

Abstract
Ex vivo and in vitro studies have revealed the remarkable amyloid inhibitory potency and specificity of iododiflunisal in relation to transthyretin [Almeida, Macedo, Cardoso, Alves, Valencia, Arsequell, Planas and Saraiva (2004) Biochem. J. 381, 351–356], a protein implicated in familial amyloidotic polyneuropathy. In the present paper, the crystal structure of transthyretin complexed with this diflunisal derivative is reported, which enables a detailed analysis of the protein–ligand interactions. Iododiflunisal binds very deep in the hormone-binding channel. The iodine substituent is tightly anchored into a pocket of the binding site and the fluorine atoms provide extra hydrophobic contacts with the protein. The carboxylate substituent is involved in an electrostatic interaction with the Nζ of a lysine residue. Moreover, ligand-induced conformational alterations in the side chain of some residues result in the formation of new intersubunit hydrogen bonds. All these new interactions, induced by iododiflunisal, increase the stability of the tetramer impairing the formation of amyloid fibrils. The crystal structure of this complex opens perspectives for the design of more specific and effective drugs for familial amyloidotic polyneuropathy patients.
Keywords: amyloid fibril, amyloid inhibitor, complex crystal structure, familial amyloidotic polyneuropathy, iododiflunisal, transthyretin
Abbreviations: NSAID, non-steroidal anti-inflammatory drug; r.m.s., root mean square; T4, thyroxine; TTR, transthyretin
INTRODUCTION
TTR (transthyretin) transports approx. 20% of the T4 (thyroxine) circulating in the plasma and is the main carrier of retinol, by forming a complex with the retinol-binding protein [1]. Early X-ray crystallography studies by Blake et al. [2] revealed that the protein is a tetramer with identical subunits. Each subunit, the monomer, has a β-sandwich structure composed of two four-stranded β-sheets (DAGH and CBEF) and a small helical region between strands E and F. The four monomers assemble, forming a central channel where two T4 molecules can be accommodated simultaneously.
TTR is one of the human proteins that forms amyloid fibrils associated with diseases such as familial amyloid cardiomyopathy, senile systemic amyloidosis and familial amyloidotic polyneuropathy. The fibrils have a characteristic green birefringence when observed under polarized light after Congo Red staining. Furthermore, their X-ray diffraction patterns always disclose a β-sheet structure organized with the sheets parallel to the fibril axis and the β-strands perpendicular to it [3].
Several models for TTR amyloid fibrils have been described in the literature. Although some researchers proposed that the TTR tetramers would associate through disulphide bridges [4], others proposed that dimers [5,6] and monomers [7] were the building blocks. Redondo et al. [8] addressed this question using two synthetic TTR variants in which the four monomers, in one case, and two dimers, in a different variant, were linked by disulphide bonds. ‘In vitro’ fibrillogenesis assays led to the conclusion that these mutants were unable to polymerize, even at low pH. However, on the reduction of the disulphide bonds with 2-mercaptoethanol, these variants were capable of assembling into fibrils.
It is now accepted that most probably the dissociation of the TTR tetramer into monomers occurs before fibril formation. An assembly mechanism, where two edge strands (C and D) are displaced and the fibril is formed by end-to-end alignment of the non-native monomers, was proposed based on EPR measurements of various spin-labelled single-cysteine mutants of TTR [9]. Interestingly, the crystal structure of the highly amyloidogenic L55P-TTR variant revealed a structural change of the edge strand D, leading to a long loop between strands C and E [10]. Furthermore, the displacement of strand D is in agreement with the observation that the protein's sulphur-containing amino acids became oxidized on fibrillization [11], since these residues are in the vicinity of the displaced strand and probably become more accessible to the solvent.
Since TTR amyloid fibril formation requires the dissociation of the tetramer into monomeric species before aggregation, small molecules capable of binding in the T4 binding channel, which runs through the centre of the molecule and induces the formation of new intersubunit interactions, should stabilize the native state of the protein and thereby prevent the early steps of amyloid fibril formation. In fact, several compounds have already been shown to have an inhibitory effect on amyloid fibril formation in vitro, due to binding in the TTR channel [12]. Among the most promising inhibitors are NSAIDs (non-steroidal anti-inflammatory drugs) such as diclofenac, diflunisal and flufenamic acid [13]. Other proposed approaches for the stabilization of TTR's native structure include the sulphonation of the single cysteine residue, situated at position 10 of the polypeptide chain, by the addition of sulphite [14]. It was suggested that the sulphonated cysteine residue forms a new hydrogen bond with the main chain nitrogen of Gly57, positioned in the β-strand D, contributing to the inhibition of tetrameric dissociation [15].
