

Abstract
Drug-induced gene expression profiles can identify potential mechanisms of toxicity. We focus on obtaining signatures for cardiotoxicity of FDA-approved tyrosine kinase inhibitors (TKIs) in human induced-pluripotent-stem-cell-derived cardiomyocytes, using bulk transcriptomic profiles. We use singular value decomposition to identify drug-selective patterns across cell lines obtained from multiple healthy human subjects. Cellular pathways affected by cardiotoxic TKIs include energy metabolism, contractile, and extracellular matrix dynamics. Projecting these pathways to published single cell expression profiles indicates that TKI responses can be evoked in both cardiomyocytes and fibroblasts. Integration of transcriptomic outlier analysis with whole genomic sequencing of our six cell lines enables us to correctly reidentify a genomic variant causally linked to anthracycline-induced cardiotoxicity and predict genomic variants potentially associated with TKI-induced cardiotoxicity. We conclude that mRNA expression profiles when integrated with publicly available genomic, pathway, and single cell transcriptomic datasets, provide multiscale signatures for cardiotoxicity that could be used for drug development and patient stratification.
Subject terms: Data processing, Data integration, Cardiovascular biology
Using a new computational pipeline for identification of drug-selective transcriptomic responses and FAERS data, the authors identified potential pathways and genomic variants indicative of cancer drug cardiotoxicity in iPSC-derived cardiomyocytes.
Introduction
Adverse side-effects of therapeutically useful drugs continue to be a substantial problem1. Post-approval pharmacovigilance studies often reveal adverse events that lead to warning labels mandated by the FDA2. Early indications of a potential for adverse events will be useful in drug development3,4 and the use of whole genome sequence data from individuals can predict who might be susceptible to adverse events. These assertions are based on the premise that various functions at the molecular and cellular levels drive adverse events in different cell types5. Preclinical studies at the molecular level are useful, as has been demonstrated by HERG channel protein interacting drugs and the potential for arrhythmias6,7. Drug-related adverse events are often organ-selective. Many efficacious antineoplastic drugs, such as tyrosine kinase inhibitors (TKI) that are used for targeted therapy, are associated with cardiac insufficiencies and development of heart failure8,9. We have shown that drug-induced transcriptomic profiles in adult human heart cells can be associated with clinical adverse event propensity as assessed from FDA pharmacovigilance data10. However, systematic mapping of the molecular pathways, cell origins and genomic determinants associated with drug therapy-related cardiotoxicity have not been conducted yet. The goal of this study was to conduct experiments to identify such transcriptomic signatures and by integrating signatures with publicly available data generate hypotheses for multiscale understanding of cardiotoxicity in future studies.
Human induced pluripotent stem cell (hiPSC)-derived cardiomyocytes11 have been useful for understanding cardiotoxicity12. Hence, transcriptional profiles in human cardiac cells can form starting points for studies focused on mechanism-based drug signatures that could be used for prediction of cardiotoxicity potential. Here, we have used six hiPSC-derived cardiomyocyte cell lines from healthy individuals to study 52 FDA-approved and two experimental drugs and identify drug-selective transcriptomic signatures across the different human subject lines by singular value decomposition (SVD)-based analysis of total transcriptomic responses. Single cell analyses of these hiPSC-derived “ventricular cardiomyocytes” indicated that they are composed of multiple clusters. They include a cluster very similar to adult cardiomyocytes. Additional clusters show varying levels of similarity to different cell types in the human heart, including fibroblasts. Projections of drug-affected pathways inferred from bulk transcriptomic data of hiPSC-derived cardiomyocytes on to single cell expression profiles of human hearts from healthy and heart failure patients indicate that TKIs could affect both cardiomyocytes as well as other cell types to produce adverse events, and that the drug-induced pathways are similar to those altered in hiPSC-derived cardiomyocytes from patients with failing hearts13,14. Using outlier analyses and Whole Genome Sequencing (WGS) from the cell lines used for the transcriptomic studies, we correctly re-identified the genomic variant casually associated with anthracycline-induced cardiotoxicity (AIC) that was originally discovered in genome-wide association studies (GWAS)15. We used this approach to identify and predict potential effects of genomic variants associated with TKI action as identified by transcriptomic signatures. These findings indicate that integration of experimental data with public data sets can be a powerful driver of multiscale understanding of organ level adverse events associated with drug therapy.
Results
To identify pathway activities and potential genomic variants associated with TKI-induced cardiotoxicity (TIC), we treated six hiPSC-derived cardiomyocyte cell lines from six healthy individuals (Supplementary Fig. 1) (Supplementary Data 1) with one of 27 TKIs, 4 anthracyclines and 26 other cardiac and non-cardiac-acting drugs for 48 hours, using therapeutically relevant drug concentrations (Supplementary Data 2A, Supplementary Table 116). Untreated control data was generated at the same time and has been previously published as part of characterization of these cell lines16. In this study, we used the untreated control data from each experiment to identify the drug-treatment-induced differentially expressed genes (DEGs). All drugs (except endothelin and TNF-alpha) are FDA-approved. The group of TKIs contained 23 small molecule TKIs and 4 monoclonal antibodies against TKs. They could be separated into 10 cardiotoxic and 17 non-cardiotoxic TKIs based on the results of clinical studies (Supplementary Data 3) and FAERS data analysis (Supplementary Fig. 2). Bulk transcriptomic analysis of control16 and drug-treated cell lines generated 266 lists of DEGs (Supplementary Data 4) (Supplementary Data 5 shows averaged DEGs). All lists of DEGs, each representing the response observed for one sample, i.e., a unique cell line/drug combination (Supplementary Fig. 3A), were subjected to pairwise correlation and hierarchical clustering (Supplementary Fig. 3B).
Singular value decomposition to reveal drug-selective expression responses
To identify drug-selective expression responses we used SVD to search for shared gene expression components in multiple cell lines treated with the same drug (Fig. 1A, Supplementary Fig. 4). Clustering of the unprocessed transcriptomic data only grouped a few samples treated with the same drugs into the same cluster (Fig. 1B, Supplementary Fig. 3B). In contrast, the identity of the cell line, or the amplitude of the drug response, i.e., the number of significant DEGs, mainly determined the clustering results (Supplementary Fig. 5A). To determine if we could identify drug-selective clustering, we calculated one F1 score for each drug that documents how close all cell lines treated with that drug cluster together (Supplementary Fig. 5B). With possible values larger 0 up to 1, the median F1-score of 0.116 documents very low drug-selective clustering efficiencies (Fig. 1C).
Fig. 1. Singular value decomposition identifies drug-selective gene expression responses.

266 samples, each representing a unique combination of one out of three to six hiPSC-derived cardiomyocyte cell lines treated with one out of 54 drugs, were subjected to bulk RNAseq analysis. 266 lists of differentially expressed genes (DEGs) were calculated using the negative-binomial test implemented in the ‘exactTest’ functionality of the edgeR package. A Our computational pipeline uses singular value decomposition (SVD) to identify drug-selective gene expression responses that are components of the complete responses. Flow chart is used with permission from Mount Sinai Health System, licensed under CC BY. See methods section and Supplementary Fig. 4 for details. B Pairwise correlation analysis followed by hierarchical clustering reveals that most drug responses are dominated by cell-line-selective effects hiding drug-selective effects. Heatmap colors describe drugs used for treatment, as documented in E. See Supplementary Fig. 5A for larger dendrogram. C We used F1 score statistics to document the clustering efficiency, i.e., how close samples treated with the same drug cluster together. Low clustering efficiencies quantitatively describe the finding that only a few complete DEG responses are dominated by drug-selective effects. D Projection of the complete DEG responses into each of the identified 54 drug-selective subspaces greatly increases the clustering efficiencies for all 54 drugs. Numbers of treated cell lines are shown below the bars. Orange: Small molecule kinase inhibitors (KI), red: monoclonal antibodies against KIs, purple: anthracyclines, blue: cardiac-acting drugs, turquoise: non-cardiac-acting drugs. E Pairwise correlation of 266 merged drug-selective responses, followed by hierarchical clustering, documents that SVD allows identification of components induced in all cell lines treated with the same drug. Clusters that contain drugs with similar mechanisms are labeled with gray bars. White insets indicate drugs in those clusters that are not part of the outlined mechanisms. White circles indicate outlier samples that were identified by our pipeline and cluster as outliers in the merged dataset as well. See Supplementary Fig. 13 for a larger dendrogram. #: count of, Ø: without, &: and.
SVD (Supplementary Fig. 6A) of all 266 samples identified 266 eigenarrays (Supplementary Fig. 6B) whose linear combination using sample-specific coefficients gives the complete gene expression profiles. The first eigenarray correlates (Supplementary Fig. 6C) with the amplitude of the response (Supplementary Fig. 6D), and its most prominent genes (Supplementary Data 6) enrich for muscle contraction (Supplementary Fig. 6E, Supplementary Data 7). These results indicate that the first eigenarray describes a general response to perturbation that masks drug-selective effects. Removal of the first eigenarray (Supplementary Data 8) disrupts the clustering by the number of significant DEGs (Supplementary Fig. 7). Since the clustering is still dominated by cell-line-selective effects, the removal did not markedly improve the drug-selective clustering efficiencies (Supplementary Fig. 8).
We ranked the remaining eigenarrays by their ability to separate samples of each drug or cell line from all other samples (Supplementary Fig. 9A). Our algorithm allowed a clear separation of cell-line and drug-selective effects (Supplementary Fig. 9B). For each drug, we combined unique sets of drug-selective eigenarrays to drug-selective subspaces with the goal of optimizing the drug’s clustering efficiency (Supplementary Fig. 10A), while preserving a maximum of the original information, as quantified by cosine similarity. Changing the relative contribution of F1 scores (Supplementary Fig. 10B) and cosine similarities (Supplementary Fig. 10C), we screened the potential subspaces for outlier responses where one cell line showed a significantly different response to the drug of interest than all other cell lines (Supplementary Figs. 10D, 11, 12). Such outlier responses were linked to our genomic analysis at a later stage. For 24 drugs, we could identify subspaces with outlier responses (Supplementary Fig. 10E), for the other 30 drugs, we selected subspaces based on high relative contribution of the F1 score (95%).
Drug-selective gene expression profiles are similar for drugs with similar mechanisms
Projection of gene expression profiles into the final drug-selective subspaces generated drug-selective gene expression profiles (Supplementary Data 9) (averaged DEGs in Supplementary Data 10) with high clustering efficiencies (Fig. 1D, Supplementary Fig. 10F). Drug-selective DEGs were combined into a new matrix. Clustering of this matrix identifies clusters that mostly contained samples that were treated with the same drug and documented preservation of outlier characteristics (Fig. 1E, Supplementary Fig. 13). Twelve of the 24 identified outlier samples are clearly separated from the cell lines treated with the same drug (closed circles in Fig. 1E and Supplementary Fig. 13). Another seven outlier samples are grouped together with the samples treated with the same drug in a larger cluster (open circles in Supplementary Fig. 13), but are separated from those, once that cluster is subclustered. Only four identified outliers reside in the same cluster (open rectangles) as the other cell lines treated with the same drug. Comparison of Figs. 1B and 1E in terms of the breadth of the colored bars allows for visualization of this drug-selective clustering.
Our algorithm identified drug-selective gene expression profiles for each drug independently. Nevertheless, drugs with similar mechanisms and overlapping targets still cluster together (Fig. 1E, Supplementary Fig. 13), indicating the potential biological validity of our approach. All eight TKIs targeting EGFR signaling, either by inhibiting the receptor (EGFR, ERBB2) or its intracellular ERK signaling cascade (MAP2K1, MAP2K2)17 are part of the same cluster. One of the four non-TKI drugs in this cluster, phenylephrine, and isoprenaline which is part of a slightly bigger cluster, stimulate adrenergic signaling that can cross-activate ERK signaling in the heart18,19, as documented for phenylephrine in the perfused rat heart19. Two additional non-TKI drugs in this cluster, verapamil and amiodarone, influence adrenergic signaling as antagonists20–22 and verapamil has been shown to antagonize ERK signaling in rat cardiomyocytes23. Similarly, the JAK inhibitors ruxolitinib and tofacitinib and the proteasome inhibitors bortezomib and carflizomib are part of two independent clusters that contain no other drugs. Two of three antidiabetics, rosiglitazone and saxagliptin are part of a smaller cluster that first merges with a cluster containing decitabine, a drug with hyperglycemia as a major side effect24 and then with a cluster containing cyclosporine and olmesartan. Cyclosporine increases insulin resistance in humans25 and olmesartan −/+ amlodipine ameliorates it in rats26 and patients27, respectively. The anthracyclines epirubicin, daunorubicin and doxorubicin are part of larger cluster, while the anthracycline idarubicin clusters together with their identified outlier samples.
