

Abstract
The lack of comparable metabolic state assays severely limits understanding the metabolic changes caused by genetic or environmental perturbations. The present study reports the application of a novel derivatization method for metabolome analysis of yeast, coupled to data-mining software that achieve comparable throughput, effort and cost compared with DNA arrays. Our sample workup method enables simultaneous metabolite measurements throughout central carbon metabolism and amino acid biosynthesis, using a standard GC-MS platform that was optimized for this purpose. As an implementation proof-of-concept, we assayed metabolite levels in two yeast strains and two different environmental conditions in the context of metabolic pathway reconstruction. We demonstrate that these differential metabolite level data distinguish among sample types, such as typical metabolic fingerprinting or footprinting. More importantly, we demonstrate that this differential metabolite level data provides insight into specific metabolic pathways and lays the groundwork for integrated transcription–metabolism studies of yeasts.
Keywords: fingerprint, footprint, metabolomics, redox metabolism, Saccharomyces cerevisiae, yeast
Abbreviations: AMDIS, automated mass spectra deconvolution and identification system; FDA, Fisher discriminant analysis; MCF, methyl chloroformate; PCA, principal component analysis; PPP, pentose phosphate pathway; TCA, tricarboxylic acid
INTRODUCTION
The metabolome is an experimentally accessible feature of the cell that manifests important and extensive phenotypic information [1,2]. Interaction networks of transcription factor regulation [3–5] and protein-binding signals [6] infer active signal transduction pathways when integrated with differential transcript levels [7–9]. Databases documenting our understanding of signalling pathways continue to evolve [10]. However, understanding how these active pathways mediate a macroscopic phenotype is difficult, and generally we may not draw accurate metabolic conclusions using only this kind of data. For instance, increases in mRNA levels do not always correlate with increases in protein levels [11], and once translated a protein may or may not be enzymatically active [12].
Flux balance analysis represents a powerful in silico attempt to relate genetic information to metabolism by optimizing the stoichiometrically feasible set of reaction fluxes [13–15]. Cellular responses to simple perturbations, such as enzyme-deletion mutants, may be simulated by removing the corresponding reaction from the stoichiometric matrix. However, currently, flux balance analysis represents a limited, qualitative approach which does not substitute for experimental characterizations.
Metabolic fingerprinting and footprinting has succeeded in experimental characterization of genetic mutants on the basis of intracellular and extracellular aggregate MS metabolite data respectively [16,17]. Although limited insight to the function of orphan genes may be gained, these techniques fail to indicate adjustments occurring in specific metabolic pathways, and thus make difficult a straightforward integration of the results with corresponding transcriptome data.
The experimental challenges to metabolic phenotype characterization directly stem from the diverse roles of intracellular metabolites in the overall conversion of nutrients to cell mass. In comparison with proteins or nucleic acids, metabolite pools turnover more rapidly and display a wider range of chemical characteristics. No single experimental technique can assay the entire metabolome, from ionic inorganic species to hydrophilic carbohydrates, volatile alcohols and ketones, amino and non-amino organic acids, hydrophobic lipids and complex natural products. Classically, metabolite analysis targets one or a few similar metabolites with a specific enzymatic assay or chromatographic procedure. These targeted analyses do not provide the broad metabolic state characterization required to fully understand the interplay between different pathways operating within the cell.
For volatile compounds, gas chromatography (GC) coupled to MS allows high analysis throughput at relatively low cost. GC-MS separates complex mixtures with high efficiency [18,5], and accurately identifies compounds by deconvoluting overlapping chromatographic peaks by the utilization of an AMDIS (automated mass spectra deconvolution and identification system) [19,20]. However, most naturally occurring metabolites are not sufficiently volatile to be analysed directly on a GC system. Chemical derivatization of the metabolites is therefore required, and high analysis throughput by GC-MS relies on fast and efficient derivatization techniques.
Recently, we reported one novel chemical derivatization protocol enabling GC-MS detection of amino and non-amino organic acids through conversion into volatile derivatives [21]. Of the approx. 600 metabolites documented by Förster et al. [22] in genome-wide metabolic pathway reconstruction for yeast, approx. 40% are amines, amino acids and organic acids (not including fatty acids) which play crucial roles in central carbon metabolism and amino acid biosynthesis. Unlike silylation, which is an often used method for derivatization of metabolites, our protocol offers instantaneous reaction without heating or water exclusion, much lower reagent costs, easy separation of the derivatives from the reactive mixture, which causes less damage to the GC-capillary column, and the whole process involves only a few steps which is easier for automation.
Although this derivatization hinted at the opportunity for high-throughput application, the procedure did not possess the sensitivity to detect most intracellular metabolites at physiological concentrations nor did we have a large library to identify the peaks. We also did not possess the experimental throughput or analysis software to fully scale up this technology. In the present work, we report on a far more sensitive analysis method and the high-throughput implementation of this method for simultaneous metabolite measurements across central carbon metabolism and amino acid biosynthesis.
EXPERIMENTAL
Yeast strains
Two Saccharomyces cerevisiae strains were used: CEN.PK113-7D (MATaMAL2-8c SUC2) as reference strain, and the mutant CEN.MS1-10CT1 (gdh1(209,1308)::loxP gdh2::PGKp-GDH2-KanMX3).
Flask culture
Both S. cerevisiae strains were cultivated aerobically and anaerobically in triplicate, using shake flasks containing glucose (20 g·l−1), (NH4)2SO4 (5.0 g·l −1), MgSO4.7 H2O (0.5 g·l −1), KH2PO4 (3.0 g·l −1), vitamins and trace elements [23]. Aerobic cultivations were performed using a rotary shaker at 30 °C and 200 rev./min, in shake flasks containing 150 ml of medium and cotton plugs. Anaerobic cultivations were carried out under moderate shaking (130 rev./min) at 30 °C in shake flasks containing 150 ml of medium with tight rubber plugs. The flasks were flushed with nitrogen prior to cultivation and the medium was supplemented with ergosterol (10 mg·l −1) according to Verduyn et al. [24].
