

Abstract
Background:
Although most individuals effectively control herpesvirus infections, some suffer from severe and/or recurrent infections. A subset of these patients possess defects in NK cells, lymphocytes which recognize and lyse herpesvirus-infected cells; however, the genetic etiology is rarely diagnosed. PLCG2 encodes a signaling protein in NK cell and B cell signaling. Dominant-negative or gain-of-function variants in PLCG2 cause cold urticaria, antibody deficiency, and autoinflammation. However, loss-of-function variants and haploinsufficiency have not been reported to date.
Objective:
We aimed to identify the genetic cause of NK cell immunodeficiency in two families, and herein describe the functional consequences of two novel loss-of-function variants in PLCG2.
Methods:
We employed whole exome sequencing in conjunction with mass cytometry, microscopy, functional assays, and a mouse model of PLCG2 haploinsufficiency to investigate two families with NK cell immunodeficiency.
Results:
We identified novel heterozygous variants in PLCG2 in two families with severe and/or recurrent herpesvirus infections. In vitro studies demonstrated that these variants were loss-of-function due to haploinsufficiency with impaired NK calcium flux and cytotoxicity. In contrast to previous PLCG2 variants, B cell function remained intact. Plcg2+/− mice also displayed impaired NK cell function with preserved B cell function, phenocopying human disease.
Conclusions:
PLCG2 haploinsufficiency represents a distinct syndrome from previous variants characterized by NK cell immunodeficiency with herpes virus susceptibility, expanding the spectrum of PLCG2-related disease.
Clinical Implication:
We identified PLCG2 heterozygous variants in 2 families with severe and/or recurrent herpesvirus infections. This report demonstrates the impact of PLCG2 haploinsufficiency.
Keywords: NK cell, PLCG2, haploinsufficiency, immunodeficiency, herpes virus
Capsule Summary
Dominant-negative and gain-of-function variants in PLCG2 have been previously reported to cause cold urticaria, antibody deficiency, and autoinflammation. PLCG2 haploinsufficiency is a distinct and novel immunodeficiency leading to NK cell dysfunction and is associated with severe and/or recurrent herpesvirus infections.
Introduction
Nearly all individuals will encounter herpesviruses such as herpes simplex virus 1 (HSV1) or cytomegalovirus (CMV) in their lifetime (1). Although most will present with only limited recurrences, some patients will continue to have unusually severe and/or recurrent herpesvirus infections (2). A subset of these patients possess defects in natural killer (NK) cells, innate lymphocytes which recognize and lyse herpesvirus-infected cells (3–5). NK cell deficiency (NKD) can result from either aberrant NK cell development (classical NKD) or reduced NK cell function (functional NKD), typically evaluated by measuring NK cell killing and CD107 degranulation after incubation with K562 target cells (3). Despite these diagnostic tools, there is minimal understanding of the genetics underlying functional NKD with only one previously reported genetic cause (FCGR3A) (6, 7), and most patients do not receive a definitive diagnosis. Herein, we present a novel syndrome of functional NK cell immunodeficiency caused by heterozygous loss-of-function variants in PLCG2.
NK cells recognize herpesvirus-infected cells using an array of germline-encoded activating receptors, such as CD16, NKG2D and 2B4 (8). Many activating receptors signal via a pathway involving Src and Syk kinases, LAT, and PLCG2-induced secondary messengers (8). PLCG2 (encoding phospholipase C-γ2) is recruited to LAT, phosphorylated, and subsequently cleaves PIP2 into IP3 and DAG. This critical process initiates an increase in cytosolic calcium via both the release of intracellular calcium stores as well as extracellular influx, leading to the polarized mobilization of cytotoxic granules towards the target cell (9). PLCG2 is expressed in hematopoietic cells and is related to the ubiquitously-expressed PLCG1. While PLCG1 is sufficient for many signaling pathways including mitogen and TCR signaling, PLCG2 is uniquely necessary for NK cell and B cell signaling, as demonstrated by their profound disruption in Plcg2−/− mice (10–12). Although PLCG2 is traditionally thought of as a NK- and B- cell signaling molecule, it also plays a role in several other cell types including platelets (13), neutrophils (14), monocytes, and macrophages (15).
Variants in PLCG2 have been previously reported in a separate domain (the C-terminal SH2 domain, cSH2), causing the syndromes PLAID and APLAID. PLAID is caused by exonic deletions compromising the cSH2 domain and results in gain-of-function at sub-physiologic temperatures (16–18). At normal temperatures, PLAID acts in a dominant-negative manner to dysregulate immune signaling. While NK cell degranulation is reduced in PLAID, B cell and mast cell dysregulation underlie the predominant clinical phenotypes in these patients, including antibody deficiency and cold urticaria (16–18). APLAID is caused by a constitutive gain-of-function variant (S707Y) and results in autoinflammation and B cell immunodeficiency (perhaps as a result of substrate depletion) (19). Herein, we demonstrate that heterozygous loss-of-function variants in PLCG2 result in a clinical phenotype distinct from PLAID and APLAID, extending the spectrum of disease seen with human variants in PLCG2.
Methods
Data Availability Statement
With the exception of whole-exome sequencing data, the distribution of which may compromise research participant privacy and/or consent, the data that support the findings of this study are available from the corresponding author upon reasonable request.
Patients and Sample Collection
All human samples were obtained using written informed donor consent and were used with the approval of either the Washington University School of Medicine Institutional Review Board or the Baylor College of Medicine Institutional Review Board for the Protection of Human Subjects. All samples were obtained in compliance with the Declaration of Helsinki. Peripheral blood mononuclear cells (PBMC) were isolated by density centrifugation over Ficoll-Paque according to manufacturer’s instructions prior to liquid nitrogen cryopreservation in fetal bovine serum and DMSO.
Animal Studies
Plcg2 mice were generated by Jim Ihle (St. Jude Children’s Research Hospital) (10) and backcrossed onto a C57BL/6 background for 10 generations by Marco Colonna (Washington University) (11). Mice were maintained under specific pathogen–free conditions and used between 8 and 14 weeks of age. Mouse experiments were performed in both male and female mice with equivalent results. All experiments were approved and conducted in accordance with Washington University Animal Care and Use Committee guidelines for animal care and use.
Exome Sequencing
For kindred A, exome sequencing was performed as previously described (48). Briefly, genomic DNA was extracted from whole blood or saliva and coding regions enriched using SureSelect All Exon (Agilent Technologies) followed by next-generation sequencing (Illumina) at St. Louis Children’s Hospital through the Genome Technology Access Center at Washington University. For kindred B, whole exome sequencing was performed on genomic DNA by Baylor Genetics as previously described (49, 50). After quality control, alignment and variant calling, exome data was analyzed using institution-specific pipelines to identify potential variants. Medium or high impact variants (i.e., non-synonymous changes, early stops, frameshifts and splice site variants) with minor allele frequencies less than 0.01 in ExAC (29) were prioritized and potential variants were cross-referenced between kindreds. Both kindreds were negative for variants in known genes associated with immunodeficiency and immunodysregulation (51), including HLH variants (52) and NK cell associated variants in MCM4, GATA2, IRF8, FCGR3A (CD16) and KLRC2 (NKG2C) (3, 53–56).
Transfection Experiments
293T cells (ATCC CRL-3216) were cultured in Dulbecco’s modified eagle media supplemented with 10% fetal bovine serum. Cells were transfected with a mammalian expression vector driven under MND promoter containing N-terminal FLAG tagged PLCG2 (WT or relevant variant) using Lipofectamine LTX (Invitrogen) and incubated for 24 hours. Cells were then stimulated with 100μM pervanadate and either lysed with RIPA buffer or fixed using 1.6% formaldehyde 15 minutes post-stimulation. Western blot analysis of protein expression was performed using anti-FLAG (Sigma, clone M2) or Actin (Santa Cruz Biotechnology, clone C-11). PLCG2 phosphorylation was analyzed by flow cytometry as described below using PE anti-PLCG2 Y759 (clone K86-689.37, BD).
NK Cell Cytotoxicity Assays
PBMCs isolated as described above were thawed and allowed to rest for 1 hour in Roswell Park Memorial Institute (RPMI) media supplemented with 20% FBS, pyruvate, non-essential amino acids, glutamine and HEPES. Cells were then seeded into 96 well U-bottom plates along with CellTrace Violet (Invitrogen) labeled K562 (ATCC, CCL-243) tumor target cells at a PBMC: K562 ratio of 50:1. After incubation for 4 hours, 7-AAD (BD) was added, and target cell death was quantified using flow cytometry. Mouse NK cell assays were performed similarly except that NK cells were isolated from spleen using EasySep Mouse NK Cell Enrichment (Stem Cell Technologies) and then mixed with CellTrace Violet labeled YAC-1 (ATCC, TIB-160) or RMA-S cells (kind gift from Wayne Yokoyama).
Microscopy
For analysis of NK-target cell conjugates, fixed cell confocal microscopy was performed using patient NK cells and K562 erythroblast target cells. 3 X 106 PBMC from patient or heathy donor were incubated with K562 target cells for 45 minutes then fixed, permeabilized and stained with anti-perforin Alexa Fluor 488 (clone dg9), anti-tubulin biotin followed by Pacific Blue conjugated streptavidin, and phalloidin Alexa Fluor 568. Images were acquired on a Zeiss AxioPlanII with a Yokogawa CSU-10spinning disk and Hamamatsu ORCA-ER camera. Excitation lasers (405 nm, 488 nm, 561 nm, 647 nm) were merged through a Spectral Applied Research laser merge. Images were taken throughout the volume of cell conjugates using 0.5μm steps. Acquisition and analysis were performed using Volocity software (PerkinElmer). For measurement of actin accumulation, analysis was performed as described previously (57). Briefly, area and intensity of F-actin at the immunological synapse were measured for a defined area. Cortical F-actin intensity from both NK and target cells was subtracted from this measurement to generate a quantitative measure of specifically accumulated actin at the synapse. For MTOC polarization, MTOC were defined as the highest intensity staining of α-tubulin and the distance between this and the center of the immunological synapse was measured for 30 conjugates each from patient and healthy donor. Granule convergence was analyzed as previously described (58). Distance between individual lytic granules and the MTOC were measured and the mean of these was calculated for each cell.
