

Abstract
BACKGROUND
CEP290-associated inherited retinal degeneration causes severe early-onset vision loss due to pathogenic variants in CEP290. EDIT-101 is a clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) gene-editing complex designed to treat inherited retinal degeneration caused by a specific damaging variant in intron 26 of CEP290 (IVS26 variant).
METHODS
We performed a phase 1–2, open-label, single-ascending-dose study in which persons 3 years of age or older with CEP290-associated inherited retinal degeneration caused by a homozygous or compound heterozygous IVS26 variant received a subretinal injection of EDIT-101 in the worse (study) eye. The primary outcome was safety, which included adverse events and dose-limiting toxic effects. Key secondary efficacy outcomes were the change from baseline in the best corrected visual acuity, the retinal sensitivity detected with the use of full-field stimulus testing (FST), the score on the Ora–Visual Navigation Challenge mobility test, and the vision-related quality-of-life score on the National Eye Institute Visual Function Questionnaire–25 (in adults) or the Children’s Visual Function Questionnaire (in children).
RESULTS
EDIT-101 was injected in 12 adults 17 to 63 years of age (median, 37 years) at a low dose (in 2 participants), an intermediate dose (in 5), or a high dose (in 5) and in 2 children 9 and 14 years of age at the intermediate dose. At baseline, the median best corrected visual acuity in the study eye was 2.4 log10 of the minimum angle of resolution (range, 3.9 to 0.6). No serious adverse events related to the treatment or procedure and no dose-limiting toxic effects were recorded. Six participants had a meaningful improvement from baseline in cone-mediated vision as assessed with the use of FST, of whom 5 had improvement in at least one other key secondary outcome. Nine participants (64%) had a meaningful improvement from baseline in the best corrected visual acuity, the sensitivity to red light as measured with FST, or the score on the mobility test. Six participants had a meaningful improvement from baseline in the vision-related quality-of-life score.
CONCLUSIONS
The safety profile and improvements in photoreceptor function after EDIT-101 treatment in this small phase 1–2 study support further research of in vivo CRISPR-Cas9 gene editing to treat inherited retinal degenerations due to the IVS26 variant of CEP290 and other genetic causes. (Funded by Editas Medicine and others; BRILLIANCE ClinicalTrials.gov number, NCT03872479.)
INHERITED RETINAL DEGENERATIONS ARE mendelian disorders in which pathogenic variants of more than 280 genes cause dysfunction and death of the photoreceptor cells of the retina, resulting in visual impairment.1 Inherited retinal degenerations affect persons across the age spectrum and are a leading cause of blindness worldwide.2,3 Pathogenic variants of the gene encoding centrosomal protein 290 (CEP290) cause CEP290-associated inherited retinal degeneration, a severe condition associated with visual impairment during the first decade of life. Pathogenic variants in CEP290 are the most common cause of childhood genetic retinal blindness, historically known as Leber’s congenital amaurosis.4 The most common of the variants known to cause CEP290-associated inherited retinal degeneration is the c.2991+1655A→G (p.Cys998X) variant in intron 26 of the CEP290 gene (IVS26 variant), which is present in up to 77% of persons with this disease in the United States.5 This intronic mutation results in the inclusion of a cryptic exon in the CEP290 messenger RNA that disrupts the expression of CEP290, which is critical for the formation and function of photoreceptor sensory cilia (Fig. S1 in the Supplementary Appendix, which is available with the full text of this article at NEJM.org).6–8 No treatment has been approved for CEP290-associated inherited retinal degeneration.9 Supportive interventions are the standard of care and include the use of glasses, magnifiers, canes, and Braille, as well as modifications to the home.10
CEP290-associated inherited retinal degeneration is characterized by disorganized outer segments of rod and cone photoreceptors and early death of rods in the midperipheral retina, with the retention of cones in the macula (most central part of the retina).11–13 The optic nerves and occipital cortices may also remain structurally intact despite the profoundly reduced input from rod and cone photoreceptors.14 This suggests that there may be an opportunity to intervene and target spared photoreceptors in order to restore vision. Experimental approaches to treatment include the use of antisense oligonucleotides to suppress the inclusion of the cryptic exon by the IVS26 variant and the delivery of the CEP290 gene in miniaturized versions because its coding sequence is too large for adeno-associated virus (AAV)–mediated gene augmentation.15–17
EDIT-101 is a gene-editing therapy mediated by clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) that is designed to permanently remove the CEP290 IVS26 variant.18 EDIT-101 uses an AAV5 vector with high tropism for photoreceptors that comprises DNA encoding a Staphylococcus aureus Cas9 (SaCas9) nuclease expressed with the photoreceptor-specific GRK1 promoter and two highly specific guide RNAs (Fig. S2). In the BRILLIANCE study, we assessed the safety and efficacy of a single escalating dose of EDIT-101 in patients with CEP290-associated inherited retinal degeneration caused by a homozygous or compound heterozygous CEP290 IVS26 variant.
METHODS
STUDY OVERSIGHT
We conducted this phase 1–2, open-label, single-ascending-dose study in accordance with the Good Clinical Practice guidelines of the International Council for Harmonisation and the principles of the Declaration of Helsinki. The trial protocol (available at NEJM.org) was approved by the institutional review board at each of the five study sites (see the Supplementary Appendix). Editas Medicine was the study sponsor, and an independent data and safety monitoring committee provided oversight. The authors vouch for the accuracy and completeness of the data and for the fidelity of the study to the protocol.
PARTICIPANTS AND TREATMENT
Eligible participants were at least 3 years of age at the time of screening and had CEP290-associated inherited retinal degeneration with homozygosity for the CEP290 IVS26 variant or compound heterozygosity for the CEP290 IVS26 variant and another variant. Additional inclusion criteria are listed in the Supplementary Appendix. Informed consent was obtained from all the participants or their legal representative before the initiation of study activities.
Participants received oral prednisone (0.5 mg per kilogram of body weight per day) beginning 3 days before the injection of EDIT-101. Participants then underwent a standard pars plana vitrectomy and received a single subretinal injection of up to 300 μl of EDIT-101 in the eye with the worse vision (study eye). EDIT-101 was administered to adult participants at a low dose (6×1011 vector genomes [vg] per milliliter; cohort 1), an intermediate dose (1×1012 vg per milliliter; cohort 2), or a high dose (3×1012 vg per milliliter; cohort 3), and the study drug was administered to pediatric participants at the intermediate dose (cohort 4). An excerpt from a representative surgical procedure is shown in the video. Prednisone therapy was administered for 4 weeks after surgery, followed by a 15-day period during which the dose was changed or tapered according to the investigator’s judgment.19 Participants were then monitored every 3 months for 1 year, followed by less frequent monitoring for 2 years (Fig. S3).
Owing to circumstances unrelated to the study, the sponsor paused enrollment. We report here the results of an interim analysis.