Extracellular sampling
Three extracellular samples were removed from each flask when the cells had reached a D600=6.0. We filtered 3 ml of the culture medium using Millipore membrane (0.45 μm) and then freez-dried the filtrate under low temperature (− 56 °C) using a Christ Alpha 1-4 freeze dryer.
Intracellular sampling, quenching and extraction
Five cellular samples were removed from each flask when the cells had reached a D600=6.0, the cell metabolism was quenched and the intracellular metabolites were extracted according to the procedure described by Koning and van Dam [25]. The metabolites were concentrated by freeze-drying at a low temperature (− 56 °C) using a Christ Alpha 1-4 freeze dryer.
Sample derivatization
After dissolving the freeze-dried solids from both intracellular and extracellular samples in 200 μl of sodium hydroxide solution (1%), we performed derivatization analysis as described by Villas-Bôas et al. [21].
GC-MS analysis
We used a Hewlett-Packard system HP 6890 gas chromatograph coupled to a HP 5973 quadrupole mass selective detector (EI) operated at 70 eV. The column used for all analysis was a J&W DB1701 (Folsom, CA, U.S.A.), 30 m×250 μm (internal diameter)×0.15 μm (film thickness). The MS was operated in scan mode (start after 5 min; mass range, 38–550 a.m.u. at 2.88 s/scan).
We modified the analysis parameters from the original protocol described by Villas-Bôas et al. [21] in order to improve the sensitivity of the method. The samples were injected under pulsed splitless mode (39 kPa for 0.45 min, 14 ml·min−1 split flow after 0.45 min). The oven temperature was initially held at 45 °C for 2 min. Thereafter, the temperature was raised with a gradient of 9 °C·min−1 until it reached 180 °C. This temperature, 180 °C, was held for 5 min. Next, the temperature was raised with a gradient of 40 °C·min−1 until it reached 220 °C. The temperature was again held for 5 min. Lastly the temperature was raised with a gradient of 40 °C·min−1 until it reached 240 °C, which was held for 11.5 min. The flow through the column was held constant at 0.8 ml of He·min−1. The injection volume was 2.5 μl. The temperature of the inlet was 120 °C, the interface temperature was 230 °C, and the quadrupole temperature was 150 °C.
Data normalization
As a preface to addressing normalization, direct comparisons of GC-MS metabolite levels and microarray fluorescence transcript levels provide improved context of our data set. In each case, we analyse a data matrix of sample repetitions and genes or metabolites. In this data matrix, Xk (i, j) describes the level of gene transcript or metabolite j in sample repetition i. However, the values in the data matrix elements correspond to disparate signal types: our method counts ions, the microarray scanner measures transcript dye fluorescence. This signal-type disparity leads to two observations. First, our data matrix contains elements identically equal to zero for metabolites below detection limits. By contrast, transcript absence or presence classification requires comparison with the overall sample distribution. Secondly, our data matrix normalization includes only direct metabolite signals and does not reflect the overall sample distribution characteristics. By contrast, normalized transcript levels reflect the location within the overall distribution. In other words, many investigators describe transcript levels in the context of a log normal distribution, whereas our normalized metabolite levels need no mapping to a particular distribution.
We designed the data normalization protocol to minimize sample variability within classes (e.g. aerobic), which we found also maximizes sample variability between classes (e.g. aerobic and anaerobic). Within-class and between-class matrices, often used in a FDA (Fisher discriminant analysis), provided an appropriate framework to directly compare various normalization protocols [26]. For each class and normalization protocol, we constructed a data matrix Xk. In this normalized data matrix, the Xk (i, j) element corresponds to metabolite j of sample repetition i for the class k. A Python script generated the Xk matrix in MATLAB format based upon three inputs: a metabolite library of 60 detected and identified metabolites (columns of Xk); a sample-class key linking samples (rows of Xk) to their respective classes (k); and the AMDIS analysis report, cataloguing relevant data for each identified GC peak (elements of Xk). The Python script and three inputs, in addition to the raw GC-MS data, may be found at http://www.cmb.dtu.dk/additional_material_for_publications/. Within a class, different normalization protocols resulted in different values for data matrix elements.
For the different normalization protocols, each data matrix element corresponded to a primary value specific to a particular metabolite peak, possibly scaled by secondary values specific to that sample. Specific to the particular metabolite peak, we used as the primary value either the raw total peak signal or the metabolite amount, calculated through an AMDIS algorithm weighting several factors, such as purity. Specific to the sample, we used as the possible secondary value a combination of overall ion count, internal standard (EDTA) peak level, and the measured biomass weight. Of course, different normalization protocols produced data matrices of different magnitudes. To enable comparison, we scaled the primary values as percentages of the row (sample) and then used a weighting vector that incorporated the secondary values without changing the overall data matrix magnitude.
For each class and normalization protocol, we constructed a within-class variance matrix Wk for each data matrix Xk using the mean metabolite values xk. By combining classes in an overall data matrix X, we then constructed an overall within-class matrix W and between-class variance matrix B. The magnitudes (norms) ∥W∥ and ∥B∥ provide metrics for the within-class and between-class sample variability, respectively, for a given normalization protocol. Specifically, we constructed the within-class and between-class data matrices as follows:
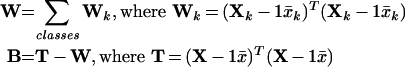 |
(1) |
We compared the within-class and between-class sample variability for each normalization protocol and made two observations. Firstly, we compared the primary values. We observed that the total peak signal provides superior within-class sample variability (56% improvement) and a superior between-class, within-class sample variability ratio (20% improvement) with respect to the AMDIS algorithm for peak amount. Second, we compared combinations of the secondary values specific to each sample. We observed for each case that a combination of overall ion count, internal standard peak level, and measured biomass weight significantly sacrificed within-class sample variability. On account of these two observations, we initially selected the peak total signal as the value for the metabolite levels contained in our data matrix Xk and did not directly incorporate a secondary value.