In summary, our SVD pipeline uncovers similar expression profiles induced by similar drugs. Since it does not actively search for such similarities, but only aims at maximizing the overlap between DEGs induced by the same drug, this finding serves as internal validation for the extraction of drug-selective components by our computational pipeline.
The grouping of drugs with similar mechanisms into higher-level clusters explains eleven non-outlier samples that are separated from the other samples treated with the same drug (closed rhombuses), leaving only four samples that are not part of drug-selective clusters for no obvious biological reason (open rhombuses) (Supplementary Fig. 13).
SVD-based resolution of drug-dependent DEGs enables identification of subcellular processes relevant to TKI-induced cardiotoxicity
Complete and decomposed DEGs were subjected to pathway enrichment analysis using the Molecular Biology of the Cell Ontology (MBCO)28 (Supplementary Fig. 14A). MBCO subcellular processes (SCPs) are organized in three to four levels where higher-level SCPs (i.e., levels with lower numbers) describe more general, and lower-level SCPs more detailed cell biological processes. Predicted up- or downregulated SCPs of each drug/cell line combination and SCP level were ranked by significance (Supplementary Data 11, 12 and 13). We refer to these ranks as enrichment ranks in the following.
Overall comparison of the enrichment results before and after decomposition documents a large increase in the number of overlapping SCPs in different cell lines treated with the same drug (Supplementary Figs. 14B, C, D, E, 15A).
In addition, our SVD decomposition allowed identification of SCPs that were masked in the complete dataset. For example, we could document downregulation of SCPs involved in single protein degradation as an effect selective for anthracyclines (Supplementary Fig. 15B), results that we did not obtain using the complete DEGs (Supplementary Fig. 15C). For a discussion of these SCPs, their potential link to supportive treatments and examples that demonstrate agreement with prior knowledge from small-scale experiments see Supplementary Note 2.
Subcellular processes and cell types indicative of cardiotoxic response to TKI treatment
To identify signatures associated with TIC we searched for SCPs that are almost exclusively up- or downregulated at higher enrichment ranks by cardiotoxic TKIs as compared to non-cardiotoxic TKIs, using F1 score and Area under the Curve (AUC) statistics (Fig. 2A, Supplementary Figs. 16 and 17, Supplementary Data 14). SCPs were ranked by decreasing AUC. The top 10, 10, 25 and 10 level-1, -2, -3 and -4 SCPs were grouped by functional similarities (Fig. 2B, Supplementary Fig. 18) and integrated into the MBCO hierarchy (Fig. 2C, Supplementary Fig. 19). In addition, we mapped identified SCPs back to the cardiotoxic TKIs that up- or downregulate them (Supplementary Fig. 20).
Fig. 2. Potential subcellular processes indicative of TKI-induced cardiotoxicity.

Up- and downregulated genes among the top 600 drug-selective gene expression profiles were subjected to pathway enrichment analysis using MBCO and Fisher’s Exact test. Significantly up- or downregulated SCPs (nominal p-value ≤ 0.05) were ranked separately by significance for each sample and SCP level. A To screen for SCPs that are selectively induced or repressed by cardiotoxic TKIs, we calculated how many cardiotoxic and non-cardiotoxic TKIs upregulate an SCP of interest in any cell line at any rank cutoff from 1 to 30. Definition of cardiotoxic TKIs as true positives allowed calculation of an F1 score (beta = 0.25) at each analyzed enrichment rank and quantification of the Area under the Curve (AUC). Similarly, we calculated F1 scores and an AUC by analyzing TKIs that downregulate the same SCP. To filter for mixed effects, we subtracted half of the other AUC from each AUC. SCPs were ranked by decreasing AUCs. Flow chart is used with permission from Mount Sinai Health System, licensed under CC BY. B Top up- (red) or downregulated (dark blue) 25 level-3 SCPs predicted for the cardiotoxic TKIs were grouped based on the higher-level functions. White numbers indicate AUC ranks. C The same analysis was applied to level-1, -2 and -3 SCPs, except that we only focused on the AUC obtained for enrichment ranks 1 to 20, due to a smaller number of SCPs within these levels. We also repeated the whole analysis, screening for SCPs selectively induced or repressed by non-cardiotoxic TKIs. Identified SCPs up- and downregulated for cardiotoxic (red and dark blue, respectively) and non-cardiotoxic TKIs (light blue and orange, respectively) for all levels were integrated into the MBCO hierarchy. Selected branches are shown. See Supplementary Fig. 19 for all predictions.
Besides being elevated or decreased above or below a threshold that determines an SCP’s association with a cardiotoxic response, an SCP’s activity might already be beyond that threshold at baseline level, i.e., before TKI treatment. Candidates for this group could be SCPs that are regulated by non-cardiotoxic TKIs. A non-cardiotoxic TKI might change the baseline SCP activity from sufficient to insufficient for a cardiotoxic response. Consequently, our second focus was the identification of SCPs that non-cardiotoxic TKIs would up- or down-regulate to potentially suppress cardiotoxicity (Supplementary Figs. 18, 19, 21).
Single cell RNA sequencing (RNAseq) of four of our six cardiomyocyte cell lines identified at least two major subtypes mapping to adult cardiomyocytes and an epicardium-derived cell type that is similar to fibroblasts16 (Supplementary Fig. 22A–D, Supplementary Data 15, 16A, B). The SCPs we identified by bulk transcriptomics often describe canonical functions of cell types other than cardiomyocytes. We determined how the top SCPs in our data map to our subtypes (Supplementary Fig. 23, Supplementary Data 16C–F), and to the major cell types of the adult human heart analyzed by others using single cell transcriptomics29 (Fig. 3A, Supplementary Fig. 23, Supplementary Data 17, 18A–D).
Fig. 3. SCPs can be mapped to cellular subtypes and known cardiomyopathy disease mechanisms.
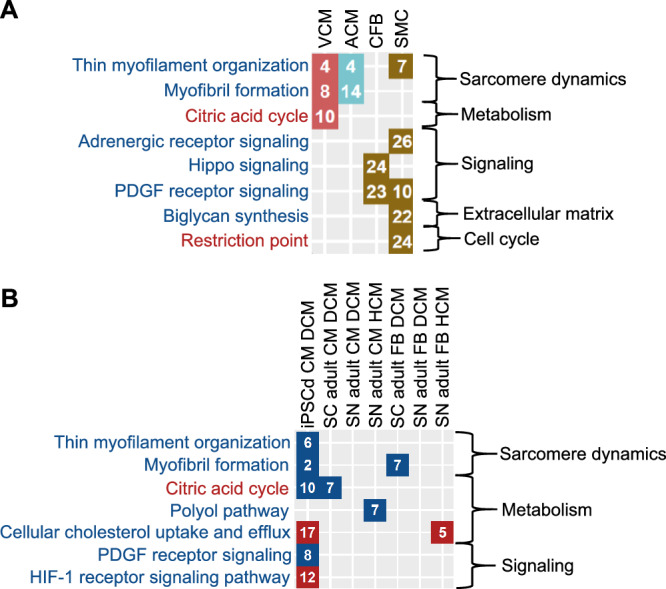
A We subjected marker genes for ventricular and atrial cardiomyocytes (VCM, red fields, and ACM, turquoise fields, respectively), cardiac fibroblasts (CFB, brown fields) and smooth muscle cells (SMC, brown fields) obtained from single nucleus RNAseq of the adult human heart29 to pathway enrichment analysis using MBCO and Fisher’s exact test. Significant SCPs of each cell type (nominal p-value ≤ 0.05) were ranked by significance (numbers in the diagram). Names of SCPs whose higher and lower activities favor a cardiotoxic response are colored red and blue, respectively. B DEGs in heart cells obtained by single cell (SC)13 or nucleus (SN)14 RNAseq from patients with DCM or HCM as well as in hiPSC-derived cardiomyocytes obtained from an infant patient with DCM32 were subjected to pathway enrichment analysis using MBCO and Fisher’s exact test. Significantly up- (red fields) or downregulated (blue fields) SCPs of each cell type (nominal p-value ≤ 0.05) were ranked by significance (numbers in the diagram). Names of SCPs are colored as described in (A).
Many of the identified SCPs described well-known biology involved in inherited and acquired cardiomyopathies that develop independently of chemotherapy and map to multiple cell types of the adult human heart. Our results suggest that cardiotoxic TKIs might mimic these mechanisms by early (within 48 hours) transcriptional regulation.
Four SCPs involved in muscle contraction and sarcomere renewal were simultaneously identified for the cardiotoxic and non-cardiotoxic drugs (Fig. 2B, Supplementary Fig. 18). Another two SCPs were identified for each group independently. These SCPs that span all four SCP levels (Fig. 2C, Supplementary Fig. 19) are regulated by the two TKI toxicity groups in opposite directions. The difference in directionality suggests insufficient sarcomere renewal as relevant for TIC. All cardiotoxic TKIs, except vandetinib and trastuzumab downregulate (Supplementary Fig. 20), while 13 of 17 non-cardiotoxic TKIs upregulate (Supplementary Fig. 21) at least one of these SCPs, respectively. The SCPs map to cardiomyocyte clusters identified by single cell RNAseq of our cell lines and of the adult human heart29 (Fig. 3A, Supplementary Fig. 23). In agreement with our data, proper heart functioning depends on a continuous turnover of cardiomyocyte sarcomere proteins30. About half of hypertrophic and dilated cardiomyopathies are associated with mutations in sarcomeric proteins31. Further supporting this line of reasoning, we found downregulation of muscle contraction-related SCPs in single cell RNAseq from hiPSC-derived cardiomyocytes from an infant with DCM32 (Supplementary Data 19A–C) and from adult cardiomyocytes13,14 (Supplementary Data 20A, B) (Fig. 3B, Supplementary Fig. 24). These SCPs might serve as a starting point for supportive therapy. Four of the six regulated genes of the SCP ‘Thin myofilament organization’ (Supplementary Data 11C) are involved in blocking the binding of the myosin head to the thin myofilament during muscle contraction. This mechanism is targeted by the new drug mavacamten that was recently approved by the FDA to treat obstructive Hypertrophic Cardiomyopathy (HCM)33.
Our data associates the upregulation of SCPs involved in the citric acid cycle and mitochondrial energy generation with a cardiotoxic response (Figs. 2B, C, Supplementary Figs. 18, 19). They are almost exclusively upregulated by pazopanib (Supplementary Fig. 20), a TKI with a high rate of cardiotoxicity (>10%) (Supplementary Data 3). In contrast, many studies document an overall reduction in oxidative phosphorylation during heart failure34 and identified SCPs are downregulated in adult Dilated Cardiomyopathy (DCM)13,14 and HCM13,14 cardiomyocytes as well as in hiPSC-derived DCM cardiomyocytes32, as predicted from single cell and nucleus RNAseq datasets (Fig. 3B, Supplementary Fig. 24, Supplementary Data 21A, B, C). In support of our findings, compensatory upregulation of oxidative phosphorylation was suggested for patients belonging to a large DCM subgroup that is caused by truncating titin variants35–37.
