

Summary
Stemformatics.org has been serving the stem cell research community for over a decade, by making it easy for users to find and view transcriptional profiles of pluripotent and adult stem cells and their progeny, comparing data derived from multiple tissues and derivation methods. In recent years, Stemformatics has shifted its focus from curation to collation and integration of public data with shared phenotypes. It now hosts several integrated expression atlases based on human myeloid cells, which allow for easy cross-dataset comparisons and discovery of emerging cell subsets and activation properties. The atlases are designed for external users to benchmark their own data against a common reference. Here, we use case studies to illustrate how to find and explore previously published datasets of relevance and how in-vitro-derived cells can be transcriptionally matched to cells in the integrated atlas to highlight phenotypes of interest.
Keywords: computational stem cell biology, gene expression, Cell Atlas, myeloid cell, pluripotent stem cell
Graphical abstract
Highlights
-
•
Stemformatics.org hosts over 430 high-quality expression datasets
-
•
Integrated atlases show the comparison between in-vitro- and in-vivo-derived blood cells
-
•
Helps users compare data generated across laboratories, methods, or cell sources
In this article, Wells and colleagues describe the Stemformatics reference atlases, which provide fast and intuitive comparisons of a user’s own stem cell data against primary human myeloid cells. The toolkit supports comparison to tissue-resident or disease-associated myeloid cells, including heatmaps of major cellular pathways.
Introduction
Stem cell research increasingly relies on molecular profiles to identify and benchmark cell types derived in a dish. Likewise, it is common to use previously published data as a resource to inform, or benchmark new methods for generating specific cell types from pluripotent sources (Cahan et al., 2021), but it can be hard to find high-quality data in a format that is readily comparable to your own. In part, this is because reviewing the quality of published data requires expertise not readily accessible to every stem cell laboratory. In addition, the technologies providing readouts for stem cell phenotypes are changing rapidly, especially within the omics field. This further complicates approaches to compare data derived between studies when data formats may differ substantially. Stemformatics addresses this gap between published observations and reusable data by providing a resource to easily find high-quality, curated data from primary and pluripotent cell sources. It has been designed for users who are not computationally proficient, to enable easy exploration of published data, as well as tools to assist users upload and compare their own datasets against curated atlases compiled of many published datasets.
Stemformatics was first introduced to the stem cell community in 2011 as the collaboration platform for Stem Cells Australia (Wells et al., 2013). It hosted the Project Grandiose stem cell reprogramming consortium in 2014 (Hussein et al., 2014; Tonge et al., 2014) and was updated in 2019 to allow for cross-dataset comparisons at the level of an individual gene (Choi et al., 2019). The focus of the site has since moved from the collection and curation of relevant datasets to the integration of data into cohesive atlases. To support this integration, we have assigned uniform nomenclature to all sample metadata imported into the resource (Tables 1 and S2). This feature facilitates comparisons between similar samples that have been generated by different laboratories, to give biological insight into shared phenotypes. For example, the Stemformatics-integrated atlases allow users to identify genes whose expression is characteristic of cells with the same lineage, derivation source, or activation state.
Table 1.
Most highly represented cell types in Stemformatics
| Cell type | Tissue of origin | Parental cell type | Number of samples (number of datasets) |
|---|---|---|---|
| Monocyte | Blood | – | 1,706 (20) |
| Umbilical cord blood | – | 61 (3) | |
| iPSC | – | Fibroblast | 423 (32) |
| – | PBMC | 352 (4) | |
| – | 12 assorted cell types | 285 (38) | |
| ESC | – | – | 646 (95) |
| Dendritic cell | Blood | – | 203 (18) |
| – | Monocyte | 171 (5) | |
| Umbilical cord blood | 4 assorted cell types | 67 (3) | |
| 8 assorted tissues | 97 (10) | ||
| Macrophage | – | Monocyte | 423 (10) |
| – | iPSC | 46 (6) | |
| 6 assorted tissues | – | 37 (6) | |
| MSC | Bone marrow | – | 448 (43) |
| Fibroblast | Skin | – | 196 (33) |
| 8 assorted tissues | – | 153 (26) | |
| Other | 6,302 (182) |
Stemformatics reprocesses all hosted data from the source files, to standardize the data formats and assess uniform quality control metrics. Approximately 30% of public datasets reviewed by Stemformatics failed reprocessing or reannotation, because of ambiguities in the sample tables or because of poor-quality primary data (Choi et al., 2019). All these properties mean Stemformatics has been built on the FAIR (Findable, Accessible, Interoperable, and Reusable) guiding principles (FAIR Principles) and provides a unique resource to the stem cell research community. This is a key procedure to the FAIR data principles by which Stemformatics operates: not only reusing public data but also adding domain knowledge and value to those published studies. In our previous publication (Choi et al., 2019), we drew some comparisons between Stemformatics and other sites, and some further data portals have since appeared, including DISCO (Li et al., 2022), which provides visualization tools for many single-cell RNA sequencing (scRNA-seq) datasets. However, Stemformatics is the only resource of its kind where (A) induced pluripotent stem cells (iPSCs) and in-vitro- and in-vivo-derived blood cells can be explored and compared together at the transcriptional level (Choi et al., 2019; Wells et al., 2013), (B) bulk RNA sequencing (RNA-seq) data can be projected onto reference data for benchmarking, and (C) each cell in the integrated atlas can be traced back easily to its primary data source.
