

RESUME
La détection d’un taux élevé des immunoglobulines E (IgE) sériques est d’abord évocatrice d’allergie, d’atopie ou de parasitose. Cependant, certaines valeurs très élevées peuvent être le signe de maladies plus sévères. Nous proposons une stratégie diagnostique basée sur des données cliniques et biologiques pour identifier les différentes maladies héréditaires de l’immunité qui présentent également des niveaux d’IgE sériques anormalement élevés.
ABSTRACT
The detection of a high serum immunoglobulin E (IgE) level is first suggestive of allergy, atopy or parasitosis. However, some very high values can be a sign of more severe diseases. We propose a diagnostic strategy based on clinical and biological data to identify the various hereditary immune diseases that also present with abnormally high serum IgE levels.
introduction
Les immunoglobulines E (IgE), identifiées en 1966 par Ishizaka et initialement nommées IgND, sont des anticorps naturellement présents dans l’organisme (1,2).
Ce sont des protéines monomériques d’une masse moléculaire de 190 KDa, qui circulent dans le sang.
Leur demi- vie dans le plasma est courte, allant de 2 à 4 jours.
Cependant, ces IgE peuvent persister durant de longues périodes lorsqu’elles sont recrutées sur les FcR des mastocytes dans les tissus.
Les IgE sont les principaux médiateurs de la réponse allergique chez l’Homme.
Les valeurs totales d’IgE augmentent de la petite enfance à l’adolescence, avec un pic à l’âge de 10-15 ans, dont les valeurs de référence sont comprises entre 0 et 100 à 150kUI/l, puis diminuent tout au long de l’âge adulte ( 3,4).
Le taux d’IgE est significativement plus élevé chez les hommes par rapport aux femmes (5,6).
Le taux élevé des immunoglobulines E sériques (IgE) est une situation fréquente en consultation, notamment d’allergologie.
En effet, les terrains atopiques et les maladies associées comme l’asthme allergique, l’eczéma, la rhinite allergique, l’aspergillose broncho-pulmonaire allergique (ABPA) sont habituellement associés à une augmentation des IgE totales allant de 2 à 10 fois plus élevés que la normale (7,8).
Le taux élevé d’IgE est une caractéristique mais non une spécificité des maladies allergiques, une augmentation peut être retrouvée dans un grand nombre de situations pathologiques variées telles que(9) :
• Affections parasitaires: helminthes, ascariose, filariose, schistosomiases, toxocarose, nématodes, la gale.
• Infections virales: cytomégalovirus (CMV), mononucléose infectieuse, virus d’Epstein-Barr (EBV), virus de l’immunodéficience humaine (VIH).
• Affections néoplasiques: carcinomes bronchiques, myélome à IgE
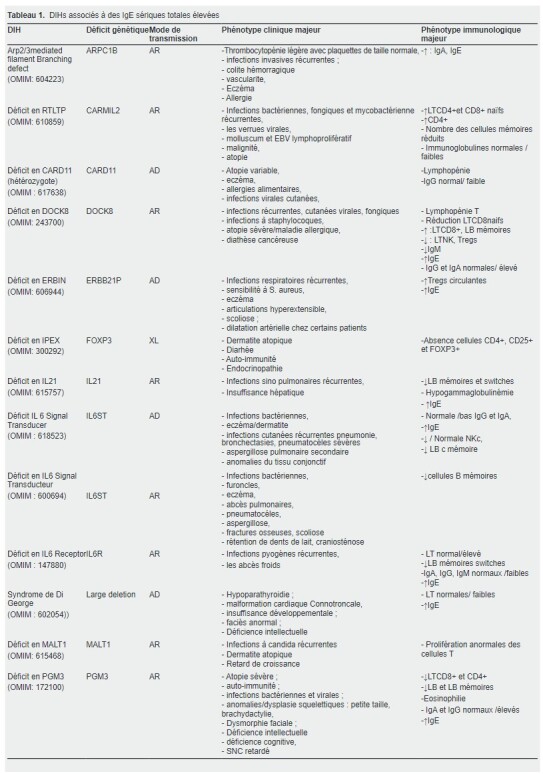
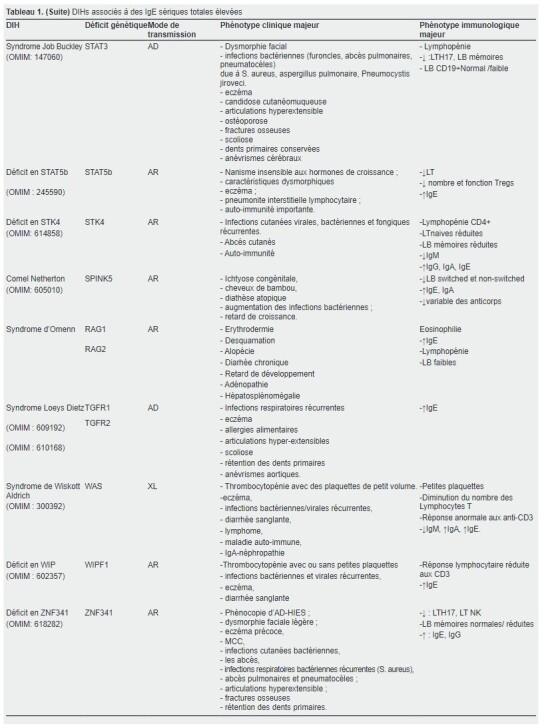
Une élévation extrême des IgE sériques a été également décrite en association avec la survenue de certains déficits immunitaires rares d’origine génétique (Tableau 1) (10, 11 ).
Nous avons conduit une recherche bibliographique sur Pub-Med, en utilisant les mots clés ″" Hyperimmunoglobulinemia E″, ″" HyperIgE″, ″" High IgE″ et ″" Primary Immune Deficiencies and high IgE″ pour collecter les données les plus récentes concernant les différents types de déficits immunitaires héréditaires(DIHs) associés à des immunoglobulines E (IgE) très élevés.
Les DIHs, longtemps connus sous le nom de déficits immunitaires primitifs (DIPs), englobent plus de 400 maladies héréditaires.
Ils sont organisés selon la dernière classification de l’Union International des Sociétés d’Immunologie (IUIS) en dix catégories en fonction du phénotype immunologique et le mode de transmission.