Increasing evidence links ferroptosis, an iron-dependent accumulation of lipid peroxides that triggers cell death, to heart failure38. Some identified SCPs in our data might converge on ferroptosis as an endpoint. The SCP ‘Desaturation of fatty acids’ was associated with a cardiotoxic response (Fig. 2B, C, Supplementary Figs. 18, 19) due to its upregulation by pazopanib (Supplementary Fig. 20). Upregulated genes mapping to this SCP (Supplementary Data 11C) can generate polyunsaturated fatty acids (PUVAs)39,40 that are precursors of lipid peroxides38. A lower activity of the level-2 SCP ‘Cellular antioxidant systems’ and its level-1 parent ‘Cellular redox homeostasis’, pathways offering protection against ferroptosis38, is associated with a cardiotoxic response (Supplementary Figs. 18 and 19). They were downregulated by the cardiotoxic TKIs vandetinib and bevacizumab (Supplementary Fig. 20). In addition, downregulation of ‘Cellular iron storage’ by the anthracyclines doxorubicin and daunorubicin identified by us (Supplementary Figs. 14D, 15B) and others41 suggests stimulation of ferroptosis by an increase of intracellular free iron41. In agreement, supportive therapy with iron chelators protects against AIC42, though the protective effect might involve mechanisms that are unrelated to iron homeostasis43.
Potential clinical relevance of our findings is additionally indicated by identification of SCPs involved in cholesterol metabolism and natriuretic peptide signaling (Fig. 2B, Supplementary Fig. 18). Our SCPs agree with recommended treatment schemes of cardiomyopathy and drug-induced cardiomyopathy involving statins44 and neprilysin45,46, respectively. Prediction of extracellular collagen-crosslinking (Supplementary Fig. 18) can be linked to histopathological observations that correlate with disease progression47,48. Involvement of identified signaling pathways in TIC i.e., PDGF, HIF-1alpha, Oncostatin M and Hippo signaling (Fig. 2B, Supplementary Fig. 18) is supported by their involvement in drug-independent cardiomyopathy. These and other findings regarding the predicted SCPs are discussed in further detail in Supplementary Note 2.
Taken together the data from our experiments, when integrated with data from the literature, indicate the TKIs are likely to have their cardiotoxic effects by regulating SCPs in multiple cell types of the heart and mimicking mechanisms involved in drug-independent cardiomyopathy and heart failure.
Subcellular processes associated with anthracycline toxicity
To document which pathway activities are specifically associated with AIC in distinction to the cardiotoxicity induced by very cardiotoxic TKIs (frequency >10%), we used the F1 score and AUC algorithm to compare both groups of drugs (Supplementary Fig. 25). Results were integrated into the MBCO hierarchy (Supplementary Fig. 26). As already described, the observed downregulation of ‘Cellular iron storage’ agrees with the central role of ferroptosis in AIC41. Observed upregulation of ‘Nucleotide excision repair’ agrees with the documented involvement of both nucleotide excision repair and homologous recombination in recognition and repair of anthracycline-DNA adducts49. Upregulation of histone genes and other genes involved in chromatin remodeling could be the consequence of anthracycline-induced histone eviction50. As suggested by our data, dysregulation of mitochondrial dynamics is another mechanism involved in AIC41. Similarly, dysregulation of proteasomal degradation and autophagy has been reported previously51, as well as activation of cell death pathways52.
TKI transcriptomic signatures are similar in cardiomyoctes cocultured with and without endothelial cells
Our experimental protocol raises the possibility that the signatures we obtain are the consequence of our single cell type culture conditions that are missing other cell types of the human heart. One cell type thought to be relevant for cardiotoxicity is the endothelial cell type of the blood vessels53. To determine if our signatures in cardiomyocytes may be influenced by other cell types, we repeated the stimulation of two selected cell lines with pazopanib and dabrafenib in absence or presence of human coronary arterial endothelial cells (HCAECs) (Supplementary Fig. 27A, Supplementary Data 2B). Cardiomyocytes in the presence of endothelial cells showed increased beating rates. Drug treatment at therapeutic doses did not affect the morphology or viability of either endothelial cells (Supplementary Fig. 27B) or cardiomyocytes. Cardiomyocytes and endothelial cells were cocultured in coculture plates with porous membranes for 48 hours and then treated with drugs for another 48 hours. This set up allows to obtain the cardiomyocytes separately for extraction and bulk transcriptomics under the same conditions used for the original experiments.
Pazopanib is a multi-tyrosine kinase inhibitor whose antineoplastic effect is associated with inhibition of angiogenesis and interference with vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) signaling in human endothelial cells54. Endothelial dysfunction caused by VEGF blockage is discussed as a potential mechanism involved in pazopanib-induced cardiomyopathy55, suggesting that pazopanib’s high cardiotoxicity (>10%, Supplementary Data 3) could at least partly depend on interactions between endothelial cells and cardiomyocytes. As a second drug, we selected the B-Raf inhibitor dabrafenib, as a representative for TKIs with less evidence for cross-cellular effects56,57.
Eight lists of DEGs of the new data (Supplementary Data 22A) were merged with the original 266 lists (Supplementary Data 4). The merged 274 lists of DEGs were projected into the drug-selective subspaces identified from the original 266 lists (Supplementary Data 22B, C), followed by pairwise correlation analysis and hierarchical clustering. Both, pazopanib and dabrafenib (Supplementary Fig. 28A) treated cell lines clustered closely together and there was only a slight change in the overall F1 scores (Supplementary Fig. 28B). A distinct clustering of DEGs generated in presence or absence of the endothelial cells was not observed for either TKI. Clustering of the new combined final drug-selective DEG matrix that now contained 274 instead of 266 lists of DEGs showed only minor rearrangements within a larger cluster of nine drugs that contained pazopanib and dabrafenib (Supplementary Fig. 28C and D). Predicted pathways (Supplementary Fig. 29, Supplementary Data 23–25) and cardiotoxic pathways induced or repressed by pazopanib (Supplementary Fig. 30A) and dabrafenib (Supplementary Fig. 30B) showed only minor differences between the new and old datasets. Overall, this analysis suggests that the effect of endothelial cocultures on TKI-induced signatures in cardiomyoctyes is minimal.
Candidates for genomic variants associated with drug-induced cardiotoxicity
Using whole genome sequencing of the six cell lines16, our next analysis step focused on the identification of potential genomic variants that might be associated with a higher or lower risk for drug-induced cardiotoxicity (DIC) or more specifically TIC (Fig. 4A, Supplementary Fig. 31). Cardiotoxic responses to TKI treatment are observed in less than 10% to 20% of the treated patients (Supplementary Data 3). Consequently, we hypothesized that the population-wide frequency of a potential monogenic allele associated with DIC should be in a similar range. Relevance for cardiac tissue was assumed for those variants that map to gene coding regions or are part of cis-expression (e) or -splicing (s) QTLs in the adult human heart58. We hypothesized that potentially cardiotoxic or -protective variants could interfere with a drug’s pharmacokinetics (PK) or -dynamics (PD) or with pathways that are targeted by that drug.
Fig. 4. Identification of genomic variants that are potentially associated with a cardiotoxic response.

Whole genome sequencing of our six cell lines16 was used to identify alleles in our cell lines at known variant positions. A See text and methods for details of our pipeline for identification of potential genomic variants involved in PK/PD or induced SCP activities for a drug of interest. Flow chart is used with permission from Mount Sinai Health System, licensed under CC BY. B Clustering of DEGs within the daunorubicin-selective subspace reveals an outlier response in cell line MSN09 after daunorubicin treatment. C The identified variant rs2229774 in cell line MSN09 maps to the coding region of the transcription factor RARG regulating the expression of TOP2B and ABCB8, both involved in PK/PD of daunorubicin and doxorubicin that induce an outlier response in this cell line. D Enrichment significance ranks for “WNT-Beta-catenin signaling pathway” obtained by analysis of upregulated genes after daunorubicin or doxorubicin treatment of indicated cell lines. The cell line MSN09 (purple) contains the rs2229774 mutation in the RARG gene. Field colors change from bright to dark yellow with increasing ranks. ‘>’ indicates that the SCP was not predicted or predicted with a rank > 99. E In total, we identified 213 and 201 potential variants associated with TIC or AIC by interference with PK/PD mechanisms, respectively. Variants mapping to multiple gene classes are split equally among them to prevent double counting. Drug names are colored according to their class (orange: Small molecule kinase inhibitors (KI), red: monoclonal antibodies against KIs, purple: anthracyclines). F We compared the overlap of identified SCP genes associated with a cardiotoxic or non-cardiotoxic response to genes associated with inherited DCM or HCM, either within the HuGE Phenopedia database or identified in GWAS. TWAS: transcription-wide-association studies. G Variants that meet our population-wide criteria were mapped to up- and downregulated level -2, -3 and -4 SCPs that we predicted as indicative for TIC. Variants that map to identified SCPs of multiple levels are only counted for the lowest level SCPs (higher level numbers) to prevent double counting. Drug names are colored as described in (E). H Up- and downregulated SCPs associated with a cardiotoxic response were mapped back to the cardiotoxic TKIs that induce them. Numbers in brackets show identified variants for each SCP gene. Blue indicates that the SCP is a level-3 SCP.
Variants of the first interference group should induce a different transcriptomic response, allowing us to link cell-line-specific variants to identified 24 outlier responses. Since, based on statistical likelihoods, either none or only one of our six healthy volunteers should suffer from cardiotoxicity induced by a drug of interest, we focused on variants that are overexpressed in an outlier cell line for a drug of interest, if compared to the other five lines. Mechanistic knowledge is added by considering only variants that map to genes with potential involvement in a drug’s PK/PD.
Variants of the second group should map to pathways that either contain drug target proteins or are induced or repressed by the drug of interest on the transcriptional level. Using the results of our transcriptomic analysis, we focused on the second subgroup with a particular interest in those SCPs that are associated with a cardiotoxic or non-cardiotoxic response.
Identification of genomic variants interfering with a drug’s PK/PD
Based on outlier responses to daunorubicin and doxorubicin (Fig. 4B, Supplementary Figs. 11 and 12), our PK/PD algorithm allowed re-identification of the variant rs2229774 (Fig. 4C, Supplementary Data 25) within the coding region of the RARG gene15, one out of three genomic variants with the strongest evidence for AIC59. The suppressive activity of the transcription factor RARG on the expression of the anthracycline target TOP2B is reduced by this variant, leading to increased AIC60. Pathway enrichment analysis suggests missing upregulation of WNT receptor signaling as a potential mechanism of rs2229774-triggered AIC (Fig. 4D, Supplementary Fig. 14D). In agreement, inhibition, or activation of WNT signaling in mice and cell assays enhances or mitigates AIC, respectively61–63.
In total, our outlier-based screening approach identified 128 and 111 potential variants that map to 100 and 88 genes involved in the PK/PD of three anthracyclines and three cardiotoxic TKIs, respectively (Fig. 4E, Supplementary Fig. 32A, Supplementary Data 26). Identified variant candidates could trigger or protect from AIC or TIC. Three of the variants map to drug target proteins of ponatinib and trametinib (Supplementary Fig. 32B), TKIs with medium (1–10%) and high ( > 10%) rates of TIC, respectively (Supplementary Data 3).
Considering all 24 drugs with documented outlier responses for this analysis we identified 464 variants (Supplementary Fig. 32A) mapping to 288 genes (Supplementary Fig. 32C) (Supplementary Data 26).
SCP-based identification of genomic variants associated with TKI-induced cardiotoxicity
Identified level-2, -3 and -4 SCPs associated with the response to cardiotoxic and non-cardiotoxic TKIs enrich for genes that are mapped to inherited DCM or HCM, either within the HuGE Phenopedia database64 or obtained from GWAS65,66 (Fig. 4F). We left out level-1 SCPs from this analysis, since they were predicted based on genes mapping to a sub-function of the general cellular functions the level-1 SCPs describe. It needs to be considered that some of these variants could contribute to drug cardiotoxicity by modulating propensity for cardiac insufficiency without primarily effecting cardiomyocytes. Nevertheless, the significant overlap supports our hypothesis that TKIs induce cardiotoxicity by mimicking mechanisms involved in inherited or acquired drug-independent cardiomyopathy, as discussed above and in Supplementary Note 2.
To identify a second set of potential variants interfering with TIC we mapped all variants that met our population-wide requirements to identified SCPs. In total, we identified 634, 410 and 80 non-overlapping variants mapping to level-2, -3 and -4 SCPs that are up- or downregulated by cardiotoxic TKIs (Fig. 4G) (see Supplementary Fig. 32D for variants mapping to SCPs regulated by non-cardiotoxic TKIs). Mapping identified SCPs back to the drugs that induce them (Supplementary Fig. 20) enables identification of variants that might interfere with cardiotoxicity of a drug of interest (Supplementary Data 27). For example, we predicted 13 variants mapping to four genes of the SCP ‘Thin myofilament organization’ (Fig. 4H) that might interfere with the cardiac response to sunitinib and ponatinib downregulating this SCP in six (enrichment ranks 2 × 3, 4, 2 × 7) and five (5 × 1) of six treated cell lines, respectively (Supplementary Fig. 20), but also to lapatinib, dabrafenib and bevacizumab. Two of those genes are involved in the same function that is targeted by mavacamten, as described above.