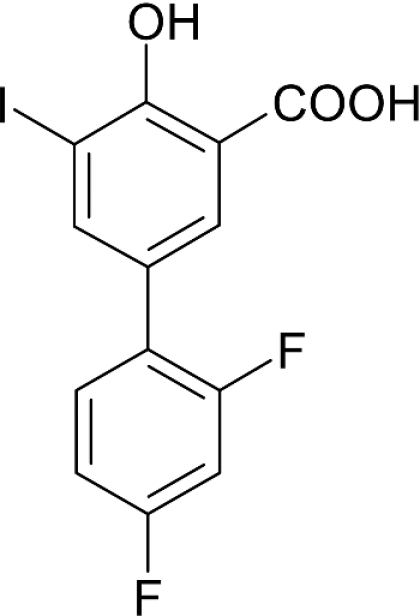
In the course of a screening programme, we have identified an iodinated substance of a difluorophenyl derivative of salicylic acid, which we have termed iododiflunisal (2′,4′-difluoro-4-hydroxy-5-iodo-[1,1′-biphenyl]-3-carboxylic acid) (Figure 1). The compound shows unique features when compared with other proposed TTR stabilizers like diflunisal, flufenamic acid and diclofenac. Among these well-studied drugs, iododiflunisal is the only one that shows an evident stabilizing effect on TTR tetramers in plasma under semi-denaturing conditions [16]. Moreover, the biological action of iododiflunisal in plasma seems to be very selective owing to the fact that it preferentially displaces T4 from TTR over albumin and T4 binding globulin. In addition, it prevents the oligomerization of TTR Y78F, as evaluated by transmission electron microscopy [16]; TTR Y78F is a mutant protein that in solution reveals epitopes similar to those present in fibrils, as assessed by monoclonal antibodies specific to TTR amyloid fibrils, and has high propensity to form amyloid fibrils [17].
Figure 1. Chemical structure of iododiflunisal, a difluorophenyl derivative of iodinated salicylic acid.
To identify the critical molecular features and understand the molecular interactions that give rise to iododiflunisal amyloid inhibitory properties, elucidation of the three-dimensional structure of the TTR–iododiflunisal complex at the atomic level was attempted. Accordingly, in the present study, we report the crystal structure of TTR complexed with iododiflunisal and identify the key interactions between the protein and the drug, as well as the newly obtained interactions between the monomers in the protein tetramer.
EXPERIMENTAL
Co-crystallization of the TTR–iododiflunisal complex
Recombinant TTR was produced in a bacterial expression system using Escherichia coli BL21 [18] and purified as described elsewhere [19]. Iododiflunisal was identified in the course of a screening program for the synthesis of TTR amyloid inhibitors performed at CSIC (IIQAB, Barcelona, Spain) and at the University of Oviedo (Oviedo, Asturias, Spain). Iododiflunisal was prepared by electrophilic aromatic iodination of diflunisal and used after HPLC purification and characterization by NMR and MS.
The protein (12.6 mg·ml−1) was incubated with iododiflunisal [molar ratio: iododiflunisal (99.9% purity)/TTR=10] in 0.165 M sodium citrate buffer (pH 7.0) containing 0.25% (v/v) 1,2,3-heptanetriol for 2 days at 4 °C. Crystals of the complex, suitable for X-ray diffraction, were obtained by hanging-drop vapour-diffusion techniques at 14 °C. Crystals belonging to space group P21212 were grown within 1 week by mixing 3 μl of the TTR–iododiflunisal complex with 3 μl of reservoir solution containing 200 mM citrate buffer, 2.4 M ammonium sulphate and 6% (v/v) glycerol (pH 5.0). Crystals with maximal dimensions of 0.5×0.3×0.1 mm3 were transferred to reservoir solutions containing increasing concentrations of glycerol (10–20%) and flash frozen in liquid nitrogen.
Data collection, processing and refinement
The X-ray diffraction data were collected using synchrotron radiation on ID14-3 beam line at the ESRF (European Synchrotron Radiation Facility, Grenoble Cedex, France).
Crystals were diffracted to a maximum resolution of 1.7 Å (10−10 m). Determination of the crystal orientation and integration was performed with MOSFLM [20], and the scaling and merging of the reflections were performed using programs SCALA and TRUNCATE [21].
The structure of the TTR complex was determined by molecular replacement with AMoRe [22] using T119M-TTR as the starting model (PDB accession no. 1F86; [23]), after the removal of water molecules and mutation of residue 119 to alanine. Several cycles of refinement were performed with the program CNS [24], alternating with manual model building using the program Turbo-FRODO [25] in an SGI graphic workstation, until the protein model was completely fitted to the Fourier map. The refinement was monitored using Rfree, the cross-validation R-factor, calculated from a set of 5% of the reflections, which was kept aside from the initial refinement. The refinement was obtained using SHELXL [26]. Water molecules were added at the position of positive peaks (>3σ) on the difference Fourier maps where good hydrogen bond geometry existed. The position of iododiflunisal could be clearly identified in the Fo–Fc electron density map (where Fo and Fc are the observed and calculated structure factor amplitudes respectively) and the occupancies of the ligand atoms were refined using SHELXL. The last refinement cycles of the TTR–iododiflunisal model were performed with REFMAC5 [27] from the CCP4 suite [21]. The stereochemistry of the refined model was checked with PROCHECK [28] and all the residues included in the protein model were in the allowed regions of the Ramachandran plot. The Matthews coefficient is 2.18 Å·Da−1 for the crystals of the complex corresponding to a solvent content of 43%.