Les déficits immunitaires héréditaires affectent le fonctionnement du système immunitaire, rendant les individus plus vulnérables aux infections récurrentes, aux maladies auto-immunes et à d’autres complications graves, certains types sont plus graves que d’autres et peuvent entraîner des complications potentiellement mortelles (12).
En raison de la diversité de ces troubles génétiques, un diagnostic précoce et précis revêt une importance cruciale.
Ce diagnostic requiert une approche multidisciplinaire impliquant des immunologistes, des généticiens, des pédiatres et d’autres professionnels de santé.
Un diagnostic précoce assure une prise en charge optimale permettant une intervention rapide et appropriée, diminuant ainsi la fréquence et la gravité des infections, ainsi que des complications associées.
En plus des traitements spécifiques, ce diagnostic permet la mise en place d’une surveillance médicale adaptée, un soutien émotionnel et éducatif, et donc une meilleure qualité de vie.
Dans notre travail ; nous nous sommes concentrés sur les DIHs avec des taux élevés en IgE sériques, un groupe nommé et classifié par IUIS comme HIES, qui renferme dix entités pathologiques (déficits en STAT3, ZNF341, IL6ST, IL6R, CARD11, ERBIN, PGM3, TGFBR1/2 et SPINK5) (13, 14).
Cependant de nombreux autres DIHs conduisent à des IgE élevés mais ne sont pas classés par l’IUIS dans les HIES.
Ces maladies incluent les syndromes de Wiskott Aldrich, d’Omen, de Di-George, IPEX, et les déficits en ARPC1B, MALT1, STAT5B, STK4, CARMIL2, IL21, IL21 R, WIP et DOCK8.
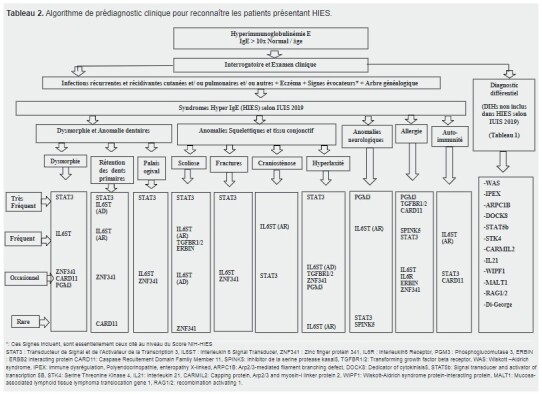
Nous proposons un arbre décisionnel qui facilitera le prédiagnostic des DIHs avec une hyperimmunoglobulinémie E et la prise en charge des patients(Tableau 2).
Syndrome d’Hyper-IgE (HIES) entité hétérogène
Les HIES sont un groupe hétérogène et complexe de déficits immunitaires rares, appartenant aux déficits immunitaires combinés ou syndromiques, impliquant à la fois les réponses humorales et cellulaires (15).
Ils se manifestent dès la naissance ou pendant la petite enfance par une triade classique de symptômes tels que des infections bactériennes, virales ou fongiques précoces, récidivantes, et essentiellement pulmonaires et/ ou cutanées, de l’eczéma également précoce et chronique, avec des taux d’IgE sériques généralement au-dessus de 2000 UI/ml (un taux 10 fois supérieur à la limite normale) associé à une éosinophilie dans certains cas.
Bien que certaines manifestations cliniques soient similaires dans tous les types de HIES, comme un taux élevé d’IgE, de l’eczéma et des infections pulmonaires et/ou cutanées récurrentes, il existe une variabilité phénotypique et immunologique (16,17) et une diversité pathologique pouvant toucher le système squelettique, vasculaire, neurologique et des tissus conjonctifs (18).
Il existe deux transmissions génétiques connues, une forme autosomique dominante dite AD-HIES, où on retrouve le déficit en STAT3 (Transducteur du signal et activateur de la transcription 3), déficit en CARD11 (Caspase Recruitment Domain Family Member 11), déficit IL6ST (Interleukin 6 Signal transducer), déficit en ERBIN (ERBB2 interacting protein) et syndrome de Loeys Dietz(TGFBR1/2).
L’autre forme est autosomique récessive (AR-HIES), et phénotypiquement variable selon le gène muté.
Cinq déficits AR, incluant des mutations dans PGM3 (Phosphoglucomutase 3), dans SPINK5 (Inhibiteur de la sérine peptidase Kasal 5) responsable de la maladie de Comel Netherton, dans IL6ST (Interleukin 6 Signal transducer), dans ZNF341 (Zinc finger protein 341) et dans IL6R (Interleukin 6 Receptor) ont été rapportés.
Les patients suspectés d’HIES sont pré-diagnostiqués à l’aide du score NIH (National Institut of Health) établit par Bodo Grimbacher en 1999 ( 19 ).
Ce score est composé de 20 critères biologiques et cliniques dont le taux d’IgE totales, le nombre d’éosinophile, l’intensité de l’eczéma, l’ensemble d’infections évocatrices de ce syndrome(abcès, candidose, dilation des bronches, pneumonie),la détection d’anomalies osseuses et dentaires tel la rétention des dents primaires, une scoliose, des fractures pathologiques, une dysmorphie faciale, un palais ogival et le développement de lymphome.
Des points sont attribués en fonction de la sévérité des différents signes cliniques du patient
Ainsi, tout score NIH> 40 points indique que le patient souffre probablement d’un HIES, entre 20 et 40 points une possibilité d’HIES, et < 20 points une improbabilité de ce syndrome.
Cependant, ce score se révèle souvent inadapté chez les nourrissons et les jeunes enfants en raison du manque de symptômes cliniques lié à leur âge.
Syndrome de Job et phénocopies
Le syndrome de Job représente la forme historique d’HIES.
La première description de ce syndrome date de 1966 chez deux sœurs présentant des abcès staphylococciques froids récurrents, de l’eczéma et des infections respiratoires, rappelant le personnage biblique dont le corps était couvert de furoncles ( 20 )
Six ans plus tard, Buckley et al. ont rapporté chez deux adolescents souffrant d’infections pyogènes récurrentes (des abcès cutanés, pulmonaires et auriculaires récurrents), un retard de croissance, une dermatite chronique accompagnée d’une dysmorphie faciale, une hyperéosinophilie, en plus des taux élevé des IgE, d’où la dénomination « Syndrome Job-Buckley » ( 21 ).
