

Abstract
Antiphospholipid syndrome (APS) is a systemic autoimmune disorder characterized by increased risk for arterial, venous and/or microvascular thrombosis and various obstetrical complications, including recurrent miscarriages, premature births, and preeclampsia, in association with the persistent presence of phospholipid antibodies (aPLs). The clinical spectrum of APS ranges from mild clinical manifestations to the development of a catastrophic event involving multiorgan failure and high mortality due to disseminated thrombosis. APS diagnosis requires the detection of serum autoantibodies targeting cardiolipins and/or the plasma protein β-2-glycoprotein I (β2GPI). However, the full spectrum of specific autoantigens primarily recognized by aPLs remain unknown, which has hampered therapeutic development. On page XXX of this issue, Muller-Calleja et al. (1) report the identification of a cell surface antigenic complex composed of endosomal lysobiphosphatidic acid (LBPA) presented by the endothelial protein C receptor (EPCR), which is specifically recognized by aPLs and promotes immune dysregulation and thrombosis in mice.
The pathogenic mechanisms leading to the generation of aPLs and to the various vascular and organ complications characteristic of APS are incompletely characterized and pleiotropic, involving events such as endothelial, platelet and myeloid cell activation, as well as dysregulation and crosstalk of the complement and coagulation cascades (2). aPLs characteristically display reactivity toward various anionic phospholipids (cardiolipins) and plasma proteins. β2GPI has high avidity for phospholipid surfaces and is considered a major target of aPLs (2).
Less is understood about the antigen specificity and pathogenicity of aPLs that primarily target phospholipids. In addition to activating the extrinsic pathway of coagulation (3), phospholipid reactive aPLs induce translocation of endosomal Toll-like receptor 7 (TLR7) and TLR8 from the endoplasmic reticulum to the endosomes in myeloid cells, thereby sensitizing them to proinflammatory signaling and interferon (IFN) responses driven by danger signals (4). APS can occur in isolation (primary) or in association with other systemic autoimmune diseases such as systemic lupus erythematosus (SLE). Both SLE and primary APS have been associated with dysregulation of the type I IFN pathway, which has several putative pathogenic roles (5, 6).
EPCR is expressed by endothelial and myeloid cells, placental trophoblasts and various differentiated and progenitor cells that may be targeted by aPLs. It is a CD1d-like transmembrane glycoprotein with a tightly bound phospholipid in its antigen presenting groove. Phospholipid-bound EPCR promotes anticoagulant protein C activation, with downstream antithrombotic effects (7). However, EPCR can bind other ligands besides protein C and this may determine pro- or anti-inflammatory effects (8). Furthermore, EPCR activation of proteinase-activated receptor 2 (PAR-2) triggers TLR4-mediated type I IFN responses in mouse myeloid cells, independent of coagulation (9). Therefore, in context-dependent manners, EPCR may be pro- or antithrombotic and trigger innate immune responses that are relevant to autoimmunity.
Previous reports identified in vitro reactivity of aPLs against LBPA, a phospholipid that is mostly expressed in late endosomes (10). Building on these observations, Muller-Calleja et al. report that phospholipid-reactive human monoclonal aPLs induce inflammatory gene responses in myeloid, endothelial, and embryonic cells in an EPCR-dependent manner, and that EPCR can serve as a receptor for aPL endosomal trafficking. The ability of EPCR to bind aPLs requires LBPA in the antigen-presenting groove.
To assess whether this pathway was pathogenic in vivo, Muller-Calleja et al. used mouse models of aPL-induced fetal loss and thrombosis and found that EPCR-deficient mice were protected from these complications. Additionally, in a mouse model of SLE, aPLs were also prothrombotic and induced IFN responses in myeloid cells in an EPCR-LBPA-dependent manner. Furthermore, immunization of non-autoimmune-prone mice with aPLs induced the appearance of cardiolipin-reactive antibodies and expansion of EPCR-LBPA reactive B1 cells, a subset of B cells that has been linked to innate immune responses and autoimmunity. Supporting that engagement of the EPCR-LBPA complex promotes inflammation, administration of an antibody blocking the EPCR-LBPA pathway to a mouse model of SLE inhibited the synthesis of aPLs and other SLE-associated autoantibodies and mitigated renal inflammation and damage.
Although Muller-Calleja et al. used only one SLE mouse model, these results suggest that the EPCR-LBPA pathway may play a broader role in immune dysregulation not only driven by aPLs but, potentially, to other pathways associated with SLE-like immune dysregulation, including the type I IFN pathway. It remains unclear how EPCR-LBPA leads to the thrombotic phenotype observed in mice and whether the type I IFN pathway is also involved in this complication, given previous studies linking IFN-mediated transcriptional modulation to platelet activation (11). It also remains to be determined what makes EPCR preferentially bind LBPA over other phospholipids, to allow preferential attachment to aPLs.