Details of the crystallographic data collection and refinement statistics are shown in Table 1.
Table 1. Data collection and refinement statistics.
| Data collection | |
| Space group | P21212 |
| Unit cell | |
| a (Å) | 43.3 |
| b (Å) | 85.8 |
| c (Å) | 64.9 |
| α=β=γ (°) | 90 |
| Resolution (Å) | 1.70 |
| No. of observations (total/unique) | 244185/27386 |
| Multiplicity (overall/last shell) | 3.7/2.9 |
| Rmerge (%)* (overall/last shell) | 7.3/12.0 |
| Completeness (%) (overall/last shell) | 98.6/93.0 |
| Data refinement | |
| Resolution range | 20–1.7 |
| R-factor†/Rfree (%) | 19.9/22.1 |
| No. of reflections (working/free) | 25568/1372 |
| No. of water molecules | 169 |
| Average B-factor (Å2) | |
| Protein (main chain/side chain) | 17.1/22.1 |
| Iododiflunisal | 32.1 |
| Water | 34.8 |
| R.m.s. bonded Bs (Å2) | 1.448 |
| R.m.s. deviations from standard geometry | |
| R.m.s. bond length (Å) | 0.009 |
| R.m.s. angle (°) | 1.280 |
| Ramachandran plot | |
| Most favoured (%) | 93.0 |
| Allowed (%) | 7.0 |
* Rmerge=∑|I−〈I〉|/∑〈I〉, where I is the observed intensity and 〈I〉 is the average intensity of multiple observations of symmetry-related positions.
† R-factor=∑||/Fo|−|Fc||/∑|Fo|, where |Fo| and |Fc| are observed and calculated structure factor amplitudes respectively.
RESULTS AND DISCUSSION
Overall structure of the TTR–iododiflunisal complex
The crystals belong to the orthorombic space group P21212, with unit cell dimensions a=43.3 Å, b=85.8 Å and c=64.9 Å. The asymmetric unit contains two monomers, A and B, which form a dimer. The two dimers that form the tetrameric protein are related by a crystallographic 2-fold axis that runs along the hormone-binding channel.
The final protein model includes residues 10–124 from monomer A and 10–125 from monomer B, since the first nine residues from the N-terminal and last three (monomer A) and two (monomer B) residues from the C-terminal could not be located in the electron density map. A total of 169 water molecules were identified and included in the final model. The side chains of residues Asn27 (A), Arg34 (A), Glu72 (A), Ser115 (A) and Asn27 (B) were refined in two well-defined conformations.
Monomers A and B are chemically identical and adopt generally similar conformations. The r.m.s. (root mean square) deviation between main chain atom positions of all residues of monomers A and B is 0.5 Å. The largest differences are observed in the loop fragment 98–103, which is characterized by high thermal motion. On the other hand, when we compare the main chain atom positions of the complexed and native (PDB accession no. 1TTA [29]) protein structures, the r.m.s. deviation of 0.5 Å indicates that the overall protein structure is conserved on ligand binding. Small alterations occur in the hormone binding channel, resulting from the binding of iododiflunisal, and will be analysed in detail below.
Monomers A and B and their symmetry-related pair form a cavity in the dimer interface that can accommodate simultaneously two T4 molecules. The two hormone-binding sites AA′ and BB′ have similar topologies. They are funnel-shaped and mainly hydrophobic, with hydrophilic contributions from residues Lys15 and Glu54 located at the entrance of the binding site. The initial electron density map clearly indicated the position of the iododiflunisal molecules, which occupy the binding sites available in the protein for its natural ligand. Owing to the crystallographic 2-fold axis that runs along the channel, two symmetry-related positions for iododiflunisal were observed in each binding site (Figure 2). Therefore the occupancy of the two iododiflunisal molecules was set initially to 0.5 and was refined as a free variable to a final value of 0.35. Similar occupancy for the ligands was also found in other X-ray structures of TTR in complex with binding molecules. For example, in the models of TTR complexed with flurbiprofen, dibenzofuran-4,6-dicarboxylic acid, o-trifluoromethylphenyl anthranilic acid and N-m-trifluoromethylphenylphenoxazine-4,6-dicarboxylic acid, the ligand occupancies range from 0.28 to 0.41 [12]. This deviation from the maximum value of occupancy could be explained in light of the crystal structures of TTR bound to its native ligand. Thus the crystal structure of the TTR mutant TTR T119M (Thr119→Met) bound to T4 [23] was refined to a final occupancy value of 0.25 for the T4 atoms. In the present study, it has been described that a negative co-operativity operates while the second molecule tries to reach into the complex already formed by the entrance of a first T4 molecule. This effect can be measured in terms of the association constants for binding of the first and the second T4 molecules, which are in the region of 108 and 106 M−1 respectively [2]. It is possible that this same influence also occurs for other ligands.
