

Abstract
Major histocompatibility complex (MHC) presented peptides (pMHC) give insight into T cell immune responses, a critical step towards developing a new generation of targeted immunotherapies. Recent instrumentation advances have propelled mass-spectrometry to being arguably the most robust technology for discovering and quantifying naturally-presented pMHC from cells and tissues. However, sample preparation has remained a major limitation due to time-consuming and labor-intensive workflows. We developed a high-throughput and automated platform with enhanced speed, sensitivity and reproducibility relative to prior studies. This pipeline is capable of processing up to 96 samples in 6 hours or less yielding high-quality pMHC mixtures ready for mass spectrometry. Here, we describe our efforts to optimize purification and mass spectrometer parameters, ultimately allowing us to identify as many as almost 5,000 pMHC I and 7,400 pMHC II from as little as 2.5×107 cells, respectively. We believe this platform will facilitate and accelerate immunopeptidome profiling and benefit clinical research for immunotherapies.
Graphical Abstract
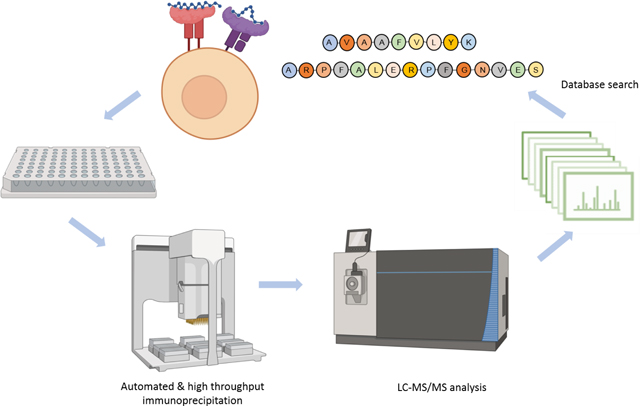
Introduction
Peptide ligands presented by Major Histocompatibility Complexes (pMHC) hold tremendous promise for treatment modalities in cancer, infectious disease, autoimmunity, and allergy due to their central role in immune surveillance. One common method for identifying disease-relevant pMHCs combines next-generation sequencing with in silico binding affinity predictions to implicate putative non-self epitopes worthy of further scrutiny. Despite considerable improvements in pMHC predictions in recent years, empirical observations1 continually reveal gaps between theoretical and naturally presented antigens.
Direct peptide sequencing using liquid chromatography and tandem mass spectrometry (LC-MS/MS) circumvents these limitations. LC-MS/MS-based pMHC identification holds the potential to reveal post-translational modifications (PTMs), alternative splicing, and presentation by poorly-characterized MHC alleles. These kinds of pMHC could be particularly specific to disease states, yet evade accurate binding affinity prediction. Lastly, since binding affinity alone predicts neither antigen presentation nor T cell recognition of pMHC, a wider view of pMHC repertoires may be necessary for rationally designing disease-targeting therapies2. Despite these advantages, however, sample preparation protocols for MHC immunoprecipitation, peptide elution, and analysis via LC-MS/MS are both time and labor intensive, making this strategy difficult to scale for large, clinical studies.
In the nearly three decades since pMHC discovery using LC-MS/MS was first described 3–5, tremendous technological advances have demonstrated the immense potential this approach has for clinical applications. For example, our prior study interrogated MHC I and MHC II antigen presentation in 17 patients with untreated mantle cell lymphoma (MCL) and additionally from two MCL cell lines, revealing over 24,000 pMHC I and 12,500 pMHC II, including tumor-specific “ideotypic” pMHC from all patients we tested 6–10. A prior study also shows success with this technique, analyzing clinically relevant neoepitopes presented on primary tissue from 25 metastatic melanoma patients11. Recently this approach has been combined with a quantification platform to profile immunopeptidome response to CDK4/6 inhibition and interferon-γ in melanoma cell lines12.