STUDY OUTCOMES
The primary outcome of the study was safety, which included treatment-related adverse events, procedure-related adverse events, and dose-limiting toxic effects. Key secondary efficacy outcomes included the change from baseline in the best corrected visual acuity (lower values indicate better visual acuity); the retinal sensitivities to blue light, red light, and white light as measured with dark-adapted full-field stimulus testing (lower values indicate better retinal sensitivity to light)20,21; the score on the Ora–Visual Navigation Challenge (VNC) mobility test (higher scores indicate better vision-related mobility); and the vision-related quality-of-life score as assessed with the use of the National Eye Institute Visual Function Questionnaire–25 (NEI VFQ-25) or the Children’s Visual Function Questionnaire (higher scores indicate better visual function).
On the basis of guidance from the Food and Drug Administration,22 a change from baseline of at least 0.3 log10 of the minimum angle of resolution (logMAR) in the best corrected visual acuity (corresponding to ≥15 letters on the Early Treatment Diabetic Retinopathy Study [ETDRS] chart) was considered to be clinically meaningful. The best corrected visual acuity of participants with reduced acuity (<1.6 logMAR) who could not read letters on the ETDRS chart was assessed with the use of the Berkeley Rudimentary Vision Test.23 For this test, a best corrected visual acuity of 3.9 logMAR is equivalent to light perception; 3.5, to black–white discrimination; and 3.2, to white field projection.
A change from baseline of at least 0.6 log cd-sec per square meter in the retinal sensitivity detected with full-field stimulus testing was considered to be visually meaningful (clinically meaningful with respect to vision) on the basis of previous studies of CEP290-associated inherited retinal degeneration and RPE65-associated inherited retinal degeneration13,16,20,24–26 and a previous natural history study (ClinicalTrials.gov number, NCT03396042).27 The estimated loss of dark-adapted cone-mediated sensitivity was calculated as the difference between the sensitivity deter-mined with the use of the red stimulus in the study participants and the mean sensitivity measured at the cone-plateau phase of the dark-adaptation function in healthy persons.16,28,29
Visual function navigation was assessed with the use of the VNC, a four-course, multiluminance mobility test (see the Supplementary Methods and Fig. S4). A change from baseline of at least 3 points in the score on the VNC was considered to be visually meaningful on the basis of the natural history study. Vision-related quality of life was assessed in the adult participants with the use of the NEI VFQ-25.30 Vision-related quality of life in was assessed in the pediatric participants with the use of the Children’s Visual Function Questionnaire.31 Improvement in the score on each questionnaire was defined as an increase of at least 4 points from baseline.30
Additional secondary outcomes included pupillary responses as assessed with a hand-held pupillometer and the thickness of the photoreceptor-cell layer in the fovea (central macula) as measured with in vivo microscopy with optical coherence tomography (OCT). Assessment of the kinetic visual field and measurements of contrast sensitivity and color vision were also attempted (see the Supplementary Methods).
STATISTICAL ANALYSIS
Because CEP290-associated inherited retinal degeneration is an ultrarare disease, no power analysis was performed to determine the sample size. The widths of the confidence intervals were not adjusted for multiplicity and were not used for hypothesis testing. All computations were performed with the use of SAS and R software.
RESULTS
PARTICIPANTS
Fourteen participants were treated with EDIT-101 (Fig. S5). The median age among adults was 37 years (range, 17 to 63), and the two enrolled children were 9 and 14 years of age (Table 1). One of the adult participants turned 18 years of age after consent was initially provided, and the participant provided consent as an adult before receiving treatment. All the participants were non-Hispanic and White (as reported by the participant or their legal representative), and 36% were male. The study participants were representative of the population with CEP290-associated inherited retinal degeneration, which is largely of European descent and has a wide age spectrum (Table S1). Two participants were homozygous for the CEP290 IVS26 variant.
Table 1.
Characteristics of the Participants at Baseline.*
| Participant | Sex | Age | Zygosity (Second Allele)† | Best Corrected FST Sensitivity | Red-Light Visual Acuity‡ | ||
|---|---|---|---|---|---|---|---|
| Study Eye | Control Eye | Study Eye | Control Eye | ||||
| yr | logMAR | log cd-sec/m 2 | |||||
| Cohort 1 | |||||||
| C1P1 | Female | 50 | CH (c.1445T→A) | 3.5 | 3.5 | −2.0 | Limited data |
| C1P2 | Male | 42 | CH (c.5587–1G→C) | 3.9 | 3.9 | Limited data | Limited data |
| Cohort 2 | |||||||
| C2P1 | Female | 54 | Homozygous | 2.7 | 2.3 | −1.7 | −1.8 |
| C2P2 | Male | 20 | CH (c.508A→T) | 1.4 | 1.4 | −2.1 | −2.4 |
| C2P3 | Female | 19 | CH (c.7344_7345delTT) | 0.6 | 0.5 | −3.2 | −3.2 |
| C2P4 | Female | 63 | CH (c.5668G→T) | 0.9 | 0.6 | −3.9 | −3.8 |
| C2P5 | Female | 17§ | CH (c.3123dupT) | 3.9 | 3.9 | −0.5 | −0.5 |
| Cohort 3 | |||||||
| C3P1 | Female | 28 | CH (c.4723A→T) | 2.3 | 1.6 | −1.5 | −1.9 |
| C3P2 | Female | 38 | CH (c.5668G→T) | 1.0 | 0.9 | −3.7 | −3.7 |
| C3P3 | Female | 36 | CH (c.4661_4663delAAG) | 3.9 | 3.9 | −0.6 | −0.6 |
| C3P4 | Male | 35 | CH (c.1666delA) | 2.0 | 0.7 | −1.6 | −2.2 |
| C3P5 | Female | 59 | CH (c.5788_5792delAAAGA) | 2.9 | 2.3 | −1.3 | −1.8 |
| Cohort 4 | |||||||
| C4P1 | Male | 14 | Homozygous | 3.9 | 3.9 | −1.3 | −1.2 |
| C4P2 | Male | 9 | CH (c.4723A→T) | 1.2 | 1.2 | −2.6 | −3.0 |
Cohort 1 comprised adults who received a low dose (6×1011 vector genomes [vg] per milliliter) of EDIT-101 gene therapy in the worse (study) eye; cohort 2, adults who received an intermediate dose (1×1012 vg per milliliter); cohort 3, adults who received a high dose (3×1012 vg per milliliter); and cohort 4, children who received an intermediate dose. The contralateral (control) eye was untreated. Participant identifiers are defined according to cohort (C) number and participant (P) number. FST denotes full-field stimulus testing.
The second allele is provide for participants with compound heterozygosity (CH).
A best corrected visual acuity of 3.9 log10 of the minimum angle of resolution (logMAR) is equivalent to light perception; 3.5 logMAR, to black–white discrimination; and 3.2 logMAR, to white field projection.