En 2007, les mutations dominantes négatives (DN) dans STAT3 ont été rapportées comme la première cause génétique de ce syndrome.
Plus récemment, au moins deux autres gènes ont été impliqués incluant ZNF341et IL6ST.
Déficit AD en STAT3:
Le déficit AD dans STAT3 représente la cause la plus fréquente de syndromes d’hyper-IgE, contribuant à environ 90 % des cas.
Les patients avec ce trouble présentent des pathologies multi-systémiques, immunologiques et non immunologiques.
Les signes principaux sont l’eczéma, les infections récurrentes et les anomalies du tissu conjonctif, qui peuvent inclure des fractures osseuses secondaires à un traumatisme mineur, une chute tardive des dents de lait, une hyperflexibilité articulaire, des anomalies vasculaires, de la scoliose ( 22, 23 ) ainsi qu’une dysmorphie faciale qui apparaît généralement à l’adolescence ( 24 ).
L’HIES se manifeste généralement au cours de la période néonatale par une éruption cutanée (souvent diagnostiquée comme une pustulose éosinophile) qui évolue par la suite en une dermatite eczématoïde, qui est souvent induite par des infections staphylococciques (25,26)
Les principaux critères de diagnostic, en plus des taux sériques très élevés des IgE > 2 000 UI/ml, sont une hyperéosinophilie > 700 /µl et une diminution des cellules T et B mémoires circulantes associées à une diminution ou absence des cellules Th17 productrices d’IL17, le reste du bilan immunitaire est normal (27)
Déficit AR en ZNF341:
Il s’agit de la forme AR du syndrome de Job ; la plus récente des HIES, décrite en 2018 comme maladie phénotypiquement semblable au déficit en STAT3 (ADHIES), avec des infections récurrentes et des anomalies squelettiques ( 28 , 29 ) Le facteur de transcription ZNF341 (Zinc finger protein 341) contrôle l’expression basaleet inductible de STAT3
Les mutations dans ZNF341 conduisent à des niveaux insuffisants de STAT3.
Le déficit AR en ZNF341 est une phénocopie du déficit AD en STAT3 (30).
Déficit AR en IL6ST:
IL6ST (Interleukin 6 Signal Transducer) encode la protéine GP130, le co-récepteur de toutes les cytokines de la famille IL-6 incluant : IL-6, IL-11, IL-27, LIF, OSM, CT-1 and CNTF.
Les variants bialléliques dans le domaine extra-cellulaire de l’IL6ST provoquent une forme récessive du syndrome d’hyper-IgE (HIES) caractérisée par un taux élevé d’IgE, une éosinophilie, une réponse de phase aiguë défectueuse, une sensibilité aux infections bactériennes et des anomalies squelettiques, y compris la craniosténose.
Ces mutations bialléliques dans IL6ST associées avec un HIES sont associées à une perte de réponse sélective des cytokines IL-6 et IL-11, toutes deux dépendantes de GP130 pour leur fonction ( 31 , 32 )
Avec la découverte des défauts complets en IL-6R et IL-11RA (33,34), la découverte de ces mutations bialléliques a démontré un rôle crucial des voies de signalisation dépendantes de l’IL6 et de l’IL-11 dans la physiopathologie du déficit AD en STAT3 (35).
Déficit AD en IL6ST:
Des mutations dominantes dans l’IL6ST peuvent également entraîner un syndrome de Job typique avec des anomalies squelettiques, des infections pulmonaires et cutanées récurrentes, eczéma, des taux élevés des IgE et une éosinophilie ( 36 )
Les mutations dominantes dans IL6ST conduisent à l’accumulation à la surface cellulaire de protéines tronquées, bloquant la fonction de la protéine sauvage résiduelle.
Comme les mutations bialléliques dans IL6ST, ces mutations DN inhibent préférentiellement la signalisation de l’IL-6 et de l’IL-11 (32).
À noter que les infections pulmonaires et leurs conséquences (pneumatocèle) sont particulièrement sévères chez ces patients.
Déficit AR en IL6R:
L’interleukine 6 est une cytokine immuno-régulatrice, qui active STAT3 via sa liaison à son récepteur qui se compose de l’IL-6R (Interleukin 6 Receptor) et de la GP130 (encodée par IL6ST).
Le déficit complet en IL6R est une forme autosomique récessive des HIES, dont le spectre clinique englobe des infections récurrentes cutanées et pulmonaires, de l’eczéma, des abcès froids et une éosinophilie.
Il est important de noter qu’en comparaison des défauts dans IL6ST, STAT3 et ZNF341, les phénotypes extra-hématopoïétiques sont absents reflétant l’intégrité fonctionnelle de la voie de l’IL-11, indépendante de IL-6R (34).
Autres syndromes d’hyper-IgE dans la classification IUIS
Déficit AD en TGFBR1et TGFBR2 (Loeys Dietz):
Ces déficits AD en TGFBR1et TGFBR2 définit la maladie de Loeys Dietz.
Il s’agit d’une maladie transmise selon un mode autosomique dominant, causée par des mutations des gènes codant pour les récepteurs 1 et 2 du facteur de croissance transformant β, récepteur de type 1 ou 2(Transforming growth factor β receptor 1 ou 2 ; TGFBR1 et TGFBR2).
Outres les taux sériques très élevés d’IgE, quatre caractéristiques principales suggèrent le diagnostic de ce syndrome : tortuosité artérielle (artères torsadées ou spiralées), hypertélorisme (espacement des yeux anormalement grand), luette bifide (séparée en deux) ou large et l’anévrisme.
Les autres signes de ce syndrome se manifestent par une craniosynostose, une exotropie, une hypoplasie malaire, une micrognathie, un rétrognathisme, des anomalies cérébrales, un retard mental, une peau fine, et une hyperlaxité articulaire.
Déficit AD en CARD11:
Déficit immunitaire de transmission autosomique dominante (Caspase recruitment domain Family Member 11), les mutations CARD11 sont associées à un large éventail de manifestations cliniques et biologiques ( 39 ).
On retrouve chez ces patients une atopie sévère (dermatite atopique, allergie alimentaires, rhinite allergique, asthme)comme symptôme essentiel.