Despite these advances, the pMHC isolation method has remained relatively unchanged13–16. Although its implementation by different research groups has introduced several procedural variations, most share the common problems of requiring multiple days to perform and requiring a researcher’s focused attention on just a handful of specimens at a time. Both throughput challenges can decrease experimental reproducibility, making comparisons between specimens difficult. For MHC immunoprecipitation to become a reliable tool for immunotherapy research, a standardized high-throughput and sensitive protocol that minimizes human error must be established. Transferring immunoprecipitation procedures to a 96-well plate format17,18 was recently shown to offers substantial improvements in speed and throughput. However, as described, it still requires extensive manual intervention.
If automated liquid handling devices can be incorporated into robust, high-throughput MHC immunoprecipitation protocols, extraordinary improvements in throughput, speed, sensitivity, and reproducibility could be possible. Here, we introduce a novel pipeline for pMHC purification using the AssayMAP Bravo platform from Agilent Technologies. It employs a nearly completely automated, 96-well plate workflow which takes cell lysate as input and processes it through peptide purification. We found that the decreased amount of manual procedures coupled with efficient microchromatography cartridges led to substantial improvements in reproducibility, yield, and sample processing times relative to our prior manual procedure: The protocol described herein can routinely yield several thousands of pMHC I and II identifications from under 50 million input cells. We believe that this method could be broadly implemented to help standardize and accelerate the pace of capturing therapeutically significant data, and thus be a major driver in the creation of novel antigen-based immunotherapies.
Materials and Methods
Cell culture and lysis
Raji cells (HLA-A*03:01/A*03:01, HLA-B*15:10/ B*15:10, HLA-C*03:04/C*04:01)19 overexpressing the viral entry receptor DC-SIGN (a gift from Eva Harris, UC Berkley) were grown in RPMI 1640 media (Cytiva, South Logan, Utah) supplemented with 10% FBS (Geminibio, Sacramento, California) and Penicillin, streptomycin, and L-glutamine (Corning, Corning, New York). The cells were harvested into working aliquots as described in the text, washed twice with 1x PBS (Corning, Corning, New York), and flash frozen in liquid nitrogen. The cells were then stored at −80°C.
Raji cell pellets were lysed on ice for 20 minutes (1% CHAPS, 20mM Tris pH 8, 150mM NaCl, supplemented with cOmplete mini protease inhibitor tablet, (Roche, Basel, Switzerland); Halt protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, Massachusetts); and PMSF (Millipore Sigma, Saint Louis, Missouri, 0.1mM )). 0.8 mL of lysis buffer was used per 1×108 cells. The lysate was then cleared by two rounds of centrifugation at 16,000 × g for 20 minutes each at 4°C, with the supernatant being transferred to a fresh tube between centrifugations. To reduce variation between cell pellets, all the lysate to be used in an experiment (2×108 to 1×109 cells worth) was pooled and thoroughly mixed. This lysate was then aliquoted into a 96 well plate for introduction onto the AssayMAP.
Standard Bovine Brain Peptides Preparation
A standard complex protein mixture was prepared from bovine brain tissue (Schaub’s Meat Fish and Poultry, Palo Alto, CA) as previously described20. Briefly, 10 g of snap-frozen tissue was thawed on ice, dounce homogenized, and lysed by tip sonication in RIPA lysis buffer (0.1% SDS, 1% NP-40, 1% sodium deoxycholate, 150 mM NaCl, 25 mM Tris-HCl, pH 7.6) plus EDTA-free protease inhibitor cocktail (Roche, Basel, Switzerland). The proteins were reduced (5 mM dithiothreitol, 56°C, 30 min), alkylated (14 mM iodoacetamide, 20°C, 1 h), then quantified with the bicinchoninic acid assay and precipitated with trichloroacetic acid. The protein pellet was resuspended in 8 M urea/100 mM ammonium bicarbonate and the protein mixture was diluted to 1 M urea for digestion overnight at 37°C with trypsin at an enzyme/substrate ratio of 1:100. The resulting peptides were desalted with Sep-Pak C18 columns (Waters, Milford, MA).