The website has been completely redesigned using modern infrastructure and technologies. It now runs a separate API (application programming interface) server, which can be used to query the data directly for advanced users. The user interface server uses modern JavaScript technologies for easier development and maintenance. The system and the code have also been designed to work as a base for other data portals, suitable for research-oriented environments. This report provides an update on the new functionality on the site and examples of how the stem cell community can use the resource to benchmark their own samples or identify new biological insights from examining the behavior of their favorite gene in high-quality curated data.
Results
Stemformatics provides multiple ways to access and explore the datasets
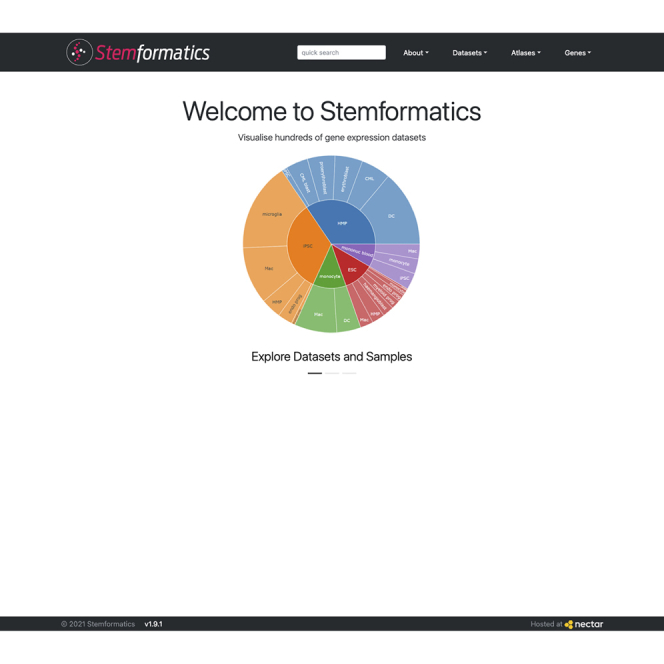
The datasets on Stemformatics are readily accessible in different ways, to encourage exploration and discovery. A quick search at the top of the page performs a search through dataset metadata, such as author or any of the key words used in the abstract (Figure 1A). The Visual Data Explorer (/datasets/explore, Figure 1B) is an interactive sunburst plot which can represent hierarchical relationships with its concentric circles, such as parental cell types which give rise to final cell types in a differentiation process. Following the links from either of these methods leads to the page which shows details for each dataset, including a principal component analysis (PCA) plot of the samples, and the sample table. The sample table contains harmonized information which enables easier comparison of datasets, such as the media used in sample derivation. Even though there are limitations on the level of sample detail present in each dataset (primarily due to missing data, but it also takes time to annotate these manually), Stemformatics makes it much easier to obtain such information in a consistent format from a single source. Another way of finding data is through collections (/datasets/collections, Figure 1C), which contain groups of related datasets. This is also where the link to all datasets can be found. Each dataset can be assigned multiple project tags to group datasets together. This feature also serves collaborative projects requiring a collection of relevant datasets. Filter page (/datasets/filter, Figure 1D) can be used to create a smaller subset of a collection based on properties of interest, such as particular sequencing technologies or cell types.
Figure 1.

Exploring datasets hosted on Stemformatics can happen from multiple starting points
A quick search menu allows for word-based searches throughout the dataset metadata (A). Visual data explorer allows for an interactive view of related samples (B). And collections allow access to datasets grouped together under a project (C). The list of datasets returned can then be further filtered – here we used the term “pluripotent” in the quick search, then narrowed down the results to RNA-seq data from the blood (D) and clicking on the individual dataset allows for PCA and gene expression plots, metadata views, and data downloads (E). In this example, the LIN28B gene is expressed in some iPSC-derived macrophages, which is distinct from their monocyte-derived macrophage counterparts. Data is viewed as a box-whisker plot (median and interquartile range shown) or as a violin plot. Users can elect to show individual data points on the graph.