Ces patients souffrent également fréquemment d’infections cutanées virales et respiratoires, et plus rarement d’auto-immunité et de lymphome.
Ils ont une immunité humorale gravement altérée se manifestant par une hypogammaglobulinémie importante et des cellules B mémoires absentes ou diminuées.
Déficit AD en ERBIN:
C’est un trouble héréditaire dominant, qui se manifeste par de l’eczéma, des infections respiratoires récurrentes, des articulations Hyperextensible, une scoliose et des anomalies vasculaires, un chevauchement des symptômes avec le déficit en STAT3.
Une altération de la signalisation de STAT3 réduit l’expression d’ERBIN et dérégule l’activation de la voie TGF-β, entraînant une augmentation des cellules Tregs circulantes ce qui explique pourquoi ce déficit partage le phénotype allergique et non immunologique du déficit en STAT3 (40).
Déficit AR en SPINK5:
Le syndrome de Comel Netherton (SN) est une maladie rare autosomique récessive, due à des mutations bialléliques du gène SPINK5 (Inhibiteur de la sérine protéase kasal 5) codant l’inhibiteur de protéase LEKT (Inhibiteur lymphoépithélial de type kasal) exprimé dans les couches épidermiques supérieures de la peau et l’épithélium.
Son expression défectueuse entraine une inflammation cutanée congénitale et des cheveux bambou, en plus de plusieurs manifestations atopiques (41).
Sur le plan clinique, ce déficit se manifeste par une triade de signes : érythrodermie ichtyosiforme congénitale, trichorrhexis invaginata (cheveux bambou), atopie ainsi qu’un retard staturopondéral ( 42 )
Biologiquement, des IgA faibles et également une diminution des lymphocytes B mémoires et naïfs sont observés tandis que les lymphocytes T sont normaux.
Le tableau clinique peut être sévère chez les nouveau-nés avec des complications engageant le pronostic vital conduisant à une létalité postnatale élevée.
Les manifestations cutanées ainsi que les anomalies capillaires persistent tout au long de la vie.
Déficit AR en PGM3:
Il s’agit de mutations bi-alléliques délétères touchant le gène codant la protéine PGM3 (Phosphoglucomutase3) qui intervient dans plusieurs étapes de la glycosylation des protéines
Ce déficit autosomique récessif provoque un trouble multisystémique caractérisé par des maladies atopiques (eczéma, allergie, asthme), infections récurrentes cutanées et pulmonaires, anomalies squelettiques ainsi que des anomalies neurologiques et hématologiques telle que les retards cognitifs et moteurs, également l’auto-immunité comme la vascularite sont observées (43)
Les résultats du laboratoire se présentent sous forme d’une éosinophilie, une cytopénie et neutropénie fréquente, avec un taux élevé des IgG et IgA, et une lymphopénie avec un rapport CD4/CD8 inversé (44 ).
Il a été proposé que la similitude entre le déficit en PGM3 et les HIES classiques est due à une glycosylation anormale de GP130, entraînant une diminution de l’expression et de la fonction de la protéine (45 ).
Autres DIHs avec IgE fréquemment élevé mais non classifiés HIES par l’IUIS
Déficit XR en WAS:
Le syndrome de Wiskott –Aldrich est une maladie à transmission récessive lié à l’X, due à des variations délétères dans WAS, un gène du chromosome X.
Ce déficit touche quasi exclusivement les garçons
Les manifestations sont principalement une microthrombocytopénie, un eczéma, des infections et un risque accru de manifestations autoimmunes et/ou oncohématologiques ( 46 ), associe à une diminution des lymphocytes T et des IgM accompagné d’une élévation des IgA.
Déficit AR en WIP:
Wiskott-Aldrich syndrome protein-interacting protein (WIP) est une immunodéficience combinée avec une thrombocytopénie congénital.
Pathologie très rare avec un phénotype clinique et biologique presque similaire au syndrome de Wiskott Aldrich (WAS), elle se manifeste par des infections virales et bactériennes récurrentes, des diarrhées sanglantes( 47 ), une thrombocytopénie avec/sans petites plaquettes en plus d’une réponse lymphocytaires anti-CD3 réduites et défectueuses alors que le nombre des lymphocytes B est faible ou normale ( 48 ).
Déficit AR en RAG1 ou RAG2 - Syndrome d’Omen (OS):
Le syndrome d’Omen est dû à des mutations dans les gènes RAG1 et RAG2 (gènes activant la recombinaison de l’ADN).
Ce déficit conduit est un déficit immunitaire combiné sévère (SCID), de transmission autosomique récessive, caractérisé par une érythrodermie, une alopécie, des diarrhées chroniques, un retard de croissance et aussi des symptômes inflammatoires tels que les adénopathies, une hépatosplénomégalie, une érythrodermie généralisée exsudative diffuse (dès les premiers mois de la vie).
Sans greffe de moelle, ces patients ont une espérance de vie très courte ( 49 ).
Déficit AD avec microdélétion de la région 22q11.2:
Reconnu sous plusieurs noms notamment Vélocardiofacial ou syndrome de Di-George ( 50 ), il s’agit d’une anomalie chromosomique congénitale caractérisé le plus souvent par des malformations cardiaques, palatines, une dysmorphie faciale, un retard du développement et une hypocalcémie, de transmission autosomique dominante avec quelques cas sporadiques qui ont étaient décrits
Déficit XR en FOXP3 - Syndrome XR IPEX:
Le Syndrome de dérèglement immunitaire - polyendocrinopathie - entéropathie lié à l’X (IPEX), est une maladie rare due à des mutations du gène FOXP3 qui entraîne un dysfonctionnement des lymphocytes T régulateurs conduisant à un désordre auto-immunitaire ( 51 )
Ces caractéristiques cliniques comprennent une hypertrophie sévère des ganglions lymphatiques, des amygdales, des végétations adénoïdes et de la rate, un diabète de type 1, de l’eczéma, des allergies alimentaires et des infections ainsi que des diarrhées persistantes (52).
Déficit AR en ARPC1B:
Un autre syndrome associé à une élévation marquée des IgE est le déficit en ARPC1B (Actin Related Protein 2/3 Complex, subnit 1B), facteur clé de l’assemblage et la maintenance du complexe protéique ARP2/3 lié à l’actine.