Mass Cytometry
Mass cytometry is a high-dimensional single cell analysis technique based on flow cytometry but differs in its use of metal tagged antibodies in lieu of fluorophores. Procedure performed as described previously (59, 60) and acquisition was performed using a CyTOF instrument (Fluidigm). Data was analyzed and viSNE was performed using Cytobank (38, 61). Briefly, for the panel in Table E1A (See Table E1A in Online Repository), PBMCs were isolated, thawed and rested as detailed above. Cells were stained with cisplatin to track cell viability. Between 1×106 and 3×106 PBMCs per time point were stained with metal conjugated extracellular antibodies (Fluidigm), seeded in 96-well U bottom plates and stimulated with either a cocktail of A) 500U/mL IFNα (Peprotech), 500ng/mL LPS (Invivogen), 50ng/mL IL-12 (Peprotech), and 500U/mL IL-2 (Proleukin), and CD16/CD3/IgM crosslinking (using surface staining antibodies followed by anti-mouse IgG, Biolegend) for 0, 3 and 15 minutes. Cells were then fixed in 1.6% formaldehyde and permeabilized in 100% methanol. After washes, intracellular staining was performed before DNA staining using Cell-ID Intercalator-Ir (Fluidigm). For the panel in Table E1B, cells were stained with cisplatin to track cell viability. Between 1×106 and 3×106 PBMCs were unstimulated or mixed with 1:1 K562 cells and 500U/mL IL-2 (Proleukin) in the presence of GolgiStop (BD), GolgiPlug (BD) and metal-conjugated CD107 antibody (Fluidigm). Cells were then stained with conjugated extracellular antibodies (Fluidigm) or antibodies conjugated to the desired metal using Maxpar Antibody Labeling Kit (Fluidigm). Cells were fixed in CytoFix/CytoPerm (BD), washed in Perm Buffer (eBioscience) and stained with intracellular antibodies before DNA staining using Cell-ID Intercalator-Ir (Fluidigm).
Flow Cytometry
Where indicated, flow cytometry was performed using either surface staining alone at room temperature for 15 minutes or surface staining in combination with methanol permeabilized intracellular staining at room temperature for 60 minutes before acquisition using a Fortessa X-20 (BD). Data analysis performed using Cytobank (61) or FlowJo. Human antibodies used in this study: APC CD56 (clone 5.1H11, Biolegend), Pacific Blue CD3 (clone OKT3, Biolegend), APC Cy7 CD14 (Clone HCD14, Biolegend), FITC NKG2D (clone 1D11, Biolegend), FITC CD16 (clone 3G8, BD), PE NKp44 (clone P-44-8, Biolegend), PE 2B4 (clone C1.7, Biolegend), PE-Cy7 CD19 (clone HIB19, Biolegend), APC IgM (clone MHM-88, Biolegend), APC-Cy7 HLADR (clone L243, Biolegend), and Alexa Fluor 488 total PLCG2 (clone K86-1161, BD). Mouse antibodies used in this study: BV786 CD45 (clone 30-F11, BD), BV421 NK1.1 (clone PK136, BD), BV510 IgM (clone R6-60.2, BD), FITC CD11b (clone M1/70, Biolegend), PE-Cy7 CD27 (clone LG3A10, Biolegend), PE CD4 (clone GK1.5, Biolegend), PerCP IgD (clone 11-26c.2a, Biolegend), APC B220 (clone RA3-6B2, BD), APC-Cy7 CD3 (clone 17A2, Biolegend), APC-R700 CD8 (clone RPA-T8, BD), FITC CD43 (clone S11, Biolegend), PE CD24 (clone 30-F1, Biolegend), PE-Cy7 CD11b (clone M1/70, BD), PE Ly49H (clone 3D10, BD), and APC-Cy7 CD19 (clone 1D3, BD).
ELISA
Human serum IgG and IgM levels were analyzed using commercially available ELISA kits (Invitrogen) according to manufacturer instructions. Briefly, ELISA plates were coated with capture antibody overnight before washing, blocking and incubation with diluted patient sera or standard. Plates were then washed again, incubated with HRP-conjugated detection antibody, washed again and incubated with TMB substrate solution. Solution was then stopped with 1M phosphoric acid and absorbance was measured at 450nm.
Calcium Flux Analysis
For human samples, NK cells were enriched using RosetteSep Human NK Cell Enrichment (Stem Cell Technologies). 1–2×106 enriched NK cells were then loaded with Indo-1 dye (Invitrogen) and labeled with PE or FITC conjugated mouse IgG antibodies against the NK cell receptors 2B4 and NKG2D. Kinetic measurements of calcium flux were obtained using a BD Fortessa X-20 at baseline and then upon antibody crosslinking using anti-mouse IgG. Mouse calcium flux analysis was similarly performed: NK cells were isolated from spleen using EasySep Mouse NK Cell Enrichment (Stem Cell Technologies), loaded with Indo-1, labelled with APC NK1.1 followed by crosslinking and acquisition as above. Patient B.II.4 was performed similarly except that expansion beforehand was required due to limited patient sample and anti-NKp44 and anti-NKG2D were used for crosslinking. NK cells were expanded as previously described (28). Briefly, 106 PBMCs from patient B.II.4 or healthy control were co-incubated with 106 irradiated (100Gy) K562-mbIL15-41BBL (Kind gift from Dario Campana, National University of Singapore) for 7 days. After 7 days, cells were removed and assessed for purity. T cells (CD3+) were present at less than <1%. 100U/mL of recombinant IL-2 (Proleukin) was added to the culture and incubated for 7 more days with partial media exchange every 2 days. After 14 days total, NK cells expanded 10 to 15-fold with >95% purity (CD56+CD3−) and were then used for cytotoxicity and calcium flux assays. For murine B cell calcium flux analysis, naive B cells were gated on in whole splenocytes (B220+CD27−) and treated as above, with the exception of using anti-mouse IgM as the crosslinking antibody. Human B cell calcium flux analysis was performed from PBMCs (gated as CD19+) with the exception of using anti-human IgM as the crosslinking antibody. For Epstein-Barr virus-transformed lymphoblastoid cell lines, lymphocytes from a healthy female control were cultured with EBV as previously published (62), cultured in RPMI 1640 media + 10% fetal bovine serum, and nucleofected with Nucleofector Kit V (Lonza) using program O-017 for 24 hours before calcium flux analysis. Successful nucleofection was confirmed by GFP fluorescence, and cells were loaded with Indo-1 dye as detailed above before crosslinking with anti-human IgM.
Statistics
Normal internal reference ranges for human NK cell cytotoxicity and mass cytometry were determined using 25 healthy controls; outliers were removed using ROUT (Robust regression and Outlier removal) and the central 95th percentile was determined. Upper and lower bounds are visualized by dashed lines. Unless normality was established after D’Agostino & Pearson omnibus test for normality, pairwise comparisons are made using Mann-Whitney U test with Bonferroni correction for multiple comparisons Where noted, comparisons between healthy controls and patients are performed using age and gender matched healthy control donors. All statistics performed using GraphPad Prism.
Molecular Dynamics, Structural Analysis, and Conservation Analysis
Structural diagrams were generated using PyMOL v2.0 (PyMOL Molecular Graphics System). Conservation analysis of the G595 and L183 residues were generated using M-Coffee with ESPript secondary structure analysis (63–65). For molecular dynamics, 134.9μs of aggregate simulation time of the SH2 domain (107 aa) with wild-type (69.4μs) and G595R mutant (65.5μs) sequences was ran with GROMACS 2016.1 at 300K using the AMBER03 force field with explicit TIP3P solvent (66–68). Salt was added to neutralize the system and create a solution concentration of 100mM (13 Na+/14 Cl− for wild-type, 16 Na+/18 Cl− for G595R). Simulations were prepared by placing the starting structure for each sequence in a dodecahedron box that extended 1.0Å beyond the protein in any dimension. Each system was then energy minimized with the steepest descent algorithm until the maximum force fell below 100 kJ/mol/nm using a step size of 0.01 nm and a cutoff distance of 1.2 nm for the neighbor list, Coulomb interactions, and van der Waals interactions. For production runs, all bonds were constrained with the LINCS algorithm and virtual sites were used to allow a 4fs time step (69). Cutoffs of 1.0 nm were used for the neighbor list, Coulomb interactions, and van der Waals interactions. Before being run in production, systems were equilibrated with position restraints for all heavy atoms for 1ns. The Verlet cutoff scheme was used for the neighbor list. The stochastic velocity rescaling (v-rescale) thermostat was used to hold the temperature at 300 K. Conformations were stored every 10ps (70). The initial structure for both simulations was a homology model of Swiss Model threading the human PLCG2 sequence onto 4EY0, a crystal structure of the close human paralog, PLCG1 (31). The human paralog was used (rather than the murine homolog) because the crystal structure contains both nSH2 and cSH2 domains and so provided more information about the course of the C-terminal amino acids of the nSH2 domain than the murine homolog structure 2DX0(30). A microstate decomposition was built using khybrid clustering with a radius of 1.5Å and 5 rounds of kmedoids updates on the entire 135μs dataset using backbone atoms (C, Cα, N, O) and Cβ (except residue 595) to produce 2314 states (71). Using the cluster centers derived in this way, the data for the wild-type and the mutant was reassigned separately and generated separate Markov state models on this shared state space. Transition probabilities were fit with the transpose method (72). The state space had near complete coverage for both sequences with the wild-type sampling 2204/2315 states and the mutant sampling 2150/2315 states. The lag time was 1.5 ns and was determined using the implied timescales test.
Genomic Analysis of PLCG2
Genomic variants filtered from the gnomAD database are compared by CADD (Combined Annotation-Dependent Depletion) Score using a previously published visualization software, PopViZ (20).
Results
We identified patients from two unrelated nonconsanguineous families with autosomal dominant immunodeficiency, characterized by recurrent infections and reduced NK cell killing. In family A (Figure 1A), patient A.I.2 is a CMV/HSV1-seronegative 52-year-old Caucasian female with a history of arthralgias, antiphospholipid syndrome, and late-onset recurrent Staphylococcal septicemia. Her daughter, A.II.3, is a 19-year-old female with a history of arthralgias and autoimmunity (positive antinuclear antibody and type 1 diabetes), as well as recurrent HSV1 gingivostomatitis requiring prophylactic valacyclovir. Family B (Figure 1B) consists of patient B.II.4, a 9-year-old Qatari male with a history of CMV myocarditis, as well as adenoviral hepatitis. There was no history of immunodeficiency or autoimmunity in any other family members including his mother, B.I.2. Clinical NK cell testing in both families revealed reduced NK cell K562 killing (Figure 1C), despite intact CD107 degranulation against K562 cells and normal cytotoxic granule contents (Tables E2 & E3). Flow cytometry of peripheral blood demonstrated normal NK cell percentages (Figure 1D) and absolute counts inconsistent with classical NKD (Tables E2 & E3). Further clinical immunology evaluation of immunoglobulin levels (IgM, IgG, IgA, and IgE), protective antibody titers, T cell mitogen stimulation, and immune subpopulation analysis was also unremarkable (Tables E2 & E3).