Since most variants identified in GWAS of drug-independent cardiomyopathy and mapping to our SCPs occur at high frequency and are not part of cis e/s-QTLs, they do not meet our population-wide requirements. Hence, we do not list them as being involved in drug-induced cardiotoxicity. Some of these variants might not primarily affect cardiomycoytes or other abundant cells in the heart. Additionally, HCM and DCM exist as both monogenic and polygenic diseases. Similarly, single or multiple variants might be related to TIC, requiring development of more sophisticated genomic algorithms. Nevertheless, identified SCPs are a good starting point to reduce the burden of multiple testing hypothesis by restricting the focus on genes with a functional implication in TIC.
Discussion
A key question that arises in identifying transcriptional signatures from short term treatment of cell lines with drugs is whether early transcriptional events in a 1–2 day time frame can be predictive of later physiological (pathophysiological) states of an organ that takes weeks or months to manifest when a drug is used clinically. While it is not possible to answer this question for multiple organs and drug classes, in the case of TKIs and heart the answer is yes. Two types of relationships support this conclusion. We previously showed that transcriptomic signatures of TKIs with differing toxicity profiles in FAERS could be associated with these profiles67. In this study, the pathway analyses of drug-induced gene expression lists across cell lines show that many TKIs with adverse event propensity selectively regulate pathways that have also been shown to be involved in different types of cardiomyopathies68. Additionally, the identity of the pathways themselves such as contraction dynamics and extracellular matrix are directly relevant to development of cardiac insufficiency. It is noteworthy that mavacamten, a drug approved in 2022 by the FDA to treat obstructive hypertrophic cardiomyopathy33 regulates the level of actin-myosin bridge formation and thus controls contractile dynamics.
Further, our previous data served as the external validation set for a computational study that integrated transcriptomic signatures with machine learning to predict cardiotoxicity69. Taken together, it appears reasonable to conclude that the pathways regulated by cardiotoxic drugs can serve as pathway signatures for cardiotoxicity. Such signatures could be used in drug development. Transcriptomic profiles of drug candidates could predict risk of cardiotoxicity from early cell-based study (Fig. 5A). Additionally, knowledge of pathway activities involved in cardiotoxic side effects may allow focused drug repurposing with the aim to reverse relevant pathway activities and not overall TKI-induced gene expression (Fig. 5B)70. Ideally, pathway activities targeted by the mitigating drug should not participate in the antineoplastic effect, allowing continuation of TKI treatment even when cardiotoxicity is a possibility, as anticipated by the concept of permissive cardiotoxicity71.
Fig. 5. Potential use of cell-based transcriptomic data for drug therapy induced adverse events.

The flowcharts summarize how integration of experimentally gathered transcriptomic data with publicly available pathway and genomic data bases can (A) help predict toxicity of drug candidates, (B) identify potential new drug targets to mitigate cardiotoxicity and (C) enable design of clinical studies to associate genomic variants with cardiotoxicity propensity. All three flow charts are used with permission from Mount Sinai Health System, licensed under CC BY.
Our method for identifying drug-selective DEGs could have wider applications since both bulk and single cell expression profiles often represent the combination of multiple DEG sets induced by different actions on the same cell type or mixtures of cell types (i.e., tissue). Our computational pipeline decomposes these mixed gene expression profiles into individual effect-selective DEG sets and identifies the DEG sets involved in the response of interest. For example, grouping patients with the same disease into subgroups based on shared disease clinical manifestations could enable identification of gene expression profiles associated with each selected subgroup that can lead to tailored treatment options. A particular advantage of our pipeline is that patient groupings do not need to be mutually exclusive, i.e., patients can be assigned to each disease subgroup independently of the other assignments.
A serendipitous finding from our studies on the hiPSC-derived cardiomyocytes is that single cell RNAseq allowed us to resolve these cells into multiple clusters16. As single cell transcriptomic data from normal29 and diseased13,14 adult human heart became available in 2022 we compared the clusters in our data to the different cell types in the adult human heart. This comparison allowed us to identify the group that is most like ventricular cardiomyocytes and to identify a group related to fibroblasts. Several of the top ranked SCPs are most likely expressed in all these cell types, since they are not specifically enriched in any of them. Thus, the integration of bulk transcriptomic and single cell transcriptomic data allows us to project the SCPs onto different cell types of the heart. These integrated analyses suggest that the cardiotoxic drugs are likely to have their pathophysiological effects by affecting multiple cell types. The effects on fibroblasts and extracellular matrix dynamics72 can readily affect the elasticity of the cardiac muscle and it has long been known that extracellular collagen-crosslinking is directly related to heart failure47,48. It appears likely that the effects of TKIs on fibroblasts can contribute in part to cardiotoxic effects. In evaluating drug candidates for cardiotoxicity, it may be necessary to consider the drugs’ effects not only on cardiomyocytes but also on cardiac fibroblasts and other cell types in the heart. Nevertheless, an initial screen with just cardiomyocytes in cell-based experiments may suffice to weed out compounds that may be cardiotoxic. Our data with cardiomyocyte and endothelial cell cocultures did not change the pazopanib- and dabrafenib-induced expression patterns, supporting this idea.
Using the common differential expression patterns for each drug across cell lines, it is possible to identify outlier responses in a particular cell line (i.e., human subject). Such responses could provide information about genomic characteristics that could be associated with adverse events since these events occur in typically 1–20% of the population. Genomic variants associated with the proteins involved in PK/PD are likely candidates. We tested the validity of this assumption. We found that the genomic variant rs2229774 within the coding region of RARG can be identified by integration of whole genome wide sequencing of our six healthy cell lines and transcriptomic outlier analysis of the anthracycline drugs. GWAS15 and follow-up studies60 had previously identified RARG to be causally linked to AIC, since RARG regulates the expression of the anthracycline target topoisomerase 2B. The variant in the RARG gene is one of three genomic variants with the strongest clinical evidence for AIC59. Based on this validation we searched for genomic variants associated with cardiotoxic TKIs. We considered PK, PD, and key pathways regulated by these drugs and developed a list of variants whose occurrence could trigger TIC in a monogenetic manner. Here, data integration identifies genomic variants that could serve as hypotheses for targeted genomic studies to identify risk determinants (Fig. 5C), even before a sufficient number of patients has developed side effects to meet minimum sample sizes required for traditional GWAS statistics. Comparison of induced pathways between regular and outlier responses could suggest targets for mitigating therapies. Our data indicates missing upregulation of WNT-signaling after anthracycline treatment in the RARG variant positive cell line, a known target for enhancement or mitigation of AIC in mice and cell assays61–63. Building on our outlier-based strategy, more sophisticated genomic algorithms could be developed to allow prediction of variants involved in polygenetic traits.
Although our study has successfully identified pathway signatures for TKI-induced cardiotoxicity, some limitations should be noted. One limitation of our study is its focus on transcriptomic responses induced by TKI treatment. For many drugs the known mechanism of action involves the direct inhibition of physiological function by binding to their protein targets. Resulting drug effects can be independent of transcriptomic effects, such as changes in protein activities or protein degradation rates. Inclusion of additional high-throughput technologies will allow more extensive characterization of drug-induced cardiotoxicity. Functional assays offer an additional opportunity to predict a drug’s cardiotoxicity73–80 and can be used to analyze if our transcriptomic findings translate into physiological effects that could be targeted by mitigating therapies. We also recognize the limited number of cell-lines (i.e., human subjects) used in our study. Although a matrix of six lines (three males, three females) and 54 drugs is substantial, we may have been able to extract more pathway and genomic information, if we could have conducted the study with 20 or 30 lines. Further, systematic coculturing of cardiomyocytes with endothelial cells, pericytes, fibroblasts and immune cell subtypes will yield a more detailed mechanistic information underlying the cardiotoxicity. Additionally, our study is at one time and one therapeutically relevant concentration and does not separate transient from permanent effects by analyzing reversal of transcriptomic changes after drug withdrawal. Testing for many of these variables was limited by resource availability.
In spite of these limitations, our study shows that integration of transcriptomic data from one cell type with pathway and genomic data in various publicly available datasets can provide reliable pathway signatures and serve as a powerful hypothesis generator for different laboratory and clinical studies to detect and avoid adverse events associated with drug therapy.
Methods
Materials
The sources of all materials are listed in Supplementary Table 1A that is taken from Schaniel et al.16, since the two studies were conducted concurrently. It is also available as detailed Protocols associated with the LINCS project (http://iyengarlab.org/dtoxs/). Supplementary Table 1B contains the sources of all materials used for the co-culture experiments.
Healthy human subject iPSC lines
Skin fibroblasts were obtained from six healthy volunteers (Supplementary Data 1), reprogrammed into human induced pluripotent stem cells (hiPSC) using the mRNA plusmicroRNA boost reprogramming method according to the manufacturer’s recommendation (Stemgent now Reprocell). Subjects were recruited and consented under Mount Sinai Institutional Review Board-approved protocol (HS# 14-00530). See related documents at our previous publication16. Reprogrammed lines, which were characterized for normal karyotype and pluripotency, were differentiated using the embryoid body formation method in PRMI 1640 containing 2 mM/L L-glutamine, 4 x 10−4 M monothioglycerol and 50 mg/mL ascorbic acid (differentiation medium) the presence of 3 ng/mL BMP4 for one day, followed by 20 ng/mL BMP and 20 ng/mL Activin A for two days. On day 3 embryoid bodies were harvested, washed and cultured in 5 ng/mL VEGF plus 5 μM XAV939 for two days, followed by culture in 5 ng/mL VEGF for three days. Medium was changed to differentiation medium and exchanged every three to four days until day 20 when embryoid bodies were dissociated and replated onto Matrigel-coated 6-well plates at a density of 1 x 106 cells/mL in differentiation medium. The following day, the medium was replaced with 4 mM lactate in DMEM without glucose (lactate medium) for four days, after which the lactate medium was titrated down in the following lactate to differentiation medium ratios: day 5, 3:1; day 6, 1:1; day 7: 1:3; day 8, 0:4. Cells were maintained for several days in differentiation medium before drug treatment for 48 h, using concentrations as described (Supplementary Data 2A).
Human Coronary Artery Endothelial Cells (HCAECs)
HCAECs were purchased from PromoCell, they were thawed and maintained as indicated. Briefly, HCAEC were maintained in Endothelial Cell Growth Medium MV supplemented with 10% Fetal Calf Serum, 0.8% Endothelial Cell Growth supplement, 10ng/ml Epidermal Growth Factor, 90µg/ml Heparin and 1 µg/ml Hydrocortisone (Endothelial Cell Growth Medium MV Kit, PromoCell). Cells were grown up to 80% confluence and then passaged using 0.04% Trypsin/0.03% EDTA and trypsin was inactivated with 0.05% Soybean Trypsin Inhibitor with 0.1% BSA (PromoCell).
Treatment of cardiomyocyte cell lines
We stimulated six cardiomyocyte cell lines with 54 different single drugs or DMSO for 48 h. The sources and concentrations of the drugs used for the treatments are in Supplementary Table 1. Typically, each cell line/drug combination had quadruplicate measurements, while 11 or 12 control replicates were generated, except for cell line MSN02 with 5 control replicates. While DMSO and drug treatments were done in parallel, data generated from the control replicates has been published previously16.
Treatment of cardiomyocytes in coculture with HCAECs
For co-cultures of HCAECs with human iPSC-derived cardiomyocytes (CMs), the HCAECs were plated on glass coverslips (18 mm diameter). Glass coverslips were first sterilized under a UV light for 15 min, then placed in a petri dish with 70% ethanol for 5 min. The glass coverslips were thoroughly rinsed 3 times with sterile filtered ddH2O and allowed to air dry under a tissue culture hood. Once the glass coverslips were dried they were coated with 1 mg/ml Poly-L-lysine hydrobromide (PLL) overnight at 4 °C. The next day, the PLL was rinsed thoroughly three times with water and the glass coverslips were allowed to air dry under a tissue culture hood.