Although we observed that secondary values, such as overall sample ion counts, introduced undesirable sample-to-sample variability, we nevertheless noted that, on the whole, clear trends emerge in the secondary values from class to class. For instance, the overall sample ion count average for aerobic samples was significantly (P=10−14) higher than the average for anaerobic samples. Among other factors specific to each sample, the overall ion count reflects the freeze-dried metabolite salt re-suspension effectiveness. However, the re-suspended metabolite sample reflects the identical composition, neglecting preferential metabolite losses. Despite significant experimental effort for sample-to-sample consistency, we nevertheless observed large increases of within-class sample variability when we incorporated secondary normalizations, such as overall sample ion count. We concluded that secondary normalizations proved useful in aggregate, but should not be incorporated directly when comparing metabolites among classes.
For this reason, rather than directly incorporating secondary values specific to each sample, we indirectly incorporated only the overall sample ion count secondary normalization averaged over the entire class to estimate absolute metabolite levels. We did not indirectly incorporate EDTA levels or biomass weights, because the mean values were not significantly statistically different between classes, as expected for cultures harvested at similar attenuances. The highly statistical significance of differing overall ion count means between classes justifies this indirect incorporation.
A summary of normalization conclusions is found in the Results section.
Data projection
For our data matrix X, we produce projections through an eigen-value decomposition of W−1B for the above definitions of the within class variance matrix W and between class variance matrix B. Specifically, the eigenvector matrix L and diagonal eigenvalue matrix Λ satisfy the following equality:
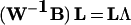 |
We use the best two or three eigenvector projections, as determined by the magnitude of the corresponding eigenvalue, to map each sample to the reduced dimensional space. For projection j and sample data vector x, we determine sample projection value as follows:
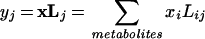 |
(2) |
RESULTS
Experimental design
To simultaneously assay the levels of many metabolites, we both increased the GC-MS sensitivity and created a library of metabolites. First, switching from a split to splitless GC-MS sample injection increased the sensitivity 100-fold and enabled the observation of hundreds of metabolite peaks. Secondly, we enlarged the MCF (methyl chloroformate) library to identify some of these hundreds of peaks.
Any metabolite containing an amino or carboxylic acid group may be potentially derivatized by our MCF method, separated by a GC column, and identified using our MS library. We selected 75 metabolites that play an important role in central carbon metabolism and amino acid biosynthesis (Scheme 1), and which were present in the genome-scale reconstructed metabolic network of yeast [22]. Table 1 lists the metabolites presently contained in our library and all the metabolite abbreviations according to Förster et al. [22] genome-wide metabolic reconstruction.
Scheme 1. Metabolite library coverage.

We generated a MCF derivatization metabolite library, covering much of central carbon metabolism and nearly all of amino acid biosynthesis pathways. We show the metabolic network of amino acid biosynthesis in S. cerevisiae during aerobic growth, highlighting (in grey) metabolites in our library. Continuous arrows indicate a one-step reaction, and broken arrows indicate a series of biochemical reactions where the numbers indicate the reaction steps in the pathway. This illustrative figure does not include all metabolites present in the library.
Table 1. List of metabolites and their respective abbreviations that are identified by GC-MS after MCF derivatization.
Metabolites and their abbreviations are listed according to Förster et al. [22] genome-wide metabolic reconstruction.
| Abbreviation | Amino acids | Abbreviation | Organic acids | Abbreviation | Others |
|---|---|---|---|---|---|
| ALA | Alanine | ADI | Adipate | HMF | 5-Hydroxymethyl-2-furfuraldehyde |
| ASN | Asparagine | AKA | 2-Oxoadipate | NADH | NAD+ |
| ASER | O-Acetyl-L-serine | AKB | 2-Oxobutyrate | NADP | NADP |
| ASP | Aspartate | AKG | 2-Oxoglutarate | THIA | Thiamine |
| CYS | Cysteine | AKV | 2-Oxovalerate | ||
| DABA | 2,4-Diaminobutyrate | BKA | 3-Oxoadipate | ||
| DAMA | D-2-Aminoadipate | CAC | Cis-aconitate | ||
| DAPA | 2,5-Diaminopimelic acid | CIT | Citrate | ||
| GABA | 4-Aminobutyrate | CITC | Citraconate | ||
| GLN | Glutamine | CITM | Citramalate | ||
| GLU | Glutamate | COU | Coumarate | ||
| GLY | Glycine | C140 | Myristate | ||
| HCYS | Homocysteine | FUM | Fumarate | ||
| HIS | Histidine | GLUT | Glutarate | ||
| HPRO | Trans-4-hydroxyproline | GLYC | Glycerate | ||
| HTRP | 5-Hydroxy-tryptophan | GLX | Glyoxalate | ||
| ILE | Isoleucine | IPPMAL | 2-Isopropylmalate | ||
| LAMA | L-2-aminoadipate | ISO | Isocitrate | ||
| LEU | Leucine | ITC | Itaconate | ||
| LLCT | Cystathionine | LAC | Lactate | ||
| MET | Methionine | MAL | Malate | ||
| NAGLU | N-acetyl-L-glutamate | MALT | Malonate | ||
| ORN | Ornithine | NAC | Nicotinate | ||
| PABA | 4-Aminobenzoate | OAA | Oxaloacetate | ||
| PGLU | Pyroglutamate | OIVAL | 3-Methyl-2-oxovalerate | ||
| PHE | Phenylalanine | PEP | Phosphoenolpyruvate | ||
| PRO | Proline | PHT | Phthalate | ||
| SAH | S-Adenosyl-L-homocysteine | PIME | Pimelate | ||
| SAM | S-Adenosyl-L-methionine | PYR | Pyruvate | ||
| SER | Serine | SUC | Succinate | ||
| TALA | β-2-Thienyl-DL-alanine | 2HB | 2-Hydroxybutyrate | ||
| THR | Threonine | 2HIB | 2-Hydroxyisobutyrate | ||
| TRP | Tryptophan | 2PG | 2-Phosphoglycerate | ||
| TYR | Tyrosine | ||||
| VAL | Valine | ||||
| 2AB | 2-Aminobutyrate | ||||
| 2PAAC | D-2-phenylaminoacetate |
Data analysis
Simultaneous GC-MS metabolite profiling produces data with high dimension. Above all, we aimed to understand the salient metabolic features distinguishing the four cell populations (two cultivation conditions for two strains) with a rigorous statistical implementation. Unlike the non-specific metabolite fingerprinting and footprinting techniques [16,17], we specifically aimed to determine differential levels of individual metabolites between known classes.
