in EcoTILLING analyses.
Acknowledgements
We acknowledge Professor Takeshi Nishino (Graduate School of Agricultural Science, Tohoku University, Japan) for kindly providing the wild B. oleracea samples. We are grateful to Professor Xuxiao Zong (Institute of Crop Science, Chinese Academy of Agricultural Sciences, China) for his assitance on the bioinformatics analysis. We thank Tianyao Zhang and Hao Li for excellent field experiments and technical assistance. This work was supported by the National Natural Science Foundation of China (Grant No. 31100911) and the National Science & Technology Pillar Program during the Twelfth Five‐Year Plan Period (Grant No. 2013BAD01B01 and 2013BAD01B03).
References
- Allender, C.J. and King, G.J. (2010) Origins of the amphiploid species Brassica napus L. investigated by chloroplast and nuclear molecular markers. BMC Plant Biol. 10, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allender, C.J. , Allainguillaume, J. , Lynn, J. and King, G.J. (2007) Simple sequence repeats reveal uneven distribution of genetic diversity in chloroplast genomes of Brassica oleracea L. and (n = 9) wild relatives. Theor. Appl. Genet. 114, 609–618. [DOI] [PubMed] [Google Scholar]
- Birky, C.W. Jr . (2001) The inheritance of genes in mitochondria and chloroplasts: laws, mechanisms, and models. Annu. Rev. Genet. 35, 125–148. [DOI] [PubMed] [Google Scholar]
- Chalhoub, B. , Denoeud, F. , Liu, S. , Parkin, I.A. , Tang, H. , Wang, X. , Chiquet, J. , Belcram, H. , Tong, C. , Samans, B. , Corréa, M. , Da Silva, C. , Just, J. , Falentin, C. , Koh, C.S. , Le Clainche, I. , Bernard, M. , Bento, P. , Noel, B. , Labadie, K. , Alberti, A. , Charles, M. , Arnaud, D. , Guo, H. , Daviaud, C. , Alamery, S. , Jabbari, K. , Zhao, M. , Edger, P.P. , Chelaifa, H. , Tack, D. , Lassalle, G. , Mestiri, I. , Schnel, N. , Le Paslier, M.C. , Fan, G. , Renault, V. , Bayer, P.E. , Golicz, A.A. , Manoli, S. , Lee, T.H. , Thi, V.H. , Chalabi, S. , Hu, Q. , Fan, C. , Tollenaere, R. , Lu, Y. , Battail, C. , Shen, J. , Sidebottom, C.H. , Wang, X. , Canaguier, A. , Chauveau, A. , Bérard, A. , Deniot, G. , Guan, M. , Liu, Z. , Sun, F. , Lim, Y.P. , Lyons, E. , Town, C.D. , Bancroft, I. , Wang, X. , Meng, J. , Ma, J. , Pires, J.C. , King, G.J. , Brunel, D. , Delourme, R. , Renard, M. , Aury, J.M. , Adams, K.L. , Batley, J. , Snowdon, R.J. , Tost, J. , Edwards, D. , Zhou, Y. , Hua, W. , Sharpe, A.G. , Paterson, A.H. , Guan, C. and Wincker, P. (2014) Early allopolyploid evolution in the post‐Neolithic Brassica napus oilseed genome. Science, 345, 950–953. [DOI] [PubMed] [Google Scholar]
- De Santis‐MacIossek, G. , Kofer, W. , Bock, A. , Schoch, S. , Maier, R.M. , Wanner, G. , Rüdiger, W. , Koop, H.U. and Herrmann, R.G. (1999) Targeted disruption of the plastid RNA polymerase genes rpoA, B and C1: molecular biology, biochemistry and ultrastructure. Plant J. 18, 477–489. [DOI] [PubMed] [Google Scholar]
- Dong, W. , Liu, J. , Yu, J. , Wang, L. and Zhou, S. (2012) Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One, 7, e35071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drescher, A. , Ruf, S. , Calsa, T. Jr , Carrer, H. and Bock, R. (2000) The two largest chloroplast genome‐encoded open reading frames of higher plants are essential genes. Plant J. 22, 97–104. [DOI] [PubMed] [Google Scholar]