Ce complexe est impliqué dans différentes activités de remodelage des filaments d’actines ; sa perturbation entraine des anomalies de l’immunité innée et adaptative, contribuant ainsi à une dérégulation immunitaire
Il se manifeste par une altération de la prolifération des lymphocytes T, une thrombocytopénie (53,54).
Sur le plan clinique, on note des manifestations auto-immunes ainsi que des tumeurs malignes, des infections bactériennes et virales récurrentes, de l’eczéma, une vascularite cutanée ainsi que des hémorragies gastriques, plus précisément des colites gastriques (55).
Déficit AR en DOCK8:
Initialement classifiée comme HIES par l’IUIS après sa découverte en 2009 (56), cette maladie est maintenant classée avec les déficits immunitaires combinés.
Cette maladie est causée par des mutations bialléliques dans le gène DOCK8 (Dédicateur des cytokines 8).
Ce gène code pour la protéine DOCK8 qui est impliquée dans l’organisation du cytosquelette d’actine.
La perte de fonction de DOCK8 entraîne un défaut de migration des lymphocytes T, NK et des cellules dendritiques.
Sur le plan clinique cette entité pathologique se caractérise, en plus des infections bactériennes et fongiques par des infections virales cutanées sévères principalement due au Molluscum contagiosum, à l’Herpès simplex et au Papillomavirus humain
On note aussi des candidoses, de l’asthme et/ou de multiples allergies alimentaires et médicamenteuses ( 57 ) ainsi que des tumeurs malignes incluant des lymphomes et des carcinomes conduisant à la mortalité à un jeune âge.
L’eczéma de la déficience en DOCK8 peut être sévère et difficile à contrôler d’où la nécessité d’une thérapie topique, ou une immunothérapie comme le dupilumab.
Le mauvais pronostic vital de cette pathologie est une indication de greffe de moelle osseuse
Déficit AR en MALT1:
Ce déficit immunitaire combiné est due à des mutations du gène MALT1 (Mucosa-associated lymphoid tissue lymphoma translocation gene 1).
Cliniquement, il se manifeste par des infections bactériennes à Candida, des bronchectasies, une dermatite atopique sévère et un retard de croissance ( 58 )
Biologiquement, on note des taux normaux des cellules T et B mais avec altération de leurs fonctions, en particulier une prolifération anormale des cellules T et une altération d’activation de la voie NFkB ( 59 ).
Déficit AR en STAT5b:
Le déficit autosomique récessif du transducteur de signal et de l’activateur de la transcription 5 b, conduit à un CID.
Les principales caractéristiques cliniques sont le retard de croissance postnatal sévère, une dermatite atopique à un jeune âge, des pneumonies interstitielles lymphocytaires, des maladies auto-immunes et des infections virales récurrentes (60).
Sur le plan immunologique, ce dérèglement immunitaire est secondaire à une signalisation anormales des cytokines et une altération de la signalisation des récepteurs de l’hormone de croissance, ce qui se traduit par une lymphopénie, un très faibles taux des cellules NK et T (CD4+/CD8+), un défaut fonctionnel des cellules Tregs en plus d’une déficience importante en IGF-I (insulin-like growth factor 1)(61,62) Ce déficit est potentiellement mortel.
Déficit AR en STK4:
Le déficit autosomique récessif de STK4 (Serine Thréonine Kinase 4), anciennement nommé MST1 (Mammalian Sterile-20-Like 1 Kinase) conduit à un CID.
Cette pathologie est rare et présente les mêmes caractéristiques cliniques du déficit en DOCK8, des infections cutanées virales (HPV, EBV, Molluscum) sévères et récurrentes, des infections fongiques et bactériennes, des abcès cutanés ainsi que de l’auto-immunité (63). Le phénotype immunologique est caractérisé par une neutropénie, des taux élevés d’IgA et IgG, des taux faibles d’IgM et un nombre faible des LT CD4+ ( 64).
Déficit AR en CARMIL2:
Il s’agit d’un déficit autosomique récessif, due à des mutations bi-alléliques du gène CARMIL2 (Capping Protein Regulator and Myosin 1 Linker 2, ou RLTPR).
Les patients souffrent de manifestations cliniques hétérogènes.
On retrouve des phénotypes atopiques comme une dermatite atopique sévère, asthme allergique, allergie alimentaire, urticaire (65 ,66), des lésions similaires à du psoriasis, des infections cutanées et respiratoires, des œsophagites à éosinophiles, des maladies inflammatoires digestives à début très précoces, et des tumeurs à EBV des muscles lisses.
Sur le plan immunologique, on note une altération d’activation, de différenciation et migration des lymphocytes T accompagné d’un nombre réduit/absent des Tregs.
Déficit AR en IL21 et IL21R
Cette pathologie appartient au groupe des déficits immunitaires communs variables (DICV).
Sa transmission due à des mutations du gène IL21et son récepteur IL21 R (Interleukin-21 Receptor).
Anomalie autosomique récessive potentiellement mortelle dont le phénotype clinique est variable, qui se caractérise par un large éventail de symptômes cliniques, qui apparaissent à jeune âge, notamment des infections récurrentes, une susceptibilité accrue aux infections virales et bactériennes, des troubles auto-immunes tels que la polyarthrite rhumatoïde et le lupus érythémateux systémique, ainsi que des complications gastro-intestinales, y compris la maladie inflammatoire de l’intestin. (67)
IL21 est une cytokine qui joue un rôle important dans l’homéostasie immunitaire.
Ce déficit se traduit sur le plan immunologique par une réduction du nombre total des lymphocytes T, une diminution du pourcentage de B mémoires, une hypogammaglobulinémie et une réponse aux anticorps spécifiques faible (68 ).
conclusion
La présence de taux d’IgE totales sériques très élevés ne doit pas limiter le diagnostic aux causes largement reconnues (atopie, parasitose, néoplasie, allergie), mais ils peuvent être également évocateurs d’immunodéficiences sous-jacentes qui partagent des facteurs en communs y compris les éruptions cutanées telles que l’eczéma, l’éosinophilie et les infections d’où la nécessité de bien interpréter la lecture des IgE et de différencier de façon précoce et appropriée chaque entité pathologique afin de les prendre en charge.