Within a metabolic network context, differential metabolite levels infer the activation and deactivation of specific metabolic pathways between cell populations by measuring the accumulation of various metabolite pools. In summary, typical metabolic fingerprinting or footprinting may classify a sample as class ‘A’ or ‘B’, whereas differential metabolic levels (finger-printing with metabolite identification) demonstrated that cells in ‘A’ contain, for instance, much higher levels of TCA (tricarboxylic acid) cycle intermediates and infers different genetic and/or allosteric regulation.
To accomplish our goal, we addressed the five questions listed below.
(i) How should we normalize raw data to assign metabolite level values?
From the raw GC-MS data, we must determine levels of individual metabolites. The GC-MS output produces information for each MS scan, and the AMDIS software analyses the scans to produce a flat text file of information for the peaks identified from the library. The normalization protocol assigns a metabolite level given the peak information. Specifically, we must determine the information about the specific peak, the corresponding sample and overall data class that we must include to accurately represent the physiological metabolite level.
We designed the data normalization protocol to minimize sample variability within the classes (e.g. wild-type aerobic). Hypothetically, we would choose the protocol giving a set of metabolites levels as {5, 4, 6} instead of {5, 2, 11}. We found that protocols minimizing within-class variance simultaneously maximized between-class variance as well. A full normalization treatment in the context of gene expression measurements is described in the Experimental section. In summary, we concluded: (i) the total peak signal provided a superior metabolite level value in comparison with the calculated AMDIS amount; (ii) within a class, samples conserve relative composition (percentages) more than secondary values, such as total sample ion counts; and (iii) incorporating the conserved relative composition and the class average overall ion counts (that differ highly significantly between classes) provides an improved estimate of an actual physiological levels in comparison with a sample percentage. For the aforementioned reasons, we construct our data matrix as follows:
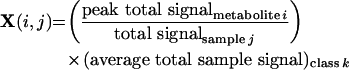 |
(3) |
(ii) What sets of metabolites were detected and identified in each sample?
In each GC-MS sample, we observed hundreds of peaks and scanned for 75 amino and non-amino organic acid metabolites in our library. We identified 38, 33, 45 and 42 peaks intracellularly and 10, 11, 29 and 29 peaks extracellularly for wild-type and mutant aerobic and wild-type and mutant anaerobic respectively. Figure 1 compares the metabolites identified in the various classes.
Figure 1. Observed metabolites.

We detected and identified a slightly different set of metabolites for each data class. Metabolites detected and identified in one class and not another resulted in infinite differential ratios and corresponding statistical confidences. The Förster et al. [22] genome-wide metabolic reconstruction abbreviations are found in Table 1. aer, aerobic; ana, anaerobic; mt, mutant; wt, wild-type.
(iii) Could our data distinguish samples among strains and cultivation conditions?
To attempt to distinguish samples among classes, we projected the metabolite level data from each sample to a lower dimensional space. Two often-used data projection methods are PCA (principal component analysis) and FDA. For each projection (metabolite level linear combination), PCA maximizes variation in the reduced dimensions, whereas FDA maximizes separation between classes [26]. For this reason, we apply FDA to visualize samples in an attempt to distinguish among classes and revealed very clear separation as shown in Figure 2, with further details described in the Experimental section. We concluded that our data successfully distinguish among data classes. Furthermore, we may classify unknown new samples. For instance, we observed that excluding five intracellular samples per class resulted in robust clustering and subsequent successful classification of the excluded samples.
Figure 2. Sample visualization.

GC-MS metabolite data from MCF derivatization successfully distinguishes among strains and cultivation conditions. Projecting intracellular metabolite data from approx. 60 samples into a 3D space reveals distinct clustering of the four data classes. For each sample, we calculated projection values as a linear combination of metabolite values determined by FDA.
(iv) How do flask-to-flask and within-flask variability compare?
In contrast to gene expression shake flask experimentation, we observe that sample-to-sample variability exceeds flask-to-flask variability (Figure 3), and we thus treat samples from different shake flasks equivalently. Foreshadowing our statistical significance analysis, we note that class-to-class (‘good’) variance exceeded sample-to-sample (‘bad’) variance for most metabolites.
Figure 3. Error structure among shake flasks.

A histogram of metabolite residuals, x−x, reveals the error structure between samples and shake flasks for wild-type aerobic intracellular metabolites. The residuals do not display a large flask bias relative to the overall sample variability. Residuals from the other data classes behaved similarly. Thus we adopt the equal-means hypothesis among shake flasks and samples from each shake flask were treated as independent repetitions.
(v) What sets of metabolites differed significantly between cell populations?