Various tools exist to explore each dataset—the gene expression plot shows the expression profile of any gene in the dataset, find correlated genes (Figure 1E), view the dataset and sample metadata, and download the expression data and sample metadata as text files.
Use case 1: Exploring gene expression pattern across multiple datasets
Stemformatics is targeted primarily at biologists, who generally have deep knowledge about particular genes and are interested in their expression patterns across different datasets. Stemformatics addresses the problem of finding well-curated data in the public domain that can be used reliably to examine patterns of gene expression in experimental series that are relevant to the stem cell biologist. In this use case, the expression of the XCR1 gene in myeloid cells is explored using Stemformatics. XCR1 is known as a key receptor for cross-presentation of antigens which play crucial roles in directing anti-tumor adaptive immune responses, and so is commonly used as a marker of type I conventional dendritic cells (cDC1s) (Kroczek and Henn, 2012). In this example, we identify tissue-source is a variable that may alter XCR1 expression, such that the use of this marker may not be suitable for the isolation of cDC1s in all human tissues. Because tissue context can impact XCR1 expression, it may also be unreliable to use as a marker of the cDC1 subset in iPSC-derived dendritic cells (DCs).
To demonstrate how to find this information in Stemformatics, we first search for cell types where this gene is highly expressed using the gene to samples function which can be found under “genes” in the top navigation menu (/genes/genetosamples). High expression is determined within a dataset – a cell type receives a higher score for this gene if its expression is greater than the median of all cell types in the same dataset. We leverage the large number of datasets in Stemformatics to show multiple datasets where the same pattern is observed for the same cell type. In this example, we find that XCR1 is highly expressed in DCs, as expected (Figure S1), and this pattern is seen in multiple datasets, increasing the confidence of this result. The gene to samples function also allows for a search within tissue types, and when we perform this search, it returns synovial fluid as one of the tissues where cDC1s have been sampled. Following up on this result, we find that the observation comes from Canavan et al. (2018), where XCR1-expressing cells were associated with activation of CD8+ T cells in an inflammatory arthritis setting.
Next, we can explore the expression of XCR1 further in the Dendritic Cell Atlas, which can be found under “atlases” in the top navigation menu (/atlas/dc). Stemformatics-integrated atlases provide a unique resource that enables direct comparisons of cells across datasets so that these can be used as reference data. This contrasts to the common approach of finding a reference dataset from a single laboratory, which may introduce additional biases because the observation is seen in a single platform or tissue of origin for example. Because each Stemformatics atlas is built from a manually curated list of many datasets, we can compare gene expression patterns between similar biological samples that have been profiled across different platforms or under a variety of other experimental conditions.
The PCA plot of the atlas is the default view and can be used to visualize the relationship of cells to each other. Cells can be colored based on predefined categories, such as cell type, tissue of origin, or parental cell source (Figure 2A). The data can be explored further by customizing the sample groups shown on the graph. This feature allows users to combine categories—for example cell type and tissue source—so as to group and view samples annotated with these terms (Figure 2B). This feature allows the user to explore more precise relationships between cellular behaviors and experimental factors, including discovery of the influence of various experimental factors on a cell’s transcriptional state. Because these states can be visualized across many independent datasets, the reproducibility of these behaviors is straight forward to assess. The user can also search for individual genes in the atlas and view its expression as a box or violin plot (Figure 2D) or as a color gradient imposed on the PCA plot (Figure 2E).
Figure 2.

Different visualizations options available on a Stemformatics atlas page
PCA plot (A) is the default view which allows for cell type and cell state comparisons. In this example, we investigate tissue specificity of cDC1 and cDC2 by creating a customized sample group which combines cell type and tissue type (B). This leads us to view that samples cluster by tissue type within each cell type (C). Querying the expression of XCR1 gene shows higher expression in blood than bone marrow within each of these cell types (D, E).
In this example, we are interested in viewing the expression of XCR1 across various subsets of DCs in a tissue-specific manner (Figure 2). Stemformatics allows users to create a custom sample group which combines common annotation classes—in this example, cell type with tissue type (Figures 2B and 2C). As a result, we observe relatively higher expression of XCR1 in cDC1s of blood vs. bone marrow origin (Figure 2D). This may assist researchers seeking to isolate these cells from tissues, indicating where commonly used markers such as XCR1 may not be suitable. This use case illustrates a key feature of Stemformatics, where multiple related datasets are linked to each other through exploratory features of the portal and through the integrated atlases, and these are powered by improved sample annotations working behind the portal.
