

Abstract
XylT (β1,2-xylosyltransferase) is a unique Golgi-bound glycosyltransferase that is involved in the biosynthesis of glycoprotein-bound N-glycans in plants. To delineate the catalytic domain of XylT, a series of N-terminal deletion mutants was heterologously expressed in insect cells. Whereas the first 54 residues could be deleted without affecting the catalytic activity of the enzyme, removal of an additional five amino acids led to the formation of an inactive protein. Characterization of the N-glycosylation status of recombinant XylT revealed that all three potential N-glycosylation sites of the protein are occupied by N-linked oligosaccharides. However, an unglycosylated version of the enzyme displayed substantial catalytic activity, demonstrating that N-glycosylation is not essential for proper folding of XylT. In contrast with most other glycosyltransferases, XylT is enzymatically active in the absence of added metal ions. This feature is not due to any metal ion directly associated with the enzyme. The precise acceptor substrate specificity of XylT was assessed with several physiologically relevant compounds and the xylosylated reaction products were subsequently tested as substrates of other Golgi-resident glycosyltransferases. These experiments revealed that the substrate specificity of XylT permits the enzyme to act at multiple stages of the plant N-glycosylation pathway.
Keywords: Arabidopsis, glycosyltransferase, Golgi apparatus, N-glycan biosynthesis, proteolytic processing, xylosyltransferase
Abbreviations: CT domain, cytoplasmic and transmembrane domain; E-64, trans-epoxysuccinyl-L-leucylamido-(4-guanidino)butane; endo H, endoglycosidase H; FucT, core α1,3-fucosyltransferase; GnGn, GlcNAcβ1-2Manα1-6(GlcNAcβ1-2Manα1-3)Manβ1-4GlcNAcβ1-4GlcNAc; GnGn-octyl, GlcNAc β 1-2Manα 1-6 (GlcNAc β 1-2Manα1-3)Manβ1-O-octyl; GnGnF3, GlcNAc β1-2Man α 1-6(GlcNAc β 1-2Manα 1-3)Manβ1-4 GlcNAcβ1-4(Fucα 1 -3)GlcNAc; GnGnX, Xylβ1-2[GlcNAcβ1-2Manα1-6(GlcNAcβ1-2Manα1-3)]Manβ1-4GlcNAcβ1-4GlcNAc; GnT I, β1,2-N-acetylglucosaminyltransferase I; GP, glycopeptide; MA-octyl, Manα1-6(Galβ1-4GlcNAcβ1-2Manα1-3); MGn-octyl, Manα1-6(GlcNAcβ1-2Manα1-3)Manβ1-O-octyl; MGnX-octyl, Xylβ1-2[Manα1-6(GlcNAcβ1-2Manα1-3)]Manβ1-O-octyl; MM-octyl, Manα1-6(Manα1-3)Manβ1-O-octyl; MMX-octyl, Xylβ1-2[Manα1-6(Manα1-3)]Manβ1-O-octyl; Man5Gn, Manα1-6(Manα1-3)Manα1-6(GlcNAcβ1-2Manα1-3)Manβ1-4GlcNAcβ1-4GlcNAc; MALDI–TOF-MS, matrix-assisted laser-desorption ionization–time-of-flight MS; PNGase F, peptide N-glycosidase F; (r)ER, (rough) endoplasmic reticulum; XylT, β1,2-xylosyltransferase
INTRODUCTION
N-glycosylation is considered to be one of the most important post-translational protein modifications. In both animal and plant cells, a highly ordered biosynthetic pathway accounts for the addition of N-linked oligosaccharides to selected asparagine residues of newly synthesized proteins and their subsequent maturation [1]. The basic steps of the N-glycosylation pathway are evolutionarily highly conserved, with both oligomannosidic and complex-type N-glycans being present in animal and plant glycoproteins. However, the structures of mature N-glycans differ between plants and mammals because of major differences in the final steps of the biosynthetic pathway. Mature mammalian N-glycans are primarily of the complex type (containing one or more antennae attached to the Man3GlcNAc2 core oligosaccharide). Mature plant N-glycans are mainly of the paucimannosidic type (Man3GlcNAc2 or Man4GlcNAc2 structures, lacking antennae but containing β1,2-xylose and/or core α1,3-fucose residues). Plant glycoproteins may also contain small amounts of complex N-glycans carrying terminal Lea structures, but generally lack sialic acid [2,3].
The enzyme that is responsible for the addition of xylose to the N-linked oligosaccharides of plant glycoproteins is XylT (β1,2-xylosyltransferase). XylT, which catalyses the transfer of xylose from UDP-xylose to the core β-linked mannose of N-linked oligosaccharides, is unique to plants and some invertebrates. The enzyme has been purified from soya beans [4], but the small amounts of purified protein obtained prevented a detailed investigation of its physicochemical properties and structural features. To circumvent these limitations, we have previously cloned the cDNA encoding Arabidopsis thaliana XylT [5]. The enzyme is a type II membrane protein typical of Golgi-bound glycosyltransferases. These proteins are usually composed of a short N-terminal cytoplasmic tail, a hydrophobic transmembrane anchor and a large luminal part consisting of a putative stem region and a C-terminal catalytic domain. We have recently shown that the CT (cytoplasmic and transmembrane) regions of XylT are sufficient for the retention of a reporter protein in the Golgi apparatus and thus are responsible for proper subcellular localization of the enzyme in plant cells [6]. However, the stem region and the catalytic domain of XylT have not been delineated yet. In fact, the existence of such a stem region within XylT remains unclear, since all N-terminal deletion mutants of its luminal part that have been produced so far are devoid of enzymatic activity [7].
In the present study, we have used the baculovirus system for heterologous expression of different N-terminally truncated, soluble forms of recombinant A. thaliana XylT in insect cells. This has enabled us to identify the stem region and determine the minimal length of the catalytic domain of the enzyme. Furthermore, this approach has allowed us to obtain sufficient amounts of purified XylT for detailed analyses of the enzymatic and structural properties of the enzyme.
EXPERIMENTAL
Materials
Oligonucleotide primers were synthesized by VBC-Genomics Bioscience Research (Vienna, Austria). Restriction enzymes were purchased from Roche (Lewes, East Sussex, U.K.), New England Biolabs (Hitchin, Herts., U.K.) and Fermentas (St. Leon-Rot, Germany). Protein molecular-mass standards were obtained from New England BioLabs, UDP[14C]glucose (330 mCi/mmol), UDP-[14C]GlcNAc (288 mCi/mmol), UDP[14C]xylose (263 mCi/mmol) and GDP-[14C]fucose (277 mCi/mmol) from New England Nuclear and UDP[3H]galactose (9.4 Ci/mmol) from Amersham Biosciences. 2-Acetamido-1,2-deoxynojirimycin was generously provided by Dr A. Stütz (Institute of Organic Chemistry, Technical University of Graz, Austria). MM-octyl [Manα1-6(Manα1-3)Manβ1-O-octyl] was purchased from Toronto Research Chemicals (North York, ON, Canada). MGn-octyl [Manα1-6 (GlcNAc β 1-2 Man α1-3) Man β 1-O-octyl] and MA-octyl [Manα1-6(Galβ1-4GlcNAcβ1-2Manα1-3)Manβ1-O-octyl] were synthesized as described in [8]. GnGn-GP [where GnGn stands for GlcNAcβ1-2Manα1-6(GlcNAcβ1-2Manα1-3)Manβ1-4GlcNAcβ1-4GlcNAc and GP for glycopeptide] was prepared as described previously [9]. Purified recombinant rabbit GnT I (β1,2-N-acetylglucosaminyltransferase I) and human GnT II were obtained by the method described in [8,10]. Recombinant A. thaliana XylT and FucT (core α1,3-fucosyltransferase) produced in Pichia pastoris were purified as described previously [11]. Purified recombinant Drosophila melanogaster α-mannosidase II [12] was kindly provided by D. Kuntz (Ontario Cancer Institute, Toronto, Canada). All other reagents were purchased from Sigma unless otherwise indicated.