We wanted to use a 1:1 ratio of HCAECs to CM cells as referenced from a summary of studies that had measured the proportion of CMs to endothelial cells in the human heart81. We seeded approximately 125,000 HCAECs per coverslip in a 6 well plate. Since HCAECs are doubling in culture every two to three days, we wanted to ensure that the HCAECs did not exceed approximately 500,000 cells at the end of the experiment. We let the HCAECs settle onto the glass coverslips in the Endothelial Cell Growth Medium MV overnight and the next day we co-cultured them with CM cells using Nunc co-culture plates.
CM cells were plated at 500,000 cells per well of a 6-well plate and grown in CM media. One co-culture insert containing the coverslip with the HCAECs facing top was gently inserted into each well of that 6-well plate (see Supplementary Fig. 27A for cartoon of the co-culture). We made sure enough media covered both the CM cells and the HCAECs (about 4 ml of media per well). The co-cultures were maintained in CM media and allowed to acclimate for 48 h before we began the drug treatments, using concentrations as described (Supplementary Data 2B).
At the end of the experiment, the glass coverslips with the HCAECs were removed and placed into 4% paraformaldehyde solution for 30 min. The coverslips were then washed with PBS and cells were stained with Actin-Green. We briefly washed the coverslips with PBS and incubated them with 1% BSA in 0.25% Triton-X with 4 drops of Actin-Green for 30 mins, briefly washed them with PBS and finally mounted them with ProLong Gold with Dapi. We imaged on a Zeiss LSM 880 Confocal.
Bulk transcriptomics
After treatment, cardiomyocyte cells of each replicate were separately harvested in TRIzol Reagent (Thermo Fisher Scientific, 15596018), followed by RNA isolation according to the manufacturer’s instructions. We quantified the RNA using Qubit RNA BR Assay kit (Thermo Fisher Scientific, Q10211) on a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Q32866). The RNA 6000 Nano kit (Agilent, 5067-1511) was used to determine the RNA integrity on a 2100 Bioanalyzer Instrument (Agilent, G2939BA). We only used RNA samples with an RNA Integrity Number (RIN) of > 8 (our samples commonly had a RIN of 9-10) for library preparation. A minimum of 100 ng total RNA (ideally 300-500 ng) was subjected to library preparation, using the TruSeq Stranded mRNA Library Prep (96 Samples) (Illumina, 20020595), TruSeq RNA adapter plate (96 plex) (Illumina, 15016427) and IDT for Illumina-TruSeq RNA UD Indexes (96 Indexes, 96 Samples) (Illumina, 20022371) and according to Illumina’s TruSeq Stranded mRNA reference guide. We quantified the prepared libraries on a Qubit 2.0 Fluorometer using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Q32851). Library integrity was checked on a 2100 Bioanalyzer Instrument using the Agilent DNA 1000 Kit (Agilent, 5067-1505). We only subjected libraries with a library peak 250–450 bp and no to very low adapter contamination to sequencing. Pooled libraries were clustered in either an S1 flowcell or a full NovaSeq 6000. The cell line/drug combinations were then sequenced using a 100bp Single End configuration, and typically 24 million reads were mapped to the hg38 reference genome for each cell line/drug combination.
Curation of cardiotoxic risk profiles from FAERS database
We curated data from the FDA adverse event reporting system (FAERS) database to calculate reporting odds ratios of drugs that act on cardiac systems. We downloaded data from the FDA website from (2004_Q1 to 2019_Q4) which contains over 10 million adverse event reports in AERS (2004_Q1 to 2012_Q3) & FAERS (2012_Q4 to 2019_Q4). The quarterly data files include the following: a) demographic and administrative information including imaging reports, b) each patient identified by an id number, c) drug information from the case report, d) adverse event information from the report, f) patient outcome information from the report, g) information on the source of the report and h) information on the date of report. To parse FAERS (and AERS) data into a relational database (by default it is sqlite), we downloaded the ASCII zip file for each quarter and used the FAERS toolkit (http://github.com/kylechua/faers-toolkit). For further analysis, we used a python script to parse raw data from text files and identify the frequency of occurrence of adverse events of interest. Finally, we used a reporting odds ratios (mean and 95% confidence interval) formula for each drug of interest.
Identification of differentially expressed genes
Control transcriptomic data16 and transcriptomic data generated by treatment of six cardiomyocyte cell lines with 54 drugs was analyzed, as described previously82, except that we used human reference genome hg38 in this study. Briefly, we aligned the bulk transcriptomic data to hg38 using STAR83, followed by mapping of read counts to genes using the RefGene reference annotation and feature counts84. Pairwise correlation, followed by hierarchical clustering, identified replicates with deviating gene expression profiles that were consequently removed from the analysis (Supplementary Data 2). Drug-treated or control cell lines were subjected to differential expression analysis using the negative-binomial test implemented in the ‘exactTest’ functionality of the edgeR package85. Each of the calculated 266 lists of differentially expressed genes describes the response observed in a unique cell line/drug combination. Genes with a False Discovery Rate (FDR) of at max 10% were defined as significant. Each of these lists contained significance p-values for 16,345 genes, ranging from zero to one. We transformed these p-values into -log10(p-values) and added a negative sign in case of downregulated genes (or kept the positive sign in case of upregulated genes), to document the direction of change. P-values that were zero were replaced by 10−64 before transformation, since this is the lowest non-zero p-value that is generated by our edgeR pipeline.
Pairwise correlation and hierarchical clustering
All generated signed -log10(p-values) were integrated into a matrix where the columns represent the 266 treated cell line/drug combinations and the rows the 16,345 genes. We calculated the pairwise correlation coefficients between all rows and all columns, followed by hierarchical clustering of the rows and columns based on the pairwise correlation coefficients (distance function: (1-cor(x))/2). Rows and columns of the original matrix were rearranged based on the clustering results before visualization. To document how efficient the clustering algorithm groups cell line/drug combinations that represented different cell lines treated with the same drug into the same cluster, we calculated an F1 score for each cluster drug combination. Every cluster that could be obtained by cutting the dendrogram at any height and contained at least two cell line/drug combinations was analyzed. The F1 score is the harmonic mean of precision and recall (we selected beta = 1). Here, precision documents the fraction of cell line/drug combinations in a particular cluster that were treated with a particular drug, recall documents the fraction of all cell line/drug combinations treated with that drug that were in that cluster. For each drug, we determined the cluster with the highest F1 score and saved that F1 score for that drug.
Singular value decomposition and identification of drug-selective subspaces and gene expression profiles
The gene expression matrix filled with the signed -log10(p-values) was subjected to our computational pipeline for the identification of drug-selective responses (Supplementary Fig. 4). It is based on singular value decomposition (SVD) and implemented in the programming language R. SVD decomposes the input matrix into a matrix of orthonormal left singular vectors, a diagonal matrix of singular values and a matrix of orthonormal right singular vectors. Applied to gene expression data, the left singular vectors are often referred to as eigenarrays86, because each gene expression profile (columns in the input matrix) is a linear combination of all eigenarrays (Supplementary Fig. 6A). The right singular vectors contain the cell line/drug combination-specific coefficients for this linear combination. The eigenarray-specific singular values document how much of the initial variance can be explained by each eigenarray and need to be added as additional factors to the terms in the linear combination to reconstruct the original values.
To further characterize the eigenarrays, we correlated the cell line/drug combination-specific coefficients associated with each eigenarray with the number of significantly differentially expressed genes (DEGs) (FDR ≤ 0.1) in each cell line/drug combination, using Pearson correlation (Supplementary Fig. 6C). The first eigenarray was removed from the gene expression matrix and the new matrix subjected to the same pipeline. We used a two-tailed t-test to analyze whether the cell line/drug combination-specific coefficients of each eigenarray significantly differed between all cell line/drug combinations of the same drug and all other cell line/drug combinations (Supplementary Fig. 9A). Similarly, we analyzed whether the cell line/drug combination-specific coefficients of each eigenarray significantly differed between all cell line/drug combinations of the same cell line and all other cell line/drug combinations, using a two-tailed t-test. As a result, we obtained one p-value for each drug and eigenarray combination or cell line and eigenarray combination. P-values were transformed into -log10(p-values) and integrated into a matrix where the columns represent the eigenarrays and the rows the drugs or cell lines. The matrix was subjected to pairwise correlation analysis and hierarchical clustering, as described above.
Any combination of eigenarrays spans a subspace that contains a particular fraction of the complete DEG profiles. For each drug, we ranked all eigenarrays by significance and generated 263 potential drug-selective subspaces spanned by the top three to 265 ranked eigenarrays. We then projected all 266 complete gene expression profiles into each of these subspaces, followed by pairwise correlation analysis and hierarchical clustering. Each potential drug-selective subspace can be characterized by two different measures. The first measure is the maximum F1 score that documents how close all cell line/drug combinations treated with the drug of interest cluster together. The second measure is the median cosine similarity that documents how much of the gene expression profiles, after removal of the first eigenarray induced by the drug of interest, is still preserved in that subspace. The maximum F1 score was determined as described above for the complete dataset. For each subspace we calculated a final selection score that is the weighted mean between the maximum F1 score and median cosine similarity. The relative contribution of the F1 score and the median cosine similarity to the final selection score was defined by the F1 score weight. High and low F1 score weights favor F1 score or median cosine similarity, respectively. Varying the F1 score weights from 0.00 to 0.95 in steps of 0.05 and searching for the subspace with the highest selection score yielded 20 potential drug-selective subspaces for each drug.
Besides our interest in the characterization of drug-selective gene expression profiles, we were also interested in the identification of cell lines that showed a significantly different response from the other cell lines to treatment with the same drug. To identify cell lines with such outlier responses we screened each of the 20 potential drug-selective subspaces for clustering results that indicated an outlier response, i.e., a single cell line that does not cluster together with the other cell lines treated with the same drug. To find such outlier responses, we calculated cell line/drug combination-specific F1 scores using the same approach described above, except that the cell line/drug combination of interest has to be in that cluster. Dixon’s Q test of cell line/drug combination-specific F1 scores was used to identify outlier responses (Bonferroni-corrected p-value ≤ 0.05). Identified outliers were only accepted if the mean F1 score of all non-outlier cell line/drug combinations was larger than 0.5. If an outlier response was identified in any of the 20 potential drug-selective subspaces, we ranked the potential subspaces by Dixon’s Q test adjusted p-value, followed by decreasing mean F1 score of all non-outlier cell line/drug combinations and increasing F1 score weight. The top ranked subspace was defined as the drug-selective subspace. If no outlier was identified, we selected that subspace with the highest selection score based on an F1 score weight of 0.95.
Projection of gene expression profiles into the final drug-selective subspaces generated drug-selective gene expression profiles that were combined into a new matrix.
Pathway enrichment analysis
We subjected the complete set of DEGs, the DEGs after removal of the first eigenarray and the drug-selective DEGs to pathway enrichment analysis (Supplementary Fig. 14A). For each group of DEGs, up- and downregulated genes among the top 600 most significant DEGs (as indicated by complete or decomposed absolute -log10(p-values)) were separately subjected to enrichment analysis using the Molecular Biology of the Cell Ontology (MBCO)28 (http://mbc-ontology.org or http://github.com/SBCNY/Molecular-Biology-of-the-Cell) and Fisher’s Exact test. Only SCPs predicted with a maximum nominal p-value of 0.05 were considered. For each treatment, predicted up- or downregulated level-1, -2, -3 and -4 SCPs were separately ranked by significance, generating eight lists of ranked SCPs per treatment. Pathway enrichment analysis was implemented in the programming language C#.