The participant turned 18 years of age after consent was initially provided and provided consent as an adult before receiving treatment.
At baseline, there was a broad range in the best corrected visual acuity (from 0.6 to 3.9 [light perception] logMAR) and in the sensitivity to red light as measured with full-field stimulus testing (−0.6 to −3.9 log10 cd-sec per square meter) among the study eyes. Most participants had a severe loss of visual acuity (<1.6 logMAR), which led to the use of the Berkeley Rudimentary Vision Test to determine their spatial discrimination (Table 1).23
Baseline differences in spectral sensitivity (red–blue) as measured with full-field stimulus testing showed elevated thresholds (by at least 3 log units) and a total loss of rod photoreceptor function in all the participants.20 The thickness of the outer nuclear layer of photoreceptor cells in the fovea, as measured on OCT scans, was within normal limits or within 10 μm of the lower limit of the normal range in most participants and eyes with quantifiable images (except in Participant 2 in cohort 1 [C1P2] and Participant C3P1), a finding that was consistent with earlier reports and confirmed the dissociation between structure and function in this genetic form of inherited retinal degeneration.13
As of February 2023, the median follow-up was 376 days (interquartile range, 193 to 454) (Fig. S6). Participant C1P1 could not be followed up at the study site because of a mask mandate during the coronavirus disease 2019 pandemic. This participant is undergoing follow-up for safety at a nonstudy site, with no safety issues having been reported.
SAFETY
Adverse Events and Dose-Limiting Toxic Effects
No ocular serious adverse events related to the study treatment or dose-limiting toxic effects occurred in the study participants. Four nonocular serious adverse events occurred — one participant with a clinical history of epilepsy had two seizure events and one major depressive episode, and one participant had an unintentional injury to a digit. Most adverse events were mild (76%) or moderate (22%) in severity, and 41% were related to the procedure.
A total of 22 ocular adverse events related to the study treatment occurred in the participants (Table 2), with the most notable event being visual impairment (in Participants C3P1 and C3P4). In Participant C3P4, the impairment, which occurred after surgery, was due to a vitreous hemorrhage attributed to the study procedure and has resolved. Two adult participants who received the high dose of EDIT-101 had subretinal hyperreflective mounds on OCT imaging at 3 to 6 months after treatment (Fig. S7). Participant C3P4 did not have changes in the best corrected visual acuity when the hyperreflective mounds were present, and the participant’s condition improved without glucocorticoid therapy. Participant C3P1 had reduced vision associated with the mounds, and vision improved after a course of oral glucocorticoid treatment. Participants C2P5 and C3P5 had retinal pigment epitheliopathy in the area of the subretinal injection (Fig. S8), which was not associated with changes in photoreceptor function or vision. Participant C1P2 underwent a second surgery to treat persistent subretinal fluid and a dislocated intraocular lens.
Table 2.
Ocular Adverse Events Related to EDIT-101.*
| Adverse Event | Overall (N = 14) | Cohort 1 (N = 2) | Cohort 2 (N = 5) | Cohort 3 (N = 5) | Cohort 4 (N = 2) |
|---|---|---|---|---|---|
| number of participants (percent) | |||||
| Any ocular event | 7 (50) | 1 (50) | 3 (60) | 3 (60) | 0 |
| Anterior chamber cell | 1 (7) | 0 | 0 | 1 (20) | 0 |
| Anterior chamber inflammation | 2 (14) | 1 (50) | 1 (20)† | 0 | 0 |
| Conjunctival edema | 1 (7) | 1 (50) | 0 | 0 | 0 |
| Cystoid macular edema | 1 (7) | 0 | 0 | 1 (20)‡ | 0 |
| Conjunctival hyperemia | 1 (7) | 1 (50) | 0 | 0 | 0 |
| Photophobia | 1 (7) | 0 | 1 (20) | 0 | 0 |
| Eye inflammation | 1 (7) | 1 (50) | 0 | 0 | 0 |
| Retinal degeneration | 1 (7) | 0 | 0 | 1 (20)‡ | 0 |
| Retinal deposits | 1 (7) | 0 | 0 | 1 (20)‡ | 0 |
| Retinal drusen | 1 (7) | 0 | 1 (20)‡ | 0 | 0 |
| Retinal infiltrates | 1 (7) | 1 (50) | 0 | 0 | 0 |
| Retinal pigment epithelial tear | 1 (7) | 1 (50) | 0 | 0 | 0 |
| Retinal pigment epitheliopathy | 2 (14) | 0 | 1 (20)‡ | 1 (20)§ | 0 |
| Subretinal hyperreflective exudation | 1 (7) | 0 | 0 | 1 (20)‡ | 0 |
| Visual impairment | 2 (14) | 0 | 0 | 2 (40)¶ | 0 |
| Vitritis | 1 (7) | 1 (50)‖ | 0 | 0 | 0 |
| Vitreal cells | 1 (7) | 0 | 0 | 1 (20) | 0 |
Ocular treatment-related adverse events are presented according to system organ class and Medical Dictionary for Regulatory Activities, version 23.1, preferred term. The reported adverse events were recorded between the time of study initiation (February 2020) and the data cutoff date (August 2, 2023). One additional participant (C3P4) had an episode of subretinal deposits that was not captured in the adverse event database as of the August 2023 data-cutoff date.
Two cases of anterior chamber inflammation were reported in this participant, and one of the events remained unresolved as of August 2023.
The adverse event in this participant remained unresolved as of August 2023.
The resolution of retinal pigment epitheliopathy in this participant remained unknown as of August 2023.
Visual impairment in one of the participants remained unresolved as of August 2023.
Two cases of vitritis were reported in this participant.
Biodistribution and Immunogenicity
Viral genomes were detected in tears, nasal mucosa, and blood in 93%, 29%, and 36% of the participants, respectively. No viral genomes were detected in semen. Viral shedding resolved within 7 days after treatment in most participants and by month 3 in all the participants (Fig. S9). Preexisting immune responses to AAV5 and SaCas9 were detected in 64% and 36% of the participants, respectively. Most of the participants with preexisting immunity had an immune response after treatment. The two participants without preexisting immunity to AAV5 (C1P1 and C2P3) also had immune responses to AAV5 after treatment (Fig. S10). No SaCas9-binding antibodies were detected before or after treatment. AAV5-binding antibody and neutralizing antibody titers showed small increases (not exceeding 105) after treatment.
EFFICACY
Best Corrected Visual Acuity
The mean change from baseline in the best corrected visual acuity was −0.21 logMAR (90% confidence interval [CI], −0.45 to 0.02) in the study eyes and −0.01 logMAR (90% CI, −0.09 to 0.06) in the untreated contralateral (control) eyes. Four of 14 participants (29%) had improvement that reached the prespecified threshold for clinically meaningful improvement of at least 0.3 logMAR (Fig. 1B, Fig. S11, and Table S3). The acuity in these four participants (C1P1, C2P1, C3P5, and C4P1) was measured with the Berkeley Rudimentary Vision Test. Three of these participants had a response by month 3. Transient improvement was also observed in the control eyes in Participants C4P1 and C1P2. None of the participants with better vision (defined as a best corrected visual acuity of ≤1.4 logMAR) had improvement in the best corrected visual acuity.