Construction of baculovirus transfer vectors
DNA fragments encoding various truncated forms of A. thaliana XylT were obtained by PCR using different sense primers (Δ39, 5′-AAAACTGCAGCCTCCTCATATATACCAC-3′; Δ54, 5′-AAATACTGCAGATTCAGAAACCGTGGCCG-3′; Δ59, 5′-AAATACTGCAGCCGATCTTACCTTCTTACCTCCC-3′; Δ76, 5′-AAAACTGCAGACTGGCTCCTGCGAAGG-3′; Δ79, 5′-TT-ATACTGCAGTGCGAAGGTTACTTCGGG-3′; Δ178, 5′-AATACTGCAGCGTTTTGGTGGAGGAGAAGG-3′), and a universal antisense primer (5′-GCCGGAATTCTTAGCAGCCAAGGC-TCTTCA-3′). The PCR products were cleaved with PstI and EcoRI at the underlined sites and ligated into pVTBacHis-1 baculovirus transfer vector [13], digested with the same enzymes. In these constructs, the truncated XylT proteins are placed downstream of the melittin signal peptide, a His6 epitope and an enterokinase cleavage site.
After the final cloning step, all expression constructs were subjected to DNA sequencing to rule out any artifactual mutation. DNA sequencing was performed in a thermocycler using the BigDye Terminator 3.1 Cycle Sequencing kit and a Prism 3100 genetic analyzer (Applied Biosystems, Foster City, CA, U.S.A.). Sequence data were obtained using gene-specific or vector-derived sense and antisense oligonucleotides as the primers.
Heterologous expression of A. thaliana XylT in insect cells
Spodoptera frugiperda Sf9 and Sf21 cells (both obtained from A.T.C.C., Manassas, VA, U.S.A.) were grown in IPL-41 (Sigma, St. Louis, MO, U.S.A.) medium containing 5% (v/v) heat-inactivated fetal bovine serum (Invitrogen). Each recombinant baculovirus transfer vector (1 μg) was co-transfected with 200 ng of BaculoGold viral DNA (BD PharMingen, Erembodegem, Belgium) into Sf9 cells using Lipofectin® (Invitrogen) according to the manufacturer's instructions. After maintaining for 5 days at 27 °C, the supernatant containing recombinant baculovirus was used for the infection of Sf21 cells (we prefer this cell line to Sf9 cells for the high-yield production of recombinant proteins in the baculovirus system). Cells and conditioned media were harvested and subjected to enzyme analysis and immunoblotting [10].
Tunicamycin treatment of baculovirus-infected Sf21 cells
Two Sf21 cultures (1.8×106 cells each) were incubated with the respective recombinant baculovirus for 90 min at room temperature (23 °C). The virus-containing supernatant was then removed and replaced with fresh culture medium. After maintaining for 5 h at 27 °C, one culture received 5 μg/ml tunicamycin (Roche), whereas the other was treated with solvent (DMSO; final concentration, 0.1%) alone. After 24 h, a second aliquot of tunicamycin and/or DMSO was added. Incubation was continued for a further 40 h before cell harvest and analysis as above.
Purification of recombinant A. thaliana XylT
Culture supernatants (200 ml) of Sf21 cells infected with the respective baculovirus were cleared by centrifugation and dialysed twice against 2 litres of dialysis buffer (10 mM sodium phosphate buffer, pH 7.0, 40 mM NaCl and 0.02% NaN3). The supernatant containing 20 mM imidazole and 10% (v/v) glycerol was loaded on to a 5 ml column of chelating Sepharose (Amersham Biosciences) charged with Ni2+ ions, equilibrated in the same buffer. After successive washes with 40 and 80 mM imidazole, the enzyme was eluted with 250 mM imidazole in dialysis buffer. Protein-containing eluate fractions were pooled and dialysed twice against 1 litre of 50 mM Mes buffer (pH 7.0) containing 150 mM NaCl and 0.02% (w/v) NaN3. After concentrating by ultrafiltration and addition of proteinase inhibitors [1 mM PMSF, 5 μg/ml E-64 and 5 μg/ml leupeptin, where E-64 stands for trans-epoxysuccinyl-L-leucylamido-(4-guanidino)butane], the purified enzyme was stored at 4 °C.
Assay of XylT activity
Standard XylT activity assays were performed in a total volume of 20 μl of a buffer containing 0.1 mM MGn-octyl and 0.1 mM UDP-[14C]xylose (3000–4000 c.p.m./nmol) as substrates and containing 50 mM Mes (pH 7.0), 0.5% Triton X-100, 0.5 mg/ml BSA, 0.5 mM dithiothreitol, 1 mM MnCl2, 10 μM 2-acetamido-1,2-deoxynojirimycin (to inhibit β-hexosaminidases), 0.5 mM PMSF, 2.5 μg/ml E-64 and 2.5 μg/ml leupeptin. 2-Acetamido-1,2-deoxynojirimycin and the proteinase inhibitors PMSF, E-64 and leupeptin were omitted for assays using purified enzyme. After incubation at 37 °C for 30 min, reactions were stopped by the addition of 0.5 ml of 20 mM sodium tetraborate containing 2 mM EDTA. The radioactive reaction product was isolated by anion-exchange chromatography and quantified by liquid-scintillation counting. One unit of XylT activity corresponds to the formation of 1 μmol of reaction product/min.
XylT was also assayed using many other acceptor substrates under the same assay conditions.
Preparation of GnGn-octyl [GlcNAcβ1-2Manα1-6(GlcNAcβ1-2Manα1-3)Manβ1-O-octyl]
Synthesis of GnGn-octyl was performed in a total volume of 200 μl of buffer (100 mM Mes, pH 6.3, 1 mg/ml BSA and 20 mM MnCl2), containing 0.5 mM MGn-octyl and 10 mM UDP-GlcNAc, and 12 m-units of purified recombinant human GnT II [8]. The reaction was performed overnight at room temperature. The reaction product was purified by sequential ion-exchange chromatography with Dowex 1×8 and Dowex 50×8 eluted with water. As a final preparation step, the substrate was applied to a Sep-Pak C18 reverse-phase cartridge (Whatman Biosystems, Maidstone, Kent, U.K.) and eluted with methanol. Synthesis of GnGn-octyl was monitored by TLC using silica gel plates (Merck) and CH2Cl2/methanol/water (65:35:6, by vol.) as the chromatography solvent. The chromatogram was sprayed with 0.2% orcinol in 20% (v/v) H2SO4 and heated for 10 min at 110 °C. The yield of purified product was measured by amino sugar analysis [14].
Preparation of MGnX-octyl [Xylβ1-2[Manα1-6(GlcNAcβ1-2Manα1-3)]Manβ1-O-octyl]
Preparative synthesis of MGnX-octyl was accomplished in a total volume of 200 μl of 50 mM Mes buffer (pH 7.0), 1 mM MnCl2 and 1 mg/ml BSA, containing 3 mM UDP-xylose and 0.48 mM MGn-octyl. After the addition of 6 m-units of purified recombinant A. thaliana XylT, the sample was incubated for 16 h at 23 °C. The product was analysed, purified and quantified as described above for GnGn-octyl.
Preparation of MMX-octyl {Xylβ1-2[Manα1-6(Manα1-3)]Manβ1-O-octyl}
To obtain MMX-octyl, 200 μl of 100 mM sodium citrate buffer (pH 5.0) containing 0.30 mM MGnX-octyl was incubated for 16 h at 37 °C with 0.3 unit of jack-bean β-N-acetylglucosaminidase. The reaction was monitored by TLC as described above. Once the conversion of MGnX-octyl into MMX-octyl was complete, the reaction was stopped by heating to 95 °C for 5 min.
Preparation of Man5Gn [Manα1-6(Manα1-3)Manα1-6(GlcNAcβ1-2Manα1-3)Manβ1-4GlcNAcβ1-4GlcNAc]-GP
α-Amylase from Aspergillus oryzae was purified as described in [15], dissolved in 0.15 M Tris/HCl (pH 7.8), containing 1 mM CaCl2 and digested for 48 h at 37 °C with Pronase (proteinase type XXV) at a final enzyme/substrate ratio of 1:25. Man5 [Manα1-6 (Manα 1-3) Manα1-6 (Manα1-3)Manβ1-4GlcNAcβ1-4GlcNAc]-GP was then isolated by gel filtration on a Sephadex G-50 column (1 cm×50 cm) eluted with 5% (v/v) acetic acid, and characterized by MALDI–TOF-MS (matrix-assisted laser-desorption ionization–time-of-flight MS) and amino sugar analysis.