- Fior, S. , Li, M. , Oxelman, B. , Viola, R. , Hodges, S.A. , Ometto, L. and Varotto, C. (2013) Spatiotemporal reconstruction of the Aquilegia rapid radiation through next‐generation sequencing of rapidly evolving cpDNA regions. New Phytol. 198, 579–592. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick, L.M. and Keegstra, K. (2001) A method for isolating a high yield of Arabidopsis chloroplasts capable of efficient import of precursor proteins. Plant J. 27, 59–65. [DOI] [PubMed] [Google Scholar]
- Guo, Y.M. , Chen, S. , Li, Z.Y. and Cowling, W.A. (2014) Center of origin and centers of diversity in an ancient crop, Brassica rapa (Turnip Rape). J. Hered. 105, 555–565. [DOI] [PubMed] [Google Scholar]
- Heinze, B. , Koziel‐Monte, A. and Jahn, D. (2014) Analysis of variation in chloroplast DNA sequences. Methods Mol. Biol. 1115, 85–120. [DOI] [PubMed] [Google Scholar]
- Henry, R.J. , Rice, N. , Edwards, M. and Nock, C.J. (2014) Next‐generation technologies to determine plastid genome sequences. Methods Mol. Biol. 1132, 39–46. [DOI] [PubMed] [Google Scholar]
- Hernández‐León, S. , Gernandt, D.S. , Pérez de la Rosa, J.A. and Jardón‐Barbolla, L. (2013) Phylogenetic relationships and species delimitation in pinus section trifoliae inferrred from plastid DNA. PLoS One, 8, e70501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson, A. , Idaghdour, Y. , Gbeha, E. , Grenier, J.C. , Hip‐Ki, E. , Bruat, V. , Goulet, J.P. , de Malliard, T. and Awadalla, P. (2014) High‐resolution genomic analysis of human mitochondrial RNA sequence variation. Science, 344, 413–415. [DOI] [PubMed] [Google Scholar]
- Hu, Z.Y. , Hua, W. , Huang, S.M. and Wang, H.Z. (2011) Complete chloroplast genome sequence of rapeseed (Brassica napus L.) and its evolutionary implications. Genet. Resour. Crop Evol. 58, 875–887. [Google Scholar]
- Hua, W. , Li, R.J. , Zhan, G.M. , Liu, J. , Li, J. , Wang, X.F. , Liu, G.H. and Wang, H.Z. (2012) Maternal control of seed oil content in Brassica napus: the role of silique wall photosynthesis. Plant J. 69, 432–444. [DOI] [PubMed] [Google Scholar]
- Kikuchi, S. , Bédard, J. , Hirano, M. , Hirabayashi, Y. , Oishi, M. , Imai, M. , Takase, M. , Ide, T. and Nakai, M. (2013) Uncovering the protein translocon at the chloroplast inner envelope membrane. Science, 339, 571–574. [DOI] [PubMed] [Google Scholar]
- Kode, V. , Mudd, E.A. , Iamtham, S. and Day, A. (2005) The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 44, 237–244. [DOI] [PubMed] [Google Scholar]
- Li, D. , Qi, X. , Li, X. , Li, L. , Zhong, C. and Huang, H. (2013a) Maternal inheritance of mitochondrial genomes and complex inheritance of chloroplast genomes in Actinidia Lind.: evidences from interspecific crosses. Mol. Genet. Genomics, 288, 101–110. [DOI] [PubMed] [Google Scholar]
- Li, R. , Ma, P.F. , Wen, J. and Yi, T.S. (2013b) Complete sequencing of five araliaceae chloroplast genomes and the phylogenetic implications. PLoS One, 8, e78568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , Chen, B. , Xu, K. , Wu, J. , Song, W. , Bancroft, I. , Harper, A.L. , Trick, M. , Liu, S. , Gao, G. , Wang, N. , Yan, G. , Qiao, J. , Li, J. , Li, H. , Xiao, X. , Zhang, T. and Wu, X. (2014a) Genome‐wide association study dissects the genetic architecture of seed weight and seed quality in rapeseed (Brassica napus L.). DNA Res. 21, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , Li, Y. , Song, J. , Xu, H. , Xu, J. , Zhu, Y. , Li, X. , Gao, H. , Dong, L. , Qian, J. , Sun, C. and Chen, S. (2014b) High‐accuracy de novo assembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy. New Phytol. 204, 1041–1049. [DOI] [PubMed] [Google Scholar]