Understanding our data set in terms of metabolic pathways required determination of the pathway elements that significantly differ between two cell populations. A simple metabolite average ratio does not reflect that data characteristics. In other words, a 5-fold increase in a metabolite level with respect to a wild-type cell population may not be meaningful, because both levels fall within the observed variability for that metabolite. Furthermore, the ratio uncertainty increased as one level approaches zero. As such, we must provide an error model to assign statistical significance to each ratio.
For gene transcript levels, investigators often employ maximum likelihood parameter estimation that accounts for chip-to-chip and flask-to-flask variability [27]. As we demonstrated in Figure 3, our error displayed a less complicated structure. For this reason, we applied a simple Student's t test between sample repetitions in two classes and calculated the P value, the probability that the two-metabolite signal distributions possess an identical mean. A P value near zero indicated a differential metabolite level. Thus, for each metabolite in each cell population, we calculated a mean ratio and a P value to estimate a statistical confidence in this ratio. This data is shown in Figure 1.
Specific observations
Differential metabolite level data offer many avenues for analysis and interpretation. As an implementation proof-of-concept, exhaustive treatment of this data lies outside the scope of the present work. However, we have provided brief observations about striking characteristics of our data set. These observations offer a promising glimpse of future systematic integration of differential metabolite levels of yeasts with reaction fluxes measurements and/or gene expression. Throughout the observations, we will comment on statistically significant data contained in Figure 1 and on the spreadsheet at http://www.cmb.dtu.dk/additional_material_for_publications/.
Anaerobic versus aerobic cultivations
Figure 1 demonstrates that anaerobic cultivations possess higher levels of both intracellular and secreted metabolites on the whole. In the anaerobic cultivations, over half of the intracellular metabolites were detected extracellularly (excluding glutarate and 2-hydroxybutyrate that were detected only extracellularly). More importantly, increased levels of intracellular metabolites often resulted in increased levels extracellularly, and vice versa. Thus our analysis supports the footprinting approach reported by Allen et al. [16] which considered the extracellular metabolite levels as a rough, first-order approximation for intracellular metabolites.
In addition, despite no homologous sequences for lactate biosynthetic enzymes in yeast, we observed lactate at higher levels anaerobically for both intracellular and extracellular samples. Martins et al. [28] described the methylglyoxal catabolism in wild-type strains of S. cerevisiae that forms D-lactate. The authors observed an intracellular accumulation of D-lactate. Lactate dehydrogenases (DLD1 and CYB2) involved in lactate catabolism in S. cerevisiae are repressed by glucose and induced by lactate. Our results showed that lactate is also secreted to the extracellular medium at significant levels, both under aerobic and anaerobic cultivations.
We also detected some unexpected compounds, e.g. glyoxylate was identified both during aerobic and anaerobic growth at considerably high levels. The glyoxylate cycle is normally found to be inactive during growth on glucose as the sole carbon source due to glucose repression [29]. In our data set, the glyoxylate pathway could be unrepressed when the cell samples were collected (D600 of 6.0, mid- to late-exponential phase) or an alternative pathway for glyoxylate biosynthesis in S. cerevisiae could be activated. This observation underscores the broadness of our experimental technique by generating hypotheses not contained in existing metabolic models.
Similarly, we detected myristic acid at high extracellular levels during anaerobic growth (Figures 1 and 4). In yeast food products, no information exists about this important nutritional metabolite. In clinical trials, myristate has been shown to reduce cardiovascular disease risk [30,31] and lowers the cholesterol-binding plasma low-density lipoprotein C levels, in which myristate plays an important compositional role. Myristate is also present in the flavour components of essential oils [32] and spices [33]. As a saturated fatty acid, myristate is involved in fatty-acid acylation of proteins in higher eukaryotes [34,35]. Proteins with N-terminal myristoyl-glycine residues have been also found in S. cerevisiae, and they are related to the biosynthesis of membrane proteins [34]. Extracellular myristate can be a good indicator of oxygen depletion during S. cerevisiae cultivations. Although we are missing a definitive conclusion, we postulated that higher extracellular myristate levels result from the reduced biomass formation rate, requiring less acylation of proteins for membrane synthesis.
Figure 4. Metabolic array comparing cell populations.

Based upon the metabolite level ratios and statistical significances (P values), we visualized differences among the cell populations. Uppercase metabolite abbreviations indicate intracellular detection, whereas lowercase metabolite abbreviations indicate extracellular detection. The Förster et al. [22] genome-wide metabolic reconstruction abbreviations are found in Table 1.
In addition, we noted that our method successfully detected the highly unstable metabolite adenosyl-L-methionine, an intermediate in sulphur amino acid metabolism (Figures 1 and 4), in anaerobic cultivations. In addition to photo and oxygen sensitivity, this metabolite degrades rapidly at 0 °C. Although the long heating periods of the traditional silylation procedures precluded detection of this molecule, we observed this labile metabolite due to the low temperature (− 40 °C) sample preparation and gentle MCF derivatization conditions.
Wild-type versus mutant
We compare the metabolite profiles of the mutant and wild-type separately aerobically and anaerobically. The mutant was a redox-engineered strain with a deleted NADPH-dependent glutamate dehydrogenase (encoded by GDH1) and overexpressed NADH-dependent glutamate dehydrogenase (encoded by GDH2). The NADPH-dependent glutamate dehydrogenase has been identified as the major enzyme in the cell responsible for nitrogen assimilation during growth with ammonium as the sole nitrogen source [36], and accounts for a considerable fraction of the NADPH consumption associated with biomass formation [37]. On the other hand, the overexpressed NADH-dependent glutamate dehydrogenase is an alternative pathway for ammonium assimilation, although it normally serves a catabolic function in wild-type strains.
We observed two distinct and significant (P value<0.01) patterns in the metabolite profiles of this mutant compared with its parental strain: (i) aerobically, many metabolites were present at higher levels in the mutant (Figure 1 and Scheme 2), and (ii) anaerobically, many metabolites were present at lower levels in the mutant (Figure 1).