Standard Nonspecific Peptides Preparation
A standard complex nonspecific peptide mixture was prepared from the human WT33 cell line (28.4 million cells, gift from Wysocka Lab at Stanford). Cells were lysed with urea lysis buffer (8M urea, 150 mM NaCl, and 5 mM dithiothreitol, 50 mM Tris, pH 8) plus EDTA-free protease inhibitor cocktail (Roche, Basel, Switzerland), sonicated, and the cell debris was precipitated by centrifugation (16,000 × g at 4°C for 15 min, two rounds). The supernatant was alkylated with 14 mM iodoacetamide (45 min at room temperature in dark). Proteins were precipitated with methanol/chloroform and resuspended in 8M urea/100mM ammonium bicarbonate. The sample was then diluted to 4 M urea and pH adjusted to 2 with hydrochloric acid. Pepsin (Promega, Madison, WI) was added for a final enzyme/substrate ratio of 1:25. This was then incubated for 30 min at room temperature, and quenched with the addition of sodium hydroxide to a final pH=8. The resulting peptides were acidified and desalted with Sep-Pak C18 columns (Waters, Milford, MA).
Automated MHC Immunoprecipitation
MHC class I and MHC class II molecules were isolated from Raji cells with the pan-MHC I antibody W6/3221 (BioXCell, West Lebanon, New Hampshire) and MHC II DR antibody L24322 (Leinco, Fenton, Missouri). AssayMAP Bravo protein A cartridges (Agilent, Santa Clara, California) were primed and washed with Tris-buffered saline (TBS) (Thermo Fisher Scientific, Waltham, Massachusetts), and then used to capture antibodies from stock aliquots23 (Supplemental Figure 1A, Table 1). Raji cell lysate was then pre-cleared using fresh protein A cartridges (Supplemental Figure 1B, Table 1). The pre-cleared lysate was added to W6/32 or L243 antibody coupled cartridges to capture MHC complexes. The cartridges were then washed with 1x TBS and eluted with 10% acetic acid. Note that Table 1 shows the optimized parameters in the protocol determined by the experiments described in this report. The detailed parameters for each optimization experiment are included in Supplemental Table 1.
Table 1.
AssayMAP Bravo software parameters for the four deck layouts shown in Supplemental Table 1.
| Antibody binding | |||
|---|---|---|---|
|
| |||
| Step | Volume | Flow Rate | Buffer |
| Prime | 100 μL | 300 μL/min | 1X PBS |
| Equilibrate | 50 μL | 10 μl/min | 1X PBS |
| Load Sample | 125 μL | 3 μL/min | Antibody in PBS |
| Cup Wash 1 | 25 μL | 1X TBS | |
| Internal Cartridge Wash 1 | 50 μL | 10 μL/min | 1X TBS |
|
| |||
| Pre-clearing cell lysate | |||
|
| |||
| Step | Volume | Flow Rate | Buffer |
| Prime | 100 μL | 300 μL/min | 1X PBS |
| Equilibrate | 50 μL | 10 μL/min | 1X PBS |
| Load Sample | 800 μL | 45 μL/min | Cell Lysate |
|
| |||
| Immunoprecipitation | |||
|
| |||
| Step | Volume | Flow Rate | Buffer |
| Prime | 100 μL | 300 μL/min | 1X PBS |
| Equilibrate | 50 μL | 10 μL/min | 1X PBS |
| Load Sample | 800 μL | 10 μL/min | Pre-cleared lysate |
| Cup Wash 1 | 25 μL | 1x TBS | |
| Internal Cartridge Wash 1 | 100 μL | 10 μL/min | 1x TBS |
| Stringent Syringe Wash | 50 μL | 10% Acetic Acid | |
| Elute | 100 μL | 5 μL/min | 10% Acetic Acid |
|
| |||
| Peptide desalting | |||
|
| |||
| Step | Volume | Flow Rate | Buffer |
| Prime | 100 μL | 300 μL/min | 50% ACN, 0.1% FA |
| Equilibrate | 50 μL | 10 μL/min | 0.1% FA |
| Load Sample | 100 μL | 5 μL/min | Previous eluate |
| Cup Wash | 25 μL | 0.1% FA | |
| Internal Cartridge Wash | 50 μL | 10 μL/min | 0.1% FA |
| Stringent Syringe Wash | 50 μL | 50% ACN, 0.1% FA | |
| Elute | 40 μL | 5 μL/min | 50% ACN, 0.1% FA |
MHC Peptide Desalting
Peptides eluted from immunoprecipitated MHC complexes were captured by underivatized polystyrene-divinylbenzene reverse phase S (RP-S) cartridges (Agilent, Santa Clara, California) at 10 μL/min. The cartridges were then washed with 0.1% formic acid and eluted with 40 μL of 50% acetonitrile in 0.1% formic acid (Supplemental Figure 1D, Table 1). The resulting peptides were dried by vacuum centrifugation (Centrivap, Labconco Corporation, Kansas City, MO) and stored at −20°C.