Figure 1: Familial NK Cell Deficiency Associated with Novel Heterozygous PLCG2 Variants.
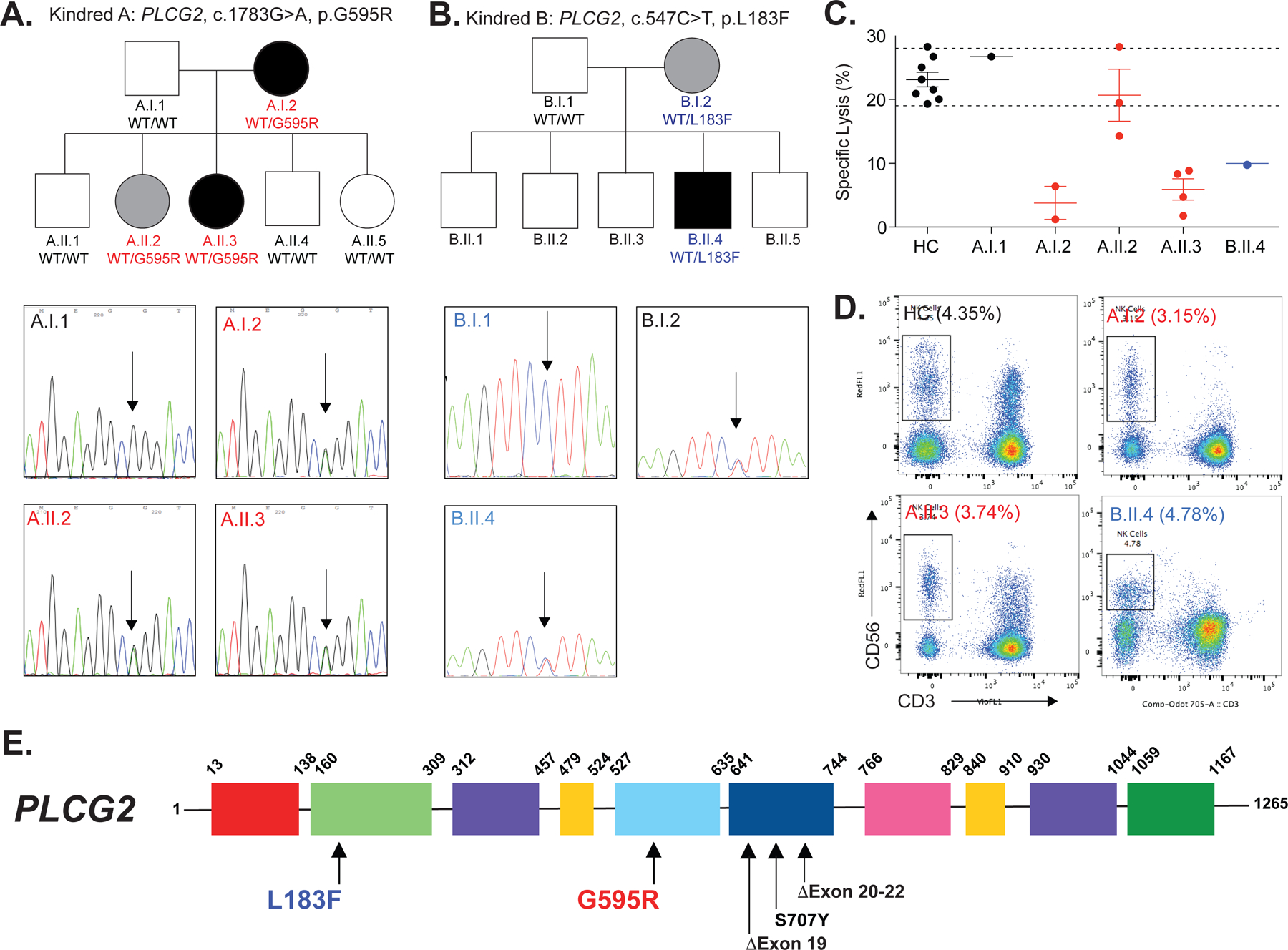
(A) Pedigree of family A; affected heterozygotes are shown in black symbols while unaffected or unevaluated heterozygotes are shown in gray or white, respectively. WT, wild-type allele. Sanger-sequencing chromatograms are shown for patients and unaffected relatives. Arrow denotes site of heterozygosity. (B) Pedigree and Sanger sequencing of family B is displayed as in subFigure A. (C) NK cell killing against K562 cells is quantified after incubation for four hours at a peripheral blood mononuclear cell (PBMC) to K562 ratio of 50:1. Upper and lower internal reference ranges are displayed with dashed lines. See Online Repository for generation of reference ranges, Table E3. Each point represents a unique biologic replicate, either a separate blood draw (patients) or a separate individual (controls). (D) Flow cytometry evaluation of NK cells (CD3− CD56+) in healthy control (HC) versus patients A.I.2, A.II.2, A.II.3 and B.II.4. Percentage of NK cells in the lymphocyte gate is displayed. Internal normal NK cell reference range, 2.8% to 15.5%. (E) The location of variants, including previously reported PLAID (Exon 19 or 20–22 deletions) and APLAID (S707Y) variants are displayed with the domain structure of PLCG2. PH, Pleckstrin homology; nSH2, N-terminal Src Homology 2; cSH2, C-terminal SH2; SH3, Src Homology 3. Except where limited by patient sample availability (B.II.4 in subFigures C and D), all data is representative of two or more independent experiments. All error bars represent standard deviation from the mean.
The two index patients and their parents underwent Trio whole-exome sequencing (WES) which revealed novel heterozygous PLCG2 missense variants in both families. These results were validated with Sanger Sequencing. Patients A.I.2 and A.II.3 possessed heterozygous variants (c.1783G>A, p.G595R) in PLCG2, located in the N-terminal SH2 domain (nSH2) (Figure 1E). A CADD (Combined Annotation Dependent Depletion) score of 26 was calculated, indicating this variant is likely to be highly deleterious (20). An additional healthy HSV1-seropositive 17-year-old sister (A.II.2) with the G595R variant was identified by Sanger sequencing; however, her borderline-normal NK cell killing suggests incomplete penetrance, a common feature of autosomal dominant immune syndromes (21) and is frequently seen in Inborn Errors of Immunity (IEI). Patient B.II.4 and his mother (B.1.2) possessed a different heterozygous variant in PLCG2 (c.547C>T, p.L183F), located in the EF hand domain of PLCG2 (Figure 1E), which is critical for membrane targeting and enzymatic activity in other Phospholipase C isozymes (22, 23). A CADD score of 24.4 was calculated for this variant, indicating its highly probable deleteriousness as well. His mother, B.I.2, also possessed this L183F variant but was reportedly healthy and unfortunately, she was not available for further evaluation. The locations of these variants and other reported PLCG2 variants are diagrammed in Figure 1E. Given the two family members with apparent incomplete penetrance, we have included an Excel document (Table E4A–F: Abridged WES Data) in the Online Repository. Tabs A–F include WES data for both families; 3 family members each. We have thoroughly re-evaluated these lists and have not identified a second known disease-causing gene that would modify the phenotype in affected individuals.
As immunodeficiencies commonly arise from aberrant immune cell development or signaling, mass cytometry (CyTOF, see Methods and Tables E1A & E1B) was employed to analyze these processes in the peripheral blood. Consistent with clinical studies, NK cell abundance, as well as the distributions of immunomodulatory CD56Bright and cytotoxic CD56Dim NK cells, were intact (Figure 2A). Family A demonstrated reduced B cells with preserved naïve to class-switched memory B cell percentages, suggesting a defect in B cell output but not activation (Figure 2B). In support of this, serum immunoglobulins, seroconversion, and IgM-induced calcium flux were normal (Figure 2C–D, Tables E2 & E3). T cell development (See Figure E1A in the Online Repository) and T cell calcium flux (Figure E1B) were unperturbed. Furthermore, clinical T cell functional assessment was normal (Table E3). Although total number of T cells were normal, the distribution of T follicular-helper cells (TFH) was altered with increased TFH2 cells and decreased TFH1 cells in both families (Figure E2A), a pattern seen previously in human autoimmunity (24). Although autoimmunity and NK cell function has been correlated in various disease contexts previously, the link between these is not well established (25). The number of monocytes and dendritic cells, but not other myeloid cells (i.e., granulocytes), was reduced in both families as well (Figure E2B, Table E2). Human monocyte activation with macrophage colony stimulating factor (MCSF) is dependent on PLCG2 induced calcium flux, possibly contributing to the observed monocytopenia and bacterial susceptibility in patient A.1.2 (15) (Table E2).
Figure 2: Analysis of NK Cells and B Cells in PLCG2 Haploinsufficiency.