Synthesis of Man5Gn-GP was performed in a final volume of 300 μl of buffer (0.1 M Mes, pH 6.3, 1 mg/ml BSA and 20 mM CoCl2), containing 3 mM Man5-GP and 10 mM UDP-GlcNAc, and 30 m-units of purified recombinant rabbit GnT I [10] for 16 h at 23 °C. Product formation was monitored by MALDI–TOF-MS. The reaction product was purified by sequential ion-exchange chromatography with Dowex 1×8 and Dowex 50×8 eluted in water. As a final preparation step, the sample was fractionated on a Superdex Peptide PE (7.5 mm×300 mm) gel-filtration column eluted with 0.1 M NH4HCO3, using an Äkta purifier chromatography system (Amersham Biosciences). To detect Man5Gn-GP in the fractions, aliquots were spotted on to TLC plates and stained with orcinol/H2SO4. Positive fractions were combined and freeze-dried. The yield of purified product was determined by amino sugar analysis.
Preparation of GnGnX (Xylβ1-2GnGn)-GP
GnGn-GP (3 mM) was incubated in 400 μl of 25 mM Mes (pH 7.0) containing 9 mM UDP-xylose, 0.5 mM PMSF, 0.5 μg/ml leupeptin and 0.5 μg/ml pepstatin A for 24 h at 23 °C with 25 m-units of recombinant A. thaliana XylT produced in P. pastoris [11]. Conversion into GnGnX-GP was monitored by MALDI–TOF-MS analysis. The reaction product was purified by gel filtration on a Sephadex G-50 (1 cm×50 cm) column eluted with 5% acetic acid. To detect GnGnX-GP in the fractions, aliquots were spotted on to TLC plates and stained with orcinol/H2SO4. Positive fractions were combined, passed through a C18 reverse-phase cartridge and concentrated in vacuo. The yield of purified product was determined by amino sugar analysis.
Preparation of GnGnF3 [GlcNAcβ1-2Manα1-6(GlcNAcβ1-2Manα1-3)Manβ1-4GlcNAcβ1-4(Fucα1-3)GlcNAc]-GP
GnGn-GP (1.5 mM) was incubated in 400 μl of 25 mM Mes (pH 7.2), containing 6 mM GDP-fucose, 10 mM MnCl2, 0.5 mM PMSF, 0.5 μg/ml leupeptin and 0.5 μg/ml pepstatin A for 24 h at 23 °C with 19 m-units of recombinant A. thaliana FucT produced in P. pastoris [11]. Conversion into GnGnF3-GP was monitored by MALDI–TOF-MS. The reaction product was purified and characterized as described above.
MALDI–TOF-MS analysis of XylT glycosylation sites
In-gel digestion of XylT was performed as described previously [16,17]. Briefly, 3 μg of purified two-chain XylT was separated by SDS/PAGE and stained with Coomassie Blue. Individual bands were excised from the gel, minced and washed twice with 50% (v/v) acetonitrile to remove the residual SDS and Coomassie dye. The slices were then dried in vacuo, subjected to reduction with 10 mM dithiothreitol dissolved in 0.1 M NH4HCO3 for 60 min at 56 °C, followed by alkylation in 0.1 M NH4HCO3 containing 55 mM iodoacetamide for 45 min at room temperature in the dark. The gel pieces were then washed and dried as above before incubation in 12 μl of 25 mM NH4HCO3 buffer (pH 8.0) containing 2.5 mM CaCl2, 0.1% n-octyl glucoside and 50 ng of bovine trypsin overnight at 37 °C. The tryptic peptides thus generated were recovered from the gel particles by two extractions with 40 μl each of 66% acetonitrile containing 0.1% (v/v) trifluoroacetic acid, assisted by ultrasonication. The combined extracts were concentrated in vacuo, dissolved in 10 μl of 5% acetonitrile containing 0.1% trifluoroacetic acid and subjected to MALDI–TOF-MS analysis.
Further fragmentation of the tryptic peptides was achieved by subsequent digestion with 50 ng of endoproteinase Glu-C (Roche) by the same procedure as for the trypsin treatment.
Size-exclusion chromatography of XylT
Purified recombinant A. thaliana XylT was subjected to gel-filtration analysis on a BioSil TSK 250 (7.5 mm×300 mm; Bio-Rad Laboratories, Glattbrugg, Switzerland) column connected to an Äkta purifier chromatography system and eluted with 25 mM Tris/HCl (pH 7.5) and 150 mM NaCl. The column was calibrated with a series of molecular-mass standards. Fractions were tested for the presence of XylT by means of its enzymatic activity.
N-terminal sequencing of XylT
Purified recombinant A. thaliana XylT (5 μg) was separated by SDS/PAGE and electrophoretically transferred on to a PVDF membrane. Polypeptides detected by Coomassie Blue staining were excised and subjected to N-terminal sequence analysis using an ABI 476A protein sequencer (Applied Biosystems).
MALDI–TOF-MS
For MALDI–TOF-MS analysis, 1 μl of the sample was mixed with 1 μl of matrix [1% (w/v) α-cyano-4-hydroxycinnamic acid in 70% acetonitrile or 2% (w/v) 2,5-dihydroxybenzoic acid in 30% acetonitrile]. Aliquots of 1 μl each were applied to a flat sample plate and dried immediately under mild vacuum. MALDI–TOF-MS spectra were acquired on a DYNAMO linear TOF mass spectrometer (Thermo BioAnalysis, Hemel Hempstead, U.K.) as described in [18].
Western-blot analysis
Cell lysates and culture supernatants of the infected Sf21 cells were subjected to SDS/PAGE (12% polyacrylamide) under reducing conditions. The separated proteins were blotted on to Hybond-C membranes (Amersham Biosciences) and detected with a mouse monoclonal antibody raised against the enterokinase recognition sequence (Invitrogen). Detection of bound antibodies was performed as described previously [10].
Enzymatic deglycosylation of XylT
XylT-containing samples were denatured in the presence of 0.5% (w/v) SDS and 0.05 M 2-mercaptoethanol in a final volume of 10 μl for 5 min at 95 °C. After adding 10 μl of buffer (0.3 M sodium phosphate, pH 7.2, and 15 mM EDTA) and 5 μl of 7.5% (w/v) Triton X-100, the samples were incubated with 2 units of PNGase F (peptide N-glycosidase F; Roche) overnight at 37 °C. The reaction was stopped by the addition of 120 μl of methanol. The samples were then incubated for 30 min at room temperature and centrifuged at 15000 g for 10 min. Pellets were washed with 1 ml of acetone, dried and analysed by SDS/PAGE and silver staining. Alternatively, XylT bands were detected by means of immunoblotting.
Treatment with 2 m-units of endo H (endoglycosidase H; Roche) was performed by the same procedure, but using 0.15 M sodium citrate (pH 5.0) and 10 mM PMSF as the buffer.
Other methods
Kinetic parameters were determined by non-linear regression analysis [19]. Statistical evaluation of these data was performed with Student's t test. Inhibition constant (Ki) values were estimated from Dixon plots. The metal content of purified recombinant A. thaliana XylT was determined by means of inductively coupled plasma MS as reported in [20]. Total cellular protein content was determined by the Bradford method with the Bio-Rad Protein Assay kit (Bio-Rad Laboratories) using BSA as a standard. The protein content of purified protein samples was determined with the BCA (bicinchoninic acid) Protein Assay kit (Pierce, Rockford, IL, U.S.A.). Densitometric analysis of the silver-stained SDS/polyacrylamide gels and Western blots was performed using ImageQuaNT v4.2 software (Molecular Dynamics, Palo Alto, CA, U.S.A.). Detection of 14C-labelled compounds on TLC plates was performed with a STORM PhosphorImager (Molecular Dynamics). The activities of GnT I, GnT II and FucT were assayed essentially as described previously [8,10,21].