Use case 2: Benchmarking in-vitro-derived cells against a high-quality reference atlas
RNA-seq has become a common way to benchmark new in-vitro-derived cell types. However, it is also common to limit the comparison of these newly derived cells to those cell types that you are interested in achieving. The advantage of using a reference atlas such as Stemformatics is that the analysis is not accidently prejudiced by the selection of the comparator, which may otherwise be driven experimentally by cost or perceived relevance. Here, we use the Stemformatics atlases to assess the similarity of pluripotent stem cell-derived myeloid cells against previously published human pluripotent stem cell-derived data, as well as examples drawn from blood-derived and tissue-isolated myeloid cells.
In this example, we benchmark data from Monkley et al. (2020), who describe a new method for generating iPSC-derived myeloid cells. Here, iPSC-derived macrophages and DCs were profiled using bulk RNA-seq. The Stemformatics website makes it very easy to benchmark a bulk gene expression dataset against an atlas, where the expression matrix and sample table can be simply uploaded as tab separated text files. Two independent methods are used simultaneously on each query and results are presented in interactive plots (Figure 3). The first method used by Stemformatics is to project the query data onto the PCA space of the reference atlas. The projection of these data shows their iPSC-derived cells remain transcriptionally close to the other in-vitro-derived macrophages in the atlas, whereas their peripheral blood mononuclear cells (PBMCs) are close to the in vivo sample sources in the atlas. Interestingly, even their PBMC-derived cells assume the in vitro identity after being cultured for several days, an observation consistent with other cultured cells which were used to construct the atlas. Our analysis also confirms that the identity of the DCs derived in this study is like monocyte-derived macrophages and DCs. The authors reached a similar conclusion by generating a list of differentially expressed (DE) genes between cell types of interest, then running pathway analysis on these genes. The gene list was assessed against the myeloid literature. This is a common approach which contains some limitations: DE genes need to be run between 2 cell types and it can be unclear which pairs of cell types are best for this to define cell identity; pathway analysis can present generic ontology terms; and comparisons with a single or a few reference datasets may not be generalizable.
Figure 3.

Results of projecting an external data onto the Stemformatics atlas demonstrate its utility as a benchmarking tool
Monkley et al. is a bulk RNA-seq dataset containing iPSCs and in-vivo-derived myeloid cells, which has been projected onto the Stemformatics Dendritic Cell Atlas. Two independent methods of classifications are applied simultaneously on the website, leading to the visualization in PCA space of projected cells (bottom left – projected cells are diamond shapes) and heatmap of Capybara scores (bottom right). The user can dynamically change the reference sample group after the projection has been made, to easily compare their samples against various cell phenotypes in the atlas.
In order to provide users with a correlation score against the reference data, Stemformatics implements Capybara (Kong et al., 2022) to produce a score (between 0 and 1) for each query sample against the reference samples (Figure 3) and presents them in a heatmap. The scores are calculated against all sample groups in the atlas, not just cell type, hence concordance with tissue type or sample source (in vitro, ex vivo, in vitro) are also shown. The results for this dataset are consistent with what we observed with PCA projections, showing high concordance between their in-vitro-derived cells and the “in-vitro-derived” tissue type in the atlas.
Use case 3: Accessing data using the API
Our final example is aimed at computational biologists interested in accessing curated data and metadata for analysis of cell identity or cell differentiation series. The API server provides a convenient way for computational biologists and bioinformaticians to access the data and perform analyses without going to the front-end website. An example use-case may be to search for all RNA-seq datasets containing a particular cell type (for example, human microglia) to download the corresponding counts per million matrices for downstream analyses. In R, this code may look like this (note: each dataset in Stemformatics has a unique 4-digit number as an identifier):
#R example
library(httr)
library(jsonlite) response = GET("https://api.stemformatics.org/search/samples?query_string=microglia&field=cell_type,dataset_id")
data = content(response) # data is a list
datasetIds = unique(sapply(data, function(x) getElement(x,'dataset_id'))) # get a list of dataset ids
# Loop through each dataset id and fetch the expression matrix, and write it to file
for (datasetId in datasetIds) {
df = read.csv(paste0("https://api-dev.stemformatics.org/datasets/", datasetId, "/expression?as_file = true"),
sep = '\t', row.names = 1)
write.table(cbind(id = rownames(df),df), file = paste0(datasetId, ".tsv"), sep = "\t", quote = F, row.names = F)
}
A full list of available API calls is documented at /datasets/api on the main website, together with examples of data returned.