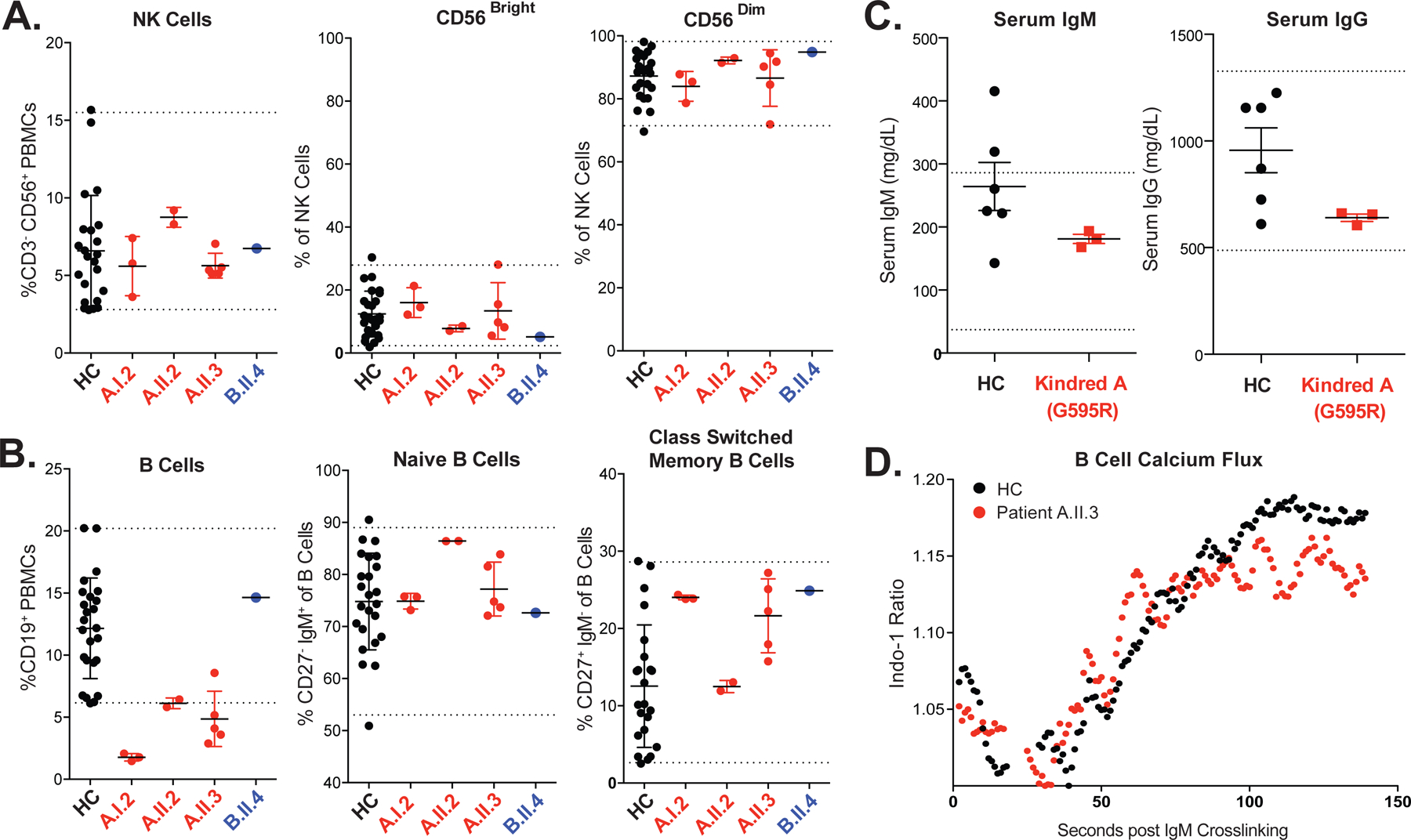
(A) Mass cytometry was performed to quantify total (CD3−CD56+), CD56Bright and CD56Dim NK cells in the peripheral blood of HC (healthy controls), G595R patients A.I.2, A.II.2, A.II.3 and L183F patient B.II.4. Internal reference ranges are shown as visualized by dashed lines (see Methods). NK cell reference range, 2.8% to 15.5%. Each patient data point represents a unique biological replicate from a different blood draw. (B) B cells (HLADR+ CD19+) are quantified and displayed as a percentage of PBMCs. Memory formation and class-switching is assessed by quantification of CD27+ IgM− cells within the B cell compartment. Normal B cell reference range, 6.2% to 20.2%. Each patient data point represents a unique biological replicate from a different blood draw. (C) ELISA quantification of serum IgG and IgM obtained from HC versus patients A.I.2, A.II.2 and A.II.3 (pooled above). Reference ranges courtesy of Mayo Clinic; IgG 487–1,327 mg/dL and IgM 37–286 mg/dL. Error bars represent standard deviation from the mean. (D) Indo-1 analysis of calcium flux in primary B cells (gated CD19+ from peripheral blood mononuclear cells) after crosslinking with anti-IgM.
CyTOF analysis of NK cell signaling revealed hypophosphorylation of PLCG2 in both families (Figure 3A). Family A demonstrated a reduction in the magnitude of PLCG2 phosphorylation while family B displayed a slower accumulation of phospho-PLCG2. PLCG2 hypophosphorylation was also confirmed by flow cytometry in patients A.I.2 and A.II.3; however, patient A.II.2 had near normal PLCG2 phosphorylation (Figure E3) consistent with her borderline-normal NK cell killing (Figure 1C). Upstream Btk/Itk, ZAP70/Syk, and Lck phosphorylation was intact, suggesting an intrinsic defect in PLCG2 (Figures 3B and E4). MAPKAPK2, activated by PKC downstream of PLCG2-induced DAG, was similarly hypophosphorylated (Figure E4) (26). Total PLCG2 protein levels in family A were analyzed to establish whether hypophosphorylation was the result of functional inhibition or reduced protein expression. Total PLCG2 protein levels were normal in both CD56Dim and CD56Bright NK cells (Figure 3C), as well as in monocytes, T cells, and B cells (Figure E5). This suggests that the G595R variant compromises function and not protein expression. Analysis of PLCG2 protein levels in patient B.II.4 was not feasible due to limited samples. The G595R patients revealed differential PLCG2 expression between immune cell subsets, including an approximately 50% lower PLCG2 expression in monocytes and CD56Dim NK cells than in T cells, B cells, and CD56Bright NK cells (Figure E5).
Figure 3: Loss-of-Function Variants in PLCG2 and Haploinsufficiency Cause NK Cell Dysregulation.
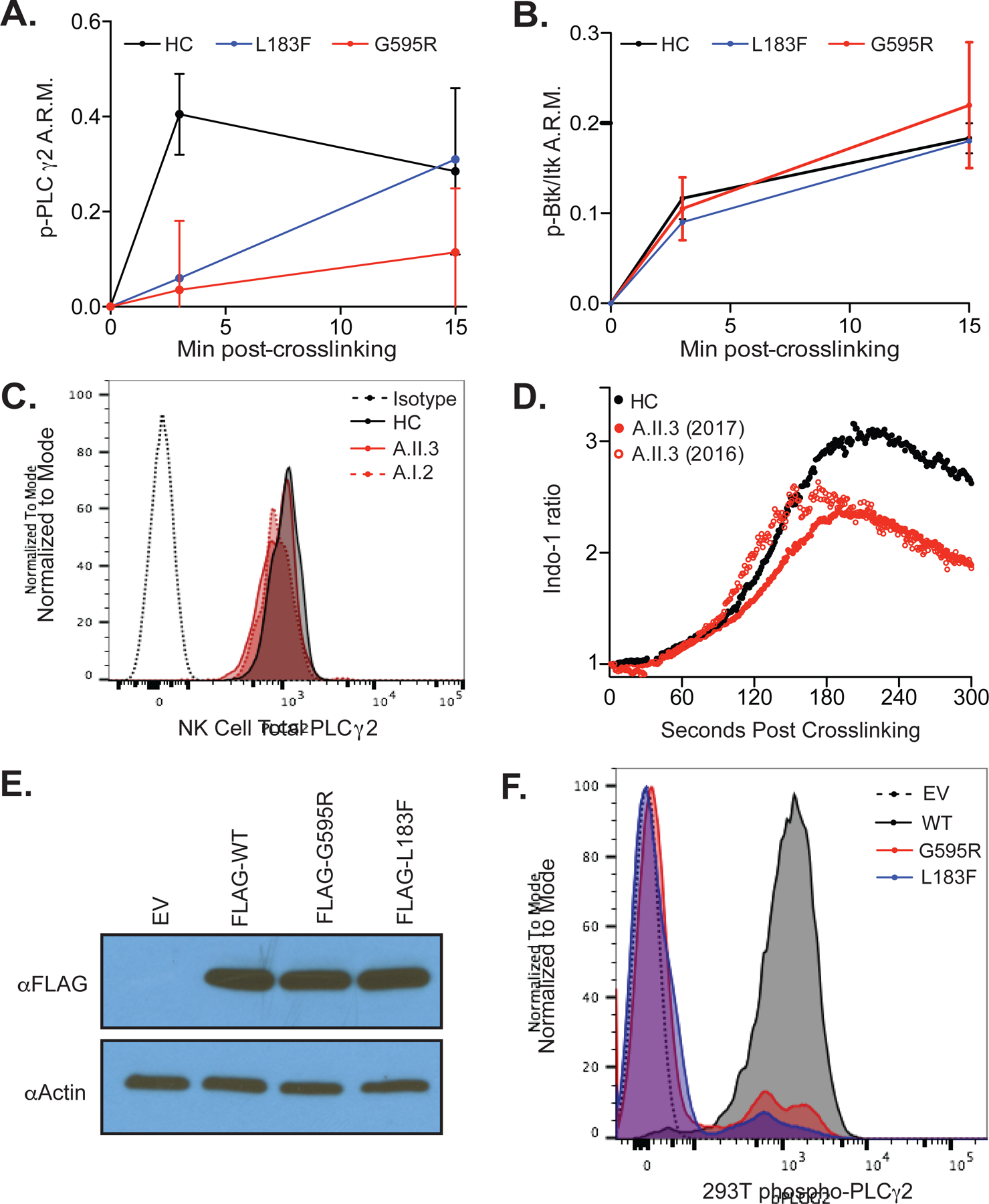
(A) PLCG2 phosphorylation in CD56Dim NK cells after CD16 crosslinking is quantified by CyTOF, normalized to time 0 using an arcsinh transformation in three healthy controls (HC; two females, one male), two G595R patients (A.II.3 and A.I.2) and one L183F patient (B.II.4). A.R.M., Arcsinh ratio of mean. Error bars represents standard deviation from the mean. (B) Btk/Itk phosphorylation is shown as in subfigure A. (C) Total PLCG2 levels in CD56Dim NK cells is quantified by flow cytometry in a healthy control versus G595R patients. Isotype, Isotype control (dotted black line). HC, healthy control (solid black line). A.II.3, G595R patient (solid red line). Patient A.I.2, G595R patient, (dotted red line). (D) Indo-1 calcium flux analysis in G595R patient A.II.3 was assessed in naïve enriched human CD56Dim NK cells after crosslinking with NKG2D and 2B4. Open and closed red circles represent two unique blood samples acquired one year apart. (E) Western blot analysis for PLCG2 protein expression in HEK293T cells transfected with wildtype or mutant FLAG-tagged PLCG2 or EV, empty vector. (F) Phosphorylation of FLAG-tagged wildtype or mutant PLCG2 after 15 minutes of pervanadate stimulation in 293T cells is quantified using phospho-flow cytometry. EV, empty vector. Except where limited by patient sample availability (B.II.4 in subfigures A and B), all data is representative of two or more independent experiments.