Figure 2. Cα trace of TTR in complex with iododiflunisal.

Monomers A (pink) and B (blue) form the crystallographic asymmetric unit. The 2-fold symmetry-related monomers A′ and B′ are displayed in grey and green respectively. (A) Omit Fo–Fc maps are contoured at 6σ over the hormone-binding site. The two strong density features found in each binding site correspond to the position of the iodines of the two symmetry-related iododiflunisal molecules. (B) Close view of the iododiflunisal binding site AA′ with omit maps contoured at 6σ (orange) and 3σ (red).
The position of the iodine atoms was unambiguously determined in the omit (Fo–Fc) electron density map contoured at 6σ, as indicated in Figure 2. In the final model, the symmetry-related positions assume a ‘V’ form, with the iodine-substituted rings placed away from the channel axis. A similar organization was found in the TTR–diflunisal complex [30].
Protein–ligand interactions
The crystallographic structure of the TTR–T4 complex [31] allowed a careful characterization of the hormone-binding sites, which are composed of three symmetry-related hydrophobic small depressions, termed halogen-binding pockets [32]. The innermost pocket (P3) is formed by the side chains of Ala108, Leu110, Ser117 and Thr119. It is essentially hydrophobic with nucleophilic contributions from the Ser115 Oγ hydroxy and from the carbonyl groups of Ser117, Thr118 and Ala108. The central pocket (P2) is also hydrophobic and is composed of the methyl and methylene groups of Leu110, Ala109, Lys15 and Leu17. The outmost pocket (P1) is located between the side chains of Ala108, Thr106, Met13 and Lys15. The Nζ of Lys15 is located close to the P1 pocket, allowing potential electrostatic interactions with the ligands.
In the TTR–iododiflunisal complex, the fluorine-substituted phenyl ring is positioned deep in the interior of the binding cavity, with the two fluorine atoms accommodated in the P3 and P3′ pockets (Figure 3). Those atoms are involved in extensive hydrophobic interactions with Ala108, Leu110, Ser117, Thr118 and Thr119 of monomers AA′ and BB′, which contribute to the overall stability of the tetrameric structure.
Figure 3. Stereo view of the interactions between the two symmetrically equivalent iododiflunisal molecules and TTR residues forming the AA′-binding site.

Binding of iododiflunisal induces the rotation of the side chains of Ser117 and Ser117′ with the consequent formation of short hydrogen bonds between the hydroxy groups of Ser117 (A)–Ser117 (B) and Ser117 (A′)–Ser117 (B′). Nζ of Lys15 is involved in weak hydrogen bond interactions with the CO2H and the OH groups of the iodine-substituted ring. For the hydroxy group, the interaction is mediated by a water molecule. The iodine atom is involved in a series of hydrophobic contacts with the side chains of Thr106, Ala108, Thr119 and Val121.
The iodine-substituted phenyl ring is positioned close to the entrance of the hormone-binding channel. The iodine atom establishes close hydrophobic interactions with side chains of residues belonging to the two monomers that form each binding site: Leu17, Thr106′, Ala108′, Thr119′ and Val121′ (interatomic distances between the iodine and protein atoms are listed in Table 2).
Table 2. Interatomic distances between the iodine substituent of iododiflunisal and TTR at the two binding sites.
| Distance to the iodine substituent (Å) | ||
|---|---|---|
| Residue | Atom | Binding sites: AA′/BB′ |
| Leu17 | Cδ1 | 3.84/3.81 |
| Thr106′ | O | 4.04/3.93 |
| Thr106′ | Cγ2 | 4.20/4.00 |
| Ala108′ | Cβ | 3.24/3.36 |
| Thr119′ | O | 3.98/4.00 |
| Thr119′ | Cγ2 | 3.31/3.38 |
| Val121′ | N | 4.06/3.96 |
| Val121′ | Cβ | 3.90/4.08 |
| Val121′ | Cγ2 | 2.69/3.29 |
The phenolic hydroxy and carboxylate groups are involved in hydrophilic interactions with Nζ of Lys15. A close inspection shows slight differences between the positions of the ligand in the two binding sites, namely in the position of the iodine-substituted phenyl ring. In one of the binding sites, the CO2H group interacts with lysine residues from both monomers, contributing further to the stability of the tetrameric form of the protein. In the other binding site, the carboxylate substituent forms a weak hydrogen bond interaction with Nζ of Lys15 and the hydroxy group is involved in a water-mediated polar interaction with the same Nζ.
Small differences between the positions of the ligand may be associated with the expected negative co-operativity of ligand binding, which was proved by isothermal titration calorimetry for diflunisal and some diflunisal analogues [30].