Figure 1. Change from Baseline in Key Efficacy Outcomes.
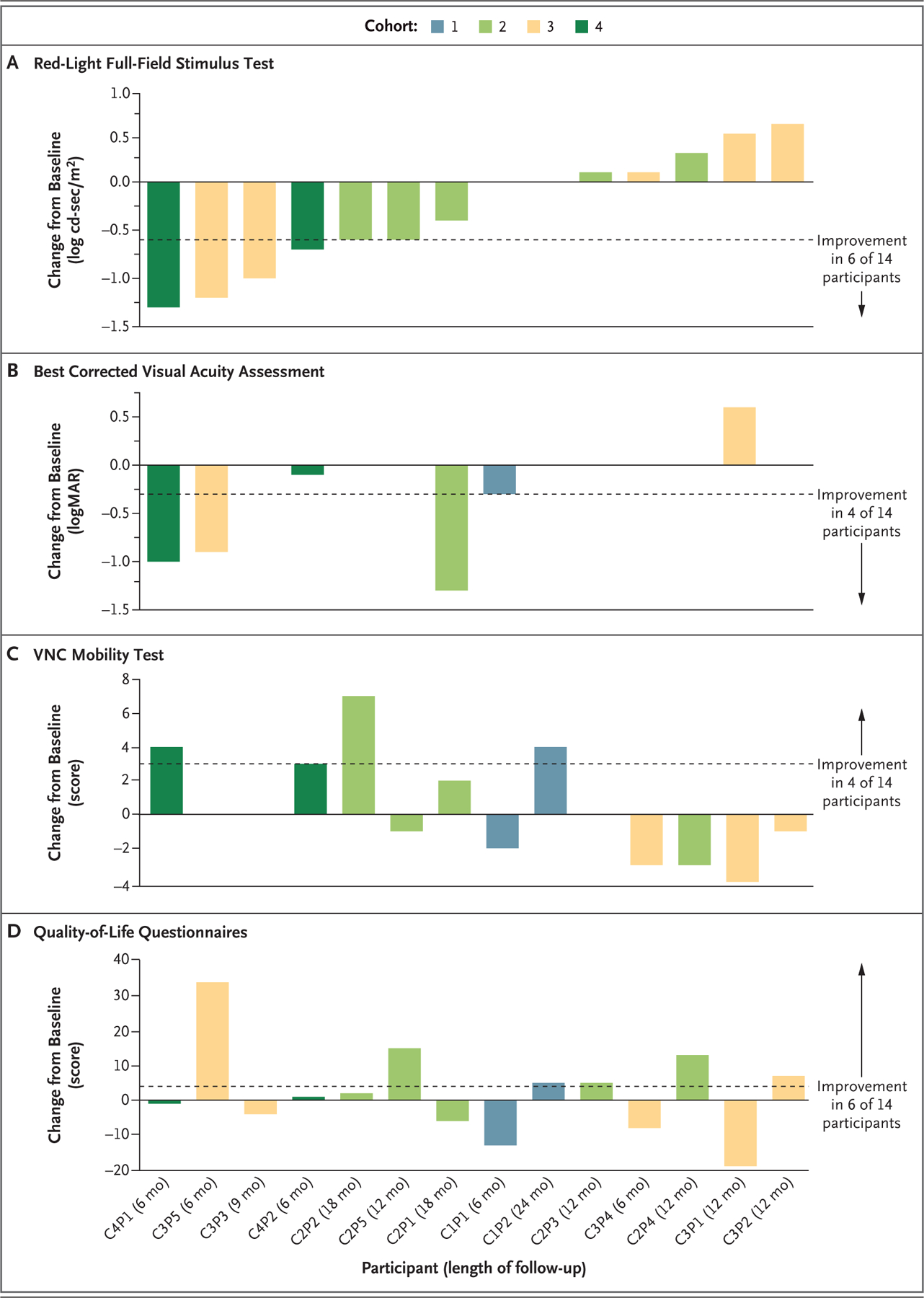
Changes from baseline to the latest follow-up assessment in the sensitivity to red light as measured with full-field stimulus testing (Panel A), the best corrected visual acuity (Panel B), the score on the Ora–Visual Navigation Challenge (VNC) mobility test (Panel C), and the vision-related quality-of-life score on the National Eye Institute Visual Function Questionnaire–25 (for adults) or the Children’s Visual Function Questionnaire (for children) (Panel D) are shown for 14 participants who received EDIT-101 gene-editing therapy and had a follow-up duration of at least 6 months. Cohort 1 comprised adults who received a low dose (6×1011 vector genomes [vg] per milliliter) of EDIT-101 in the worse (study) eye; cohort 2, adults who received an intermediate dose (1×1012 vg per milliliter); cohort 3, adults who received a high dose (3×1012 vg per milliliter); and cohort 4, children who received the intermediate dose. The contralateral (control) eye was untreated. Improvements in the full-field red light–sensitivity threshold and the best corrected visual acuity are shown as negative values, and improvements in the score on the VNC mobility test and the score on the vision-related quality-of-life questionnaires are shown as positive values. The dashed lines indicate the thresholds for meaningful improvement. The direction of improvement and the number of participants with improvement for each metric are indicated to the right of each panel. Participant identifiers (defined according to cohort [C] number and participant [P] number) and lengths of follow-up are shown on the x axis. Data from the red-light full-field stimulus test for Participant C1P2 were not available. logMAR denotes log10 of the minimum angle of resolution.
Full-Field Stimulus Testing
We assessed the retinal sensitivities to light with the use of dark-adapted full-field stimulus testing. The mean change from baseline in the sensitivity threshold (more negative values indicate improved thresholds) was −0.32 log cd-sec per square meter (90% CI, −0.63 to −0.01) in the study eyes and 0.19 log cd-sec per square meter (90% CI, 0.05 to 0.34) in the control eyes in response to stimulation with red light. In response to blue light, the mean change was −0.34 log cd-sec per square meter (90% CI, −0.72 to 0.03) in the study eyes and 0.06 log cd-sec per square meter (90% CI, −0.08 to 0.19) in the control eyes. For the response to white light, the mean change was −0.23 log cd-sec per square meter (90% CI, −0.58 to 0.11) in the study eyes and 0.05 log cd-sec per square meter (90% CI, −0.11 to 0.21) in the control eyes.