The catalytic activity of PLCG2 is initiated by phosphorylation, leading to calcium flux and granule movement and polarization. Consistent with PLCG2 hypophosphorylation, calcium flux in patient A.II.3 was stably reduced in CD56Dim NK cells after NKG2D and 2B4 receptor crosslinking (Figure 3D). Notably, CD56Bright NK cells, which express higher levels of PLCG2 protein (Figure E5), demonstrated comparable calcium flux with NKG2D and 2B4 crosslinking in patient versus control samples (Figure E6A). Calcium flux in CD16-crosslinked NK cells was also normal, consistent with the ability of CD16 to signal through both PLCG1 and PLCG2 (Figure E6B) (27). Limited patient sample required expansion of patient B.II.4 NK cells using K562-mbIL15-41BBL cells and IL-2 before analysis (28). Expanded patient B.II.4 NK cells (with A.ll.3 and A.l.2 as controls) also showed partially reduced calcium flux (Figure E6C), although this expansion process largely restored NK cell cytotoxicity (Figure E6D) in comparison with Figure 1C.
To establish that G595R and L183F are loss-of-function variants, wildtype or mutant FLAG-PLCG2 was expressed in 293T cells (which do not natively express PLCG2) and analyzed for protein expression and pervanadate-induced phosphorylation. Although both mutants expressed PLCG2 protein at normal levels (Figure 3E), FLAG-PLCG2G595R and FLAG-PLCG2L183F were hypophosphorylated compared to FLAG-PLCG2Wildtype (Figure 3F). These data demonstrate that the G595R and L183F variants are loss-of-function variants and contribute to PLCG2 haploinsufficiency.
Bioinformatic and structural analyses of G595R and L183F suggest that these residues are intolerant to mutation. The G595R and L183F variants occur at highly-conserved sites in the nSH2 domain and EF-hand domain of PLCG2, respectively (Figure E7A). Only two other individuals in ExAC are reported to have missense variants at G595R, while no missense variants in L183F have been reported (29). Although no structure exists for the EF hand domain, nSH2 structures from murine PLCG2 (30) and human PLCG1(31) facilitated analysis of the G595R variant with molecular dynamics (MD), which has been used to understand the structural effects of variants previously (32, 33). Simulations of wildtype and G595R sequences were analyzed by this approach and revealed conformational disturbances in the nSH2 βD-βE loop, potentially compromising the LAT phosphotyrosine binding site (Figure E7B, E7C). In support of the βD-βE loop being critical in SH2 function, a structurally-analogous variant (G60R) in the loop of SHP-2 has been previously reported as pathogenic and this loop serves as a protein-protein interaction site in the nSH2 of PLCG1 (34, 35).
Cytotoxic granule movement was analyzed by microscopy in NK cells conjugated to K562 target cells (Figure 4A). The microtubule organizing center (MTOC) to granule distance (MGD) and MTOC to synapse distances (MSD) were quantified. Both distances were increased in patient A.II.3, indicating dysregulated cytotoxic granule movement (Figure 4B). Synaptic actin accumulation, regulated independently of PLCG2, was unchanged (Figure 4B). In T cells, calcium flux kinetics and DAG localization influence the path and directionality of granule movement, respectively (36, 37). The observed defect in NK killing despite intact CD107 degranulation (Figure 4C) suggests that defects in both of these processes may lead to delayed or adirectional degranulation. Due to limited patient samples, we were not able to analyze granule movement in the B.II.4 index patient; however, normal CD107a degranulation (in response to PMA and Ionomycin) was observed (Figure E8A and Table E3) in B.ll.4. As well, a defect in NK cell killing was observed (Figure 1C), which suggests that a similar process is occurring in B.ll.4. Although methods to monitor DAG are limited, the defect in MAPKAPK2 phosphorylation downstream of PKC implies that this branch of PLCG2 signaling is dysregulated in both (A.II.3 and B.II.4) patients (Figure E4). Additionally, activated NK cell IFN-γ secretion was unchanged in either family A or B (Figures E8B & E8C), suggesting that NK cell cytotoxicity is the primary pathway affected by these variants.
Figure 4: PLCG2 Haploinsufficiency Alters Cytotoxic Granule Mobility, NK Cell Maturation, and the Adaptive NK Cell Response.
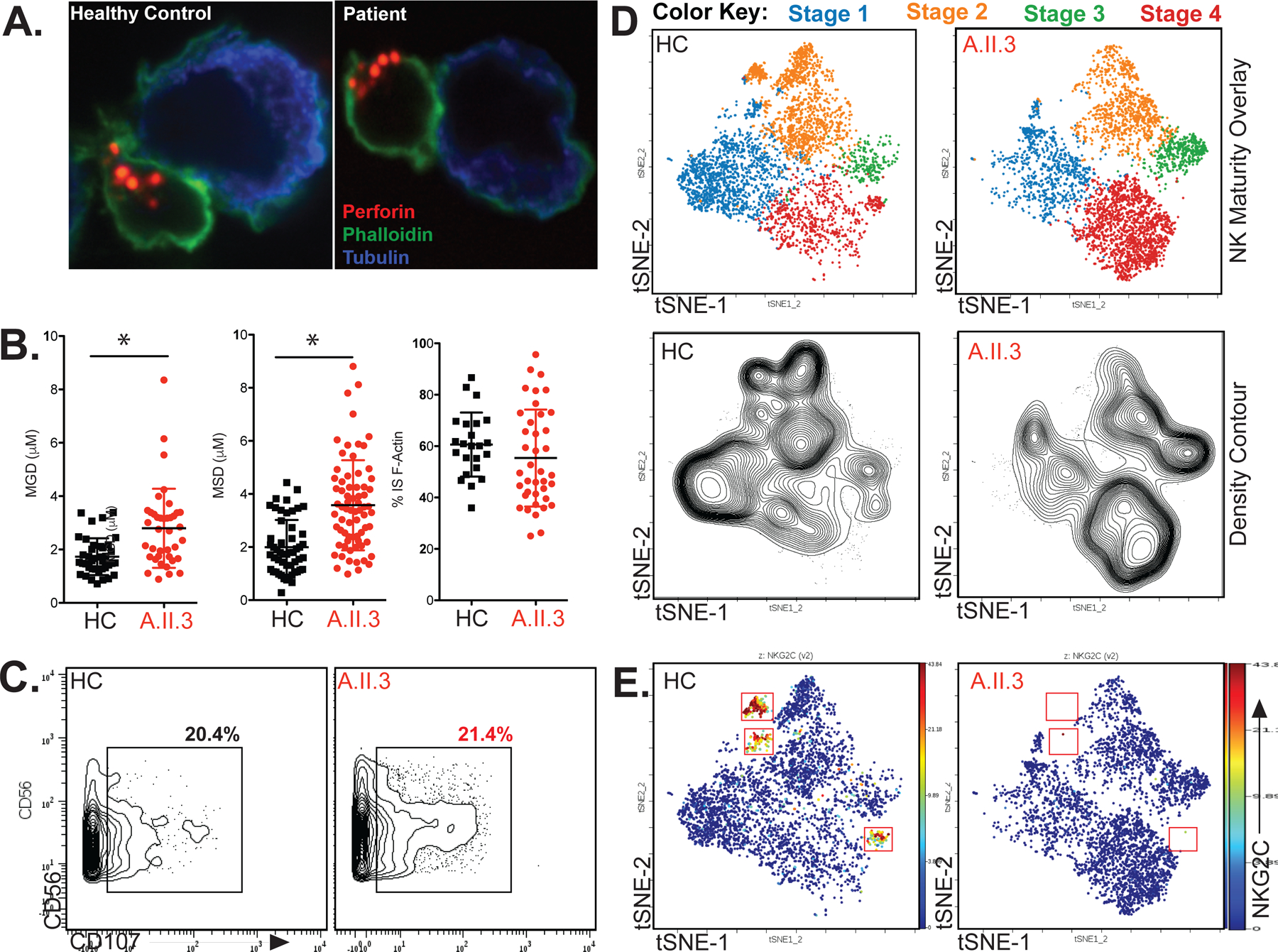
(A) Representative immunofluorescence of cytotoxic granule microscopy upon K562 target conjugation in healthy versus patient A.II.3 NK cells. (B) Quantification of microtubule organizing center (MTOC) to granule distance (MGD), MTOC to synapse distance (MSD), and synaptic actin accumulation in healthy control (HC) versus patient A.II.3. * P<0.001, Mann-Whitney U Test. (C) CD107 Degranulation against K562 target cells is quantified by CyTOF after 1:1 incubation with healthy control or patient A.II.3 PBMCs for 6 hours. (D) viSNE on NK cells (CD3− CD56+) overlaid with maturity subpopulations identified by traditional bivariate gating (top) with density visualized by contour (bottom) in both gender/age-matched healthy control (HC) and patient A.II.3. Stage 1, NKG2A−CD57−; stage 2, NKG2A+CD57−; stage 3, NKG2A+CD57+; stage 4, NKG2A− CD57+. tSNE, t-distributed stochastic neighbor embedding. (E) Similar graphical representation as in subFigure D is shown for the adaptive NK cell response marker NKG2C. All data is representative of two or more independent experiments using two patient blood samples drawn more than 1 year apart. All error bars represent standard deviation from the mean.
CyTOF was also used to examine NK cell development and receptor expression (panel in Table E1B). Clustering of NK cells with visual stochastic neighbor embedding (viSNE) enabled visualization of this high-dimensional data, whereby each point represents a cell and groups represent subpopulations which may be identified by marker expression (38). Activating and inhibitory receptor expression were comparable between patient A.II.3 and control (Figure E9); however, patient NK cell density was increased in the viSNE region corresponding to CD57+ maturation stages 3 and 4, indicating increased NK cell maturity (Figure 4D). This phenotype was also noted by flow cytometry in patient B.II.4 (Figure E10). CD57+ acquisition is typically cytokine-driven and associated with increased cytotoxicity, suggesting either persistently-elevated cytokine levels (perhaps from increased viral burden) or a potential compensatory mechanism to increase NK cell killing (39). Additionally, a distinct subpopulation of NKG2C+ NK cells was absent in patient A.II.3 (Figure 4E). In most individuals, NKG2C+ NK cells expand during CMV infection and persist thereafter, referred to as the adaptive NK cell response (40–42). The absence of this population despite CMV seropositivity in the G595R index patient, A.II.3, suggests this process may be impacted.