Identification of SCPs associated with cardiotoxic and non-cardiotoxic responses
To identify SCPs that are associated with cardiotoxic and non-cardiotoxic TKI responses, we counted, at each significance rank cutoff, how many cardiotoxic or non-cardiotoxic small molecule kinase inhibitors and monoclonal antibodies up- or downregulate an SCP of interest with a significance rank below or equal to the current cutoff (Supplementary Fig. 16). Counting results were used to calculate precision and recall of each SCP at each rank to be either up- or downregulated by either the cardiotoxic or non-cardiotoxic TKIs. F1 scores were calculated with a high emphasis on the precision (beta = 0.25). To ensure consistent results across multiple ranks, we calculated the area under the F1 score curve (AUC) between the ranks 1 and 20 for SCPs of the levels 1, 2 and 4 and 1 and 30 for SCPs of level 3. Different rank cutoffs were selected due to the different number of SCPs of each level. Consequently, we obtained four AUCs for each SCP (up- or downregulated by cardiotoxic or non-cardiotoxic TKIs). To enable cross-comparison of AUCs across different levels, we normalized the AUCs by calculation of the percent of the maximal available area covered by each AUC. To emphasize identification of SCPs that are either up- or downregulated by cardiotoxic or non-cardiotoxic TKIs, we introduced a penalty on each AUC by subtracting half of the AUC calculated for the same group but based on the DEGs changing in the opposite direction. Final AUCs of the same toxicity group were ranked (independently of the direction of change) and the level-1, -2, -3 and -4 SCPs associated with the top 10, 10, 25 and 10 ranked AUCs further investigated. Different rank cutoffs were selected due to the different number of SCPs of each level. This pipeline was implemented in C# and R.
The same pipeline was used for the identification of SCPs associated with anthracyclines or highly cardiotoxic TKIs (cardiotoxicity frequency > 10%).
Integration of additional datasets obtained from cardiomyocytes with or without endothelial cocultures
Eight lists of DEGs that were generated from bulk transcriptomic sequencing of cell lines MSN08 and MSN09 after stimulation with dabrafenib, pazopanib or vehicle control in presence or absence of endothelial co-cultures were merged with the 266 original lists of DEGs. Merged 274 lists of DEGs were projected into the drug-selective subspaces identified from the original 266 lists of DEGs, as described above. F1 scores, drug-selective DEGs and up- or downregulated SCPs were calculated as described above.
Single cell and nucleus RNAseq dataset analysis
Single cell RNAseq data of four hiPSC-derived cell lines that were also used in this study were obtained from Schaniel et al.16 (GSE175761). We reanalyzed the data using an updated Seurat package (version 4.1.3)87. Cells with feature counts below 500 and expression of mitochondrial genes of more than 50% were removed during quality control. The Seurat functionality ‘SCTransform’ was used to normalize, scale, identify the top 2000 features, and regress out mitochondrial gene counts in each individual dataset. The four datasets were integrated using the Seurat ‘IntegrateData’ pipeline. We used the top 30 principal components for dimensionality reduction. After identification of cell neighborhoods using ‘FindNeighbors’ we clustered the combined integrated Seurat object using a resolution of 0.085, since this resolution allowed identification of six different clusters that can be mapped to reasonable cardiac cell types. Cluster marker genes were calculated using the Wilcoxon Rank Sum test implemented in the ‘FindMarkers’ functionality, the ‘RNA’-assay and log-normalized ‘data’-slot and a significance cutoff of an adjusted p-value ≤ 0.05. To annotate clusters to cell types we subjected the top 500 significant marker genes to pathway enrichment analysis using Fisher’s Exact test and cell type genes identified in single cell and single nucleus RNAseq of the adult88 or the developing human heart89. To identify cluster-specific SCPs we subjected the marker genes to enrichment analysis using MBCO28, using the right-tailed Fisher’s exact test.
Supplementary Fig. 22A shows UMAPs with cluster-cell type annotations that are similar to our previous annotations published Supplementary Fig. S4D in Schaniel et al.16. Supplementary Fig. 22B shows a similar distribution of cells among the different clusters in each of the four cell lines as Supplementary Fig. S4E in our previous publication. Cell type annotations in current Supplementary Fig. 22A match those in our previous publication shown in Supplementary Fig. S4G16.
Cell type marker genes in the adult human heart29 used above were calculated using a published Seurat object and FindMarkers as described above. The top 500 marker genes of each cell type were subjected to enrichment analysis using MBCO28 and the right-tailed Fisher’s exact test.
Up- and downregulated genes between hiPSC-derived cardiomyocytes from juvenile patients with dilated cardiomyopathy and healthy controls32 were subjected to enrichment analysis using MBCO28 and the right-tailed Fisher’s exact test.
Up- and downregulated genes among the top 600 most significant DEGs in each cell type obtained by single cell13 or single nucleus RNAseq14 from patients with dilated and/or hypertrophic cardiomyopathy were subjected to enrichment analysis using MBCO28 and the right-tailed Fisher’s exact test as well.
Whole genome sequencing
Whole genome sequencing of the six cell lines used in this study has been previously published16 and is deposited on dbGAP under the accession id phs002088.v2.p1. Here, we reanalyzed the data as described previously in ref. 16, except that we used the hg38 reference genome. Sequencing reads were aligned to hg38 reference using bwa-mem2. Aligned bam-files were called to g.vcf files using deepvariant and a joint vcf of calls were generated using GLnexus. Jointly the vcf file was annotated using ANNOVAR to provide RefSeq gene-based annotations, gnomAD allele frequencies, CADD 1.6 scores, SpliceAI scores, ClinVar annotations, dbSNP annotations, GTEx annotations for atrial and ventricular expression and splice QTLs downloaded from the GTEx portal. Variants reported in the NHGRI-EBI Catalog of human genome-wide association studies were annotated.
Identification of genomic variants potentially associated with drug-induced cardiotoxicity
Building on our transcriptomic analysis, we implemented a pipeline in C# and R that searches for potential variants of drug-induced cardiotoxicity (Supplementary Fig. 31). We searched for variants that either interfere with a drug’s pharmacokinetics (PK) or -dynamics (PD), or map to SCPs identified to be associated with a cardiotoxic or non-cardiotoxic response by our transcriptomic analysis.
In both cases, we initially filtered variants based on population-wide statistics. Cardiotoxic responses to kinase inhibitor treatment are normally observed in 1% to 20% of the treated patients (Supplementary Table 4). Consequently, the population-wide frequency of an allele in a genomic variant associated with drug-induced cardiotoxicity should be in a similar range or even lower, so we focused on variant alleles with frequencies of 10% or less. For each variant, we only considered the least frequent allele, minor allele in the following (no excluded allele would have meet our population-wide frequency cutoff of 10%). Relevance for cardiac tissue should be assigned for those variants that either map to gene coding regions or are part of a cis-expression (e) or -splicing (s) QTL in the adult heart that we obtained from the GTEx Portal (GTEX v8)58. For further analysis, we only considered variants that met these filter criteria.
For the identification of variants that interfere with PK/PD of a given drug we integrated the results of our transcriptomic outlier analysis with the WGS results of our cell lines. With cardiotoxic responses in 1–20% of treated patients (Supplementary Data 3), either none or only one of our six healthy volunteers that donated skin fibroblasts for hiPSC generation and cardiomyocyte differentiation should suffer from cardiotoxicity induced by a drug of interest. Consequently, we considered only those variants in each cell line that show a higher count for the minor allele (as identified in the population-wide analysis) in that cell line compared to the other five cell lines. For the last selection step, we hypothesized that interference with the drug’s PK/PD induces a different transcriptomic response, allowing us to link drug-related outlier responses identified in our transcriptomic analysis to variants overrepresented in our cell lines. To link the remaining variants to a drug’s PK/PD, we curated all human drug target proteins, transporters and enzymes of drugs with an identified outlier response from the Drugbank database21 (2024 January). In addition to the genes directly involved in each drug’s PK/PD, we were also interested in mechanistic regulators of those genes, i.e., transcription factors or kinases. We downloaded the libraries ‘Chea_2022’90,91, ‘Encode_tf_chip_seq_2015’92, ‘Transfac_and_jaspar_pwms’93, ‘TRRUST_transcription_factors_2019’94, ‘Kea_2015’95 from the enrichR website96 (2024 January) and curated all human, mouse and rat transcription factors and kinases that regulate any target proteins, transporters or enzymes. Mouse and rat TFs as well as their target genes were mapped to human homologues, using ‘HOM_AllOrganism.rpt’ downloaded from the Mouse Genome Informatics Web Site97 and ‘homologene.data’ downloaded from the NCBI website98 (2024 January). Only variants mapping to genes involved in a drug’s PK/PD, either directly or as potential mechanistic regulators, and identified in a cell line with an outlier response to that drug were suggested as potential variants.
For the identification of variants with cardiotoxic or cardioprotective effects by interference with transcriptionally regulated SCPs that are associated with a cardiotoxic or non-cardiotoxic response, we mapped all variants identified in the population-wide analysis to identified SCPs. To prevent double counting we removed any variants mapping to genes that are annotated to multiple SCPs in parent-child relationships from all ancestor SCPs before visualizing the results as stacked bar diagrams.
Comparison of identified variant genes with prior knowledge
To compare our predictions with genes involved in inherited cardiomyopathies, we extracted all genes annotated to “Cardiomyopathy, Hypertrophic” or “Cardiomyopathy, Dilated” in the HuGE Phenopedia64 (downloaded on 2020 June 04) and genes associated with DCM65 or HCM66 in GWAS. In the DCM study65, we focused on those genes annotated to the MRI phenotypes ‘LVEF’, ‘LVESV’ and ‘LVESVi’, since DCM was the disease most strongly associated with these phenotypes, as stated by the authors. We compared both the genes listed in the column ‘Nearest Gene’ and ‘TWAS Gene’ of Suppl. data file 3 with our results. In case of the HCM66 study, we used all genes listed in the column ‘Locus Name’ in their Suppl. Data 2.
We merged all predicted genes that contain potential variants and map to level-2, -3 and -4 SCPs of the MBCO ontology28 identified to be up- or downregulated by cardiotoxic or non-cardiotoxic drugs at higher enrichment ranks as by the other TKI group. Here, we ignored genes annotated to level-1 SCPs, since level-1 SCPs cover multiple subfunctions only a few of which might contribute to the prediction of the level-1 SCPs by our algorithm. Consequently, level-1 SCPs might contain genes that are unrelated to the subfunctions that enabled their prediction. Since our algorithm can only identify genes annotated to any level-2, -3 and -4 SCPs, we selected those genes as the background set of genes for a Fisher’s exact test. For statistical accuracy, we removed all genes associated with inherited DCM and HCM in the datasets described above that were not part of the background gene list. A right-tailed Fisher’s exact test was used to calculate the significance of the overlap between the genes annotated to identified SCPs and the published gene lists.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This research was supported by a grant 5U54HG008098 to RI, EAS and from NIH Common Fund for the Drug Combination Signatures for Prediction and Mitigation of Toxicity Center as part of the LINCS program. The PredicTox Knowledge environment (/database) was developed with support from the United States FDA (FDA BAA Contract No. 75F40119C10021) to RI at Icahn School of Medicine at Mount Sinai and to the Critical Path Institute. We thank Zaara Suhail for her assistance with the Actin staining of the HCAEC cells. We thank Matthew Miyamoto for sharing the differentially expressed genes in iPSC-derived cardiomyocytes of a juvenile DMC patient upon request32. We thank Jill Gregory for help with the flowchart figures.
Author contributions
Jens Hansen - conceptualization, conducted all detailed computational analyses, wrote and revised paper. Yuguang Xiong - initial analysis of transcriptomic data, quality control, identification of DEGs. Mustafa M. Siddiq - conducted cardiomyocyte-endothelial cell co-culture experiments. Priyanka Dhanan - expanded iPSCs, conducted differentiation, characterized and maintained cardiomyocytes. Bin Hu - characterized and maintained cardiomyocytes, conducted drug treatment and first steps of RNA extraction. Bhavana Shewale - expanded iPSCs, conducted differentiation, characterized and maintained cardiomyocytes. Arjun S. Yadaw - analyzed FDA FAERS-data and developed clinical scores for cardiotoxicity. Gomathi Jayaraman - cell culture and library preparation for RNAseq. Rosa E. Tolentino - cell culture and library preparation for RNAseq. Yibang Chen - library preparation for RNAseq and quality control. Pedro Martinez - web site development and implementation. Kristin G. Beaumont - supervision of sequencing experiments. Robert Sebra - supervision of sequencing experiments. Dusica Vidovic - data curation and integration into LINCS website. Stephan C. Schürer - data curation and integration into LINCS website. Joseph Goldfarb - experimental design, and quality control, manuscript development. James M. Gallo - experimental design for drug treatment. Marc R. Birtwistle - supervision of overall project, obtained funding. Eric A. Sobie - supervision of overall project, obtained funding. Evren U. Azeloglu - supervision of experiments. Seth I. Berger - initial genomic analyses, identification of variants in cell lines. Angel Chan - participated in analyzing computational outputs and defining outcomes from clinical perspective. Christoph Schaniel - developed iPSC lines and participated in overall project management. Nicole C. Dubois - supervised expansion of iPSCs, differentiation, enrichment of cardiomyocytes and overall project management. Ravi Iyengar – conceptualization, obtained funding, led overall project management, wrote and revised paper with input from all authors, has overall responsibility for the project.