Figure 1A shows the change from baseline in the retinal sensitivities detected by means of full-field stimulus testing with red light for all the participants. The sensitivity threshold improved by at least 0.6 log cd-sec per square meter in the study eyes in 6 of 14 participants (43%), a difference that is considered to be visually meaningful (Fig. S12). A visually meaningful improvement in the sensitivity threshold did not occur in any control eyes. Of the 6 participants with a visually meaningful improvement in the study eye, the threshold in Participants C3P5 and C4P1 improved by more than 1 log unit, which is close to the maximum possible cone-mediated improvement (see “Structural–Functional Relationships” below), and the threshold in 4 participants (C2P2, C3P3, C3P5, and C4P1) improved by month 3. All 6 of these participants, which included both children, received an intermediate or high dose of EDIT-101. We observed a moderate, positive correlation between changes in the best corrected visual acuity and changes in the sensitivity threshold in response to red light stimulation (Kendall’s tau=0.50) (Table S4).
Visual Function Navigation
The mean change from baseline in the score on the VNC mobility test was 0.4 points (90% CI, −1.1 to 1.9) in the study eyes and 0.7 points (90% CI, −0.5 to 2.0) in the control eyes. An improvement from baseline of at least 3 points in the score (visually meaningful difference) was observed in 4 of 14 participants (29%) and occurred at month 6 or later. Of the 4 participants with a visually meaningful improvement, Participants C1P2 and C2P2 had greater improvement in the study eye, and Participants C4P1 and C4P2 had equal improvement in both eyes (Fig. 1C and Fig. S13). Two participants with a visually meaningful improvement navigated courses that were more complex than the courses at baseline, and the visually meaningful improvement in one participant was sustained for at least 2 years. Both pediatric participants had an improvement of at least 3 points. Three of the 6 participants with a visually meaningful improvement from baseline in retinal sensitivity to red light also had a visually meaningful improvement from baseline in the score on the VNC mobility test. We observed a moderate correlation between the sensitivity threshold in response to red light and the VNC score (r = −0.62; 95% CI, −0.11 to −0.87) (Table S4).
Vision-Related Quality of Life
The mean change from baseline in the vision-related quality-of-life score was 2.3 (90% CI, −3.9 to 8.6). The score in approximately 43% of the participants (6 of 14) was at least 4 points higher at follow-up than at baseline (meaningful improvement) (Fig. 1D and Fig. S14). It has recently been recognized that the NEI VFQ-25 may not effectively capture the effect of visual dysfunction on the vision-related quality of life in patients with inherited retinal degenerations.32 Additional patient-reported outcome tools have recently been developed to improve the assessment of the effects of genetic therapies for inherited retinal degenerations on patient experience.32,33 These new patient-reported outcome tools were not available at the start of the BRILLIANCE study.
Retinal Structure and Pupillary Responses
The thickness of the outer nuclear layer of foveal photoreceptors was measured when possible. Values at baseline and the latest follow-up visit in the study eyes and control eyes are shown in Table S2. The thickness of the outer nuclear layer was within the normal range in the study eye in 7 of 14 participants.13,34 Estimates of the change from baseline are also provided whenever the follow-up images could be reasonably aligned with the baseline images. These images showed no obvious changes in thickness after treatment, a finding that supports the safety of the subfoveal injections.
The study eyes in Participants C2P1, C3P3, C4P1, and C4P2 had improved pupillary responses with larger constriction amplitudes and faster kinetics than at baseline. This includes the study eyes in 3 of 6 participants (C3P3, C4P1, and C4P2) that had meaningful improvements in cone photoreceptor sensitivity as assessed with the use of full-field stimulus testing, which provides objective evidence of the beneficial changes in photoreceptor function. No control eyes had improved pupillary responses. The control eye in Participant C2P5 had a smaller pupillary response (change from baseline in the amplitude of the transient pupillary light reflex) during follow-up (Fig. S15).
Structural–Functional Relationships
Further analyses were performed in two of the six participants (C4P1 and C4P2) with correspondence between two or more efficacy outcomes. Although retinal imaging in patients with CEP290-associated inherited retinal degeneration can be challenging because of nystagmus or unstable fixation, cross-sections of OCT images for both participants showed normal retinal structure at the foveal center, with clear retention of photoreceptor cells — findings that are typical of CEP290-associated inherited retinal degeneration (Fig. 2A).13
Figure 2. Structural–Functional Relationships.
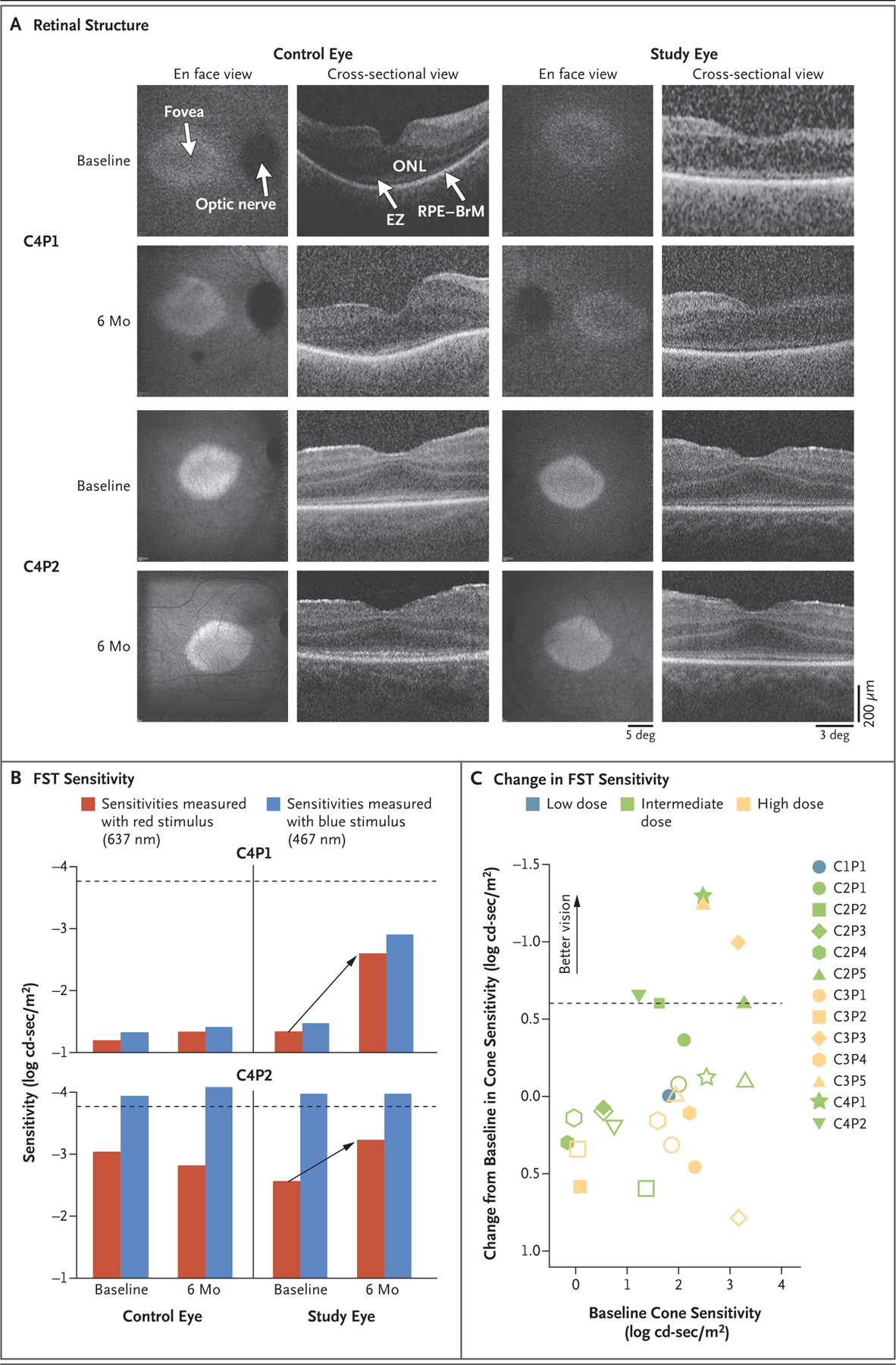