- Liu, S. , Liu, Y. , Yang, X. , Tong, C. , Edwards, D. , Parkin, I.A. , Zhao, M. , Ma, J. , Yu, J. , Huang, S. , Wang, X. , Wang, J. , Lu, K. , Fang, Z. , Bancroft, I. , Yang, T.J. , Hu, Q. , Wang, X. , Yue, Z. , Li, H. , Yang, L. , Wu, J. , Zhou, Q. , Wang, W. , King, G.J. , Pires, J.C. , Lu, C. , Wu, Z. , Sampath, P. , Wang, Z. , Guo, H. , Pan, S. , Yang, L. , Min, J. , Zhang, D. , Jin, D. , Li, W. , Belcram, H. , Tu, J. , Guan, M. , Qi, C. , Du, D. , Li, J. , Jiang, L. , Batley, J. , Sharpe, A.G. , Park, B.S. , Ruperao, P. , Cheng, F. , Waminal, N.E. , Huang, Y. , Dong, C. , Wang, L. , Li, J. , Hu, Z. , Zhuang, M. , Huang, Y. , Huang, J. , Shi, J. , Mei, D. , Liu, J. , Lee, T.H. , Wang, J. , Jin, H. , Li, Z. , Li, X. , Zhang, J. , Xiao, L. , Zhou, Y. , Liu, Z. , Liu, X. , Qin, R. , Tang, X. , Liu, W. , Wang, Y. , Zhang, Y. , Lee, J. , Kim, H.H. , Denoeud, F. , Xu, X. , Liang, X. , Hua, W. , Wang, X. , Wang, J. , Chalhoub, B. and Paterson, A.H. (2014) The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5, 3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz, K.A. , Wang, W. , Zdepski, A. and Michael, T.P. (2011) Isolation and analysis of high quality nuclear DNA with reduced organellar DNA for plant genome sequencing and resequencing. BMC Biotechnol. 11, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei, J. , Li, Q. , Qian, L. , Fu, Y. , Li, J. , Frauen, M. and Qian, W. (2011) Genetic investigation of the origination of allopolyploid with virtually synthesized lines: application to the C subgenome of Brassica napus . Heredity, 106, 955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen, H.L. (1996) The hows and whys of cytoplasmic inheritance in seed plants. Am. J. Bot. 83, 383–404. [Google Scholar]
- Nagaharu, U. (1935) Genome‐analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. J. Jpn. Bot. 7, 389–452. [Google Scholar]
- Nagy, S. , Poczai, P. , Cernák, I. , Gorji, A.M. , Hegedűs, G. and Taller, J. (2012) PICcalc: an online program to calculate polymorphic information content for molecular genetic studies. Biochem. Genet. 50, 670–672. [DOI] [PubMed] [Google Scholar]
- Nikiforova, S.V. , Cavalieri, D. , Velasco, R. and Goremykin, V. (2013) Phylogenetic analysis of 47 chloroplast genomes clarifies the contribution of wild species to the domesticated apple maternal line. Mol. Biol. Evol. 30, 1751–1760. [DOI] [PubMed] [Google Scholar]
- Nock, C.J. , Waters, D.L. , Edwards, M.A. , Bowen, S.G. , Rice, N. , Cordeiro, G.M. and Henry, R.J. (2011) Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 9, 328–333. [DOI] [PubMed] [Google Scholar]
- Pfalz, J. , Liere, K. , Kandlbinder, A. , Dietz, K.J. and Oelmüller, R. (2006) pTAC2, ‐6, and ‐12 are components of the transcriptionally active plastid chromosome that are required for plastid gene expression. Plant Cell, 18, 176–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash, S. , Wu, X.M. and Bhat, S.R. (2011) History, evolution, and domestication of Brassica crops. In Plant Breeding Reviews, Vol. 35 ( Janick, J. ed.), pp. 19–84. USA: Wiley‐Blackwell press. [Google Scholar]
- Provan, J.W. , Powell, W. and Hollingsworth, P.M. (2001) Chloroplast microsatellites: new tools for studies in plant ecology and evolution. Trends Ecol. Evol. 16, 142–147. [DOI] [PubMed] [Google Scholar]