To establish that PLCG2 haploinsufficiency is sufficient to cause NKD, a mouse model of haploinsufficiency was validated by comparing wildtype (Plcg2+/+) and Plcg2+/− mice. While Plcg2−/− mice have been previously described with severe B cell and NK cell defects, defects in Plcg2+/− mice have not been previously reported (10, 11). Subpopulation analysis was performed using flow cytometry and viSNE. As expected, major perturbations were seen in Plcg2−/− mice, including altered B cell development; however, B cell and NK cell development were intact in Plcg2+/− mice (Figure 5A–B). Similar to our patients, NK cell maturation was increased in Plcg2+/− mice (Figure 5C). Calcium flux analysis was performed in both B cells and NK cells. Although IgM-induced calcium flux was normal in Plcg2+/− B cells, NK1.1-induced calcium flux was attenuated in Plcg2+/− NK cells (Figure 5D). Correlating with reduced calcium flux, NK cell killing of RMA-S target cells was inhibited in Plcg2+/− mice (Figure 5E). Similar decreased killing was observed with YAC-1 target cells (Figure E11). Also similar to our human patients, heterozygous Plcg2 mice displayed normal degranulation against YAC-1 targets (Figure 5F). This combination of enhanced NK cell maturation, decreased NK cell calcium flux (while maintaining normal calcium flux in B cells), and decreased NK cell killing in the context of normal levels of NK cell degranulation phenocopies our patients and demonstrates that one-copy loss of PLCG2 is sufficient to cause functional NK defects.
Figure 5: Heterozygous Plcg2 Mice Phenocopy Human PLCG2 Haploinsufficiency.
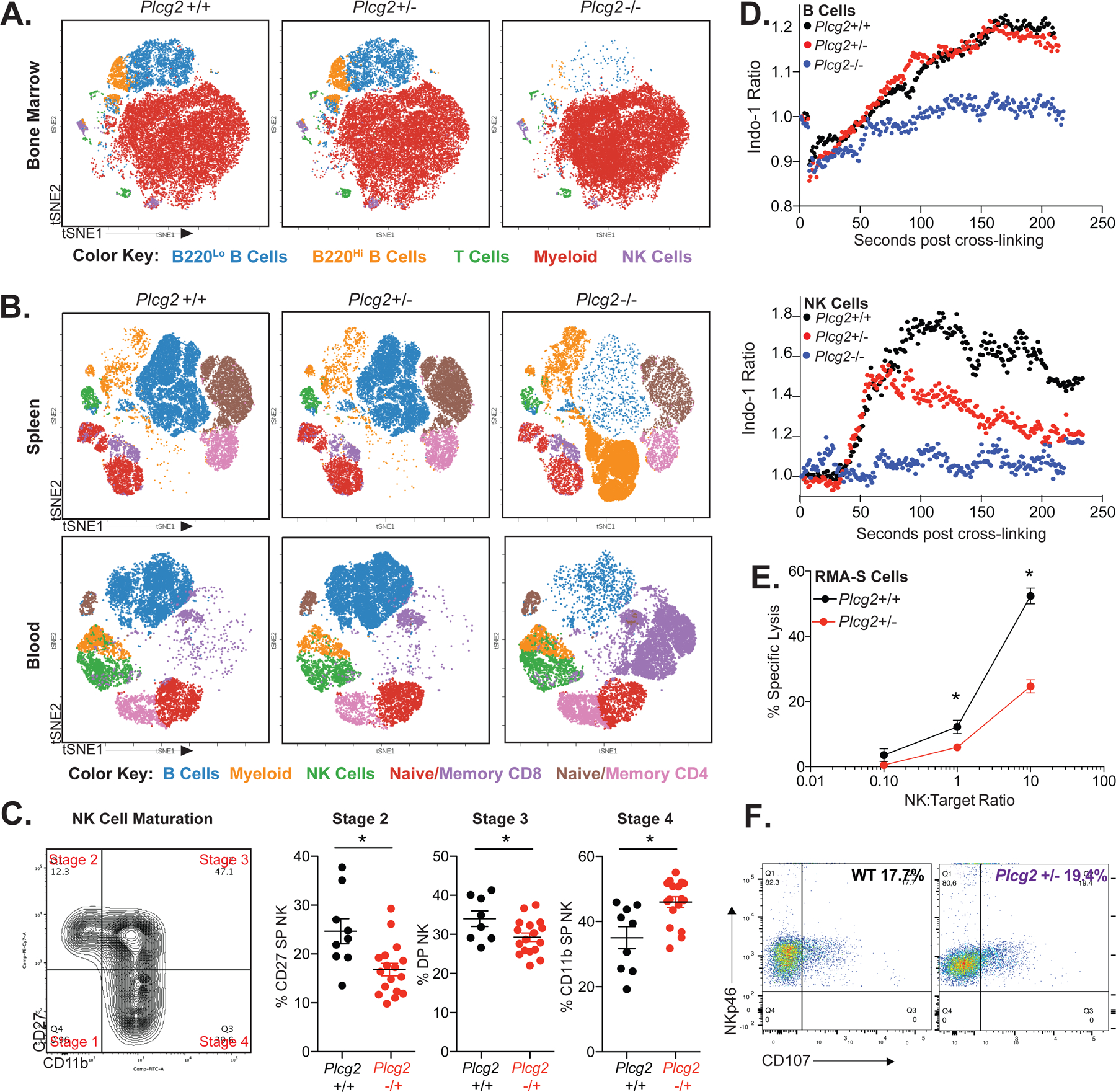
Analysis of mouse immune cell subpopulations in the bone marrow (A), spleen and peripheral blood (B) of Plcg2 wildtype (+/+), heterozygous (+/−), and homozygous (−/−) littermates using flow cytometry and displayed using viSNE clustering as in Figure 4D. Color key for cell types identified by traditional bivariate is located beneath each subFigure. tSNE, t-distributed stochastic neighbor embedding. (C) Analysis of splenic murine NK cell maturity in wildtype littermate control versus heterozygous Plcg2 mice using CD27 and CD11b expression. DP, double positive. SP, single positive. Example bivariate gating of murine NK cell maturation is shown. Each point represents a unique biologic replicate. * P<0.05, Mann Whitney U Test. (D) Indo-1 calcium flux analysis of littermate Plcg2 +/+, +/− and −/− mice is displayed after crosslinking with anti-IgM antibody (B cells gated from whole splenocytes) or anti-NK.1.1 antibody (NK cells enriched from spleen). (E) NK cell killing against RMA-S target cells was analyzed in littermate wildtype control versus heterozygous Plcg2 mice using enriched splenic NK cells at NK to target ratios of 1:10, 1:1 and 10:1. Similar results were seen with the use of YAC-1 cells (Figure E9). (F) CD107 Degranulation staining of purified NK cells from WT (wildtype, black) and Plcg2 +/− mice (purple), after 6 hours of incubation mixed 1:1 with YAC-1 targets. Pairwise comparisons at each time point performed using t-test, * P<0.05, after test for normality. All data is representative of two or more independent experiments. All error bars represent standard deviation from the mean.
Together, these data demonstrate the G595R and L183F variants are loss-of-function variants resulting in functional PLCG2 haploinsufficiency and NK cell functional defects without significant perturbance of B cells.
Discussion
The heterozygous loss-of-function variants presented herein result in PLCG2 haploinsufficiency, NK cell dysfunction, and herpesvirus infections. We acknowledge this haploinsufficient phenotype may be more complex in its pathophysiology. We have examined possible contributions by other cell types that may impact the PLCG2 loss of function seen in our patients. Despite the role of PLCG2 in B cells, these cells are functionally intact in PLCG2 haploinsufficiency. Based on the differential regulation of PLCG2 expression among lymphocytes, we propose a threshold model wherein cell types with homeostatically low levels of PLCG2 (i.e., CD56Dim NK cells and monocytes) are uniquely susceptible to further reductions in PLCG2 function. In contrast, T cells are likely shielded from PLCG2 haploinsufficiency by virtue of their high PLCG2 expression and use of alternative pathways (i.e., PLCG1 in T cells). We have shown that despite the presence of the G595R PLCG2 variant, T cells have normal CD4 and CD8 numbers and normal T cell calcium flux. This model suggests that PLCG2 may serve as a rate-limiting checkpoint against erroneous cytotoxicity in NK cells, which express lower levels of PLCG2, requiring strong PLCG2 activation for accurate and directional degranulation. Extrapolating this model further, monocytopenia was also observed in all four of the patients (Figure E2B), and monocytes express the lowest levels of PLCG2 of all cell types examined in our study (Figure E5A). While the role of monocytopenia to the observed clinical phenotypes is not clear, this may also contribute to certain features of disease, including herpesvirus and bacterial susceptibility.
Despite a number of similarities (Tables E2 & E3), family A and B each possess unique features as well. Notably, B cell output was reduced in family A (including A.II.2), but not family B although this may be influenced by other genetic background effects. Families A and B also differed in the nature of their phosphorylation defect. Family A had reduced magnitude of PLCG2 phosphorylation while family B displayed a slower increase of phospho-PLCG2. An analysis of each domain’s function provides insight into this difference. The L183F variant in family B lies within the EF-hand domains, which facilitates PLCG2 catalysis and targeting to the membrane (22, 23). Upon reaching the membrane, the nSH2 domain (affected by G595R in family A) binds to phosphorylated LAT, enabling assembly of the NK cell signalosome and interaction of PLCG2 with its kinase (Btk/Itk) (43, 44). Therefore, PLCG2 with diminished membrane localization (e.g, variants in the EF-hand domain) would be capable of normal signalosome interaction, but diffusion-limited. In contrast, PLCG2 lacking a functional nSH2 domain would be blocked from signalosome interaction and phosphorylation altogether, reducing the magnitude of calcium flux. This hypothesis is consistent with the patterns observed in our patients and suggests that PLCG2 loss-of-function variants may have domain-specific phenotypes. Nonetheless, future studies are needed to further delineate the exact mechanisms by which these variants diminish PLCG2 function.
Pathogenic variants are commonly modifiable by both genetic and environmental factors. Genetic epistasis likely plays a role in the incomplete penetrance and variable expressivity observed in many autosomal dominant syndromes (21). Immunologic context, such as the cytokine environment, may also modulate cellular and clinical phenotypes. For example, IL-2 incubation partially reverses NK cell killing defects in STX11-deficient patients (45), reminiscent of the substantial restoration of patient NK cell killing after IL-15/IL-2 cytokine exposure herein (Figure E6D). The use of collateral immunologic pathways may also alter phenotypes. For example, the versatile use of either PLCG1 or PLCG2 by some NK cell receptors (i.e., CD16) may allow compensatory signaling through these pathways. Moreover, the variable nature of the adaptive immune response may compensate for innate defects to different degrees. A combination of these likely contributes to the phenotypic differences and incomplete penetrance seen in PLCG2 haploinsufficiency. This manipulability may also present an opportunity to therapeutically modify defects, for example with modulation of IL-15 signaling using ALT-803, an investigational drug previously shown to rectify NK cell cytotoxicity defects in vivo and aid CMV clearance in humans (46).