Scheme 2. Aerobic mutant growth.

We detected the black-highlighted metabolites at higher levels in the mutant (GDH1 deleted, GDH2 over-expressed). In contrast, the grey metabolites were detected at lower levels and significant P values. Continuous arrows indicate one-step reaction, and broken arrows indicate multiple biochemical reactions. The numbers indicate the reaction steps in the pathway.
During aerobic growth, the levels of all TCA cycle intermediates increased in the mutant compared with the wild-type (Figure 1 and Scheme 2), which could be correlated with the increased TCA cycle flux for this mutant [38]. The alterations in the ammonium metabolism certainly was reflected in a requirement of an increased level of 2-oxoglutarate due to the thermodynamically less favourable glutamate synthesis when NADH is used as cofactor [38]. Excluding proline, all amino acids that have a transamination reaction involving conversion of glutamate into 2-oxoglutarate were detected at higher levels in the mutant compared with the reference strain (Scheme 2). Considering the whole reaction catalysed by glutamate NADH-dependent dehydrogenase:
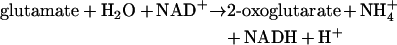 |
we expect that not only the level of 2-oxoglutarate, but also the level of ammonium ions, NADH and H+ should also increase in order to favour the reversible GDH2 reaction. Therefore the higher levels of amino acids resulted from the transamination of glutamate to 2-oxoglutarate could be explained by the requirement of higher level of ammonium, since NADH was regenerated during a higher glycolytic flux, as determined previously [38].
Oxaloacetate was detected only in the mutant samples (intracellular aerobically and extracellular anaerobically) (Figure 4). We also detected several amino acids at lower levels in the mutant samples during aerobic growth (Scheme 2). Dos Santos et al. [38] described a decrease in the PPP (pentose phosphate pathway) flux of 35–52% in the mutant compared with the wild-type during oxidative growth on glucose. This supports our observation of the lower histidine and other aromatic amino acids that are derived from pentose phosphate intermediates. Thus a decrease in the PPP flux seems to decrease the levels of these compounds. Decreasing the PPP flux, we expected that there would be an increase in the glycolytic flux, which could result in higher levels of pyruvate, lactate and oxaloacetate in comparison with the wild-type. The unstable triose phosphates formed during the glycolysis could undergo a β-elimination reaction of the phosphoryl group from the common 1,2-enediolate. This reaction leads to methylglyoxal formation [28], a possible precursor for lactate biosynthesis in yeast as discussed previously.
For the anaerobic cultivations, the metabolic profile changed dramatically with respect to the reference strain (Figure 1). This contradicts transcription results obtained with a similar mutant (Δgdh1). Under anaerobic growth, the mutant and the reference strain did not present significant transcriptional differences [39]. Certainly, in the present study, the cells appear unable to overproduce 2-oxoglutarate and ammonium to overcome the thermodynamic barriers required for glutamate biosynthesis via GDH2, because the 2-oxoglutarate and glutamate levels were much lower. Methionine was detected only in the mutant samples from the anaerobic cultivation, whereas in the wild-type samples it was detected in both cultivation conditions (aerobic and anaerobic) (Figure 4). Similar to this was the detection of extracellular oxaloacetate only in the samples from the mutant cultivated anaerobically.
DISCUSSION
We believe that our analysis method provides the first microbial metabolic state assay that achieves comparable throughput, effort and cost compared with gene expression analysis. As such, the method offers an accessible experimental and software platform for many laboratories. Our results demonstrate that the method has two important advantages: broadness and high sensitivity. The accessibility of the method highlights further potential for a wider application. With our current MS spectra metabolite library, we only identified 40% of the detected peaks. By placing the MS libraries in the public domain, we hope the library will grow and evolve with time and enable identification of the hundreds of peaks we were not able to identify presently. Larger libraries will not only benefit future metabolic state assays, but also enable the re-analysis of our existing data in the public domain and set our method to the present level of successful metabolomics methods in plant sciences [5,18,40]. Hence, the method contributes to enabling targeted and quantitative microbial metabolome analysis.
Even though our method only enables analysis of approx. 100 metabolites, this may still be sufficient for functional analysis of a large number of mutants due to several factors. (i) The method quantifies many of the precursor metabolites for cellular building blocks, and thereby even more diversified metabolism is closely linked to the part of the metabolism that can be analysed with the method. (ii) High metabolic network connectivity links many genes in short routes to central carbon metabolism and amino acid. Many metabolites participate in 10 or more reactions [41]. Due to this high degree of connectivity, a perturbation in one part of the metabolism will migrate into other parts of the metabolism, and therefore genetic perturbations often result in modifications of the central carbon metabolism, i.e. the part of the metabolic network that can be analysed with the method presented here. (iii) The method can provide quantitative information about the levels of metabolites. From the concept of metabolic control analysis, we learned that modulation of enzyme activities normally results in large perturbations in metabolite levels, but in relatively low changes in metabolic fluxes [39]. Hence, quantitative analysis of metabolites will enable quantitative linking of different parts of the metabolism.
Understanding metabolic features distinguishing cell populations requires high-throughput, i.e. analysis of a large number of metabolites in one analysis, and consistent application of an appropriate analysis technique. For this reason, we utilized robotic sample injection and developed software to integrate the above analysis techniques. The robotic sample injection provided a consistent path from sample to GC-MS output. The software provides a consistent path from GC-MS output to the metabolic features distinguishing cell populations. With negligible computational time, the software reads GC-MS output, generates data matrices and outputs spreadsheets of differential metabolite levels. The software and full data sets are provided at http://www.cmb.dtu.dk/additional_material_for_publications/.