Raji pMHC Manual Preparation
Raji MHC class-I and class-II molecules from 3×108 cells per replicate were immunoprecipitated and associated peptides were isolated as previously described with a few alterations4–6. Cell lysis was performed for 20 minutes on ice in 20 mM Tris-HCl (pH8), 150 mM NaCl, 1% (w/v) CHAPS, 0.2 mM PMSF, 1x Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Waltham, Massachusetts) supplemented with complete protease inhibitor cocktail (Roche, Mannheim, Germany). This lysate was then centrifuged twice (30 minutes, 13,200 rpm at 4°C). The supernatant was then precleared for 30 minutes using rProtein A Sepharose fast-flow beads (GE Healthcare, Uppsala, Sweden). Precleared lysate was then incubated for 5 hours at 4°C with pan-MHC-I antibody W6/32 (hybridoma kindly provided by Betsy Mellins and immunoglobulin produced and purified by Genentech) that had been coupled to rProtein A Sepharose fast-flow beads. Beads were washed 5 times with TBS (pH 7.4). Peptides were eluted from captured MHC molecules using 10% acetic acid and passed through a 10 kDa MWCO filter (pre-washed with 10% acetic acid) and concentrated using vacuum centrifugation. Following MHC I immunoprecipitation, the flow through was transferred to fresh tubes and incubated with the MHC-DR-specific antibody, L243 (hybridoma kindly provided by Ron Levy and immunoglobulin produced and purified by Genentech), coupled to rProtein A Sepharose fast-flow beads overnight at 4°C. The following wash, elution and separation steps were the same with MHC I immunoprecipitation. Peptides were desalted using C18 STAGE tips24 and then vacuum centrifuged. Clean, desalted peptides were stored at −80°C.
Raji Whole Cell Proteome Preparation
The S-Trap system (ProtiFi, Farmingdale, New York) was used to digest Raji whole cell lysate. Aliquots of 500,000 Raji cells were lysed in 50 μL of solubilization buffer (5% SDS, 50mM TEAB, pH 7.55). One microliter of Benzonase® Nuclease (25 units/μL, MilliporeSigma, Burlington, Massachusetts) was added to each sample tube and the sample tube was sonicated at 37°C in water bath to sheer DNA. The lysate was clarified by centrifugation for 20 minutes at 16,000 g, followed by reduction (5 mM dithiothreitol, 56 °C, 30 min), alkylation (14 mM iodoacetamide, 20 °C, 1 h), and quantification using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, Delaware). The alkylated lysate was acidified with 12% phosphoric acid to a final phosphoric acid concentration of 1.2%, and diluted with 300 μL of S-Trap binding buffer (90% methanol, 100mM tetraethylammonium bromide, pH 7.1). This was then passed through an S-Trap micro column using centrifugation (500 g for 30s). The S-Trap micro column was washed three times with 150 μL S-Trap binding buffer. Trypsin was added to the S-Trap column at an enzyme-to-protein ratio 1:20 and the column was incubated overnight at 37°C. The digested peptides were then eluted using 40 μL of 50 mM tetraethylammonium bromide, 0.2% formic acid, and 50% acetonitrile in 0.2% formic acid. The eluates from the three extractions were pooled, dried by vacuum centrifugation, and stored at −20°C.