Peer review
Peer review information
Nature Communications thanks Agapios Sachinidis, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
Transcriptomic and genomic data generated in this and our previous study16 have been deposited in the NCBI GEO and dbGAP databases under the accession codes GSE174773, GSE217421, GSE253490 and phs002088.v1.p1, respectively. The processed lists of DEGs and genomic variants that are used by our code deposited on Github are available at https://iyengarlab.org/dtoxs/datasets.php (‘Datasets used for prediction of transcriptomic and genomic signatures for TKI-induced cardiotoxicity’). All data sets obtained from processing are available at www.predictox.org. These include DEGs, pathways, predicted genomic variants and summary DrugTox cards. iPSC lines are available on request.
Code availability
C# and R-code have been deposited at https://github.com/DToxS/SVD-curated_transcriptomic_signatures_cardiotoxic_drugs99.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Nicole C. Dubois, Ravi Iyengar.
Contributor Information
Jens Hansen, Email: jens.hansen@mssm.edu.
Nicole C. Dubois, Email: Dubois@mssm.edu
Ravi Iyengar, Email: ravi.iyengar@mssm.edu.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-52145-4.
References
- 1.de Vries, E. N., Ramrattan, M. A., Smorenburg, S. M., Gouma, D. J. & Boermeester, M. A. The incidence and nature of in-hospital adverse events: a systematic review. Qual. Saf. Health Care17, 216–223 (2008). 10.1136/qshc.2007.023622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drozda, K., Pacanowski, M. A., Grimstein, C. & Zineh, I. Pharmacogenetic Labeling of FDA-Approved Drugs: A Regulatory Retrospective. JACC Basic Transl. Sci.3, 545–549 (2018). 10.1016/j.jacbts.2018.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith, A. F., Klotz, A. & Wormstone, I. M. Improving the drug development process by reducing the impact of adverse events: the case of cataracts considered. Drug Discov. Today21, 510–516 (2016). 10.1016/j.drudis.2016.01.001 [DOI] [PubMed] [Google Scholar]
- 4.Dorato, M. A. & Buckley, L. A. Toxicology in the drug discovery and development process. Curr. Protoc. Pharmacol.Chapter 10, Unit10 13 10.1002/0471141755.ph1003s32 (2006). [DOI] [PubMed]
- 5.Ma’ayan, A., Jenkins, S. L., Goldfarb, J. & Iyengar, R. Network analysis of FDA approved drugs and their targets. Mt Sinai J. Med. 74, 27–32 (2007). 10.1002/msj.20002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vandenberg, J. I. et al. hERG K(+) channels: structure, function, and clinical significance. Physiol. Rev.92, 1393–1478 (2012). 10.1152/physrev.00036.2011 [DOI] [PubMed] [Google Scholar]
- 7.Raschi, E., Vasina, V., Poluzzi, E. & De Ponti, F. The hERG K+ channel: target and antitarget strategies in drug development. Pharm. Res57, 181–195 (2008). 10.1016/j.phrs.2008.01.009 [DOI] [PubMed] [Google Scholar]
- 8.Jain, D. & Aronow, W. Cardiotoxicity of cancer chemotherapy in clinical practice. Hosp. Pr. (1995)47, 6–15 (2019). 10.1080/21548331.2018.1530831 [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Alvarez, A., Garcia-Albeniz, X., Esteve, J., Rovira, M. & Bosch, X. Cardiotoxicity of tyrosine-kinase-targeting drugs. Cardiovasc Hematol. Agents Med Chem.8, 11–21 (2010). 10.2174/187152510790796192 [DOI] [PubMed] [Google Scholar]
- 10.Yoshida, Y. & Yamanaka, S. Induced Pluripotent Stem Cells 10 Years Later: For Cardiac Applications. Circ. Res. 120, 1958–1968 (2017). 10.1161/CIRCRESAHA.117.311080 [DOI] [PubMed] [Google Scholar]
- 11.Sharma, A. et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci. Transl. Med.910.1126/scitranslmed.aaf2584 (2017). [DOI] [PMC free article] [PubMed]
- 12.Wang, H. et al. Adaptation of Human iPSC-Derived Cardiomyocytes to Tyrosine Kinase Inhibitors Reduces Acute Cardiotoxicity via Metabolic Reprogramming. Cell Syst.8, 412–426.e417 (2019). 10.1016/j.cels.2019.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koenig, A. L. et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat. Cardiovasc Res1, 263–280 (2022). 10.1038/s44161-022-00028-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaffin, M. et al. Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy. Nature608, 174–180 (2022). 10.1038/s41586-022-04817-8 [DOI] [PubMed] [Google Scholar]
- 15.Aminkeng, F. et al. A coding variant in RARG confers susceptibility to anthracycline-induced cardiotoxicity in childhood cancer. Nat. Genet47, 1079–1084 (2015). 10.1038/ng.3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaniel, C. et al. A library of induced pluripotent stem cells from clinically well-characterized, diverse healthy human individuals. Stem Cell Rep.16, 3036–3049 (2021). 10.1016/j.stemcr.2021.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang, L. & Fu, L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B5, 390–401 (2015). 10.1016/j.apsb.2015.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel, P. A., Tilley, D. G. & Rockman, H. A. Beta-arrestin-mediated signaling in the heart. Circ. J.72, 1725–1729 (2008). 10.1253/circj.CJ-08-0734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lazou, A., Sugden, P. H. & Clerk, A. Activation of mitogen-activated protein kinases (p38-MAPKs, SAPKs/JNKs and ERKs) by the G-protein-coupled receptor agonist phenylephrine in the perfused rat heart. Biochem. J.332, 459–465 (1998). 10.1042/bj3320459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karliner, J. S., Motulsky, H. J., Dunlap, J., Brown, J. H. & Insel, P. A. Verapamil competitively inhibits alpha 1-adrenergic and muscarinic but not beta-adrenergic receptors in rat myocardium. J. Cardiovasc. Pharm.4, 515–520 (1982). 10.1097/00005344-198205000-00025 [DOI] [PubMed] [Google Scholar]
- 21.Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res.46, D1074–D1082 (2018). 10.1093/nar/gkx1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sloskey, G. E. Amiodarone: a unique antiarrhythmic agent. Clin. Pharm.2, 330–340 (1983). [PubMed] [Google Scholar]
- 23.Cheng, W., Zhu, Y. & Wang, H. The MAPK pathway is involved in the regulation of rapid pacing-induced ionic channel remodeling in rat atrial myocytes. Mol. Med. Rep.13, 2677–2682 (2016). 10.3892/mmr.2016.4862 [DOI] [PubMed] [Google Scholar]
- 24.Ma, Y. Y. et al. Use of decitabine for patients with refractory or relapsed acute myeloid leukemia: a systematic review and meta-analysis. Hematology24, 507–515 (2019). 10.1080/16078454.2019.1632407 [DOI] [PubMed] [Google Scholar]
- 25.Sarno, G. et al. New-onset diabetes mellitus: predictive factors and impact on the outcome of patients undergoing liver transplantation. Curr. Diabetes Rev.9, 78–85 (2013). 10.2174/157339913804143234 [DOI] [PubMed] [Google Scholar]
- 26.Yanagihara, H., Ushijima, K., Arakawa, Y., Aizawa, K. & Fujimura, A. Effects of telmisartan and olmesartan on insulin sensitivity and renal function in spontaneously hypertensive rats fed a high fat diet. J. Pharm. Sci.131, 190–197 (2016). 10.1016/j.jphs.2016.06.003 [DOI] [PubMed] [Google Scholar]
- 27.Derosa, G. et al. Olmesartan/amlodipine combination versus olmesartan or amlodipine monotherapies on blood pressure and insulin resistance in a sample of hypertensive patients. Clin. Exp. Hypertens.35, 301–307 (2013). 10.3109/10641963.2012.721841 [DOI] [PubMed] [Google Scholar]
- 28.Hansen, J., Meretzky, D., Woldesenbet, S., Stolovitzky, G. & Iyengar, R. A flexible ontology for inference of emergent whole cell function from relationships between subcellular processes. Sci. Rep.7, 17689 (2017). 10.1038/s41598-017-16627-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Litvinukova, M. et al. Cells of the adult human heart. Nature588, 466–472 (2020). 10.1038/s41586-020-2797-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hnia, K., Clausen, T. & Moog-Lutz, C. Shaping Striated Muscles with Ubiquitin Proteasome System in Health and Disease. Trends Mol. Med.25, 760–774 (2019). 10.1016/j.molmed.2019.05.008 [DOI] [PubMed] [Google Scholar]
- 31.Vikhorev, P. G. & Vikhoreva, N. N. Cardiomyopathies and Related Changes in Contractility of Human Heart Muscle. Int. J. Mol. Sci.1910.3390/ijms19082234 (2018). [DOI] [PMC free article] [PubMed]
- 32.Chun, Y. W. et al. Impaired Reorganization of Centrosome Structure Underlies Human Infantile Dilated Cardiomyopathy. Circulation147, 1291–1303 (2023). 10.1161/CIRCULATIONAHA.122.060985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dalo, J. D., Weisman, N. D. & White, C. M. Mavacamten, a First-in-Class Cardiac Myosin Inhibitor for Obstructive Hypertrophic Cardiomyopathy. Ann. Pharmacother. 10.1177/10600280221117812 (2022). [DOI] [PubMed]
- 34.Lopaschuk, G. D., Karwi, Q. G., Tian, R., Wende, A. R. & Abel, E. D. Cardiac Energy Metabolism in Heart Failure. Circ. Res.128, 1487–1513 (2021). 10.1161/CIRCRESAHA.121.318241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verdonschot, J. A. J. et al. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur. Heart J.39, 864–873 (2018). 10.1093/eurheartj/ehx808 [DOI] [PubMed] [Google Scholar]
- 36.Schafer, S. et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat. Genet.49, 46–53 (2017). 10.1038/ng.3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herman, D. S. et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med.366, 619–628 (2012). 10.1056/NEJMoa1110186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang, X., Kawasaki, N. K., Min, J., Matsui, T. & Wang, F. Ferroptosis in heart failure. J. Mol. Cell Cardiol.173, 141–153 (2022). 10.1016/j.yjmcc.2022.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee, J. Y. et al. Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer. Proc. Natl Acad. Sci. USA117, 32433–32442 (2020). 10.1073/pnas.2006828117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blahova, Z., Harvey, T. N., Psenicka, M. & Mraz, J. Assessment of Fatty Acid Desaturase (Fads2) Structure-Function Properties in Fish in the Context of Environmental Adaptations and as a Target for Genetic Engineering. Biomolecules1010.3390/biom10020206 (2020). [DOI] [PMC free article] [PubMed]
- 41.Huang, J. et al. Understanding Anthracycline Cardiotoxicity From Mitochondrial Aspect. Front Pharm.13, 811406 (2022). 10.3389/fphar.2022.811406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voest, E. E., van Acker, S. A., van der Vijgh, W. J., van Asbeck, B. S. & Bast, A. Comparison of different iron chelators as protective agents against acute doxorubicin-induced cardiotoxicity. J. Mol. Cell Cardiol.26, 1179–1185 (1994). 10.1006/jmcc.1994.1136 [DOI] [PubMed] [Google Scholar]
- 43.Jones, I. C. & Dass, C. R. Doxorubicin-induced cardiotoxicity: causative factors and possible interventions. J. Pharm. Pharm.74, 1677–1688 (2022). 10.1093/jpp/rgac063 [DOI] [PubMed] [Google Scholar]
- 44.Kim, J., Nishimura, Y., Kewcharoen, J. & Yess, J. Statin Use Can Attenuate the Decline in Left Ventricular Ejection Fraction and the Incidence of Cardiomyopathy in Cardiotoxic Chemotherapy Recipients: A Systematic Review and Meta-Analysis. J. Clin. Med.1010.3390/jcm10163731 (2021). [DOI] [PMC free article] [PubMed]
- 45.McMurray, J. J. et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med.371, 993–1004 (2014). 10.1056/NEJMoa1409077 [DOI] [PubMed] [Google Scholar]
- 46.Velazquez, E. J. et al. Angiotensin-Neprilysin Inhibition in Acute Decompensated Heart Failure. N. Engl. J. Med.380, 539–548 (2019). 10.1056/NEJMoa1812851 [DOI] [PubMed] [Google Scholar]