Most of the studied small molecules affecting TTR tetramer stability are two-ring biphenyl type structures. One of the rings commonly presenting a carboxylic acid function is positioned in the protein complex towards the outer part of the binding cavity by forming electrostatic interactions with Nζ of Lys15. The second aromatic ring is positioned in the inner part of the binding channel and, since it is often substituted by halogen atoms (Cl and F), it shows additional hydrophobic interactions with the protein [30]. This orientation, usually referred to as the forward binding mode, was also found in the TTR–iododiflunisal structure; however, careful analysis of the electron density maps reveals that iododiflunisal may also bind in the reverse mode with very low occupancy. Initially, both binding modes were included in the refinement and the occupancies of iododiflunisal in the reverse binding mode was refined using SHELXL to values <0.08. Therefore this binding mode was removed from the following cycles of refinement. In fact, the main feature in the Fo–Fc electron density map corresponds solely to the iodine atom and very little indication was found for the light atoms such as carbon, oxygen and fluorine. Moreover, because of the 2-fold symmetry of the binding site and the partial superposition of the electron density corresponding to the iododiflunisal molecules bound in the forward mode, we were unable to identify clearly the reverse binding-mode conformations. Nevertheless, it is possible to observe that the iodine atom (in the reverse binding mode) is interacting with main chain atoms of Ala108, Leu110, Ser117, Thr118 and Thr119 (distance between iodine and protein atoms were in the range 3.3–4.0 Å). The partial hydrophilic character of these interactions due to the contribution from the main chain oxygen of Ser117 and from the main chain nitrogen atoms of Thr118 and Thr119 probably dictates that the forward binding mode is preferred. There is also an indication in the electron density map of an alternative conformation of the Thr119 side chain, with very low occupancy. It allows the formation of a hydrogen bond between the Oγ of Thr119 and the carboxy group of iododiflunisal bound in the reverse mode, as reported for the crystal structure of TTR–diflunisal [30].
Protein structure alterations induced by iododiflunisal
To assess the alterations in the backbone structure of TTR induced by iododiflunisal binding, we determined the Cα–Cα distance of equivalent amino acids measured across the TTR channel, i.e. across monomers A and A′ that form one of the binding sites and monomers B and B′ that form the other binding site. The Cα–Cα distances measured for the apo-TTR (PDB accession no. 1TTA) were designated by dapo TTR and are presented in Figure 4. The difference between Cα–Cα distances in the TTR–iododuflunisal complex and the apo-TTR are also shown in Figure 4 as dcomplex–dapoTTR. It is clear that there is a systematic decrease in the Cα–Cα distances in the complexed structure, indicating that the binding of iododiflunisal causes a slight collapse of the TTR-binding sites. Interestingly, a similar binding effect was already noticed in the human TTR–T4 complex [33]. These alterations are not limited to the amino acids that form the channel surface but seem to propagate to the rest of the polypeptide chain of the protein. Moreover, no significant differences were found between the AA′- and BB′-binding sites of the TTR–iododiflunisal structure.
Figure 4. Comparison of Cα–Cα distances between equivalent amino acids of the monomers forming each binding site (monomers AA′ and BB′) in apo-TTR and the TTR–iododiflunisal complex.

The Cα–Cα distances (average across AA′ and BB′) for apo-TTR are plotted in black (dapoTTR) and the differences between Cα–Cα distances in TTR–iododiflunisal and apo-TTR are plotted in white (dcomplex–dapoTTR).
Ligand binding also induces modifications on the side-chain orientations for some protein residues. In the apo-TTR structure, the hydroxy groups of Ser117 (in chains A and B) are facing the cavity and are hydrogen-bonded to a water molecule. When P3 is occupied by the fluorine atoms of iododiflunisal, there is a rotation of the side chain of Ser117, leading to a more hydrophobic surface. The newly orientated side chains of Ser117 of the two monomers of the asymmetric unit form a strong direct hydrogen bond connecting the two monomers in the dimer [Ser117 Oγ(H)–Oγ′ Ser117′=2.6 Å]. A hydrogen bond, involving the two hydroxy groups of Ser117 of monomers A and B, is also observed in the crystal structure of TTR T119M [23], which is a non-amyloidogenic variant that presents higher resistance to dissociation into monomers than the wild-type TTR [34].
Furthermore, iododiflunisal induces a rotation of the Thr119 side chain leading again to a more hydrophobic P3 pocket. As a result, the Thr119 Oγ(H)–Oγ′ Ser115′ intersubunit hydrogen bond present in the apo-TTR is replaced by a Thr119 Oγ(H)–O′ Tyr114′ bond in the TTR–iododiflunisal complex. The reorientation of the Thr119 side chain also favours the formation of a contact, mediated by a water molecule, with the carbonyl oxygen of Asp18 from another subunit of the tetramer.
If we consider the reverse binding mode that, as we said before, has very low probability of occurrence, we are able to observe just the reorientation of the Thr119 side, whereas Ser117 does not show any conformational change in relation to the position that it occupies when iododiflunisal binds in the forward binding mode.