Whereas PLAID and APLAID represent the autosomal dominant manifestations of dominant-negative and gain-of-function variants, the present patients illustrate haploinsufficiency and expand the spectrum of PLCG2-related disease. A fourth possibility, autosomal recessive loss-of-function, remains either undiscovered or is incompatible with life. While these three syndromes may be mechanistically distinct, there remains unexplained overlap (i.e., autoimmunity) that merits further investigation. Nonetheless, PLCG2 haploinsufficiency results in clinical phenotypes distinct from PLAID and_APLAID and requires a different diagnostic and therapeutic approach. PLAID, APLAID and PLCG2 haploinsufficiency are compared in Table 1.
Table 1:
Clinical Characteristics of PLAID, APLAID and PLCG2 Haploinsufficiency
| PLAID(18) | APLAID(19) | PLCG2 Haploinsufficiency | |
|---|---|---|---|
| Variant | cSH2 Deletions | S707Y | G595R, L183F |
| Mechanism of Disease | Dominant-negative or Gain-of-function* | Gain-of-function | Loss-of-function (haploinsufficiency) |
| Cold Urticaria | + | − | − |
| Inflammatory Skin Lesions and Cutaneous Granulomas | + | + | − |
| Allergic Disease | + | − | − |
| Arthralgias | − | + | + |
| Autoantibodies/Autoimmunity | + | − | + |
| Bacterial Susceptibility | + | + | + |
| Herpesvirus Susceptibility | − | − | + |
| NK Cell Count | Decreased | Normal | Normal |
| NK Cell Killing | NR | NR | Decreased |
| NK Cell Degranulation | Decreased | NR | Normal |
| NK Cell Calcium Influx | Decreased | NR | Decreased |
| B Cell Count | Decreased | NR | Normal/Decreased |
| B Cell Class Switching | Decreased | Decreased | Normal |
| IgG/IgA/IgM levels | Decreased | Normal | Normal |
| B Cell Calcium Influx | Decreased/Increased† | Increased | Normal |
| Mast Cell Degranulation | Normal/Increased† | NR | NR |
NR, not reported. PLAID, PLCG2-associated Antibody Deficiency and Immune Dysregulation. APLAID, Autoinflammation & PLCG2-associated Antibody Deficiency and Immune Dysregulation.
Temperature dependent mechanism.
At 37°C, B cell calcium is inhibited while mast cell degranulation is unchanged. At subphysiologic temperatures B cell calcium and mast cell degranulation are increased.
At present, the lack of a clear etiology complicates the management of many patients with unusually severe and/or recurrent herpesvirus infections. This study highlights a potential role for PLCG2 variants in these patients, provides insight into the regulation of human NK cell cytotoxicity, and unifies PLCG2-associated disease along a clinical spectrum that now includes PLAID, APLAID, and PLCG2 haploinsufficiency. Unlike PLAID and APLAID, which require deletions or variants at specific locations, loss-of-function variants could plausibly occur in many domains beyond the SH2 and EF hand domains, including the catalytic, SH3 and C2 domains. Of the 60,000 healthy exomes in ExAC, only 402 of 1265 residues in PLCG2 have been reported with non-synonymous variants (29), which are graphically represented by minor allele frequency and CADD score in Figure E12. The relative lack of variants suggests evolutionary pressure against deleterious variants in PLCG2, which can also be statistically assessed using publicly-available pLI scores (probability of a given gene being loss-of function intolerant) deposited in gnomAD; pLI scores above 0.9 are extremely loss-of-function intolerant and PLCG2 has a pLI score of 0.996, indicating PLCG2 is under significant evolutionary negative selection(47). This evolutionary pressure against variants in PLCG2 implicates a number of residues where novel variants may disrupt PLCG2 function. As a result, heterozygous PLCG2 variants should be considered in the differential diagnosis of patients with a number of presentations beyond cold urticaria, antibody deficiency, and autoinflammation, including but not limited to NK cell immunodeficiency.
Supplementary Material
Acknowledgements
We would like to first and foremost thank our patients for their participation, without which this work would not be possible. We would also like to thank Joshua Milner, Michael Ombrello, Olga Lubman, Daved Fremont, Olga Malkova, Stephen Oh, Catherine Miner, Molly Keppel, Deborah Lenschow, Brian Edelson, Roberta Faccio, Marco Colonna and Tarin Bigley for their helpful discussions and/or technical assistance. Additionally, we thank John Atkinson, David Hunstad, Michael Diamond, and Todd Fehniger for their critical reading of this manuscript and Ana Kolicheski and Ivan Chinn for analyzing the WES data. Critical reagents were provided by James N. Ihle at St. Jude Children’s Research Hospital (Plcg2 mice) as well Dario Campana at the National University of Singapore (K562-mbIL15-41BBL cells). Technical support was provided by the Washington University Immunomonitoring Laboratory, supported by the Andrew M. and Jane M. Bursky Center for Human Immunology and Immunotherapy. We also thank the Genome Technology Access Center (GTAC) in the Department of Genetics at Washington University for help with genomic analysis. GTAC is partially supported by a NCI Cancer Center Support grant (P30CA91842) to the Siteman Cancer Center, a ICTS/CTSA grant (UL1TR000448) from the NIH/National Center for Research Resources (NCRR), and the NIH Roadmap for Medical Research. This work was also supported in part by NIH National Research Service Award-Medical Scientist Grant T32GM02700 for J.B.A., J.R.P., and A.Y.M., NIH grants R01AI078994 and support from P30AR073752 to A.R.F., R01AI067946 and R01AI120989 to J.S.O., R01GM12400701 to G.R.B., The National Science Foundation (MCB-1552471 to G.R.B.), the Burroughs Wellcome Fund, the David & Lucile Packard Foundation, the Jeffrey Model Diagnostic and Research Center for Primary Immunodeficiencies at St. Louis Children’s Hospital, the Center for Pediatric Immunology at Washington University and St. Louis Children’s Hospital, the Jeffrey Modell Diagnostic and Research Center for Primary Immunodeficiencies at St.the Rheumatology Research Foundation, a NIH-NHGRFI/NHHLBI grant UM1HG006542 to the Baylor Hopkins Center for Mendelian Genomics (J.R.L.), as well as funding to M.A.C. from the Children’s Discovery Institute at St. Louis Children’s Hospital.
Abbreviations:
- NK Cell
Natural Killer Cell
- NKD
Natural Killer Cell Deficiency
- PLAID
PLCG2-associated Antibody Deficiency and Immune Dysregulation
- APLAID
Autoinflammation & PLCG2-associated Antibody Deficiency and Immune Dysregulation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental Materials
The supplemental materials include a list of author contributions, data for the analysis of T cells and myeloid cells in PLCG2 Haploinsufficiency, CyTOF analysis of CD56Dim NK cell signaling, PLCG2 Molecular Dynamics analysis, additional patient calcium flux assays, IFN-g upregulation in response to NK cell activation for both A and B index patients, a panel of NK activation and inhibitory receptor profiles for A.II.3, and additional NK maturation data for B.II.4. Analysis of PLCG2 variants by CADD Score and minor allele frequency, as well as Tables describing PLCG2 Haploinsufficiency patient clinical characteristics, clinical testing results, antibody lists for the CyTOF panels used herein, and Abridged WES data are included.
The authors have no competing financial interests to disclose.