References
- Ishizaka K, Ishizaka T. Identification of IgE. Journal of Allergy and Clinical Immunology. 2016 Jun;137(6):1646–1650. doi: 10.1016/j.jaci.2015.12.1343. [DOI] [PubMed] [Google Scholar]
- Johansson SGO. The discovery of IgE. Journal of Allergy and Clinical Immunology. 2016 Jun;137(6):1671–1673. doi: 10.1016/j.jaci.2016.04.004. [DOI] [PubMed] [Google Scholar]
- Lindberg R, Arroyave C. Levels of IgE in serum from normal children and allergic children as measured by an enzyme immunoassay. Journal of Allergy and Clinical Immunology. 1986 Oct;78(4):614–618. doi: 10.1016/0091-6749(86)90078-3. [DOI] [PubMed] [Google Scholar]
- Saarinen UM, Juntunen K, Kajosaari M, Björkstén F. SERUM IMMUNOGLOBULIN E IN ATOPIC AND NON-ATOPIC CHILDREN AGED 6 MONTHS TO 5 YEARS.: A Follow-up Study. Acta Paediatrica. 1982 May;71(3):489–494. doi: 10.1111/j.1651-2227.1982.tb09457.x. [DOI] [PubMed] [Google Scholar]
- Stone KD, Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. Journal of Allergy and Clinical Immunology. 2010 Feb;125(2):S73–S80. doi: 10.1016/j.jaci.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton RG, Adkinson NF. 23. Clinical laboratory assessment of IgE-dependent hypersensitivity. Journal of Allergy and Clinical Immunology. 2003 Feb;111(2):S687–S701. doi: 10.1067/mai.2003.123. [DOI] [PubMed] [Google Scholar]
- Zellweger F, Eggel A. IgE-associated allergic disorders: recent advances in etiology, diagnosis, and treatment. Allergy. 2016 Dec;71(12):1652–1661. doi: 10.1111/all.13059. [DOI] [PubMed] [Google Scholar]
- Wittig HJ, Belloit J, De Fillippi I, Royal G. Age-related serum immunoglobulin E levels in healthy subjects and in patients with allergic disease. Journal of Allergy and Clinical Immunology. 1980 Oct;66(4):305–313. doi: 10.1016/0091-6749(80)90026-3. [DOI] [PubMed] [Google Scholar]
- Garcia G, Humbert M. Infection et réponse à IgE. Revue Française d’Allergologie et d’Immunologie Clinique. 1998 Jan;38(4):241–246. [Google Scholar]
- Grimbacher B, Belohradsky BH, Holland SM. Immunoglobulin E in primary immunodeficiency diseases. Allergy. 2002 Nov;57(11):995–1007. doi: 10.1034/j.1398-9995.2002.02168.x. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Boisson B, Béziat V, Puel A, Casanova JL. Human hyper-IgE syndrome: singular or plural? Mamm Genome. 2018 Aug;29(7-8):603–617. doi: 10.1007/s00335-018-9767-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaoui A, Mokhantar K, Jeddane L, Ailal F, Elkabli H, Bousfiha AA. Quand faut-il suspecter un déficit immunitaire chez l’adulte? When to suspect an immune deficiency in adults? Tunis Med. 2022;100(8-9):585–591. [PMC free article] [PubMed] [Google Scholar]
- Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol. 2022 Oct;42(7):1508–1520. doi: 10.1007/s10875-022-01352-z. [DOI] [PubMed] [Google Scholar]
- Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022 Oct;42(7):1473–1507. doi: 10.1007/s10875-022-01289-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alyasin S, Esmaeilzadeh H, Ebrahimi N, Nabavizadeh SH, Kashef S, Esmaeilzadeh E. Phenotyping and long-term follow up of patients with hyper IgE syndrome. Allergologia et Immunopathologia. 2019 Mar;47(2):152–158. doi: 10.1016/j.aller.2018.07.009. [DOI] [PubMed] [Google Scholar]
- Al-Shaikhly T, Ochs HD. Immunology and Cell Biology. 2018 Sep doi: 10.1111/imcb.12209. http://doi.wiley.com/10.1111/imcb.12209 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Boisson B, Béziat V, Puel A, Casanova JL. Human hyper-IgE syndrome: singular or plural? Mammalian Genome. 2018 Aug doi: 10.1007/s00335-018-9767-2. http://link.springer.com/10.1007/s00335-018-9767-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadil I, Ben-Ali M, Jeddane L, Barbouche MR, Bousfiha AA. The Seven STAT3-Related Hyper-IgE Syndromes. J Clin Immunol. 2021 Aug;41(6):1384–1389. doi: 10.1007/s10875-021-01041-3. [DOI] [PubMed] [Google Scholar]
- Grimbacher B, Schäffer AA, Holland SM, Davis J, Gallin JI, Malech HL. Genetic linkage of hyper-IgE syndrome to chromosome 4. Am J Hum Genet. 1999 Sep;65(3):735–744. doi: 10.1086/302547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Schaller J, Wedgwood R, Harvard MD. JOB’S SYNDROME: Recurrent, « Cold », Staphylococcal Abscesses. The Lancet. 1966 May;287(7445):1013–1015. doi: 10.1016/s0140-6736(66)90119-x. [DOI] [PubMed] [Google Scholar]
- Buckley RH, Wray BB, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pathologica. 1972 Jan;49(1):59–70. [PubMed] [Google Scholar]
- Borges WG, Hensley T, Carey JC, Petrak BA, Hill HR. The face of Job. The Journal of Pediatrics. 1998 Aug;133(2):303–305. doi: 10.1016/s0022-3476(98)70243-4. [DOI] [PubMed] [Google Scholar]
- Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, et al. Hyper-IgE syndrome with recurrent infections--an autosomal dominant multisystem disorder. N Engl J Med. 1999 Mar 4;340(9):692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- Vogel TP, Milner JD, Cooper MA. The Ying and Yang of STAT3 in Human Disease. J Clin Immunol. 2015 Oct;35(7):615–623. doi: 10.1007/s10875-015-0187-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamlin SL, McCalmont TH, Cunningham BB, Esterly NB, Lai CH, Mallory SB, et al. Cutaneous manifestations of hyper-IgE syndrome in infants and children. The Journal of Pediatrics. 2002 Oct;141(4):572–575. doi: 10.1067/mpd.2002.127503. [DOI] [PubMed] [Google Scholar]
- Eberting CLD, Davis J, Puck JM, Holland SM, Turner ML. Dermatitis and the Newborn Rash of Hyper-IgE Syndrome. Arch Dermatol. 2004 Sep 1;140(9):1119–1125. doi: 10.1001/archderm.140.9.1119. [DOI] [PubMed] [Google Scholar]