- Qiao, J. , Ma, C. , Wimmelbacher, M. , Börnke, F. and Luo, M. (2011) Two novel proteins, MRL7 and its paralog MRL7‐L, have essential but functionally distinct roles in chloroplast development and are involved in plastid gene expression regulation in Arabidopsis. Plant Cell Physiol. 52, 1017–1030. [DOI] [PubMed] [Google Scholar]
- Qiao, J. , Li, J. , Chu, W. and Luo, M. (2013) PRDA1, a novel chloroplast nucleoid protein, is required for early chloroplast development and is involved in the regulation of plastid gene expression in Arabidopsis. Plant Cell Physiol. 54, 2071–2184. [DOI] [PubMed] [Google Scholar]
- Rana, D. , van den Boogaart, T. , O'Neill, C.M. , Hynes, L. , Bent, E. , Macpherson, L. , Park, J.Y. , Lim, Y.P. and Bancroft, I. (2004) Conservation of the microstructure of genome segments in Brassica napus and its diploid relatives. Plant J. 40, 725–733. [DOI] [PubMed] [Google Scholar]
- Rowan, B.A. , Oldenburg, D.J. and Bendich, A.J. (2009) A multiple‐method approach reveals a declining amount of chloroplast DNA during development in Arabidopsis. BMC Plant Biol. 9, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, N. , Ohshima, K. , Watanabe, A. , Ohta, N. , Nishiyama, Y. , Joyard, J. and Douce, R. (1998) Molecular characterization of the PEND protein, a novel bZIP protein present in the envelope membrane that is the site of nucleoid replication in developing plastids. Plant Cell, 10, 859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, S. , Nakamura, Y. , Kaneko, T. , Asamizu, E. and Tabata, S. (1999) Complete structure of the chloroplast genome of Arabidopsis thaliana . DNA Res. 6, 283–290. [DOI] [PubMed] [Google Scholar]
- Song, K. and Osborn, T.C. (1992) Polyphyletic origins of Brassica napus: new evidence based on organelle and nuclear RFLP analyses. Genome, 35, 992–1001. [Google Scholar]
- Tamura, K. , Peterson, D. , Peterson, N. , Stecher, G. , Nei, M. and Kumar, S. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till, B.J. , Zerr, T. , Comai, L. and Henikoff, S. (2006) A protocol for TILLING and Ecotilling in plants and animals. Nat. Protoc. 1, 2465–2477. [DOI] [PubMed] [Google Scholar]
- Van Den Berg, G. , Bryan, J. , Del Rio, A. and Spooner, M. (2002) Reduction of species in the wild potato Solanum section Petota series Longipedicellata: AFLP, RAPD and chloroplast SSR data. Theor. Appl. Genet. 105, 1109–1114. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Wang, H. , Wang, J. , Sun, R. , Wu, J. , Liu, S. , Bai, Y. , Mun, J.H. , Bancroft, I. , Cheng, F. , Huang, S. , Li, X. , Hua, W. , Wang, J. , Wang, X. , Freeling, M. , Chris Pires, J. , Paterson, A.H. , Chalhoub, B. , Wang, B. , Hayward, A. , Sharpe, A.G. , Park, B.S. , Weisshaar, B. , Liu, B. , Li, B. , Liu, B. , Tong, C. , Song, C. , Duran, C. , Peng, C. , Geng, C. , Koh, C. , Lin, C. , Edwards, D. , Mu, D. , Shen, D. , Soumpourou, E. , Li, F. , Fraser, F. , Conant, G. , Lassalle, G. , King, G.J. , Bonnema, G. , Tang, H. , Wang, H. , Belcram, H. , Zhou, H. , Hirakawa, H. , Abe, H. , Guo, H. , Wang, H. , Jin, H. , Parkin, I.A. , Batley, J. , Kim, J.S. , Just, J. , Li, J. , Xu, J. , Deng, J. , Kim, J.A. , Li, J. , Yu, J. , Meng, J. , Wang, J. , Min, J. , Poulain, J. , Wang, J. , Hatakeyama, K. , Wu, K. , Wang, L. , Fang, L. , Trick, M. , Links, M.G. , Zhao, M. , Jin, M. , Ramchiary, N. , Drou, N. , Berkman, P.J. , Cai, Q. , Huang, Q. , Li, R. , Tabata, S. , Cheng, S. , Zhang, S. , Zhang, S. , Huang, S. , Sato, S. , Sun, S. , Kwon, S.J. , Choi, S.R. , Lee, T.H. , Fan, W. , Zhao, X. , Tan, X. , Xu, X. , Wang, Y. , Qiu, Y. , Yin, Y. , Li, Y. , Du, Y. , Liao, Y. , Lim, Y. , Narusaka, Y. , Wang, Y. , Wang, Z. , Li, Z. , Wang, Z. , Xiong, Z. and Zhang, Z. (2011a) The genome of the mesopolyploid crop species Brassica rapa . Nat. Genet. 