Shown in Panel A are images of the retinal structure in the two pediatric participants (C4P1 and C4P2). En face views show near-infrared fundus autofluorescence (NIR-FAF) that originates mainly from the melanin in the retinal pigment epithelium (RPE). The NIR-FAF images obtained at month 6 have been coregistered to the images obtained at baseline to allow for comparison of the images. Cross-sectional views are optical coherence tomographic scans along the horizontal meridian crossing the fovea. Lower signals in Participant C4P1 were caused by corneal scarring and cataracts resulting from eye poking. Calibration bars are shown on the lower right. BrM denotes Bruch’s membrane, EZ ellipsoid zone, and ONL outer nuclear layer. Shown in Panel B are sensitivities determined with the use of full-field stimulus testing (FST) in the study eyes and control eyes in Participants C4P1 and C4P2. Differences in spectral sensitivities showed that sensitivities in both participants were mediated by cones. Treatment with EDIT-101 led to recovery of the cone-mediated sensitivities (arrows), which were close to the lower limit of the normal range (dashed lines) by month 6. Panel C shows the change from baseline in the full-field stimulus testing sensitivity in response to red light in 13 participants with available data. Data are from the latest follow-up examination for each participant. The dashed line indicates the cutoff for meaningful improvement. Unfilled symbols indicate control eyes.
In accordance with eligibility criteria, all the participants had detectable central photoreceptors. Images that were obtained 6 months after treatment were nearly identical to images obtained at baseline, which supports the safety of the subfoveal injection. In all the participants, the thickness of the foveal outer nuclear layer in the study eye and the control eye at baseline was either near the lower limit of the normal range (81 μm, calculated as the mean – 2SD) or within the normal limits. The thickness of the foveal outer nuclear layer in the treated eyes at the latest follow-up visit remained unchanged from the thickness at baseline.14,35,36
Retinal sensitivity as measured with full-field stimulus testing originates from the most sensitive part of the preserved retina, which is near the foveal center in most patients.13,14 At baseline, differences in retinal sensitivity to stimulation with red light and blue light showed only cone-mediated sensitivities in all participants but Participant C4P2, who had mixed photoreceptor mediation (blue light perceived by rods and red light perceived by cones) (Fig. 2B).20 The loss of sensitivity to blue light was thus driven by the total or near-total loss of rod function in all the participants.
At baseline, cone-mediated sensitivities that were estimated with the use of the red stimulus were severely low in Participant C4P1, who was homozygous for the IVS26 variant. Participant C4P2 had moderately reduced cone-mediated sensitivities. Treatment led to meaningful improvements in cone-mediated sensitivities in both participants, which were within 0.5 log units of the normal range 6 months after treatment (Fig. 2B). Sensitivities in the control eyes were similar at baseline and 6 months after treatment. When extended to all the participants, the analysis showed meaningful improvements in cone-mediated sensitivities in the treated eye in six participants (Fig. 2C). The magnitude of the recovery appeared to be related to the cone-mediated sensitivity at baseline, with a greater change observed in participants with worse sensitivities, and thus greater structural–functional dissociation (i.e., participants with preserved but highly dysfunctional cone photoreceptors), at baseline. Virtually no change in cone-mediate sensitivities occurred in participants with baseline sensitivities that were close to normal.
DISCUSSION
The results of our study support the safety of EDIT-101 to the extent that safety can be assessed in a small study. The majority of treatment-related ocular adverse events were expected; the events were typical of the study procedure and concurrent medications and in line with other AAV-mediated gene therapies.37,38 An impairment in visual acuity occurred in two participants. In one, the impairment occurred postoperatively owing to vitreous hemorrhage attributed to the study procedure and has since resolved. The impairment in the other occurred at month 6 and was associated with subretinal hyperreflective mounds on OCT imaging. Vision improved after a course of glucocorticoid treatment; recovery is ongoing. This participant has a history of similar fluctuations in vision, which were recorded during a previous natural history study (NCT03396042).27 The cause of the hyperreflective mounds, which have been observed in other studies of subretinal gene therapy, is not clear.11,38 Measurements of the thickness of the foveal photoreceptor layer on OCT images did not show changes in the thickness after treatment, which supports the safety of subfoveal injections. Follow-up over a longer period will be needed to evaluate long-term risks associated with off-target effects of gene editing.
Eleven participants (79%) had improvement in at least one of four key efficacy outcomes, whereas 6 (43%) had improvement in two or more outcomes. Four participants had a clinically meaningful improvement in the best corrected visual acuity. Nearly half the participants (6 of 14) had a visually meaningful improvement in cone photoreceptor function as assessed with the use of full-field stimulus testing, of whom all but one had an improvement in at least one other outcome. Overall, meaningful improvements in vision occurred beginning at month 3 and were sustained during subsequent visits, including up to year 2 in 1 participant. These findings support the presence of productive in vivo gene editing by EDIT-101, therapeutic levels of CEP290 protein expression, and enhanced cone photoreceptor function.