Discussion
FAIR data principles suggest that data should be not only findable, but also reusable. The Stemformatics platform provides an example of data reuse for community benefit, where we leverage data from otherwise obsolete technology platforms to build new insights into the shared behavior of cells across different laboratories, derivation methods, and activation states. The ethos of the Stemformatics team is to provide a community-facing platform that is easy for biological experts to use without requiring complex bioinformatics software. All of the infrastructure, data processing methods, and primary data sources are provided, and APIs facilitate access from external computational workflows.
By hosting many independent datasets with varied and overlapping covariates and annotating these in a consistent manner, Stemformatics provides an alternative scale of big data to scRNA-seq studies. With Stemformatics, gene networks which separate the samples are often reflecting the nuanced cell states based on the multiple covariates, such as the tissue source or activation status of the cell. We were motivated to build an integrated atlas of myeloid cells in order to compare iPSC-derived myeloid cells to their in vitro and in vivo counterparts. We wanted to explore how different sample sources or derivation methods may have an impact on the cell identity, and this was not possible when many of these features of interest were scattered all over multiple datasets. The old Stemformatics portal hosted multiple datasets in parallel, where the user could access each dataset separately. Comparing data across datasets does bring valuable biological insight, and our approach filters out platform-driven differences but reveals other technical differences in the measurements and scale of data that might otherwise compromise direct comparisons. Macrophages may be differentiated in vitro from different tissues, under different stimuli and disease conditions, for example. Observing these patterns repeatedly across many datasets increases confidence in their biological signal.
Many different methods are available for batch correcting multiple bulk transcriptome datasets to integrate them. We chose variance partition (Hoffman and Schadt, 2016), applied after rank transforming each expression matrix. This method produced robust integrated atlases with real biological clusters (Angel et al., 2020). We were then able to observe emerging properties from the combined data, such as finding that cord-blood-derived dendritic cells retained an in vitro identity, likely from lack of appropriate growth factor signaling (Elahi et al., 2022). Leveraging the scalability of this approach, we created three integrated atlases: Blood, Myeloid, and Dendritic Cell, and these are hosted on Stemformatics website (/atlas/blood, /atlas/myeloid, /atlas/dc). Each atlas comes with a set of additional annotations relevant for that system, and the web page is full of features for easy exploration and usage (Figure 2). Transparency is core to the Stemformatics user experience; so when users find a sample of interest on the atlas, a simple double click on any cell in the PCA plot will show the origin of that cell—which dataset it came from and all its annotations. The user can also look at the full list of the datasets which were used to construct an atlas. Likewise, all of the data used to construct each atlas can be downloaded as text files, including the exact colors used to render the plots.
As more scRNA-seq datasets are created, the relevance of bulk RNA-seq and microarray studies should be evaluated within the appropriate context. In the stem cell research field, in vitro models are extensively used, and these models have been developed and refined for over a decade. Hence, the data captured by older technologies hold many experimental factors which influence each model. These factors may be viewed as covariates if we are making inferences on the output of the models. It may be expected that single-cell datasets will also cover these covariates in future (see Alsinet et al. (2022) for example), but currently the bulk studies far outnumber single-cell studies, and their cheaper cost means they will continue to be produced. The former are more suitable for discovering heterogeneity within the models, and the gene networks will often reflect the cell type differences.
No single tool or reference is likely to capture the precise states of many cells; hence, it is important to understand their limitations. We recommend that multiple tools and references should be used to cross-check the results for cell type classification, rather than relying on a single source. Stemformatics atlases provide an important resource which leverages the wealth of built-up knowledge in the community. Having high levels of manual curation means Stemformatics atlases focus more deeply on particular models of interest by design (only myeloid cells for example). This should be taken into account when using Stemformatics atlases for benchmarking.
Conclusion and future directions
Stemformatics is a unique resource which empowers the stem cell and immunology research communities by hosting high-quality, curated datasets and allows users to visualize these with easy-to-use online tools. It also provides integrated expression atlases focused on myeloid cells derived from multiple sources and conditions. This makes it possible to compare iPSC-derived macrophages to their in vivo counterparts, for example. These atlases also serve as excellent benchmarking tools for related cells.
Bringing a large amount of data together across different laboratories allows for the emergence of experimental or biological variables that impact cellular phenotypes. We have previously published examples of these in the Myeloid and Dendritic Cell atlases. In future, we plan to include additional cell type atlases that allow for comparison of methods and assess the impact of genetic modification or other variables of interest to the community. Since each atlas is based on deep curation of cellular phenotypes, this relies on collaboration with domain experts in these cell types.
Recently updated website has been built on modern full stack technologies, and the API server allows for programmatic access to the data. Stemformatics has been built on FAIR principles, and all its data and code are easily accessible and can serve as a template for similar projects.