- Woellner C, Gertz EM, Schäffer AA, Lagos M, Perro M, Glocker EO, et al. Mutations in STAT3 and diagnostic guidelines for hyper-IgE syndrome. Journal of Allergy and Clinical Immunology. 2010 Feb;125:424–432.e8. doi: 10.1016/j.jaci.2009.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béziat V, Fieschi C, Momenilandi M, Migaud M, Belaid B, Djidjik R, et al. Inherited human ZNF341 deficiency. Current Opinion in Immunology. 2023 Jun;82:102326. doi: 10.1016/j.coi.2023.102326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béziat V, Li J, Lin JX, Ma CS, Li P, Bousfiha A, et al. A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Science Immunology. 2018 Jun 15;3(24):eaat4956. doi: 10.1126/sciimmunol.aat4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey-Jakobs S, Hartberger JM, Fliegauf M, Bossen C, Wehmeyer ML, Neubauer JC, et al. ZNF341 controls STAT3 expression and thereby immunocompetence. Sci Immunol. 2018 Jun 15;3(24):eaat4941. doi: 10.1126/sciimmunol.aat4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwerd T, Twigg SRF, Aschenbrenner D, Manrique S, Miller KA, Taylor IB, et al. A biallelic mutation in IL6ST encoding the GP130 co-receptor causes immunodeficiency and craniosynostosis. J Exp Med. 2017 Sep 4;214(9):2547–2562. doi: 10.1084/jem.20161810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahin T, Aschenbrenner D, Cagdas D, Köstel Bal S, Domínguez Conde C, Garncarz W, et al. Selective loss of function variants in IL6ST cause Hyper-IgE Syndrome with distinct impairments of T cell phenotype and function. Haematologica. 2018 Oct 11;:haematol.2018.194233. doi: 10.3324/haematol.2018.194233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Bastard P, Bustamante J, Casanova JL. Human autoantibodies underlying infectious diseases. J Exp Med. 2022 Mar 23;219(4):e20211387. doi: 10.1084/jem.20211387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Bastard P, Bustamante J, Casanova JL. Human autoantibodies underlying infectious diseases. J Exp Med. 2022 Mar 23;219(4):e20211387. doi: 10.1084/jem.20211387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Bastard P, Bustamante J, Casanova JL. Human autoantibodies underlying infectious diseases. J Exp Med. 2022 Mar 23;219(4):e20211387. doi: 10.1084/jem.20211387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béziat V, Tavernier SJ, Chen YH, Ma CS, Materna M, Laurence A, et al. Dominant-negative mutations in human IL6ST underlie hyper-IgE syndrome. Journal of Experimental Medicine. 2020 Jun 1;217(6):e20191804. doi: 10.1084/jem.20191804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felgentreff K, Siepe M, Kotthoff S, von Kodolitsch Y, Schachtrup K, Notarangelo LD, et al. Severe eczema and Hyper-IgE in Loeys–Dietz-syndrome — Contribution to new findings of immune dysregulation in connective tissue disorders. Clinical Immunology. 2014 Jan;150(1):43–50. doi: 10.1016/j.clim.2013.11.008. [DOI] [PubMed] [Google Scholar]
- RESERVES IUTD Orphanet: Syndrome de Loeys Dietz [Internet] [[cited 2021 Aug 17]].
- Dorjbal B, Stinson JR, Ma CA, Weinreich MA, Miraghazadeh B, Hartberger JM, et al. Hypomorphic caspase activation and recruitment domain 11 (CARD11) mutations associated with diverse immunologic phenotypes with or without atopic disease. Journal of Allergy and Clinical Immunology. 2019 Apr;143(4):1482–1495. doi: 10.1016/j.jaci.2018.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons JJ, Liu Y, Ma CA, Yu X, O’Connell MP, Lawrence MG, et al. ERBIN deficiency links STAT3 and TGF-β pathway defects with atopy in humans. J Exp Med. 2017 Jan 26;:jem.20161435. doi: 10.1084/jem.20161435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannula-Jouppi K, Laasanen SL, Ilander M, Furio L, Tuomiranta M, Marttila R, et al. Intrafamily and Interfamilial Phenotype Variation and Immature Immunity in Patients With Netherton Syndrome and Finnish SPINK5 Founder Mutation. JAMA Dermatol. 2016 Apr 1;152(4):435–442. doi: 10.1001/jamadermatol.2015.5827. [DOI] [PubMed] [Google Scholar]
- Laroussinie A, Hainaut E, Barbarot S, Drui D, Pierre P, Sonnet E, et al. Syndrome de Netherton : évaluation des axes antéhypophysaires – étude REHYNE. Annales d’Endocrinologie. 2016 Sep;77(4):277. [Google Scholar]
- Yang L, Fliegauf M, Grimbacher B. Hyper-IgE syndromes: reviewing PGM3 deficiency. Current Opinion in Pediatrics. 2014 Dec;26(6):697–703. doi: 10.1097/MOP.0000000000000158. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, et al. Autosomal recessive PGM3 mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol. 2014 May;133(5):1400–1409.:e5. doi: 10.1016/j.jaci.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ali M, Ben-Khemis L, Mekki N, Yaakoubi R, Ouni R, Benabdessalem C, et al. Defective glycosylation leads to defective gp130-dependent STAT3 signaling in PGM3-deficient patients. J Allergy Clin Immunol. 2019 Apr;143(4):1638–1640.:e2. doi: 10.1016/j.jaci.2018.12.987. doi: 10.1016/j.jaci.2018.12.987 . [DOI] [PubMed] [Google Scholar]
- Buchbinder D, Nugent DJ, Fillipovich AH. Wiskott–Aldrich syndrome: diagnosis, current management, and emerging treatments. Appl Clin Genet. 2014 Apr 3;7:55–66. doi: 10.2147/TACG.S58444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwinger W, Urban C, Ulreich R, Sperl D, Karastaneva A, Strenger V, et al. The Phenotype and Treatment of WIP Deficiency: Literature Synopsis and Review of a Patient With Pre-transplant Serial Donor Lymphocyte Infusions to Eliminate CMV. Front Immunol. 2018 Nov 2;9:2554. doi: 10.3389/fimmu.2018.02554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mousa H, Hawwari A, Al-Ghonaium A, Al-Saud B, Al-Dhekri H, Al-Muhsen S, et al. Hematopoietic stem cell transplantation corrects WIP deficiency. Journal of Allergy and Clinical Immunology. 2017 Mar;139(3):1039–1040.e4. doi: 10.1016/j.jaci.2016.08.036. [DOI] [PubMed] [Google Scholar]