All the participants had abnormal vision at baseline despite having structurally intact central photoreceptors, a structural–functional dissociation phenotype that has made CEP290-associated inherited retinal degeneration an attractive candidate for genetic interventions. We confirmed that vision at baseline was mediated by cone photoreceptors in all the participants. After treatment, improvements in cone function occurred in six participants, whereas cone-mediated sensitivities in the control eyes remained unchanged or worsened. The magnitude of improvement appeared to be related to the level of dysfunction at baseline: participants with the greatest baseline structural–functional dissociation showed the largest improvements in sensitivity. Of the six participants with improved cone-mediated sensitivities, five had improvement in at least one other outcome. Similar patterns of improvement in cone photoreceptor function but not rod photoreceptor function have been reported in patients with CEP290-associated inherited retinal degeneration treated with antisense oligonucleotides, a finding that supports the treatment potential for this condition and highlights the need to treat patients at earlier stages of the disease, when rod photoreceptors may be amenable to treatment.11,13,16
Improvements in cone function in this study and an earlier trial of antisense oligonucleotides have not consistently led to other changes in visual function attributable to cone photoreceptors, such as visual acuity.16,39 In a trial of sepofarsen, only one of five participants with light-perception vision who had improvements in cone-mediated sensitivity after treatment also had improvement in visual acuity. This participant had a history of spatial vision in early childhood, which suggests that neurodevelopmental history may influence the response to treatments such as sepofarsen and EDIT-101.16 Improvements in retinal sensitivity without changes in visual acuity were also observed in participants in a clinical trial of gene therapy for severe early-onset retinal degeneration due to pathogenic variants in GUCY2D, in which there is structural–functional dissociation similar to that in CEP290-associated inherited retinal degeneration.40,41 This discrepancy between unequivocal gains in photoreceptor function and limited improvements in spatial resolution or visual acuity warrants further study.
In preclinical models, treatment with EDIT-101 resulted in sustained CEP290 gene editing at levels that met the therapeutic threshold.18 The results of our study also suggest that the desired level of gene editing also occurs in vivo. Vision improved in participants who were heterozygous or homozygous for the CEP290 IVS26 variant, which suggests that the correction of a single variant allele can produce sufficient levels of CEP290 protein for some restoration of photoreceptor function. That each of the participants who were homozygous for the CEP290 IVS26 variant had improvement in the full-field red light–sensitivity threshold raises the possibility that targeting two alleles yields higher levels of protein expression and a potentially greater therapeutic benefit than targeting a single allele. Both pediatric participants had meaningful improvements in two or more outcomes, suggesting that earlier intervention may yield more robust results. Further study is needed to test these hypotheses.
The small sample size of this study poses limitations to the interpretation of the data, as do the absence of a sham control and the open-label design. The use of a sham control was not feasible given that the method of delivery included a subretinal injection with pars plana vitrectomy.
The results of our study suggest that EDIT-101 is safe. Preliminary evaluation of efficacy supports improved cone photoreceptor function that is consistent with on-target gene editing. These data provide proof of concept of the therapeutic potential of in vivo retinal gene editing and support further research into CRISPR-Cas9–mediated therapies for certain other inherited retinal degenerations and inherited diseases.
Supplementary Material
Acknowledgments
Supported by Editas Medicine; core grants (P30 EY014104 [to Mass Eye and Ear] and P30 EY010572) from the National Institutes of Health; the Malcolm M. Marquis, M.D., Endowed Fund for Innovation; an unrestricted grant from Research to Prevent Blindness (to the Casey Eye Institute, Oregon Health and Science University); the Irene Heinz Given and John La Porte Given Endowment; and unrestricted grants from Research to Prevent Blindness and the Paul and Evanina Mackall Foundation Trust (to the Scheie Eye Institute, Department of Ophthalmology, Perelman School of Medicine, University of Pennsylvania).
We thank the participants and their families for their continuous support of the study, the clinical study personnel at all study sites, Yan Zhao (of Mass Eye and Ear) for statistical support, and Dr. Shervonne Poleon (of Porterhouse Medical) for writing assistance with an earlier version of the manuscript.
APPENDIX
The authors’ full names and academic degrees are as follows: Eric A. Pierce, M.D., Ph.D., Tomas S. Aleman, M.D., Kanishka T. Jayasundera, M.D., Bright S. Ashimatey, O.D., Ph.D., Keunpyo Kim, Ph.D., Alia Rashid, M.D., Michael C. Jaskolka, Ph.D., Rene L. Myers, Ph.D., Byron L. Lam, M.D., Steven T. Bailey, M.D., Jason I. Comander, M.D., Ph.D., Andreas K. Lauer, M.D., Albert M. Maguire, M.D., and Mark E. Pennesi, M.D., Ph.D.
Footnotes
The authors’ full names, academic degrees, and affiliations are listed in the Appendix.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
A data sharing statement provided by the authors is available with the full text of this article at NEJM.org.
Contributor Information
E.A. Pierce, Ocular Genomics Institute, Department of Ophthalmology, Mass Eye and Ear and Harvard Medical School, Boston, Massachusetts
T.S. Aleman, Scheie Eye Institute and the Division of Ophthalmology of the Children’s Hospital of Philadelphia, Department of Ophthalmology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
K.T. Jayasundera, University of Michigan Kellogg Eye Center, Ann Arbor
B.S. Ashimatey, Editas Medicine, Cambridge, Massachusetts
K. Kim, Editas Medicine, Cambridge, Massachusetts
A. Rashid, Editas Medicine, Cambridge, Massachusetts
M.C. Jaskolka, Editas Medicine, Cambridge, Massachusetts
R.L. Myers, Editas Medicine, Cambridge, Massachusetts
B.L. Lam, Bascom Palmer Eye Institute, University of Miami, Miami
S.T. Bailey, Casey Eye Institute, Oregon Health and Science University, Portland
J.I. Comander, Ocular Genomics Institute, Department of Ophthalmology, Mass Eye and Ear and Harvard Medical School, Boston, Massachusetts
A.K. Lauer, Casey Eye Institute, Oregon Health and Science University, Portland
A.M. Maguire, Scheie Eye Institute and the Division of Ophthalmology of the Children’s Hospital of Philadelphia, Department of Ophthalmology, Perelman School of Medicine, University of Pennsylvania, Philadelphia
M.E. Pennesi, Casey Eye Institute, Oregon Health and Science University, Portland
References
- 1.Sahel J-A, Marazova K, Audo I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb Perspect Med 2014;5(2): a017111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanany M, Rivolta C, Sharon D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc Natl Acad Sci U S A 2020;117:2710–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heath Jeffery RC, Mukhtar SA, McAllister IL, Morgan WH, Mackey DA, Chen FK. Inherited retinal diseases are the most common cause of blindness in the working-age population in Australia. Ophthalmic Genet 2021;42:431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang C-H, Yang C-M, Yang C-H, Hou Y-C, Chen T-C. Leber’s congenital amaurosis: current concepts of genotype-phenotype correlations. Genes (Basel) 2021;12:1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McAnany JJ, Genead MA, Walia S, et al. Visual acuity changes in patients with Leber congenital amaurosis and mutations in CEP290. JAMA Ophthalmol 2013; 131:178–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.den Hollander AI, Koenekoop RK, Yzer S, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet 2006;79:556–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perrault I, Delphin N, Hanein S, et al. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum Mutat 2007;28:416. [DOI] [PubMed] [Google Scholar]