RESULTS
Delineation of the catalytic domain of A. thaliana XylT
Primary sequence and hydrophobicity analyses predicted that A. thaliana XylT is a type II membrane glycoprotein with a single transmembrane domain, comprising amino acids 11–30 [5]. Deletion of the N-terminal 31 residues representing the CT regions of the enzyme gives rise to soluble XylT forms that display full enzymatic activity on expression in baculovirus-infected insect cells and the yeast P. pastoris [7,11]. This demonstrated that the CT domain of XylT is dispensable for enzymatic activity. Using hydrophobic cluster analysis, a series of further N-terminal deletions covering the region of amino acids 39–79 was selected in an effort to define the boundary between the stem region and catalytic domain of the enzyme. Analysis of the predicted secondary structure of A. thaliana XylT by hydrophobic cluster analysis indicated a domain rich in proline residues (Pro60–Pro76). It has been suggested that this region may be required for XylT activity [7]. However, most of these proline residues are not found in XylT sequences from other plant species [22]. Interestingly, the proline-rich region of A. thaliana XylT is preceded by the tetrapeptide sequence K57PWP60. This KPWP motif is also present in the other XylT sequences available at present [22]. Hence, three deletion mutants were generated, two (Δ39 and Δ54 XylT) containing this motif and one (Δ59 XylT) devoid of it. Whereas these three XylT forms contain the proline-rich region, two additional truncation constructs were created that lack this domain (Δ76 and Δ79 XylT).
The XylT deletion constructs were produced by baculovirus-mediated infection of S. frugiperda Sf21 cells. Immunoblot analysis of cell extracts and culture supernatants revealed that all deletion mutants were successfully expressed in Sf21 cells, but only two forms (Δ39 and Δ54 XylT) were detected in the culture medium (Figure 1A). Cultures expressing Δ39 and Δ54 XylT displayed a total XylT activity of 2.4 and 8.8 m-units respectively with >97% of the activity being present in the supernatant. In contrast, XylT forms not detectable in the culture medium were synthesized without exhibiting any detectable activity in the cell extracts (<0.01 m-units). These results demonstrate that the presence of the KPWP motif is crucial for the production of catalytically active XylT.
Figure 1. Characterization of N-terminally truncated variants of A. thaliana XylT produced in insect cells.

(A) Heterologous expression of soluble forms of A. thaliana XylT in insect cells. Equivalent amounts of culture supernatants (sn) and protein extracts from Sf21 insect cells (c) infected with recombinant baculoviruses encoding the indicated N-terminally truncated versions of A. thaliana XylT lacking between 39 (Δ39) and 79 (Δ79) residues were separated under reducing conditions by SDS/PAGE, transferred on to a nitrocellulose membrane and probed with mouse anti-enterokinase recognition site antibodies. Migration positions of selected prestained molecular-mass standards are indicated. The detected sizes of the recombinant polypeptides were in close agreement with their theoretical molecular masses (not accounting for any N-glycans). No specific signals were obtained with lysates and conditioned media of uninfected Sf21 cells. (B) Analysis of A. thaliana XylT after endoglycosidase treatment. Protein extracts from Sf21 insect cells infected with recombinant baculoviruses encoding N-terminally truncated versions of A. thaliana XylT lacking 54 (Δ54) and 59 (Δ59) residues were incubated overnight in the presence (+) and absence (−) of PNGase F or endo H. Proteins separated by SDS/PAGE were transferred on to a nitrocellulose membrane and probed with anti-enterokinase recognition site antibodies as above.
By means of their N-terminal His6 tag, Δ39 and Δ54 XylT could be purified from Sf21 media by nickel-chelate affinity chromatography. XylT assays showed that Δ39 and Δ54 XylT have essentially identical specific activities (489 and 436 m-units/mg of XylT protein respectively). Interestingly, both forms exhibited specific activities comparable with purified recombinant A. thaliana Δ31 XylT produced in the yeast P. pastoris [11]. Moreover, the affinity of Δ39 XylT for the acceptor substrate MGn-octyl (Michaelis constant, Km=0.25 mM) is essentially the same as that of the yeast-derived enzyme (Km=0.30 mM).
Since Δ39 and Δ54 XylT display full enzymatic activity, the stem region consists of at least 24 residues (Ser31–Ser54). Given the evolutionary conservation of the KPWP motif (but not of the preceding residues Ile55 and Gln56), our results indicate that the catalytic domain of XylT encompasses amino acids Lys57–Cys534, with the stretch from Ser31 to Gln56 representing the stem region of the enzyme.
It has been reported previously that A. thaliana XylT is N-glycosylated [7]. The N-glycosylation status of intracellular Δ54 and Δ59 XylT was assessed by treatment with two different endoglycosidases, PNGase F and endo H. Whereas both oligomannosidic and complex-type N-linked oligosaccharides are sensitive to PNGase F, endo H is only capable of cleaving high-mannose N-glycans. Hence newly synthesized secretory glycoproteins acquire complete or partial resistance to endo H treatment on transfer from the rER (rough endoplasmic reticulum) to the Golgi apparatus [1]. Endoglycosidase treatment revealed that intracellular Δ54 XylT is sensitive to PNGase F but not to endo H. This suggests that the protein resides in the Golgi apparatus or in a post-Golgi compartment of the secretory pathway. In contrast, Δ59 XylT displays sensitivity to both the endoglycosidases, indicating that the protein carries oligomannosidic N-glycans as typical for ER-resident proteins [2]. This suggests that Δ59 XylT as well as shorter variants of the enzyme are retained within the rER, probably due to improper folding (Figure 1B).
Proteolytic processing of A. thaliana XylT produced in insect cells
SDS/PAGE analysis of purified Δ39 XylT under reducing conditions yielded proteins with a molecular mass of 62, 42 and 21 kDa. Interestingly, the relative amounts of smaller polypeptides increased on prolonged storage at the expense of the 62 kDa band. Furthermore, the 21 kDa band disappeared with the con-comitant appearance of polypeptides having masses in the range 15–18 kDa. This suggests that limited proteolysis occurred due to a contaminating proteinase, a problem frequently encountered when using the baculovirus expression system. This is usually attributed to a baculoviral cysteine proteinase, an enzyme efficiently inactivated by the classical cysteine proteinase inhibitors leupeptin and E64 [23]. Indeed, addition of a proteinase inhibitor cocktail, containing leupeptin and E-64, significantly retarded XylT processing during storage. In particular, conversion of the 21 kDa polypeptide into smaller fragments was completely inhibited (Figure 2A).
Figure 2. Limited proteolysis of purified recombinant A. thaliana XylT by a co-purifying proteinase.

(A) Generation of two-chain A. thaliana XylT on prolonged storage. Purified recombinant A. thaliana XylT was stored in the absence (−) or presence (+) of proteinase inhibitors (E-64, leupeptin and PMSF) for 3 months at 4 °C. Samples were analysed by SDS/PAGE under reducing conditions and silver staining. The migration positions of selected molecular-mass standards are indicated. (B) The light chain of two-chain XylT is derived from the N-terminal part of the native enzyme. Purified recombinant A. thaliana XylT stored in the absence of proteinase inhibitors for 3 months was incubated overnight in the presence (+) or absence (−) of PNGase F. Samples were then analysed by SDS/PAGE under reducing conditions and silver staining or subjected to Western-blot analysis with anti-enterokinase recognition site antibodies. *, the migration position of PNGase F.
XylT preparations containing the 62, 42 and 21 kDa polypeptides showed only a single 58 kDa band when analysed under non-reducing conditions. This suggests that the 42 and 21 kDa polypeptides are both fragments of a two-chain form of XylT, with the subunits being held together by disulphide bridge(s). Indeed, the 21 kDa polypeptide reacts with antibodies to the enterokinase cleavage site and thus represents the N-terminal frag-ment of the XylT fusion protein (Figure 2B). The N-terminal sequence of the 42 kDa protein was determined by Edman degradation and found to be RFGGGE, corresponding to XylT residues 179–184. Hence proteolytic processing of recombinant XylT occurs between amino acids Arg178 and Arg179, a cleavage sequence compatible with the enzymatic properties of the baculoviral cysteine proteinase [23]. These results demonstrate that the 40 kDa band represents the C-terminal part of XylT. Thus two-chain XylT consists of an N-terminal light chain and a C-terminal heavy chain of theoretical molecular masses of 20.5 and 39.7 kDa respectively. Interestingly, conversion of single-chain XylT into the two-chain enzyme did not affect its enzymatic activity. However, both subunits are required for the generation of catalytically active XylT since the expression of the heavy chain by itself (Δ178 XylT) did not yield an enzymatically active protein.