Experimental procedures
Resource availability
Lead contact
Further information and requests for resources and code should be directed to and will be fulfilled by the corresponding author, Christine Wells (wells.c@unimelb.edu.au).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data used by Stemformatics website are available through the website. The code behind Stemformatics is available at https://github.com/wellslab/s4m-api and https://github.com/wellslab/s4m-ui. The code which generated data for projection is at https://github.com/wellslab/stemformatics-data-projection.
System design
Stemformatics is built as two separate applications: an API server which hosts all the data and a user interface (UI) server which hosts the website (Figure 4). This is a common design paradigm for data heavy websites and allows for flexibility in both development and maintenance of the system.
Figure 4.

A schematic of the Stemformatics infrastructure, which separates the API server from the UI server and is built on modern full stack technologies, such as Flask, MongoDB, Bootstrap, and Nuxt
The API server is built on Flask-restful (Flask-RESTful). It uses pandas (Python Pandas) extensively to manipulate data frames. The dataset and sample metadata are held as collections in a MongoDB (MongoDB), while expression files are stored as text and hdf5 (HDF5 Format) files. The UI server is built on Nuxt JS (Nuxt), using BootstrapVue (BoostrapVue) to build components easily. Most of the plots are performed by Plotly (Plotly). These tools are designed for modern browsers and user interfaces.
Its open-source code base makes an excellent reference for similar efforts and is available at github.com/wellslab/s4m-api and github.com/wellslab/s4m-ui. Full stack development within a research environment comes with some challenges, which include small (or quite often one person) teams, rapid turnover of personnel, and resource issues for continued maintenance. The Stemformatics code has been designed to address some of these issues, based on a decade of experience in maintaining resources like this. The key principle is to strike a good balance between under or over-engineering the system. Under-engineering creates bloated code with highly dependent variables and states which are difficult to change and maintain. This can be avoided by breaking up complex pages or classes into independent components. Over-engineering creates code only understandable with highly specialized skills and can be avoided by greater transparency and less layers of abstraction.
Stemformatics also supports private datasets. These are datasets flagged as private in the dataset metadata, which are only accessible through an account login. We use this feature to share data with collaborators prior to publication.
Implementation of ontologies within annotation tables
We manually curated sample metadata to harmonize data groupings, expose experimental validation of cell type, and improve user experiences when searching for specific cell types (Table S2). To leverage existing knowledge in this area, we used the Ontology Lookup Service tool (EMBL-EBI-OLS) from European Molecular Biology Laboratory-European Bioinformatics Institute. For disease state, we sourced annotations from the Human Disease Ontology (EMBL-EBI-HDO). For tissue of origin, we sourced annotations from the Brenda Tissue Ontology (EMBL-EBI-BRENDA). For cell type, parental cell type, and final cell type, we used the Cell Ontology (EMBL-EBI-CO). Where known cell lines were included, we used Cellosaurus as an identifier reference (Bairoch, 2018). Harmonized sample annotation also enabled us to build the integrated atlases where many datasets from completely different studies had to be brought together.
Samples were assigned to the most specific ontology possible based on information in the original publication, such as cell markers evaluated by flow cytometry, and where such data were not available, samples were assigned to the next, more general level of the ontological hierarchy. As our resource captures in vitro reprogramming or differentiation series where intermediate cell types are not represented in standard ontologies, the nomenclature for these was assigned as “[parental cell type] transitioning to [intended final cell type]”. We used OpenRefine v3.7.4 (Open Refine) to identify and summarize facets within our metadata and to ensure consistency of formatting, spelling, and grammar across facets. All annotations are available to download directly from the Stemformatics sample tables for individual datasets, or via the API for groups of samples across different data series.
Benchmarking data using integrated atlases
The construction of the atlases has been previously described in detail (Angel et al., 2020; Elahi et al., 2022; Rajab et al., 2021), but we summarize the key steps here for convenience. First, we manually selected datasets relevant to the particular biological system and carefully annotated the samples to apply uniform nomenclature. Then we concatenated all expression matrices of the selected datasets and rank transformed them as a normalization step. Then, we used the VariancePartition package in R (Hoffman and Schadt, 2016) to assess each gene for its platform variance and filtered out genes with high platform variance as the batch correction step. The genes left behind were shown to capture the essential biological variances in the integrated data. We also tested the method for robustness by applying different normalizations, sub-sampling, and varying the cutoff values for the genes.
In order to project new data onto the Stemformatics atlases, we first fit the atlas data to a 10-dimensional PCA model using the scikit-learn package (Scikit-learn), which works out the PCA loadings. We then project the atlas data onto this 10-dimensional PCA space, of which the first 3 are shown in the user interface (the choice of 10 dimensions is somewhat arbitrary—we found very similar results with different values).