- 8.Burnight ER, Wiley LA, Drack AV, et al. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther 2014;21:662–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petit L, Khanna H, Punzo C. Advances in gene therapy for diseases of the eye. Hum Gene Ther 2016;27:563–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agarwal R, Tripathi A. Current modalities for low vision rehabilitation. Cureus 2021;13(7):e16561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aleman TS, O’Neil EC, Uyhazi KE, et al. Fleck-like lesions in CEP290-associated Leber congenital amaurosis: a case series. Ophthalmic Genet 2022;43:824–33. [DOI] [PubMed] [Google Scholar]
- 12.Parfitt DA, Lane A, Ramsden CM, et al. Identification and correction of mechanisms underlying inherited blindness in human iPSC-derived optic cups. Cell Stem Cell 2016;18:769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobson SG, Cideciyan AV, Sumaroka A, et al. Outcome measures for clinical trials of Leber congenital amaurosis caused by the intronic mutation in the CEP290 gene. Invest Ophthalmol Vis Sci 2017;58:2609–22. [DOI] [PubMed] [Google Scholar]
- 14.Cideciyan AV, Aleman TS, Jacobson SG, et al. Centrosomal-ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: implications for therapy of Leber congenital amaurosis. Hum Mutat 2007;28:1074–83. [DOI] [PubMed] [Google Scholar]
- 15.Cideciyan AV, Jacobson SG, Drack AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med 2019;25:225–8. [DOI] [PubMed] [Google Scholar]
- 16.Cideciyan AV, Jacobson SG, Ho AC, et al. Restoration of cone sensitivity to individuals with congenital photoreceptor blindness within the phase 1/2 sepofarsen trial. Ophthalmol Sci 2022;2:100133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang W, Li L, Su Q, Gao G, Khanna H. Gene therapy using a miniCEP290 fragment delays photoreceptor degeneration in a mouse model of Leber congenital amaurosis. Hum Gene Ther 2018;29:42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maeder ML, Stefanidakis M, Wilson CJ, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med 2019;25:229–33. [DOI] [PubMed] [Google Scholar]
- 19.Bennett J, Wellman J, Marshall KA, et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhoodonset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet 2016;388:661–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roman AJ, Cideciyan AV, Wu V, Garafalo AV, Jacobson SG. Full-field stimulus testing: role in the clinic and as an outcome measure in clinical trials of severe childhood retinal disease. Prog Retin Eye Res 2022;87:101000. [DOI] [PubMed] [Google Scholar]
- 21.Klein M, Birch DG. Psychophysical assessment of low visual function in patients with retinal degenerative diseases (RDDs) with the diagnosys full-field stimulus threshold (D-FST). Doc Ophthalmol 2009; 119:217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Csaky KG, Richman EA, Ferris FL III. Report from the NEI/FDA ophthalmic clinical trial design and endpoints symposium. Invest Ophthalmol Vis Sci 2008; 49:479–89. [DOI] [PubMed] [Google Scholar]
- 23.Bailey IL, Jackson AJ, Minto H, Greer RB, Chu MA. The Berkeley Rudimentary Vision Test. Optom Vis Sci 2012;89:1257–64. [DOI] [PubMed] [Google Scholar]
- 24.Aleman TS, Uyhazi KE, Serrano LW, et al. RDH12 mutations cause a severe retinal degeneration with relatively spared rod function. Invest Ophthalmol Vis Sci 2018;59:5225–36. [DOI] [PubMed] [Google Scholar]
- 25.Sumaroka A, Garafalo AV, Semenov EP, et al. Treatment potential for macular cone vision in Leber congenital amaurosis due to CEP290 or NPHP5 mutations: predictions from artificial intelligence. Invest Ophthalmol Vis Sci 2019;60:2551–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boye SE, Huang W-C, Roman AJ, et al. Natural history of cone disease in the murine model of Leber congenital amaurosis due to CEP290 mutation: determining the timing and expectation of therapy. PLoS One 2014;9(3):e92928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pierce EA, Ashimatey BS, Jayasundera T, et al. Twelve-month natural history study of CEP290-associated retinal degeneration. Ophthalmol Sci (in press) (https://www.ophthalmologyscience.org/article/S2666-9145(24)00019-8/pdf). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collison FT, Fishman GA, McAnany JJ, Zernant J, Allikmets R. Psychophysical measurement of rod and cone thresholds in Stargardt disease with full-field stimuli. Retina 2014;34:1888–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collison FT, Park JC, Fishman GA, McAnany JJ, Stone EM. Full-field pupillary light responses, luminance thresholds, and light discomfort thresholds in CEP290 Leber congenital amaurosis patients. Invest Ophthalmol Vis Sci 2015;56: 7130–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mangione CM, Lee PP, Gutierrez PR, et al. Development of the 25-item National Eye Institute Visual Function Questionnaire. Arch Ophthalmol 2001;119:1050–8. [DOI] [PubMed] [Google Scholar]
- 31.Birch EE, Cheng CS, Felius J. Validity and reliability of the Children’s Visual Function Questionnaire (CVFQ). J AAPOS 2007;11:473–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Green J, Tolley C, Bentley S, et al. Qualitative interviews to better understand the patient experience and evaluate patient-reported outcomes (PRO) in RLBP1 retinitis pigmentosa (RLBP1 RP). Adv Ther 2020;37:2884–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lacy GD, Abalem MF, Andrews CA, et al. The Michigan Retinal Degeneration Questionnaire: a patient-reported outcome instrument for inherited retinal degenerations. Am J Ophthalmol 2021;222: 60–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aleman TS, Han G, Serrano LW, et al. Natural history of the central structural abnormalities in choroideremia: a prospective cross-sectional study. Ophthalmology 2017;124:359–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ctori I, Huntjens B. Repeatability of foveal measurements using spectralis optical coherence tomography segmentation software. PLoS One 2015;10:(6) e0129005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Menke MN, Dabov S, Knecht P, Sturm V. Reproducibility of retinal thickness measurements in healthy subjects using spectralis optical coherence tomography. Am J Ophthalmol 2009;147:467–72. [DOI] [PubMed] [Google Scholar]
- 37.Bainbridge JWB, Mehat MS, Sundaram V, et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N Engl J Med 2015;372:1887–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cehajic-Kapetanovic J, Xue K, Martinez-Fernandez de la Camara C, et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat Med 2020;26:354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russell SR, Drack AV, Cideciyan AV, et al. Intravitreal antisense oligonucleotide sepofarsen in Leber congenital amaurosis type 10: a phase 1b/2 trial. Nat Med 2022; 28:1014–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobson SG, Cideciyan AV, Ho AC, et al. Safety and improved efficacy signals following gene therapy in childhood blindness caused by GUCY2D mutations. iScience 2021;24:102409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jacobson SG, Cideciyan AV, Ho AC, et al. Night vision restored in days after decades of congenital blindness. iScience 2022;25:105274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.