We believe that the true potential of this method lies in the fact that the structural information provided enables integration of the data with data on the transcriptome and the proteome comparable with the methodology applied in plant metabolomics [5]. However, even as a stand-alone method we achieved results not possible with either gene expression or metabolic fingerprinting analyses alone. First, we readily classify distinguishing characteristics of the mutant strain, which was not possible using genome-wide expression analysis [39]. Second, we demonstrated that specific metabolites could be measured, something not possible with current metabolic fingerprinting tools. We hope that in the future this method complements these analyses as a powerful tool in integrated cellular studies of micro-organisms.
Acknowledgments
We thank Ms Marie-Laure Tavernier for helping in the construction of the MS library of MCF derivatives, Dr Kristian Fog Nielsen for profitable suggestions on GC-MS method, Dr Jatin Misra for fruitful discussions on data analysis and Mrs Kianoush K. Hansen for technical support. This work has been supported by the Danish Biotechnological Instrument Center (DABIC).
References
- 1.Oliver S. G. Yeast as a navigational aid in genome analysis. Microbiology. 1997;7:405–409. doi: 10.1099/00221287-143-5-1483. [DOI] [PubMed] [Google Scholar]
- 2.Oliver S. G., Winson M. K., Kell D. B., Baganz F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998;16:373–378. doi: 10.1016/s0167-7799(98)01214-1. [DOI] [PubMed] [Google Scholar]
- 3.Lee T. I., Rinaldi N. J., Robert F., Odom D. T., Bar-Joseph Z., Gerber G. K., Hannett N. M., Harbison C. T., Thompson C. M., Simon I., et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science (Washington, D.C.) 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 4.Bar-Joseph Z., Gerber G. K., Lee T. I., Rinaldi N. J., Yoo J. Y., Robert F., Gordon D. B., Fraenkel E., Jaakkola T. S., Young R. A., Gifford D. K. Computational discovery of gene modules and regulatory networks. Nat. Biotechnol. 2003;11:1337–1342. doi: 10.1038/nbt890. [DOI] [PubMed] [Google Scholar]
- 5.Weckwerth W., Wenzel K., Fiehn O. Process for the integrated extraction, identification and quantification of metabolites, proteins and RNA to reveal their co-regulation in biochemical networks. Proteomics. 2004;4:78–83. doi: 10.1002/pmic.200200500. [DOI] [PubMed] [Google Scholar]
- 6.von Mering C., Krause R., Snel B., Cornell M., Oliver S. G., Fields S., Borket P. Comparative assessment of large-scale data sets of protein–protein interactions. Nature (London) 2002;417:399–403. doi: 10.1038/nature750. [DOI] [PubMed] [Google Scholar]
- 7.Ideker T., Thorsson V., Ranish J. A., Christmas R., Buhler J., Eng J. K., Bumgarner R., Goodlett D. R., Aebersold R., Hood L. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science (Washington, D.C.) 2001;292:929–934. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- 8.Ideker T., Ozier O., Schwikowski B., Siegel A. F. Discovering regulatory and signaling circuits in molecular interaction networks. Bioinformatics. 2002;18(suppl. 1):233–240. doi: 10.1093/bioinformatics/18.suppl_1.s233. [DOI] [PubMed] [Google Scholar]
- 9.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ideker T., Lauffenburger D. Building with a scaffold: emerging strategies for high to low-level cellular modeling. Trends Biotechnol. 2003;21:255–262. doi: 10.1016/S0167-7799(03)00115-X. [DOI] [PubMed] [Google Scholar]
- 11.Gygi S. P., Rochon Y., Franza B. R., Aebersold R. Gene expression: correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwab W. Metabolome diversity: too few genes, too many metabolites? Phytochemistry. 2003;62:837–849. doi: 10.1016/s0031-9422(02)00723-9. [DOI] [PubMed] [Google Scholar]
- 13.Famili I., Forster J., Nielsen J., Palsson O. Saccharomyces cerevisiae phenotypes can be predicted by using constraint-based analysis of a genome-scale reconstructed metabolic network. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13134–13139. doi: 10.1073/pnas.2235812100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Segre D., Vitkup D., Church G. M. Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. U.S.A. 2002;99:15112–15117. doi: 10.1073/pnas.232349399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Almaas E., Kovacs B., Vicsek T., Oltvai Z. N., Barabasi A.-L. Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature (London) 2004;427:839–843. doi: 10.1038/nature02289. [DOI] [PubMed] [Google Scholar]
- 16.Allen J., Davey H. M., Broadhurst D., Heald J. K., Rowland J. J., Oliver S. G., Kell D. B. High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nat. Biotechnol. 2003;21:692–696. doi: 10.1038/nbt823. [DOI] [PubMed] [Google Scholar]
- 17.Castrillo J. I., Hayes A., Mohammed S., Gaskell S. J., Oliver S. G. An optimized protocol for metabolome analysis in yeast using direct infusion electrospray mass spectrometry. Phytochemistry. 2003;62:929–937. doi: 10.1016/s0031-9422(02)00713-6. [DOI] [PubMed] [Google Scholar]
- 18.Roessner U., Wagner C., Kopka J., Trethewey R. N., Willmitzer L. Simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. Plant J. 2000;23:131–142. doi: 10.1046/j.1365-313x.2000.00774.x. [DOI] [PubMed] [Google Scholar]