- 47.Lopez, B., Querejeta, R., Gonzalez, A., Larman, M. & Diez, J. Collagen cross-linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage C heart failure: potential role of lysyl oxidase. Hypertension60, 677–683 (2012). 10.1161/HYPERTENSIONAHA.112.196113 [DOI] [PubMed] [Google Scholar]
- 48.Lopez, B. et al. Myocardial Collagen Cross-Linking Is Associated With Heart Failure Hospitalization in Patients With Hypertensive Heart Failure. J. Am. Coll. Cardiol.67, 251–260 (2016). 10.1016/j.jacc.2015.10.063 [DOI] [PubMed] [Google Scholar]
- 49.Spencer, D. M. et al. DNA repair in response to anthracycline-DNA adducts: a role for both homologous recombination and nucleotide excision repair. Mutat. Res. 638, 110–121 (2008). 10.1016/j.mrfmmm.2007.09.005 [DOI] [PubMed] [Google Scholar]
- 50.van der Zanden, S. Y., Qiao, X. & Neefjes, J. New insights into the activities and toxicities of the old anticancer drug doxorubicin. FEBS J.288, 6095–6111 (2021). 10.1111/febs.15583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fu, H. Y. et al. Protein Quality Control Dysfunction in Cardiovascular Complications Induced by Anti-Cancer Drugs. Cardiovasc. Drugs Ther.31, 109–117 (2017). 10.1007/s10557-016-6709-7 [DOI] [PubMed] [Google Scholar]
- 52.Sawicki, K. T. et al. Preventing and Treating Anthracycline Cardiotoxicity: New Insights. Annu Rev. Pharm. Toxicol.61, 309–332 (2021). 10.1146/annurev-pharmtox-030620-104842 [DOI] [PubMed] [Google Scholar]
- 53.Scott, S. S. et al. Intracellular Signaling Pathways Mediating Tyrosine Kinase Inhibitor Cardiotoxicity. Heart Fail Clin.18, 425–442 (2022). 10.1016/j.hfc.2022.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyamoto, S. et al. Drug review: Pazopanib. Jpn J. Clin. Oncol.48, 503–513 (2018). 10.1093/jjco/hyy053 [DOI] [PubMed] [Google Scholar]
- 55.Justice, C. N. et al. The Impact of Pazopanib on the Cardiovascular System. J. Cardiovasc Pharm. Ther.23, 387–398 (2018). 10.1177/1074248418769612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bronte, E. et al. What links BRAF to the heart function? New insights from the cardiotoxicity of BRAF inhibitors in cancer treatment. Oncotarget6, 35589–35601 (2015). 10.18632/oncotarget.5853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clerk, A. et al. Cardiomyocyte BRAF and type 1 RAF inhibitors promote cardiomyocyte and cardiac hypertrophy in mice in vivo. Biochem J.479, 401–424 (2022). 10.1042/BCJ20210615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Consortium, G. T. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science369, 1318–1330 (2020). 10.1126/science.aaz1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aminkeng, F. et al. Recommendations for genetic testing to reduce the incidence of anthracycline-induced cardiotoxicity. Br. J. Clin. Pharm.82, 683–695 (2016). 10.1111/bcp.13008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Magdy, T. et al. RARG variant predictive of doxorubicin-induced cardiotoxicity identifies a cardioprotective therapy. Cell Stem Cell28, 2076–2089.e2077 (2021). 10.1016/j.stem.2021.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liang, L. et al. Dkk1 exacerbates doxorubicin-induced cardiotoxicity by inhibiting the Wnt/beta-catenin signaling pathway. J. Cell Sci.132, 10.1242/jcs.228478 (2019). [DOI] [PubMed]
- 62.El-Ela, S. R. A., Zaghloul, R. A. & Eissa, L. A. Promising cardioprotective effect of baicalin in doxorubicin-induced cardiotoxicity through targeting toll-like receptor 4/nuclear factor-kappaB and Wnt/beta-catenin pathways. Nutrition102, 111732 (2022). 10.1016/j.nut.2022.111732 [DOI] [PubMed] [Google Scholar]
- 63.Feng, D. et al. DDX3X alleviates doxorubicin-induced cardiotoxicity by regulating Wnt/beta-catenin signaling pathway in an in vitro model. J. Biochem Mol. Toxicol.36, e23077 (2022). 10.1002/jbt.23077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu, W., Clyne, M., Khoury, M. J. & Gwinn, M. Phenopedia and Genopedia: disease-centered and gene-centered views of the evolving knowledge of human genetic associations. Bioinformatics26, 145–146 (2010). 10.1093/bioinformatics/btp618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pirruccello, J. P. et al. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat. Commun.11, 2254 (2020). 10.1038/s41467-020-15823-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harper, A. R. et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet53, 135–142 (2021). 10.1038/s41588-020-00764-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Hasselt, J. G. C. et al. Transcriptomic profiling of human cardiac cells predicts protein kinase inhibitor-associated cardiotoxicity. Nat. Commun.11, 4809 (2020). 10.1038/s41467-020-18396-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burke, M. A., Cook, S. A., Seidman, J. G. & Seidman, C. E. Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy. J. Am. Coll. Cardiol.68, 2871–2886 (2016). 10.1016/j.jacc.2016.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mamoshina, P., Bueno-Orovio, A. & Rodriguez, B. Dual Transcriptomic and Molecular Machine Learning Predicts all Major Clinical Forms of Drug Cardiotoxicity. Front Pharm.11, 639 (2020). 10.3389/fphar.2020.00639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hansen, J. et al. Systems pharmacology-based integration of human and mouse data for drug repurposing to treat thoracic aneurysms. JCI Insight, 410.1172/jci.insight.127652 (2019). [DOI] [PMC free article] [PubMed]
- 71.Porter, C. et al. Permissive Cardiotoxicity: The Clinical Crucible of Cardio-Oncology. JACC Cardio. Oncol.4, 302–312 (2022). 10.1016/j.jaccao.2022.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Porter, K. E. & Turner, N. A. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharm. Ther.123, 255–278 (2009). 10.1016/j.pharmthera.2009.05.002 [DOI] [PubMed] [Google Scholar]
- 73.Daily, N. J., Yin, Y., Kemanli, P., Ip, B. & Wakatsuki, T. Improving Cardiac Action Potential Measurements: 2D and 3D Cell Culture. J. Bioeng. Biomed. Sci.5, 10.4172/2155-9538.1000168 (2015). [DOI] [PMC free article] [PubMed]
- 74.Kawalec, P. et al. Differential impact of doxorubicin dose on cell death and autophagy pathways during acute cardiotoxicity. Toxicol. Appl. Pharm.453, 116210 (2022). 10.1016/j.taap.2022.116210 [DOI] [PubMed] [Google Scholar]
- 75.Kurokawa, Y. K., Shang, M. R., Yin, R. T. & George, S. C. Modeling trastuzumab-related cardiotoxicity in vitro using human stem cell-derived cardiomyocytes. Toxicol. Lett.285, 74–80 (2018). 10.1016/j.toxlet.2018.01.001 [DOI] [PubMed] [Google Scholar]
- 76.L’Ecuyer, T., Horenstein, M. S., Thomas, R. & Vander Heide, R. Anthracycline-induced cardiac injury using a cardiac cell line: potential for gene therapy studies. Mol. Genet Metab.74, 370–379 (2001). 10.1006/mgme.2001.3243 [DOI] [PubMed] [Google Scholar]
- 77.Li, D. L. et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation133, 1668–1687 (2016). 10.1161/CIRCULATIONAHA.115.017443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Orsolits, B., Kovacs, Z., Kriston-Vizi, J., Merkely, B. & Foldes, G. New Modalities of 3D Pluripotent Stem Cell-Based Assays in Cardiovascular Toxicity. Front Pharm.12, 603016 (2021). 10.3389/fphar.2021.603016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takasuna, K. et al. Comprehensive in vitro cardiac safety assessment using human stem cell technology: Overview of CSAHi HEART initiative. J. Pharm. Toxicol. Methods83, 42–54 (2017). 10.1016/j.vascn.2016.09.004 [DOI] [PubMed] [Google Scholar]
- 80.Toldo, S. et al. Comparative cardiac toxicity of anthracyclines in vitro and in vivo in the mouse. PLoS One8, e58421 (2013). 10.1371/journal.pone.0058421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou, P. & Pu, W. T. Recounting Cardiac Cellular Composition. Circ. Res.118, 368–370 (2016). 10.1161/CIRCRESAHA.116.308139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiong, Y. et al. A Comparison of mRNA Sequencing with Random Primed and 3’-Directed Libraries. Sci. Rep.7, 14626 (2017). 10.1038/s41598-017-14892-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics29, 15–21 (2013). 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liao, Y., Smyth, G. K. & Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res.47, e47 (2019). 10.1093/nar/gkz114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics26, 139–140 (2010). 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Alter, O., Brown, P. O. & Botstein, D. Singular value decomposition for genome-wide expression data processing and modeling. Proc. Natl. Acad. Sci. USA97, 10101–10106 (2000). 10.1073/pnas.97.18.10101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell10.1016/j.cell.2021.04.048 (2021). 10.1016/j.cell.2021.04.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tucker, N. R. et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation142, 466–482 (2020). 10.1161/CIRCULATIONAHA.119.045401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Asp, M. et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell179, 1647–1660.e1619 (2019). 10.1016/j.cell.2019.11.025 [DOI] [PubMed] [Google Scholar]
- 90.Lachmann, A. et al. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics26, 2438–2444 (2010). 10.1093/bioinformatics/btq466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Keenan, A. B. et al. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res.47, W212–W224 (2019). 10.1093/nar/gkz446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature489, 57–74 (2012). 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rauluseviciute, I. et al. JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles. Nucleic Acids Res.52, D174–D182 (2024). 10.1093/nar/gkad1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Han, H. et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res.46, D380–D386 (2018). 10.1093/nar/gkx1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lachmann, A. & Ma’ayan, A. KEA: kinase enrichment analysis. Bioinformatics25, 684–686 (2009). 10.1093/bioinformatics/btp026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res.44, W90–W97 (2016). 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Blake, J. A. et al. Mouse Genome Database (MGD): Knowledgebase for mouse-human comparative biology. Nucleic Acids Res.49, D981–D987 (2021). 10.1093/nar/gkaa1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sayers, E. W. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res.40, D13–D25 (2012). 10.1093/nar/gkr1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hansen, J. et al. DToxS/SVD-curated_transcriptomic_signatures_cardiotoxic_drugs: Multiscale Mapping of Transcriptomic Signatures for Cardiotoxic Drugs) 10.5281/zenodo.12728022 (2024). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
Transcriptomic and genomic data generated in this and our previous study16 have been deposited in the NCBI GEO and dbGAP databases under the accession codes GSE174773, GSE217421, GSE253490 and phs002088.v1.p1, respectively. The processed lists of DEGs and genomic variants that are used by our code deposited on Github are available at https://iyengarlab.org/dtoxs/datasets.php (‘Datasets used for prediction of transcriptomic and genomic signatures for TKI-induced cardiotoxicity’). All data sets obtained from processing are available at www.predictox.org. These include DEGs, pathways, predicted genomic variants and summary DrugTox cards. iPSC lines are available on request.
C# and R-code have been deposited at https://github.com/DToxS/SVD-curated_transcriptomic_signatures_cardiotoxic_drugs99.