References
- 1.Rosenthal SL, Stanberry LR, Biro FM, Slaoui M, Francotte M, Koutsoukos M, et al. Seroprevalence of herpes simplex virus types 1 and 2 and cytomegalovirus in adolescents. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 1997;24(2):135–9. [DOI] [PubMed] [Google Scholar]
- 2.Ornstein BW, Hill EB, Geurs TL, and French AR. Natural killer cell functional defects in pediatric patients with severe and recurrent herpesvirus infections. The Journal of infectious diseases. 2013;207(3):458–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mace EM, and Orange JS. Genetic Causes of Human NK Cell Deficiency and Their Effect on NK Cell Subsets. Frontiers in immunology. 2016;7:545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biron CA, Byron KS, and Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med 1989;320(26):1731–5. [DOI] [PubMed] [Google Scholar]
- 5.Yokoyama WM, Kim S, and French AR. The dynamic life of natural killer cells. Annual review of immunology. 2004;22:405–29. [DOI] [PubMed] [Google Scholar]
- 6.de Vries E, Koene HR, Vossen JM, Gratama JW, von dem Borne AE, Waaijer JL, et al. Identification of an unusual Fc gamma receptor IIIa (CD16) on natural killer cells in a patient with recurrent infections. Blood. 1996;88(8):3022–7. [PubMed] [Google Scholar]
- 7.Jawahar S, Moody C, Chan M, Finberg R, Geha R, and Chatila T. Natural Killer (NK) cell deficiency associated with an epitope-deficient Fc receptor type IIIA (CD16-II). Clin Exp Immunol 1996;103(3):408–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 2008;9(5):495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mace EM, Dongre P, Hsu HT, Sinha P, James AM, Mann SS, et al. Cell biological steps and checkpoints in accessing NK cell cytotoxicity. Immunol Cell Biol 2014;92(3):245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang D, Feng J, Wen R, Marine JC, Sangster MY, Parganas E, et al. Phospholipase Cgamma2 is essential in the functions of B cell and several Fc receptors. Immunity. 2000;13(1):25–35. [DOI] [PubMed] [Google Scholar]
- 11.Tassi I, Presti R, Kim S, Yokoyama WM, Gilfillan S, and Colonna M. Phospholipase C-gamma 2 is a critical signaling mediator for murine NK cell activating receptors. Journal of immunology (Baltimore, Md : 1950). 2005;175(2):749–54. [DOI] [PubMed] [Google Scholar]
- 12.Caraux A, Kim N, Bell SE, Zompi S, Ranson T, Lesjean-Pottier S, et al. Phospholipase C-gamma2 is essential for NK cell cytotoxicity and innate immunity to malignant and virally infected cells. Blood. 2006;107(3):994–1002. [DOI] [PubMed] [Google Scholar]
- 13.Garcia P, Gupta R, Shah S, Morris AJ, Rudge SA, Scarlata S, et al. The pleckstrin homology domain of phospholipase C-delta 1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes. Biochemistry. 1995;34(49):16228–34. [DOI] [PubMed] [Google Scholar]
- 14.Volmering S, Block H, Boras M, Lowell CA, and Zarbock A. The Neutrophil Btk Signalosome Regulates Integrin Activation during Sterile Inflammation. Immunity. 2016;44(1):73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obba S, Hizir Z, Boyer L, Selimoglu-Buet D, Pfeifer A, Michel G, et al. The PRKAA1/AMPKalpha1 pathway triggers autophagy during CSF1-induced human monocyte differentiation and is a potential target in CMML. Autophagy. 2015;11(7):1114–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Sohn H, Sun G, Milner JD, and Pierce SK. The autoinhibitory C-terminal SH2 domain of phospholipase C-gamma2 stabilizes B cell receptor signalosome assembly. Sci Signal. 2014;7(343):ra89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milner JD. PLAID: a Syndrome of Complex Patterns of Disease and Unique Phenotypes. J Clin Immunol 2015;35(6):527–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med 2012;366(4):330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Q, Lee GS, Brady J, Datta S, Katan M, Sheikh A, et al. A hypermorphic missense variant in PLCG2, encoding phospholipase Cgamma2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet 2012;91(4):713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang P, Bigio B, Rapaport F, Zhang SY, Casanova JL, Abel L, et al. PopViz: a webserver for visualizing minor allele frequencies and damage prediction scores of human genetic variations. Bioinformatics. 2018;34(24):4307–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rieux-Laucat F, and Casanova JL. Immunology. Autoimmunity by haploinsufficiency. Science (New York, NY). 2014;345(6204):1560–1. [DOI] [PubMed] [Google Scholar]
- 22.Kouchi Z, Shikano T, Nakamura Y, Shirakawa H, Fukami K, and Miyazaki S. The role of EF-hand domains and C2 domain in regulation of enzymatic activity of phospholipase Czeta. The Journal of biological chemistry. 2005;280(22):21015–21. [DOI] [PubMed] [Google Scholar]
- 23.Nomikos M, Sanders JR, Parthimos D, Buntwal L, Calver BL, Stamatiadis P, et al. Essential Role of the EF-hand Domain in Targeting Sperm Phospholipase Czeta to Membrane Phosphatidylinositol 4,5-Bisphosphate (PIP2). The Journal of biological chemistry. 2015;290(49):29519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ueno H T follicular helper cells in human autoimmunity. Curr Opin Immunol 2016;43:24–31. [DOI] [PubMed] [Google Scholar]
- 25.Fogel LA, Yokoyama WM, and French AR. Natural killer cells in human autoimmune disorders. Arthritis research & therapy. 2013;15(4):216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ueda Y, Hirai S, Osada S, Suzuki A, Mizuno K, and Ohno S. Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. The Journal of biological chemistry. 1996;271(38):23512–9. [DOI] [PubMed] [Google Scholar]
- 27.Upshaw JL, Schoon RA, Dick CJ, Billadeau DD, and Leibson PJ. The isoforms of phospholipase C-gamma are differentially used by distinct human NK activating receptors. Journal of immunology (Baltimore, Md : 1950). 2005;175(1):213–8. [DOI] [PubMed] [Google Scholar]
- 28.Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res 2009;69(9):4010–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Handa N, Takagi T, Murayama K, Terada T, Shirouzu M, and Yokoyama S. In: (RSGI) SGPI ed. RCSB PDB; 2006. [Google Scholar]
- 31.Bunney TD, Esposito D, Mas-Droux C, Lamber E, Baxendale RW, Martins M, et al. Structural and functional integration of the PLCgamma interaction domains critical for regulatory mechanisms and signaling deregulation. Structure. 2012;20(12):2062–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zimmerman MI, Hart KM, Sibbald CA, Frederick TE, Jimah JR, Knoverek CR, et al. Prediction of New Stabilizing Mutations Based on Mechanistic Insights from Markov State Models. ACS Cent Sci 2017;3(12):1311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hart KM, Ho CM, Dutta S, Gross ML, and Bowman GR. Modelling proteins’ hidden conformations to predict antibiotic resistance. Nat Commun 2016;7:12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lappalainen I, Thusberg J, Shen B, and Vihinen M. Genome wide analysis of pathogenic SH2 domain mutations. Proteins. 2008;72(2):779–92. [DOI] [PubMed] [Google Scholar]
- 35.Bae JH, Lew ED, Yuzawa S, Tome F, Lax I, and Schlessinger J. The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell. 2009;138(3):514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quann EJ, Merino E, Furuta T, and Huse M. Localized diacylglycerol drives the polarization of the microtubule-organizing center in T cells. Nat Immunol 2009;10(6):627–35. [DOI] [PubMed] [Google Scholar]
- 37.Beal AM, Anikeeva N, Varma R, Cameron TO, Vasiliver-Shamis G, Norris PJ, et al. Kinetics of early T cell receptor signaling regulate the pathway of lytic granule delivery to the secretory domain. Immunity 2009;31(4):632–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 2013;31(6):545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopez-Verges S, Milush JM, Pandey S, York VA, Arakawa-Hoyt J, Pircher H, et al. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood. 2010;116(19):3865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guma M, Angulo A, Vilches C, Gomez-Lozano N, Malats N, and Lopez-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood. 2004;104(12):3664–71. [DOI] [PubMed] [Google Scholar]
- 41.Cerwenka A, and Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nature reviews Immunology. 2016;16(2):112–23. [DOI] [PubMed] [Google Scholar]
- 42.Muntasell A, Vilches C, Angulo A, and Lopez-Botet M. Adaptive reconFigureuration of the human NK-cell compartment in response to cytomegalovirus: a different perspective of the host-pathogen interaction. European journal of immunology. 2013;43(5):1133–41. [DOI] [PubMed] [Google Scholar]
- 43.Matalon O, Fried S, Ben-Shmuel A, Pauker MH, Joseph N, Keizer D, et al. Dephosphorylation of the adaptor LAT and phospholipase C-gamma by SHP-1 inhibits natural killer cell cytotoxicity. Sci Signal. 2016;9(429):ra54. [DOI] [PubMed] [Google Scholar]
- 44.Paz PE, Wang S, Clarke H, Lu X, Stokoe D, and Abo A. Mapping the Zap-70 phosphorylation sites on LAT (linker for activation of T cells) required for recruitment and activation of signalling proteins in T cells. The Biochemical journal. 2001;356(Pt 2):461–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bryceson YT, Rudd E, Zheng C, Edner J, Ma D, Wood SM, et al. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood. 2007;110(6):1906–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mah AY, Rashidi A, Keppel MP, Saucier N, Moore EK, Alinger JB, et al. Glycolytic requirement for NK cell cytotoxicity and cytomegalovirus control. JCI Insight. 2017;2(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv. 2019:531210. [Google Scholar]
- 48.Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function ››s. Blood. 2015;125(4):591–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014;312(18):1870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369(16):1502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol 2018;38(1):96–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.George MR. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med 2014;5:69–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hughes CR, Guasti L, Meimaridou E, Chuang CH, Schimenti JC, King PJ, et al. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. The Journal of clinical investigation. 2012;122(3):814–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gineau L, Cognet C, Kara N, Lach FP, Dunne J, Veturi U, et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. The Journal of clinical investigation. 2012;122(3):821–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mace EM, Hsu AP, Monaco-Shawver L, Makedonas G, Rosen JB, Dropulic L, et al. Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset. Blood. 2013;121(14):2669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mace EM, Bigley V, Gunesch JT, Chinn IK, Angelo LS, Care MA, et al. Biallelic mutations in IRF8 impair human NK cell maturation and function. The Journal of clinical investigation. 2017;127(1):306–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Banerjee PP, and Orange JS. Quantitative measurement of F-actin accumulation at the NK cell immunological synapse. J Immunol Methods. 2010;355(1–2):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mentlik AN, Sanborn KB, Holzbaur EL, and Orange JS. Rapid lytic granule convergence to the MTOC in natural killer cells is dependent on dynein but not cytolytic commitment. Mol Biol Cell. 2010;21(13):2241–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Newell EW, Sigal N, Bendall SC, Nolan GP, and Davis MM. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012;36(1):142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science (New York, NY). 2011;332(6030):687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kotecha N, Krutzik PO, and Irish JM. Web-based analysis and publication of flow cytometry experiments. Curr Protoc Cytom 2010;Chapter 10:Unit10 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hui-Yuen J, McAllister S, Koganti S, Hill E, and Bhaduri-McIntosh S. Establishment of Epstein-Barr virus growth-transformed lymphoblastoid cell lines. J Vis Exp 2011(57). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robert X, and Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 2014;42(Web Server issue):W320–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Notredame C, Higgins DG, and Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. Journal of molecular biology. 2000;302(1):205–17. [DOI] [PubMed] [Google Scholar]
- 65.Moretti S, Armougom F, Wallace IM, Higgins DG, Jongeneel CV, and Notredame C. The M-Coffee web server: a meta-method for computing multiple sequence alignments by combining alternative alignment methods. Nucleic Acids Res 2007;35(Web Server issue):W645–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abraham MJ, Murtola T, Schulz R, Szilard P, Smith JC, Hess B, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1(2):19–25. [Google Scholar]
- 67.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem 2003;24(16):1999–2012. [DOI] [PubMed] [Google Scholar]
- 68.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 1983;79(2):926–35. [Google Scholar]
- 69.Hess B, Bekker H, Berendsen H, and Fraaojie J. LINCS: A Linear Constraint Solver for Molecular Simulations. Journal of Computational Chemistry. 1997;18(12):1463–72. [Google Scholar]
- 70.Bussi G, Donadio D, and Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys 2007;126(1):014101. [DOI] [PubMed] [Google Scholar]
- 71.Beauchamp KA, Bowman GR, Lane TJ, Maibaum L, Haque IS, and Pande VS. MSMBuilder2: Modeling Conformational Dynamics at the Picosecond to Millisecond Scale. J Chem Theory Comput 2011;7(10):3412–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bowman GR, Pande VS, and Noe F. An Introduction to Markov State Models and Their Application to Long Timescale Molecular Simulation. New York, NY: Springer Science & Business Media; 2013. [Google Scholar]
- 73.Baker NA, Sept D, Joseph S, Holst MJ, and McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98(18):10037–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
With the exception of whole-exome sequencing data, the distribution of which may compromise research participant privacy and/or consent, the data that support the findings of this study are available from the corresponding author upon reasonable request.