N-glycosylation of XylT is not required for enzymatic activity
XylT contains three potential N-glycosylation sites. Treatment with PNGase F resulted in an increased electrophoretic mobility of both subunits of two-chain Δ39 XylT (Figure 2B). Hence the light chain carries an N-linked oligosaccharide at Asn51, whereas the heavy chain is modified with N-glycan(s) at Asn301 and/or Asn479. MALDI–TOF-MS analysis of peptides released by the digestion of the heavy chain of XylT with trypsin and endoproteinase Glu-C demonstrated that both potential N-glycosylation sites of the heavy chain are at least partially occupied paucimannosidic N-glycans as typical for secretory insect glycoproteins [24]. Deviating from the normal pattern of insect N-glycans, xylosylated structures were also detected, revealing that XylT is capable of self-xylosylation (Figure 3). These results provide direct evidence that all three hypothetical N-glycosylation sites of XylT are indeed modified with N-glycans. This corrects a recent study that did suggest that Asn479 is not glycosylated. However, the latter conclusion was entirely based on indirect evidence [7].
Figure 3. All three potential N-glycosylation sites of A. thaliana XylT are modified with N-linked oligosaccharides.

The two-chain form of purified recombinant A. thaliana XylT was incubated overnight in the absence (A, C) or presence (B) of PNGase F before SDS/PAGE analysis. Bands corresponding to the XylT heavy chain were excised and subjected to trypsin treatment (A, B) or trypsin/endoproteinase Glu-C (C) treatment. Peptides thus generated were analysed by MALDI–TOF-MS. (A, B) The peptide N301FTKPVCFR309 is modified at Asn301 with N-linked oligosaccharides (A). The GP is partially modified at its cysteine residue with acrylamide instead of being carbamidomethylated, resulting in double peaks. The corresponding unglycosylated peptide was not detected in the sample. The N-glycan structures found were MM [Manα1-6(Manα1-3)Manβ1-4GlcNAcβ1-4GlcNAc], MMX {Xylβ1-2[Manα1-6(Manα1-3)]Manβ1-4GlcNAcβ1-4GlcNAc} and MMF6 [Manα1-6(Manα1-3)Manβ1-4GlcNAcβ1-4(Fucα1-6)GlcNAc]. These GP peaks are absent from the deglycosylated sample (B). (C) The peptide A459SVIIGAHGAGLTHIVSATPN479TTIFE484 is modified at Asn479 with N-linked oligosaccharides. Two main N-glycan structures could be detected (MM and MMX). The corresponding unglycosylated peptide was not detected in the sample.
Δ54 XylT lacks the N-glycosylation site at Asn51. Since this XylT variant displays full enzymatic activity, occupancy of Asn51 with an N-glycan is not necessary for the generation of catalytically active XylT. This is in agreement with a recent report, where site-directed mutagenesis was used to eliminate the N-glycosylation of Asn51 [7]. To test whether the other two N-glycosylation sites are also dispensable for XylT activity, Δ39 XylT was expressed in insect cells in the presence of tunicamycin, a well-known N-glycosylation inhibitor. The protein thus synthesized was smaller than the XylT produced by untreated cells, and was no longer sensitive to PNGase F and co-migrated with enzymatically deglycosylated control enzyme (Figure 4). This indicates that XylT N-glycosylation was completely blocked by tunicamycin. On treatment with tunicamycin, the cellular XylT content was significantly decreased (32% of the control), suggesting that unglycosylated XylT is less stable than the wild-type enzyme. However, considerable amounts of XylT activity were still detectable (19% of the untreated sample), indicating a relative specific activity of 60% when compared with control XylT. This demonstrates that N-glycosylation is not required for XylT to adopt an enzymatically competent state, although moderately lower cellular stability is observed.
Figure 4. Synthesis of unglycosylated A. thaliana XylT in insect cells.

Sf21 insect cells infected with recombinant baculoviruses encoding Δ39 XylT were cultured in the absence (–) or presence (+) of tunicamycin. Cell extracts were then incubated overnight in the presence (+) or absence (–) of PNGase F before SDS/PAGE and immunoblot analysis with anti-enterokinase recognition site antibodies. The migration positions of selected prestained molecular-mass standards are indicated.
Enzymatic and physicochemical properties of recombinant A. thaliana XylT
Recombinant Δ39 XylT was found to be optimally active at pH 6.5–7.0. The purified enzyme was found to be stable for prolonged periods at neutral pH and at temperatures of up to 30 °C. However, XylT was rapidly inactivated at pH<6.0 and >37 °C. The native molecular mass of Δ39 XylT was estimated by gel-filtration analysis to be 50 kDa, demonstrating that the enzyme resides in a monomeric state. When various nucleotide-activated monosaccharides were tested as donor substrates, enzymatic activity was only detected with UDP-xylose (Km=0.13 mM). However, UDP-glucose weakly inhibited the enzyme (Ki=4.9 mM). UDP was found to be the most effective inhibitor (Ki=42 μM), acting in a competitive manner. These properties are in good agreement with previously published results on the features of XylT isolated from soya-bean cells [4].
Purified Δ39 XylT was also used to investigate the acceptor substrate specificity of the enzyme. A series of potential acceptor substrates derived from the synthetic glycoside MM-octyl was created using different purified glycosyltransferases involved in N-glycan biosynthesis. The best acceptor substrate found was MGn-octyl, followed by its derivative GnGn-octyl. However, no activity was observed when the enzyme was assayed with MA-octyl, indicating that extension of the GlcNAcβ1-2Manα1-3Man branch with galactose prevents XylT action. Transfer of xylose to MM-octyl was also not detected (Table 1; Figure 5A). Similar features have been previously reported for purified soya-bean XylT [4].
Table 1. Acceptor substrate specificity of XylT.
Kinetic data are presented as means±S.E.M.; n.d., not detected (<0.1%). See [18] or http://www.proglycan.com for N-glycan abbreviations.
| Structure | Name | Km (app) (mM) | Relative XylT activity (%) |
|---|---|---|---|
 |
Man5-GP | – | n.d. |
 |
Man5Gn-GP | 0.46±0.09 | 35 |
 |
MGn-octyl | 0.25±0.12 | 100 |
 |
GnGn-octyl | 0.79±0.19 | 23 |
 |
MA-octyl | – | n.d. |
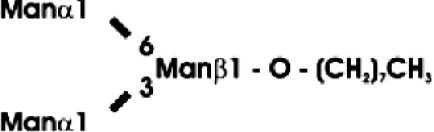 |
MM-octyl | – | n.d. |
Figure 5. Acceptor substrate specificities of XylT and GnT II.

(A) Purified recombinant A. thaliana XylT was incubated with the indicated acceptor substrates and UDP-[14C]xylose. The samples were then analysed by TLC before orcinol/H2SO4 staining (lanes 1–5) and autoradiography (lanes 6 and 7). The migration direction is indicated by an arrow. (B) Purified recombinant human GnT II was incubated with the indicated acceptor substrates and UDP-[14C]GlcNAc. The samples were then analysed by TLC before orcinol/H2SO4 staining (lanes 1–5) and autoradiography (lanes 6 and 7).
These results provide further support for the proposed sequence of glycosyltransferase action during N-glycan biosynthesis in plants [2,3]. XylT is dependent on prior action of the medial-Golgi enzyme GnT I, but modification of acceptor substrates by late-Golgi enzymes (e.g. galactosyltransferases) interferes with its activity. However, little is known about the interplay of XylT with other glycosyltransferases potentially competing for the same substrate, such as FucT. Since the synthetic glycosides developed for XylT assays are not suitable for measuring the activity of the latter enzyme, we used a newly developed set of GP substrates to address this issue. GnGn-GP was a slightly better XylT substrate when compared with the corresponding synthetic derivative, GnGn-octyl (Tables 1 and 2). We found that core α1,3-fucosylation of GnGn-GP significantly decreased its affinity for XylT. On the other hand, FucT is only slightly hampered by an earlier action of XylT (Table 2). Similar observations were made in a preliminary study ([25]; M. Bencúrová and F. Altmann, unpublished work). Therefore for efficient β1,2-xylosylation in vivo, XylT has to act before FucT within the N-glycosylation pathway of plants. This is in agreement with studies on the subcellular localization of the two enzymes in plant cells [7,26].