Comparison with other complexes of TTR bound to promising amyloid inhibitors
Several NSAIDs were proved to prevent urea-induced tetramer dissociation of TTR and to reduce dissociation of ‘in vitro’ acid-induced fibrils [12,13,30,35]. Structure–activity studies showed that diclofenac, diflunisal and flufenamic acid are among the most promising compounds against wild-type TTR amyloidogenesis [13]. Moreover, diclofenac and diflunisal are approved by the U.S. Food and Drug Administration (Rockville, MD, U.S.A.) and flufenamic acid is commonly used in many countries [13]. However, the low selectivity of these compounds towards TTR in the plasma [16] probably limits their usefulness clinically in relation to TTR amyloidosis.
Iododiflunisal was synthesized based on the results obtained with diflunisal and was proved to have increased binding selectivity as well as increased tetrameric stabilizing effect when compared with diclofenac, diflunisal and flufenamic acid [16]. The analyses of the crystal structures of TTR complexed with iododiflunisal, diflunisal, diclofenac and flufenamic acid help us to elucidate the structure–activity relationship of these four compounds.
Diflunisal binds to TTR in both forward and reverse modes with nearly identical occupancy [30]. In the forward binding mode, the fluorine substituents are positioned in the innermost pockets P3 and P3′ and the carboxylate in the outer pocket, allowing the formation of stabilizing interactions with Lys15 and Lys15′. The binding in the reverse mode was not expected [30]; in this conformation, the fluorines are positioned in the outer pocket and the carboxylate forms a hydrogen bond with Thr119 side chain. An important difference between the binding of diflunisal and iododiflunisal to TTR is that iododiflunisal has a preferred orientation in the binding pocket. This is a result of the effect of an iodine atom, which is capable of anchoring into the outer β-strand pocket (P1), forming the hydrophobic interactions that enhance the binding affinity for TTR. P1 and P1′ are in fact the most hydrophobic pockets. The polarity of the inner pockets is increased by the presence of a hydrophilic Ser117 and Thr119, and contributions of the nitrogen and oxygen atoms of the polypeptide backbone. Accordingly, a preferential binding for the outer pockets was observed in crystal structures of TTR complexed with halogen-substituted phenols [36].
Although the atomic co-ordinates of TTR complexed with diflunisal were not available, the molecular models of TTR complexed with diclofenac and flufenamic acid were deposited in the PDB and therefore can be analysed in more detail (Figure 5). In contrast with iododiflunisal and flufenamic acid, diclofenac binds in the reverse mode, which allows hydrogen bond interaction between the CO2H substituent and the side chain of Thr119. The chloro-substituted phenyl ring is positioned in the outer part of the binding cavity but it does not fit so tightly to the pocket P1, when compared with flufenamic acid and iododiflunisal.
Figure 5. The binding of iododiflunisal (A), flufenamic acid (B) and diclofenac (C) to TTR.

The electrostatic surface representation of the AA′-binding site is presented along the x-axis. The two symmetry equivalent positions of the ligands are shown in orange and blue. The PDB accession numbers for the TTR–flufenamic acid and the TTR–diclofenac complexes are 1BM7 and 1DVX respectively. For clarity, the electrostatic surfaces of residues 15–17 and 109–110 (monomer A′) were removed since they are located in front of the binding molecules.
Flufenamic acid and iododiflunisal display similar protein–ligand interactions. The carboxylate substituent of both structures is placed approximately in the same position, allowing interaction with Nζ of Lys15. The outer pocket P1 is occupied by the CO2H-substituted phenyl ring in the case of a flufenamic acid and by an iodine atom for iododiflunisal. The fluorine substituents of both compounds provide additional van der Waals contacts with the residues forming the inner pockets (P3 and P3′). The increased stabilizing activity over the TTR tetramer displayed by iododiflunisal [16], probably arises from the tight interactions between the iodine and the protein atoms in the hydrophobic pocket P3. Moreover, the long axis of the iododiflunisal molecule is slightly offset from the 2-fold z-axis, allowing the iodine and the p-fluorine substituents to occupy the binding pockets of two distinct monomers (P1 and P3′ or P1′ and P3), which enhances the intermonomer interactions and contributes to the stabilization of the native structure of the protein.
In conclusion, the fine protective properties of iododiflunisal reflect the fact that all the aromatic substituents are involved in tetramer-stabilizing interactions: new intermonomer interactions are created and others are strengthened. The preference of the molecule for the forward binding mode, which increases the contacts between the four subunits in the tetramer when compared with the reverse binding mode, is most probably due to the tight protein–iodine interactions. Furthermore, dihalogen substitutions in the hydrophobic ring and hydroxy and carboxylate substitutions in the hydrophilic ring induce the formation of new interactions, including intermonomer hydrogen bonds. Finally, ligand binding induces rotation of the side chain of Ser117, allowing the formation of new tight intermonomer hydrogen bonds. All these new interactions are responsible for a more compact structure that results in a slight collapse of the binding channel that runs through the molecule.