New data to be projected against this PCA space is first filtered to match the genes in the atlas. If less than 50% of the genes in the query data are present in the atlas, the user is shown a warning and the projection will not proceed. Then rank normalization is applied for each sample, where 0 is the lowest ranked gene and 1 is the highest. The transform function from the same scikit-learn package (Scikit-learn) is used to calculate the projected coordinates. Since we do not apply fit function again, the PCA space remains stable based on the atlas data (Angel et al., 2020). The additional advantage of this approach is that query data may be input in either raw format (e.g., RNA-seq raw counts) or in typically log normalized format, as rank is preserved.
The other advantage of PCA projection is that since each projection is independent of each other, batch effects which may be present in the query data can be ignored before the projection. The technical effects may be revealed against the annotations provided in the atlas—including platform, cell source, treatment conditions, tissue, or disease groups. Projection therefore provides an intuitive way to understand and visualize the comparison of query cells against the atlas cells and annotations available from the atlas datasets.
When the user uploads data to project them onto an atlas, Stemformatics also employs Capybara (Kong et al., 2022) to score the query data against the atlas independently of the PCA projection. Capybara uses constrained linear regression to produce the scores as a continuous variable and is particularly well suited for this analysis. We implemented a python version of its key function to work within the Stemformatics platform (code is available in our public GitHub repository – see code availability section). In addition to the reference and query expression data, groupings of atlas samples are also input into Capybara, such as cell type, sample source, and tissue of origin. The output is a matrix where rows are the query samples and columns are values of these atlas sample groupings such as individual cell types. Each value in this matrix represents the probability of the query sample matching the reference sample. This is presented as a heatmap to the user.
Note that while scRNA-seq data can be projected onto the atlases, we recommend pseudo-bulk aggregating the data first before projection. This avoids the issues created by the large numbers of zeros in the data, as well as flooding the graph with a large number of individual points which hinders website performance as well as visualization. While the projection tools we provide are very convenient, the users need to be aware that cells with very different identity to those in the atlas are not expected to show meaningful projection results. Each projection includes random value projection to highlight a possible mismatch such as this.
Acknowledgments
We would like to acknowledge other members of the Synnate team at the Hudson Institute for providing input, especially Jamie Gearing. We thank Chen Zhan for making additional changes to the website during the review process.
This work was funded by the Australian Research Council FT150100330 and the National Health and Medical Research Council Synergy grant 1186371 to C.A.W.
Author contributions
J.C. developed the methods, database, and software and wrote the manuscript. S.K.B. annotated and processed the data. P.A. developed methods. J. Bransfield and J. Barry developed software. N.F., A.M., and P.P. developed annotation guidelines. B.S. developed annotation guidelines and visualization prototypes. C.A.W. conceived the project, annotated the data, funded the project, and wrote the manuscript.
Declaration of interests
The authors declare no conflict of interest.
Published: May 23, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2024.04.012.
Contributor Information
Jarny Choi, Email: jarnyc@unimelb.edu.au.
Christine A. Wells, Email: wells.c@unimelb.edu.au.
Supplemental information
References
- Alsinet C., Primo M.N., Lorenzi V., Bello E., Kelava I., Jones C.P., Vilarrasa-Blasi R., Sancho-Serra C., Knights A.J., Park J.E., et al. Robust temporal map of human in vitro myelopoiesis using single-cell genomics. Nat. Commun. 2022;13:2885. doi: 10.1038/s41467-022-30557-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angel P.W., Rajab N., Deng Y., Pacheco C.M., Chen T., Lê Cao K.A., Choi J., Wells C.A. A simple, scalable approach to building a cross-platform transcriptome atlas. PLoS Comput. Biol. 2020;16 doi: 10.1371/journal.pcbi.1008219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A. The Cellosaurus, a Cell-Line Knowledge Resource. J. Biomol. Tech. 2018;29:25–38. doi: 10.7171/jbt.18-2902-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BoostrapVue. https://bootstrap-vue.org/.