Table 2. XylT activity is decreased by prior action of FucT.
Kinetic data are presented as means±S.E.M.; *P<0.001 and **P>0.05.
| Structure | Name | Km (app) (XylT) (mM) | Km (app) (FucT) (mM) |
|---|---|---|---|
 |
GnGn-GP | 0.40±0.15* | 0.49±0.07** |
 |
GnGnX-GP | – | 0.75±0.10** |
 |
GnGnF3-GP | 1.69±1.10* | – |
It is generally assumed that XylT acts, within the plant N-glycosylation pathway, subsequent to GnT I, α-mannosidase II and GnT II. However, preliminary evidence has been reported that XylT may also act before the latter two enzymes [27,28]. Indeed, purified recombinant A. thaliana XylT displayed significant activity with Man5Gn-GP, the physiological product of GnT I action. The affinity of XylT for Man5Gn-GP (Table 1) was in the same range as for GnGn-GP (Table 2), indicating that XylT could also act in vivo before α-mannosidase II and GnT II (Scheme 1).
Scheme 1. XylT may act at multiple stages of the plant N-glycosylation pathway.

The standard pathway is shown in black and novel steps are displayed in grey. Reactions still awaiting experimental verification are highlighted with a question mark. Man II, α-mannosidase II.
These findings prompted us to test GnT II for activity with β1,2-xylosylated substrates. The recombinant human enzyme was capable of using MGnX-octyl as acceptor, albeit less efficiently than MGn-octyl (Figure 5B). This suggests that XylT activity may precede GnT II action in vivo without interfering with subsequent maturation steps during plant N-glycan processing. However, recombinant rabbit GnT I was not capable of acting on MMX-octyl (<1% activity when compared with its standard substrate MM-octyl). Similar results have been reported previously for Acer pseudoplatanus and snail variants of the enzyme [28,29], indicating that this feature is not limited to rabbit GnT I.
A. thaliana XylT does not require metal ions as cofactors for its catalytic activity
The catalytic activity of most Golgi-bound glycosyltransferases depends strictly on the presence of an exogenous bivalent metal ion. Conflicting reports exist on the metal dependence of XylT. It has been shown for purified soya-bean XylT that this enzyme displays significant catalytic activity without the addition of any metal ion. Moreover, the enzyme was not inactivated by metal chelators. Hence it was proposed that XylT may contain a metal ion prebound to its active site [4]. In later studies, different metal ions were found to stimulate significantly the activity of recombinant A. thaliana and rice XylT [7,22]. However, these studies were performed with crude enzyme preparations. To clarify this issue, the activity of purified Δ39 XylT was determined in the presence of various bivalent metal ions (Table 3). Whereas the enzyme was active in the absence of added metal ions as well as in the presence of EDTA, the addition of 1 mM Mn2+ caused a moderate increase in XylT activity. However, higher Mn2+ concentrations did not further stimulate, but rather inhibited the enzyme. Δ39 XylT was also slightly stimulated by Mg2+, Ca2+ and, at low concentrations, Co2+ ions, whereas high concentrations of the latter as well as Ni2+ and Zn2+ ions inhibited the enzyme. These results show that A. thaliana XylT resembles the soya-bean enzyme [4] in its metal requirements and establish that XylT displays, with respect to its metal-dependence, unique features when compared with other glycosyltransferases.
Table 3. Metal dependence of XylT.
| Additions | Relative activity (%) |
|---|---|
| None | 100 |
| EDTA (10 mM) | 80 |
| CaCl2 (1 mM) | 99 |
| CaCl2 (10 mM) | 117 |
| CoCl2 (1 mM) | 99 |
| CoCl2 (10 mM) | 41 |
| MgCl2 (1 mM) | 122 |
| MgCl2 (10 mM) | 132 |
| MnCl2 (1 mM) | 180 |
| MnCl2 (10 mM) | 136 |
| MnCl2 (20 mM) | 36 |
| NiCl2 (1 mM) | 33 |
| NiCl2 (10 mM) | 1 |
| ZnCl2 (1 mM) | 5 |
One explanation for the substantial XylT activity in the absence of added metal ions would be the permanent association of a metal ion with the active site of the enzyme. Some preliminary indirect evidence for this hypothesis has been previously reported for the soya-bean enzyme [4]. Inductively coupled plasma MS was used to assess if XylT is a metalloprotein. Purified XylT contained none of the metals tested (Ca, Co, Cu, Fe, Mg, Mn, Ni and Zn) in stoichiometric amounts, their amounts not exceeding 0.01 mol metal/mol of enzyme. This spurious metal content was essentially the same as that determined for recombinant rabbit GnT I, a metal-free glycosyltransferase [30], and therefore probably reflects the presence of low levels of metal contaminants within the samples. In contrast, recombinant D. melanogaster α-mannosidase II was found to contain 0.98 mol Zn/mol of enzyme, in good agreement with its crystal structure that shows the presence of one Zn atom within the active site of the enzyme [12]. Therefore it can be concluded that XylT does not contain protein-bound metal ions.
DISCUSSION
The three-dimensional structure of XylT is as yet not known and there is little information on the minimal catalytic domain required for XylT activity. Analysis of the deduced amino acid sequence of A. thaliana XylT predicts the functional domain architecture typical for type II Golgi-bound glycosyltransferases. It has been shown previously that the putative CT region of XylT is dispensable for the enzymatic activity of the enzyme. Since removal of the N-terminal 82 amino acids led to the production of an inactive protein, it was proposed that a proline-rich domain spanning amino acids 60–76 is required for XylT activity [7]. However, we demonstrate in the present study that Δ59 XylT is catalytically inactive. The retention of this polypeptide within the ER indicates that the presence of the proline-rich region is not sufficient for proper folding of XylT. In contrast with the Δ59 variant, Δ54 XylT exhibited a similar specific activity as the full-length luminal domain of the enzyme, establishing that the 54 N-terminal amino acid residues are dispensable for the catalytic competence of XylT. Furthermore, this indicates that the highly conserved K57PWP60 sequence is essential for the generation of the active enzyme. On the basis of these results, we conclude that the minimal length of the catalytic domain of XylT encompasses residues Lys57–Cys534. Hence residues 31–56 appear to represent the stem region of the enzyme. Interestingly, the stem regions of other Golgi-resident glycosyltransferases and glycosidases contribute to the retention of these proteins in the Golgi apparatus [31]. However, the physiological significance of the XylT stem region remains unclear, since the CT domain of the enzyme is sufficient to retain a reporter protein in the Golgi [6,7].
Absence of protein N-glycosylation may prevent the proper folding of nascent polypeptide chains. For animal glycosyltransferases, it has been reported that N-glycosylation influences their folding and assists in preventing their proteolytic degradation (see e.g. [32]). It was previously proposed that N-glycosylation of A. thaliana XylT is required for the formation of the active enzyme. Using a site-directed mutagenesis approach, evidence has been provided that two of the three potential N-glycosylation sites (Asn51 and Asn301) are modified with N-linked oligosaccharides, with the presence of either N-glycan being sufficient for the generation of active XylT [7]. In the present study, we have shown that all three potential N-glycosylation sites of XylT are indeed occupied by N-glycans. In our study, carbohydrate-free XylT synthesized in the presence of the general protein N-glycosylation inhibitor tunicamycin still displayed significant enzymatic activity. We conclude that proper folding of XylT is possible in the absence of the oligosaccharide moiety, albeit at a lower efficiency.