Acknowledgments
We thank P. Moreira for excellent technical assistance. We are grateful for the financial support from FCT (Fundação para a Ciência e a Tecnologia, Lisboa, Portugal) (project POCTI/NSE/44821/2002) and acknowledge the ESRF for the use of beam line ID14-3 and for the technical assistance given by the ESRF staff. The screening programme for TTR amyloid inhibitors was supported by Fundació La Caixa (Barcelona, Spain).
References
- 1.Kanai M., Raz A., Goodman D. S. Retinol-binding protein: the transport protein for vitamin A in human plasma. J. Clin. Invest. 1968;47:2025–2044. doi: 10.1172/JCI105889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blake C. C., Geisow M. J., Oatley S. J., Rerat B., Rerat C. Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 Å. J. Mol. Biol. 1978;121:339–356. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- 3.Serpell L. C., Fraser P. E., Sunde M. X-ray fiber diffraction of amyloid fibrils. Methods Enzymol. 1999;309:526–536. doi: 10.1016/s0076-6879(99)09036-9. [DOI] [PubMed] [Google Scholar]
- 4.Terry C. J., Damas A. M., Oliveira P., Saraiva M. J., Alves I. L., Costa P. P., Matias P. M., Sakaki Y., Blake C. C. Structure of Met30 variant of transthyretin and its amyloidogenic implications. EMBO J. 1993;12:735–741. doi: 10.1002/j.1460-2075.1993.tb05707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olofsson A., Ippel H. J., Baranov V., Horstedt P., Wijmenga S., Lundgren E. Capture of a dimeric intermediate during transthyretin amyloid formation. J. Biol. Chem. 2001;276:39592–39599. doi: 10.1074/jbc.M103599200. [DOI] [PubMed] [Google Scholar]
- 6.Serag A. A., Altenbach C., Gingery M., Hubbell W. L., Yeates T. O. Identification of a subunit interface in transthyretin amyloid fibrils: evidence for self-assembly from oligomeric building blocks. Biochemistry. 2001;40:9089–9096. doi: 10.1021/bi010655s. [DOI] [PubMed] [Google Scholar]
- 7.Inouye H., Domingues F. S., Damas A. M., Saraiva M. J., Lundgren E., Sandgren O., Kirschner D. A. Analysis of x-ray diffraction patterns from amyloid of biopsied vitreous humor and kidney of transthyretin (TTR) Met30 familial amyloidotic polyneuropathy (FAP) patients: axially arrayed TTR monomers constitute the protofilament. Amyloid. 1998;5:163–174. doi: 10.3109/13506129809003842. [DOI] [PubMed] [Google Scholar]
- 8.Redondo C., Damas A. M., Saraiva M. J. Designing transthyretin mutants affecting tetrameric structure: implications in amyloidogenicity. Biochem. J. 2000;348:167–172. [PMC free article] [PubMed] [Google Scholar]
- 9.Serag A. A., Altenbach C., Gingery M., Hubbell W. L., Yeates T. O. Arrangement of subunits and ordering of beta-strands in an amyloid sheet. Nat. Struct. Biol. 2002;9:734–739. doi: 10.1038/nsb838. [DOI] [PubMed] [Google Scholar]
- 10.Sebastiao M. P., Saraiva M. J., Damas A. M. The crystal structure of amyloidogenic Leu55 –> Pro transthyretin variant reveals a possible pathway for transthyretin polymerization into amyloid fibrils. J. Biol. Chem. 1998;273:24715–24722. doi: 10.1074/jbc.273.38.24715. [DOI] [PubMed] [Google Scholar]
- 11.Gales L., Cardoso I., Fayard B., Quintanilha A., Saraiva M. J., Damas A. M. X-ray absorption spectroscopy reveals a substantial increase of sulfur oxidation in transthyretin (TTR) upon fibrillization. J. Biol. Chem. 2003;278:11654–11660. doi: 10.1074/jbc.M210798200. [DOI] [PubMed] [Google Scholar]
- 12.Klabunde T., Petrassi H. M., Oza V. B., Raman P., Kelly J. W., Sacchettini J. C. Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Biol. 2000;7:312–321. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- 13.Miller S. R., Sekijima Y., Kelly J. W. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab. Invest. 2004;84:545–552. doi: 10.1038/labinvest.3700059. [DOI] [PubMed] [Google Scholar]
- 14.Altland K., Winter P., Saraiva M. J., Suhr O. Sulfite and base for the treatment of familial amyloidotic polyneuropathy: two additive approaches to stabilize the conformation of human amyloidogenic transthyretin. Neurogenetics. 2004;5:61–67. doi: 10.1007/s10048-003-0160-1. [DOI] [PubMed] [Google Scholar]
- 15.Altland K., Winter P. Potential treatment of transthyretin-type amyloidoses by sulfite. Neurogenetics. 1999;2:183–188. doi: 10.1007/s100480050081. [DOI] [PubMed] [Google Scholar]