The study of Muller-Calleja et al. expands the repertoire of antigens potentially involved in the pathogenesis of APS. Validating these findings in large clinical cohorts will be important to assess if testing EPCR-LBPA autoantibody specificity will improve the diagnosis of primary and SLE-associated APS, and its association with various organ complications and clinical manifestations. In addition, identifying genetic drivers of abnormalities in the pathways identified in the study that are associated with APS or other autoimmune conditions, may allow for identification of subgroups of patients where this pathway could be particularly relevant.
Notably, aPLs can be generated during various acute infections, including that of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (12), as well as in cancers and with the use of certain medications. In contrast to APS, the presence of aPLs in these conditions is not necessarily linked to enhanced risk for clotting, although they may have proinflammatory effects. It remains to be confirmed whether phospholipid-reactive aPLs generated during infections signal through the EPCR-LBPA pathway. Even if this pathway is confirmed to be important in human APS, targeting EPCR may prove a double-edge sword, given its well described antithrombotic and anti-inflammatory effects. In this case, it will be important to determine if inhibiting downstream pathways , such as endosomal TLRs and/or type I IFN signaling, could result in abrogation of prothrombotic or inflammatory cascades that are triggered by this complex.
Figure . aPLs bind to EPCR/LBPA and promote thrombosis and tissue damage.
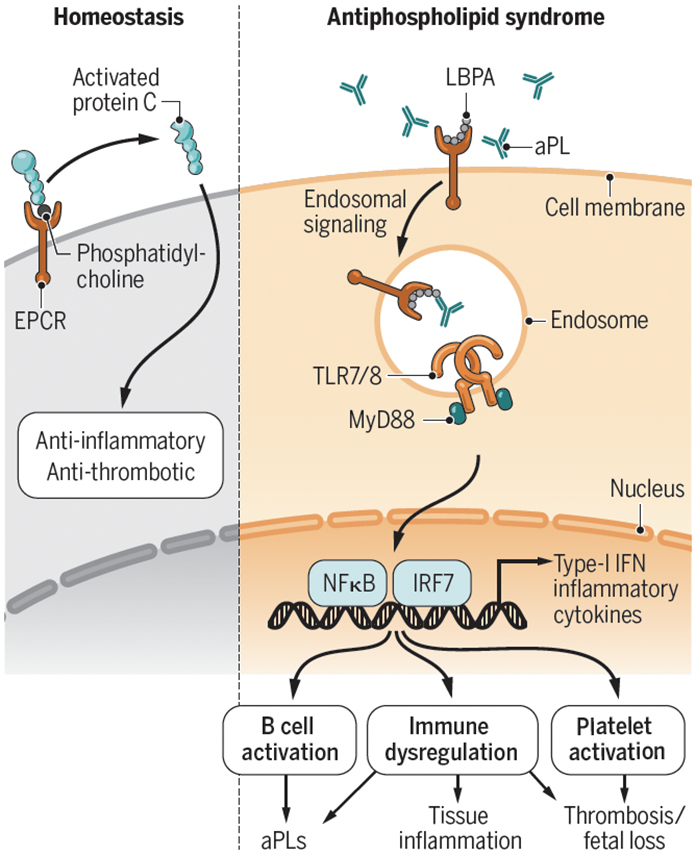
Under conditions of homeostasis, EPCR bound to phospholipids like phosphatidylcholine, activates protein C (PC) with downstream anti-thrombotic and anti-inflammatory effects. In contrast, lipid-reactive aPLs interact with EPCR bound to LBPA. This interaction promotes prothrombotic and proinflammatory responses, potentiation and activation of the endosomal TLR/type-I IFN pathway, further synthesis of autoantibodies and tissue inflammation, with effects on thrombosis and fetal loss.
aPL, phospholipid antibody; EPCR, endothelial protein C receptor; IFN, interferon; IRF7, interferon regulatory factor 7; LBPA, lysobiphosphatidic acid; MyD88, myeloid differentiation primary response protein; NFkB, nuclear factor-kb; TLR, Toll-like receptor.
ACKNOWLEDGMENTS
I am supported by NIAMS (ZIAAR041199).
References and notes
- 1.Müller-Calleja N et al. , Science 371, XXX (2021). [Google Scholar]
- 2..Garcia D, Erkan D, N. Engl. J. Med 378, 2010 (2018). [DOI] [PubMed] [Google Scholar]
- 3..Muller-Calleja N et al. , Blood 134, 1119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prinz N et al. , Blood 118, 2322 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Gupta S, Kaplan MJ, J. Clin. Invest 131, e144918 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6..Grenn RC et al. , Ann. Rheum. Dis 76, 450 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7..Dahlback B, Villoutreix BO, Arterioscler. Thromb. Vasc. Biol 25, 1311 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Kondreddy V et al. , Blood 131, 2379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9..Liang HP et al. , Blood 125, 2845 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10..Galve-de Rochemonteix B et al. , Arterioscler. Thromb. Vasc. Biol 20, 563 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Lood C et al. , Blood 116, 1951 (2010). [DOI] [PubMed] [Google Scholar]
- 12..Martirosyan A, Aminov R, Manukyan G, Front. Immunol 10, 1609 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]