Purification of A. thaliana XylT produced in insect cells leads to limited proteolysis caused by a contaminating proteinase, resulting in a two-chain form of the enzyme. The cleavage site is located in a region unique to A. thaliana XylT compared with orthologues from other plant species [22]. Interestingly, proteolytic processing does not exert any deleterious impact on the catalytic competence of the enzyme. The sequence of the cleavage site and the inhibitor profile suggest the involvement of the baculoviral cysteine proteinase [23] or of a related enzyme released by the insect cells. Indeed, activity measurements demonstrated the presence of small amounts of such a proteinase in purified XylT preparations. Remarkably, recombinant A. thaliana XylT produced in P. pastoris does not undergo proteolytic processing [11]. In this respect, it is of interest that yeasts are generally devoid of vacuolar enzymes belonging to the papain superfamily of cysteine proteinases [33].
We have used recombinant XylT to demonstrate that the enzyme acts almost equally well on two physiologically relevant substrates, MGn and Man5Gn. This suggests that XylT may act in vivo either before or after other Golgi-bound glycosyltransferases and glycosidases competing for the main XylT substrate, MGn. At least one of them, the medial-Golgi enzyme GnT II, is active on xylosylated substrates. Interestingly, preliminary evidence has been provided that XylT is also located in the medial part of the Golgi complex [7]. This suggests that plant N-glycan processing may follow more than one route, with XylT preceding GnT II or vice versa (Scheme 1).
It should be pointed out that the activity of GnT II in vivo is strictly dependent on prior action of α-mannosidase II [1]. This enzyme converts Man5Gn into MGn through a Man4Gn [Manα1-3Manα 1-6(GlcNAcβ1-2Manα1-3)Manβ 1-4GlcNAcβ 1-4GlcNAc] intermediate [12]. The occurrence of Man4GnX [Xylβ1-2[Manα1-3Manα1-6(GlcNAcβ1-2Manα1-3)]Manβ1-4GlcNAc-β1-4GlcNAc] in some plant species [16,18] suggests that α-mannosidase II has the capacity to act on xylosylated substrates. However, this processing step (Scheme 1) is yet to be verified experimentally.
It is believed that medial-Golgi retention of glycoprotein-processing glycosyltransferases and glycosidases is driven by homo- and/or hetero-typic oligomerization of the proteins in a stem region-dependent manner [31,34]. We have now shown that a soluble form of XylT encompassing the complete luminal domain of the enzyme resides exclusively in its monomeric state. These results are in agreement with the previously reported dispensability of the stem region for proper intracellular targeting of XylT [6]. This indicates that other, yet unknown, mechanisms must account for the presumed medial-Golgi localization of the enzyme.
Modification of MGn by FucT hampers any subsequent action by XylT. To ensure efficient β1,2-xylosylation during the biosynthesis of complex plant N-glycans, XylT action should therefore precede that of FucT. Indeed, preliminary evidence for a differential Golgi subcompartmentalization of these two enzymes has been reported in [26]. On the other hand, FucT seems to be unaffected by prior action of XylT [25]. No XylT activity is observed when the terminal β1,2-linked GlcNAc residue coupled with α1,3-linked mannose is either absent or modified. This indicates that XylT strictly requires the preceding action of GnT I and cannot act on complex plant N-glycans carrying Lea structures, which are produced at a late stage of glycoprotein passage through the Golgi complex [2,3].
Most glycoprotein-processing glycosyltransferases require divalent metal ions as cofactors. XylT appears to be a notable exception of this rule [4]. However, a recent study indicated that the activity of A. thaliana XylT is stimulated by the addition of metal ions [7]. Similar results were reported for the rice enzyme [22], suggesting that only soya-bean XylT is metal-independent. We now show that Mn2+, Ca2+, Mg2+ and Co2+ affect the enzymatic properties of purified A. thaliana XylT in a manner similar to that previously reported for the soya-bean enzyme [4]. Furthermore, we show that high concentrations of Mn2+ and Co2+ as well as Ni2+ and Zn2+ cause the inactivation of the enzyme. The latter results are also in line with data available on rice XylT [22]. We conclude that all XylT orthologues so far tested exhibit a similar, concentration-dependent behaviour with respect to their metal ion requirements. However, at least two XylT variants clearly display significant activity in the absence of any added metal ion, indicating that this reflects a general, unique feature of this enzyme.
Metal-dependent glycosyltransferases require the metal cofactor for efficient binding of their nucleotide-activated donor substrates (see e.g. [30]). Hence it has been proposed that a protein-bound metal is present in the active site of XylT [4]. Our results clearly show that this is not the case. Thus it appears that, for further insight into the unusual enzymatic properties of XylT, elucidation of the three-dimensional structure of the enzyme is required. In this context, it should be noted that the expression system described in the present study allows the production of essentially homogeneous XylT in amounts sufficient for crystallization experiments.
Acknowledgments
We are grateful to M. Bencúrová for P. pastoris strains and advice on the production of glycosyltransferases in yeast. We also thank R. Prohaska and F. Gauster (Department of Medical Biochemistry, University of Vienna) for N-terminal protein sequencing, D. Kuntz for providing recombinant D. melanogaster α-mannosidase II, and T. Dalik (Department für Chemie, Universität für Bodenkultur Wien) for preparation of glycopeptides and amino sugar analyses. This project was supported in part by the Austrian Science Fund (project no. 13828).
References
- 1.Kornfeld R., Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 2.Lerouge P., Cabanes-Macheteau M., Rayon C., Fitchette-Lainé A. C., Gomord V., Faye L. N-glycoprotein biosynthesis in plants: recent developments and future trends. Plant Mol. Biol. 1998;38:31–48. [PubMed] [Google Scholar]
- 3.Wilson I. B. Glycosylation of proteins in plants and invertebrates. Curr. Opin. Struct. Biol. 2002;12:569–577. doi: 10.1016/s0959-440x(02)00367-6. [DOI] [PubMed] [Google Scholar]
- 4.Zeng Y., Bannon G., Thomas V. H., Rice K., Drake R., Elbein A. Purification and specificity of β1,2-xylosyltransferase, an enzyme that contributes to the allergenicity of some plant proteins. J. Biol. Chem. 1997;272:31340–31347. doi: 10.1074/jbc.272.50.31340. [DOI] [PubMed] [Google Scholar]
- 5.Strasser R., Mucha J., Mach L., Altmann F., Wilson I. B., Glössl J., Steinkellner H. Molecular cloning and functional expression of β1,2-xylosyltransferase cDNA from Arabidopsis thaliana. FEBS Lett. 2000;472:105–108. doi: 10.1016/s0014-5793(00)01443-5. [DOI] [PubMed] [Google Scholar]
- 6.Dirnberger D., Bencúr P., Mach L., Steinkellner H. The Golgi localization of Arabidopsis thaliana β1,2-xylosyltransferase in plant cells is dependent on its cytoplasmic and transmembrane sequences. Plant Mol. Biol. 2002;50:273–281. doi: 10.1023/a:1016061815748. [DOI] [PubMed] [Google Scholar]
- 7.Pagny S., Bouissonnie F., Sarkar M., Follet-Gueye M. L., Driouich A., Schachter H., Faye L., Gomord V. Structural requirements for Arabidopsis β1,2-xylosyltransferase activity and targeting to the Golgi. Plant J. 2003;33:189–203. doi: 10.1046/j.0960-7412.2002.01604.x. [DOI] [PubMed] [Google Scholar]