- 16.Almeida M. R., Macedo B., Cardoso I., Alves I., Valencia G., Arsequell G., Planas A., Saraiva M. J. Selective binding to transthyretin and tetramer stabilization in serum from patients with familial amyloidotic polyneuropathy by an iodinated diflunisal derivative. Biochem. J. 2004;381:351–356. doi: 10.1042/BJ20040011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Redondo C., Damas A. M., Olofsson A., Lundgren E., Saraiva M. J. Search for intermediate structures in transthyretin fibrillogenesis: soluble tetrameric Tyr78Phe TTR expresses a specific epitope present only in amyloid fibrils. J. Mol. Biol. 2000;304:461–470. doi: 10.1006/jmbi.2000.4220. [DOI] [PubMed] [Google Scholar]
- 18.Furuya H., Saraiva M. J., Gawinowicz M. A., Alves I. L., Costa P. P., Sasaki H., Goto I., Sakaki Y. Production of recombinant human transthyretin with biological activities toward the understanding of the molecular basis of familial amyloidotic polyneuropathy (FAP) Biochemistry. 1991;30:2415–2421. doi: 10.1021/bi00223a017. [DOI] [PubMed] [Google Scholar]
- 19.Almeida M. R., Damas A. M., Lans M. C., Brouwer A., Saraiva M. J. Thyroxine binding to transthyretin Met 119. Comparative studies of different heterozygotic carriers and structural analysis. Endocrine. 1997;6:309–315. doi: 10.1007/BF02820508. [DOI] [PubMed] [Google Scholar]
- 20.Leslie A. G. W. Molecular data processing. In: Moras D., Podjarny A. D., Thierri J. C., editors. Crystallographic Computing 5: From Chemistry to Biology. Oxford: Oxford University Press; 1992. pp. 50–61. [Google Scholar]
- 21.Collaborative Computational Project Number 4 (1994) The CCP4 suite: programs for protein crystallography Acta Crystallogr. D Biol. Crystallogr. 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 22.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr. A Found. Crystallogr. 1994;50:157–163. [Google Scholar]
- 23.Sebastiao M. P., Lamzin V., Saraiva M. J., Damas A. M. Transthyretin stability as a key factor in amyloidogenesis: X-ray analysis at atomic resolution. J. Mol. Biol. 2001;306:733–744. doi: 10.1006/jmbi.2000.4415. [DOI] [PubMed] [Google Scholar]
- 24.Brunger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., et al. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 25.Roussel A., Cambillau C. Partner Directory. Mountain View, CA: Silicon Graphics; 1991. TurboFRODO in Silicon Graphics Geometry. [Google Scholar]
- 26.Sheldrick G. M., Schneider T. R. SHELXL: high-resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 27.Murshudov G. N. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 28.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 29.Hamilton J. A., Steinrauf L. K., Braden B. C., Liepnieks J., Benson M. D., Holmgren G., Sandgren O., Steen L. The x-ray crystal structure refinements of normal human transthyretin and the amyloidogenic Val-30–>Met variant to 1.7-Å resolution. J. Biol. Chem. 1993;268:2416–2424. [PubMed] [Google Scholar]
- 30.Adamski-Werner S. L., Palaninathan S. K., Sacchettini J. C., Kelly J. W. Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J. Med. Chem. 2004;47:355–374. doi: 10.1021/jm030347n. [DOI] [PubMed] [Google Scholar]
- 31.Wojtczak A. Structures of human transthyretin complexed with thyroxine at 2.0 Å resolution and 3′,5′-dinitro-N-acetyl-L-thyronine at 2.2 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 1996;52:758–765. doi: 10.1107/S0907444996003046. [DOI] [PubMed] [Google Scholar]
- 32.Paz P. D. L., Burridge J. M., Oatley S. J., Blake C. C. F. Multiple modes of binding of thyroid hormones and other iodothyronines to human plasma transthyretin. In: Beddel C. R., editor. The Design of Drugs to Macromolecular Targets, chapter 5. New York: John Wiley; 1992. pp. 119–172. [Google Scholar]
- 33.Wojtczak A., Neumann P., Cody V. Structure of a new polymorphic monoclinic form of human transthyretin at 3 Å resolution reveals a mixed complex between unliganded and T4-bound tetramers of TTR. Acta Crystallogr. D Biol. Crystallogr. 2001;57:957–967. doi: 10.1107/s0907444901006047. [DOI] [PubMed] [Google Scholar]
- 34.Alves I. L., Hays M. T., Saraiva M. J. Comparative stability and clearance of [Met30]transthyretin and [Met119]transthyretin. Eur. J. Biochem. 1997;249:662–668. doi: 10.1111/j.1432-1033.1997.00662.x. [DOI] [PubMed] [Google Scholar]
- 35.Hammarstrom P., Wiseman R. L., Powers E. T., Kelly J. W. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713–716. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- 36.Ghosh M., Meerts I. A., Cook A., Bergman A., Brouwer A., Johnson L. N. Structure of human transthyretin complexed with bromophenols: a new mode of binding. Acta Crystallogr. D Biol. Crystallogr. 2000;56:1085–1095. doi: 10.1107/s0907444900008568. [DOI] [PubMed] [Google Scholar]