- Hsu CC, Lee JYY, Chao SC. Omenn syndrome: a case report and review of literature. Dermatologica Sinica. 2011 Jun;29(2):50–54. [Google Scholar]
- Michaelovsky E, Frisch A, Carmel M, Patya M, Zarchi O, Green T, et al. Genotype-phenotype correlation in 22q11.2 deletion syndrome. BMC Med Genet. 2012 Dec;13(1):122. doi: 10.1186/1471-2350-13-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CB, Tahboub F, Plesec T, Kay M, Radhakrishnan K. A Review of Autoimmune Enteropathy and Its Associated Syndromes. Dig Dis Sci. 2020 Nov;65(11):3079–3090. doi: 10.1007/s10620-020-06540-8. [DOI] [PubMed] [Google Scholar]
- Barzaghi F, Hernandez LCA, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, et al. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: An international multicenter retrospective study. J Allergy Clin Immunol. 2018 Mar;141(3):1036–1049.e5. doi: 10.1016/j.jaci.2017.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahr WHA, Pluthero FG, Elkadri A, Warner N, Drobac M, Chen CH, et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nature Communications. 2017 Apr 3;8:14816. doi: 10.1038/ncomms14816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigida I, Zoccolillo M, Cicalese MP, Pfajfer L, Barzaghi F, Scala S, et al. T-cell defects in patients with ARPC1B germline mutations account for combined immunodeficiency. Blood. 2018 Nov 29;132(22):2362–2374. doi: 10.1182/blood-2018-07-863431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpi S, Cicalese MP, Tuijnenburg P, Tool ATJ, Cuadrado E, Abu-Halaweh M, et al. A combined immunodeficiency with severe infections, inflammation, and allergy caused by ARPC1B deficiency. Journal of Allergy and Clinical Immunology. 2019 Jun;143(6):2296–2309. doi: 10.1016/j.jaci.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su HC. DOCK8 (Dedicator of cytokinesis 8) deficiency. Curr Opin Allergy Clin Immunol. 2010 Dec;10(6):515–520. doi: 10.1097/ACI.0b013e32833fd718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskenvuo M, Kainulainen L, Vanto T, Lukkarinen H, Lähteenmäki P, Ruuskanen O. [Severe atopy and allergy--rare hyper-IgE syndrome caused by the DOCK8 mutation as underlying condition]. Duodecim. 2015;131(6):541–544. [PubMed] [Google Scholar]
- Jabara HH, Ohsumi T, Chou J, Massaad MJ, Benson H, Megarbane A, et al. A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. Journal of Allergy and Clinical Immunology. 2013 Jul;132(1):151–158. doi: 10.1016/j.jaci.2013.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon ML, Rozmus J, Fung SY, Hirschfeld AF, Del Bel KL, Thomas L, et al. Combined immunodeficiency associated with homozygous MALT1 mutations. Journal of Allergy and Clinical Immunology. 2014 May;133(5):1458–1462.:e7. doi: 10.1016/j.jaci.2013.10.045. [DOI] [PubMed] [Google Scholar]
- Hwa V. Human growth disorders associated with impaired GH action: Defects in STAT5B and JAK2. Molecular and Cellular Endocrinology. 2021 Jan;519:111063. doi: 10.1016/j.mce.2020.111063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa V, Nadeau K, Wit JM, Rosenfeld RG. STAT5b deficiency: Lessons from STAT5b gene mutations. 2011 Feb;25(1):61–75. doi: 10.1016/j.beem.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Nadeau K, Hwa V, Rosenfeld RG. STAT5b Deficiency: An Unsuspected Cause of Growth Failure, Immunodeficiency, and Severe Pulmonary Disease. The Journal of Pediatrics. 2011 May;158(5):701–708. doi: 10.1016/j.jpeds.2010.12.042. [DOI] [PubMed] [Google Scholar]
- Abdollahpour H, Appaswamy G, Kotlarz D, Diestelhorst J, Beier R, Schäffer AA, et al. The phenotype of human STK4 deficiency. Blood. 2012 Apr 12;119(15):3450–3457. doi: 10.1182/blood-2011-09-378158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orange JS, Chinen J, editors. Encyclopedia of Medical Immunology: Immunodeficiency Diseases [Internet] 2020. [cited 2021 Aug 28]. Available from: http://link.springer.com/10.1007/978-1-4614-8678-7. [Google Scholar]
- Wang Y, Ma CS, Ling Y, Bousfiha A, Camcioglu Y, Jacquot S, et al. Dual T cell– and B cell–intrinsic deficiency in humans with biallelic RLTPR mutations. J Exp Med. 2016 Oct 17;213(11):2413–2435. doi: 10.1084/jem.20160576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévy R, Gothe F, Momenilandi M, Magg T, Materna M, Peters P, et al. Human CARMIL2 deficiency underlies a broader immunological and clinical phenotype than CD28 deficiency. J Exp Med. 2023 Feb 6;220(2):e20220275. doi: 10.1084/jem.20220275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlarz D, Ziętara N, Milner JD, Klein C. Human IL-21 and IL-21R deficiencies: two novel entities of primary immunodeficiency. Current Opinion in Pediatrics. 2014 Dec;26(6):704–712. doi: 10.1097/MOP.0000000000000160. [DOI] [PubMed] [Google Scholar]
- Salzer E, Kansu A, Sic H, Májek P, Ikincioğullari A, Dogu FE, et al. Early-onset inflammatory bowel disease and common variable immunodeficiency–like disease caused by IL-21 deficiency. Journal of Allergy and Clinical Immunology. 2014 Jun;133(6):1651–1659.:e12. doi: 10.1016/j.jaci.2014.02.034. [DOI] [PubMed] [Google Scholar]