- 8.Mucha J., Svoboda B., Kappel S., Strasser R., Bencúr P., Fröhwein U., Schachter H., Mach L., Glössl J. Two closely related forms of UDP-GlcNAc:alpha6-D-mannoside beta-1,2-N-acetylglucosaminyltransferase II occur in the clawed frog Xenopus laevis. Glycoconj. J. 2003;19:187–195. doi: 10.1023/A:1024201824354. [DOI] [PubMed] [Google Scholar]
- 9.Staudacher E., Kubelka V., März L. Distinct N-glycan fucosylation potentials of three lepidopteran cell lines. Eur. J. Biochem. 1992;207:987–993. doi: 10.1111/j.1432-1033.1992.tb17134.x. [DOI] [PubMed] [Google Scholar]
- 10.Mucha J., Svoboda B., Fröhwein U., Strasser R., Mischinger M., Schwihla H., Altmann F., Hane W., Schachter H., Glössl J., et al. Tissues of the clawed frog Xenopus laevis contain two closely related forms of UDP-GlcNAc:alpha3-D-mannoside beta-1,2-N-acetylglucosaminyltransferase I. Glycobiology. 2001;11:769–778. doi: 10.1093/glycob/11.9.769. [DOI] [PubMed] [Google Scholar]
- 11.Bencúrová M., Rendić D., Fabini G., Kopecky E.-M., Altmann F., Wilson I. B. Expression of eukaryotic glycosyltransferases in the yeast Pichia pastoris. Biochimie. 2003;85:413–422. doi: 10.1016/s0300-9084(03)00072-5. [DOI] [PubMed] [Google Scholar]
- 12.Van den Elsen J. M., Kuntz D. A., Rose D. R. Structure of Golgi α-mannosidase II: a target for inhibition of growth and metastasis of cancer cells. EMBO J. 2001;20:3008–3017. doi: 10.1093/emboj/20.12.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarkar M., Pagny S., Ünligil U., Joziasse D., Mucha J., Glössl J., Schachter H. Removal of 106 amino acids from the N-terminus of UDP-GlcNAc: alpha-3-D-mannoside beta-1,2-N-acetylglucosaminyltransferase I does not inactivate the enzyme. Glycoconj. J. 1998;15:193–197. doi: 10.1023/a:1006928624913. [DOI] [PubMed] [Google Scholar]
- 14.Altmann F. Determination of amino sugars and amino acids in glycoconjugates using precolumn derivatization with o-phthalaldehyde. Anal. Biochem. 1992;204:215–219. doi: 10.1016/0003-2697(92)90164-3. [DOI] [PubMed] [Google Scholar]
- 15.Altmann F., Kornfeld G., Dalik T., Staudacher E., Glössl J. Processing of asparagine-linked oligosaccharides in insect cells. N-acetylglucosaminyltransferase I and II activities in cultured lepidopteran cells. Glycobiology. 1993;3:619–625. doi: 10.1093/glycob/3.6.619. [DOI] [PubMed] [Google Scholar]
- 16.Kolarich D., Altmann F. N-glycan analysis by matrix-assisted laser desorption/ionization mass spectrometry of electrophoretically separated nonmammalian proteins: application to peanut allergen Ara h 1 and olive pollen allergen Ole e 1. Anal. Biochem. 2000;285:64–75. doi: 10.1006/abio.2000.4737. [DOI] [PubMed] [Google Scholar]
- 17.Katayama H., Nagasu T., Oda Y. Improvement of in-gel digestion protocol for peptide mass fingerprinting by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2001;15:1416–1421. doi: 10.1002/rcm.379. [DOI] [PubMed] [Google Scholar]
- 18.Wilson I. B., Zeleny R., Kolarich D., Staudacher E., Stroop C. J., Kamerling J. P., Altmann F. Analysis of Asn-linked glycans from vegetable foodstuffs: widespread occurrence of Lewis a, core alpha1,3-linked fucose and xylose substitutions. Glycobiology. 2001;11:261–274. doi: 10.1093/glycob/11.4.261. [DOI] [PubMed] [Google Scholar]
- 19.Hernández A., Ruiz M. T. An EXCEL template for calculation of enzyme kinetic parameters by non-linear regression. Bioinformatics. 1998;14:227–228. doi: 10.1093/bioinformatics/14.2.227. [DOI] [PubMed] [Google Scholar]
- 20.Hann S., Koellensperger G., Obinger C., Furtmüller P. G., Stingeder G. SEC-ICP-DRCMS and SEC-ICP-SFMS for determination of metal-sulphur ratios in metalloproteins. J. Anal. At. Spectrom. 2004;19:74–79. [Google Scholar]
- 21.Staudacher E., Dalik T., Wawra P., Altmann F., März L. Functional purification and characterization of a GDP-fucose: β-N-acetylglucosamine (Fuc to Asn linked GlcNAc) α1,3-fucosyltransferase from mung beans. Glycoconj. J. 1995;12:780–786. doi: 10.1007/BF00731239. [DOI] [PubMed] [Google Scholar]
- 22.Léonard R., Kolarich D., Paschinger K., Altmann F., Wilson I. B. A genetic and structural analysis of the N-glycosylation capabilities of rice and other monocotyledons. Plant Mol. Biol. 2004;55:631–644. doi: 10.1007/s11103-004-1558-3. [DOI] [PubMed] [Google Scholar]
- 23.Brömme D., Okamoto K. The baculovirus cysteine proteinase has a cathepsin B-like S2-subsite specificity. Biol. Chem. Hoppe-Seyler. 1995;376:611–615. doi: 10.1515/bchm3.1995.376.10.611. [DOI] [PubMed] [Google Scholar]
- 24.Altmann F., Staudacher E., Wilson I. B., März L. Insect cells as hosts for the expression of recombinant glycoproteins. Glycoconj. J. 1999;16:109–112. doi: 10.1023/a:1026488408951. [DOI] [PubMed] [Google Scholar]
- 25.Bencúrová M., Hemmer W., Focke-Tejkl M., Wilson I. B., Altmann F. Specificity of IgG and IgE antibodies against plant and insect glycoprotein glycans determined with artificial glycoforms of human transferrin. Glycobiology. 2004;14:457–466. doi: 10.1093/glycob/cwh058. [DOI] [PubMed] [Google Scholar]
- 26.Fitchette-Lainé A.-C., Gomord V., Chekkafi A., Faye L. Distribution of xylosylation and fucosylation in the plant Golgi apparatus. Plant J. 1994;5:673–682. [Google Scholar]
- 27.Johnson K. D., Chrispeels M. J. Substrate specificities of N-acetylglucosaminyl-, fucosyl-, and xylosyltransferases that modify glycoproteins in the Golgi apparatus of bean cotyledons. Plant Physiol. 1987;84:1301–1308. doi: 10.1104/pp.84.4.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tezuka K., Hayashi M., Ishihara H., Akazawa T., Takahashi N. Studies on synthetic pathway of xylose-containing N-linked oligosaccharides deduced from substrate specificities of the processing enzymes in sycamore cells (Acer pseudoplatanus L.) Eur. J. Biochem. 1992;203:401–413. doi: 10.1111/j.1432-1033.1992.tb16564.x. [DOI] [PubMed] [Google Scholar]
- 29.Mulder H., Dideberg F., Schachter H., Spronk B. A., de Jong-Brink M., Kamerling J. P., Vliegenthart J. F. In the biosynthesis of N-glycans in connective tissue of the snail Lymnaea stagnalis incorporation of GlcNAc by β2GlcNAc-transferase I is an essential prerequisite for the action of β2GlcNAc-transferase II and β2Xyl-transferase. Eur. J. Biochem. 1995;232:272–283. doi: 10.1111/j.1432-1033.1995.tb20809.x. [DOI] [PubMed] [Google Scholar]
- 30.Ünligil U. M., Zhou S., Yuwaraj S., Sarkar M., Schachter H., Rini J. M. X-ray crystal structure of rabbit N-acetylglucosaminyltransferase I: catalytic mechanism and a new protein superfamily. EMBO J. 2000;19:5269–5280. doi: 10.1093/emboj/19.20.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nilsson T., Hoe M. H., Slusarewicz P., Rabouille C., Watson R., Hunte F., Watzele G., Berger E. G., Warren G. Kin recognition between medial Golgi enzymes in HeLa cells. EMBO J. 1994;13:562–574. doi: 10.1002/j.1460-2075.1994.tb06294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baboval T., Koul O., Smith F. I. N-glycosylation site occupancy of rat α-1,3-fucosyltransferase IV and the effect of glycosylation on enzymatic activity. Biochim. Biophys. Acta. 2000;1475:383–389. doi: 10.1016/s0304-4165(00)00094-5. [DOI] [PubMed] [Google Scholar]
- 33.Jones E. W. Three proteolytic systems in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1991;266:7963–7966. [PubMed] [Google Scholar]
- 34.Opat A. S., Houghton F., Gleeson P. A. Medial Golgi but not late Golgi glycosyltransferases exist as high molecular weight complexes. J. Biol. Chem. 2000;275:11836–11845. doi: 10.1074/jbc.275.16.11836. [DOI] [PubMed] [Google Scholar]