- Cahan P., Cacchiarelli D., Dunn S.J., Hemberg M., de Sousa Lopes S.M.C., Morris S.A., Rackham O.J.L., Del Sol A., Wells C.A. Computational Stem Cell Biology: Open Questions and Guiding Principles. Cell Stem Cell. 2021;28:20–32. doi: 10.1016/j.stem.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canavan M., Walsh A.M., Bhargava V., Wade S.M., McGarry T., Marzaioli V., Moran B., Biniecka M., Convery H., Wade S., et al. Enriched Cd141+ DCs in the joint are transcriptionally distinct, activated, and contribute to joint pathogenesis. JCI Insight. 2018;3 doi: 10.1172/jci.insight.95228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Pacheco C.M., Mosbergen R., Korn O., Chen T., Nagpal I., Englart S., Angel P.W., Wells C.A. Stemformatics: visualize and download curated stem cell data. Nucleic Acids Res. 2019;47:D841–D846. doi: 10.1093/nar/gky1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elahi Z., Angel P.W., Butcher S.K., Rajab N., Choi J., Deng Y., Mintern J.D., Radford K., Wells C.A. The Human Dendritic Cell Atlas: An Integrated Transcriptional Tool to Study Human Dendritic Cell Biology. J. Immunol. 2022;209:2352–2361. doi: 10.4049/jimmunol.2200366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMBL-EBI-BRENDA. The BRENDA Tissue Ontology. https://www.ebi.ac.uk/ols/ontologies/bto.
- EMBL-EBI-CO. Cell Ontology. https://www.ebi.ac.uk/ols/ontologies/cl.
- EMBL-EBI-HDO. Human Disease Ontology. https://www.ebi.ac.uk/ols/ontologies/doid.
- EMBL-EBI-OLS. Ontology Lookup Service. https://www.ebi.ac.uk/ols/index.
- FAIR Principles. https://www.go-fair.org/fair-principles/.
- Flask-RESTful. https://flask-restful.readthedocs.io/en/latest/.
- HDF5 Format. https://www.hdfgroup.org/solutions/hdf5/.
- Hoffman G.E., Schadt E.E. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinf. 2016;17:483. doi: 10.1186/s12859-016-1323-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein S.M.I., Puri M.C., Tonge P.D., Benevento M., Corso A.J., Clancy J.L., Mosbergen R., Li M., Lee D.S., Cloonan N., et al. Genome-wide characterization of the routes to pluripotency. Nature. 2014;516:198–206. doi: 10.1038/nature14046. [DOI] [PubMed] [Google Scholar]
- Kong W., Fu Y.C., Holloway E.M., Garipler G., Yang X., Mazzoni E.O., Morris S.A. Capybara: A computational tool to measure cell identity and fate transitions. Cell Stem Cell. 2022;29:635–649.e11. doi: 10.1016/j.stem.2022.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroczek R.A., Henn V. The Role of XCR1 and its Ligand XCL1 in Antigen Cross-Presentation by Murine and Human Dendritic Cells. Front. Immunol. 2012;3:14. doi: 10.3389/fimmu.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Zhang X., Ang K.S., Ling J., Sethi R., Lee N.Y.S., Ginhoux F., Chen J. DISCO: a database of Deeply Integrated human Single-Cell Omics data. Nucleic Acids Res. 2022;50:D596–D602. doi: 10.1093/nar/gkab1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MongoDB. https://www.mongodb.com/.
- Monkley S., Krishnaswamy J.K., Göransson M., Clausen M., Meuller J., Thörn K., Hicks R., Delaney S., Stjernborg L. Optimised generation of iPSC-derived macrophages and dendritic cells that are functionally and transcriptionally similar to their primary counterparts. PLoS One. 2020;15 doi: 10.1371/journal.pone.0243807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuxt. https://nuxtjs.org/.
- Open Refine. https://openrefine.org.
- Plotly. https://plotly.com/.
- Python Pandas. https://pandas.pydata.org/.
- Rajab N., Angel P.W., Deng Y., Gu J., Jameson V., Kurowska-Stolarska M., Milling S., Pacheco C.M., Rutar M., Laslett A.L., et al. An integrated analysis of human myeloid cells identifies gaps in in vitro models of in vivo biology. Stem Cell Rep. 2021;16:1629–1643. doi: 10.1016/j.stemcr.2021.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scikit-learn. https://scikit-learn.org/0.15/modules/generated/sklearn.decomposition.PCA.html.
- Tonge P.D., Corso A.J., Monetti C., Hussein S.M.I., Puri M.C., Michael I.P., Li M., Lee D.S., Mar J.C., Cloonan N., et al. Divergent reprogramming routes lead to alternative stem-cell states. Nature. 2014;516:192–197. doi: 10.1038/nature14047. [DOI] [PubMed] [Google Scholar]
- Wells C.A., Mosbergen R., Korn O., Choi J., Seidenman N., Matigian N.A., Vitale A.M., Shepherd J. Stemformatics: visualisation and sharing of stem cell gene expression. Stem Cell Res. 2013;10:387–395. doi: 10.1016/j.scr.2012.12.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used by Stemformatics website are available through the website. The code behind Stemformatics is available at https://github.com/wellslab/s4m-api and https://github.com/wellslab/s4m-ui. The code which generated data for projection is at https://github.com/wellslab/stemformatics-data-projection.