- 19.Halket J. M., Przyborowska A., Stein S. E., Mallard W. G., Down S., Chalmers R. A. Deconvolution gas chromatography/mass spectrometry of urinary organic acids – potential pattern recognition and automated identification of metabolic disorders. Rapid Commun. Mass Spectrom. 1999;13:279–284. doi: 10.1002/(SICI)1097-0231(19990228)13:4<279::AID-RCM478>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 20.Stein S. E. An integrated method for spectrum extraction and compound identification from gas chromatography/mass spectrometry. J. Am. Soc. Mass Spectrom. 1999;10:770–781. [Google Scholar]
- 21.Villas-Bôas S. G., Delicado D. G., Åkesson M., Nielsen J. Simultaneous analysis of amino and nonamino organic acids as methyl chloroformate derivatives using gas chromatography-mass spectrometry. Anal. Biochem. 2003;322:134–138. doi: 10.1016/j.ab.2003.07.018. [DOI] [PubMed] [Google Scholar]
- 22.Förster J., Famili I., Fu P., Palsson B. Ø., Nielsen J. Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res. 2003;13:244–253. doi: 10.1101/gr.234503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verduyn C., Postma E., Scheffers W. A., van Dijken J. P. Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on regulation of respiration and alcoholic fermentation. Yeast. 1992;8:501–517. doi: 10.1002/yea.320080703. [DOI] [PubMed] [Google Scholar]
- 24.Verduyn C., Postma E., Scheffers W. A., van Dijken J. P. Energetics of Saccharomyces cerevisiae in anaerobic glucose-limited chemostat cultures. J. Gen. Microbiol. 1990;136:405–412. doi: 10.1099/00221287-136-3-405. [DOI] [PubMed] [Google Scholar]
- 25.Koning W., van Dam K. A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal. Biochem. 1992;204:118–123. doi: 10.1016/0003-2697(92)90149-2. [DOI] [PubMed] [Google Scholar]
- 26.Stephanopoulos G., Hwang D., Schmitt W. A., Misra J., Stephanopoulos G. Mapping physiological states from microarray expression measurements. Bioinformatics. 2002;18:1054–1063. doi: 10.1093/bioinformatics/18.8.1054. [DOI] [PubMed] [Google Scholar]
- 27.Ideker T., Thorsson V., Siegel A. F., and Hood L. E. Testing for differentially-expressed genes by maximum-likelihood analysis of microarray data. J. Comput. Biol. 2000;7:805–817. doi: 10.1089/10665270050514945. [DOI] [PubMed] [Google Scholar]
- 28.Martins A. M., Cordeiro C. A., Ponces Freire A. M. In situ analysis of methylglyoxal metabolism in Saccharomyces cerevisiae. FEBS Lett. 2001;499:41–44. doi: 10.1016/s0014-5793(01)02519-4. [DOI] [PubMed] [Google Scholar]
- 29.Fernandez E., Fernandez M., Moreno F., Rodicio R. Transcriptional regulation of the isocitrate lyase encoding gene in Saccharomyces cerevisiae. FEBS Lett. 1993;333:238–242. doi: 10.1016/0014-5793(93)80661-d. [DOI] [PubMed] [Google Scholar]
- 30.Khosla P., Sundram K. Effects of dietary fatty acid composition on plasma cholesterol. Prog. Lipid Res. 1996;35:93–132. doi: 10.1016/0163-7827(95)00014-3. [DOI] [PubMed] [Google Scholar]
- 31.Loison C., Mendy F., Serougne C., Lutton C. Dietary myristic acid modifies the HDL-cholesterol concentration and liver scavenger receptor BI expression in the hamsters. Br. J. Nutr. 2002;87:199–210. doi: 10.1079/BJNBJN2002521. [DOI] [PubMed] [Google Scholar]
- 32.Kajiwara T., Hatanaka A., Kawai T., Ishihara M., Tsuneya T. Study of flavor compounds of essential oil extracts from edible Japanese kelps. J. Food Sci. 1988;53:960–962. [Google Scholar]
- 33.Kostrzewa E., Karwowska K. The evaluation of aromatic and flavor properties of pimento extracts. Prace Instytutow i Laboratoriow Badawczych Przemyslu Spozywczego. 1975;25:67–74. [Google Scholar]
- 34.Towler D., Glaser L. Protein fatty acid acylation: enzymatic synthesis of an N-myristoylglycyl peptide. Proc. Natl. Acad. Sci. U.S.A. 1986;83:2812–2816. doi: 10.1073/pnas.83.9.2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Towler D. A., Adams S. P., Eubanks S. R., Towery D. S., Jackson-Machelski E., Glaser L., Gordon J. I. Purification and characterization of yeast myristoyl CoA:protein N-myristoyltransferase. Proc. Natl. Acad. Sci. U.S.A. 1987;84:2708–2712. doi: 10.1073/pnas.84.9.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dickinson J. R. Nitrogen metabolism. In: Dickinson J. R., Schweizer M., editors. The Metabolism and Molecular Physiology of Saccharomyces cerevisiae. London: Taylor and Francis; 1998. pp. 57–77. [Google Scholar]
- 37.Bruinenberg P. M., van Dijken J. P., Scheffers W. A. A theoretical analysis of NADPH production and consumption in yeasts. J. Gen. Microbiol. 1983;129:953–964. doi: 10.1099/00221287-129-4-965. [DOI] [PubMed] [Google Scholar]
- 38.Dos Santos M. M., Thygesen G., Kötter P., Olsson L., Nielsen J. Aerobic physiology of redox-engineered Saccharomyces cerevisiae strains modified in the ammonium assimilation for increased NADPH availability. FEMS Yeast Res. 2003;4:59–68. doi: 10.1016/S1567-1356(03)00155-7. [DOI] [PubMed] [Google Scholar]
- 39.Bro C., Regenberg B., Nielsen J. Genome-wide transcriptional response of a Saccharomyces cerevisiae strain with an altered redox metabolism. Biotechnol. Bioeng. 2004;85:269–276. doi: 10.1002/bit.10899. [DOI] [PubMed] [Google Scholar]
- 40.Weckwerth W., Loureiro M. E., Wenzel K., Fiehn O. Differential metabolic networks unravel the effects of silent plant phenotypes. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7809–7814. doi: 10.1073/pnas.0303415101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stephanopoulos G. Metabolic engineering. Biotechnol. Bioeng. 1998;58:119–120. [PubMed] [Google Scholar]