43, 1035–1059. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Torres, M.J. , Pierce, G. , Lemke, C. , Nelson, L.K. , Yuksel, B. , Bowers, J.E. , Marler, B. , Xiao, Y. , Lin, L. , Epps, E. , Sarazen, H. , Rogers, C. , Karunakaran, S. , Ingles, J. , Giattina, E. , Mun, J.H. , Seol, Y.J. , Park, B.S. , Amasino, R.M. , Quiros, C.F. , Osborn, T.C. , Pires, J.C. , Town, C. and Paterson, A.H. (2011b) A physical map of Brassica oleracea shows complexity of chromosomal changes following recursive paleopolyploidizations. BMC Genom. 12, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Shi, C. and Gao, L.Z. (2013) Plastid genome sequence of a wild woody oil species, Prinsepia utilis, provides insights into evolutionary and mutational patterns of Rosaceae chloroplast genomes. PLoS One, 8, e73946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, N. , Li, F. , Chen, B. , Xu, K. , Yan, G. , Qiao, J. , Li, J. , Gao, G. , Bancroft, I. , Meng, J. , King, G.J. and Wu, X. (2014) Genome‐wide investigation of genetic changes during modern breeding of Brassica napus . Theor. Appl. Genet. 127, 1817–1829. [DOI] [PubMed] [Google Scholar]
- Wu, J. , Li, F. , Xu, K. , Gao, G. , Chen, B. , Yan, G. , Wang, N. , Qiao, J. , Li, J. , Li, H. , Zhang, T. , Song, W. and Wu, X. (2014) Assessing and broadening genetic diversity of a rapeseed germplasm collection. Breed Sci. 64, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, Z. and Merchant, S. (1996) The plastid‐encoded ccsA gene is required for heme attachment to chloroplast c‐type cytochromes. J. Biol. Chem. 271, 4632–4639. [DOI] [PubMed] [Google Scholar]
- Yamagishi, H. and Bhat, S.R. (2014) Cytoplasmic male sterility in Brassicaceae crops. Breed Sci. 64, 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamani‐Nour, S. , Clemens, R. and Möllers, C. (2013) Cytoplasmic diversity of Brassica napus L., Brassica oleracea L. and Brassica rapa L. as determined by chloroplast microsatellite markers. Genet. Resour. Crop Evol. 60, 953–965. [Google Scholar]
- Zeng, C.L. , Wang, G.Y. , Wang, J.B. , Yan, G.X. , Chen, B.Y. , Xu, K. , Li, J. , Gao, G.Z. , Wu, X.M. , Zhao, B. and Liu, L. (2012) High‐throughput discovery of chloroplast and mitochondrial DNA polymorphisms in Brassicaceae species by ORG‐EcoTILLING. PLoS One, 7, e47284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.J. , Ma, P.F. and Li, D.Z. (2011) High‐throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS One, 6, e20596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhelyazkova, P. , Sharma, C.M. , Förstner, K.U. , Liere, K. , Vogel, J. and Börner, T. (2012) The primary transcriptome of barley chloroplasts: numerous noncoding RNAs and the dominating role of the plastid‐encoded RNA polymerase. Plant Cell, 24, 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 List of plant materials for B. napus, B. rapa and B. oleracea.
Table S2 Total cpDNA variants in the current B. napus, B. rapa and B. oleracea populations. The information regarding genome locus, allelic frequency and related changes in coding amino acids for each variant is given.
Table S3 Primers used in EcoTILLING analyses.