LC-MS/MS method
Peptide samples were analyzed on either an LTQ Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) or a Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) (see Supplemental Table 1 for details) equipped with Dionex Ultimate 3000 LC systems (Thermo Fisher Scientific, San Jose, CA). Peptides were separated by capillary reverse phase chromatography on a 20 cm reversed phase column (100 μm inner diameter, packed in-house with ReproSil-Pur C18-AQ 3.0 m resin (Dr. Maisch GmbH)).
pMHC analyzed on a Fusion Lumos mass spectrometer:
pMHC were resuspended in 12 μL of 0.1% formic acid and 4 μL per injection was introduced into the Fusion Lumos mass spectrometer (Tune application 3.3.2782.32) using a two-step linear gradient with 4–25 % buffer B (0.1% (v/v) formic acid in acetonitrile) for 80 min followed by 25–45 % buffer B for 10 min. Data were acquired in top speed data dependent mode with a duty cycle time of 3 s. Full MS scans were acquired in the Orbitrap mass analyzer with a resolution of 120 000 (FWHM) and m/z scan range of 340–1540. Precursor ion mass range was tailored to the known mass distributions for MHC I and MHC II peptides8. For MHC I, precursors within the mass range of 700–1800 Da were selected for MS2 analysis, and for MHC II the range was extended to 700–2760 Da. A decision tree was designed combining charge state and m/z values measured from precursor scans to determine the mass ranges that could trigger subsequent MS2 scans (Supplemental Table 2). Selected precursor ions were subjected to fragmentation using higher-energy collisional dissociation (HCD) with quadrupole isolation, isolation window of 1.6 m/z, and normalized collision energy of 30%. HCD fragments were analyzed in the Orbitrap mass analyzer with a resolution of 15,000 (FWHM). Fragmented ions were dynamically excluded from further selection for a period of 15 seconds. The AGC target was set to 400,000 and 50,000 for full FTMS scans and FTMS2 scans, respectively. The maximum injection time was set to 50 ms for full FTMS scans and various values for FTMS2 scans for optimization purposes.
pMHC samples analyzed on a LTQ Orbitrap Elite mass spectrometer (size filtration comparison only):
pMHC were resuspended in 12 μL of 0.1% formic acid and 4 μL was introduced into the LTQ Orbitrap Elite mass spectrometer with three complementary acquisition methods as previously described8. Briefly, peptides were separated using a two-step linear gradient with 4–25% buffer B (0.2% (v/v) formic acid, 5% DMSO, and 94.8% (v/v) acetonitrile) for 120 min followed by 25–40% buffer B for 30 min. Three injections were made per sample to utilize multiple fragmentation modes (HCD (higher-energy collisional dissociation) or CID (collision-induced dissociation)). The first two injections excluded singly-charged species, whereas the third CID injection included them. Acquisition was executed in data-dependent mode with the full MS scans acquired in the Orbitrap mass analyzer with a resolution of 60,000 and m/z scan range 340–1,600. The top ten most intense ions were then selected for sequencing and fragmented in the Orbitrap mass analyzer at a resolution of 15,000 (full width at half maximum). Data-dependent scans were acquired from precursors with masses ranging from 700 to 1,800 Da for all MHC class 1 samples and from 700 to 2,750 Da for MHC class II samples. Precursor ions were fragmented with a normalized collision energy of 35% and an activation time of 5 ms for CID and 30 ms for HCD. Repeat count was set to 2 and fragmented m/z values were dynamically excluded from further selection for a period of 30 s. The minimal signal threshold was set to 500 counts. The AGC target was set to 1,000,000 and 50,000 for full FTMS scans, and FTMSn scans, respectively. The maximum injection time was set to 250 ms for full both FTMS scans and FTMSn scans.
Standard bovine brain peptide samples analyzed on a LTQ Orbitrap Elite mass spectrometer (for RP-S sample loading condition experiment):
trypsin-digested bovine brain peptides were resuspended in 0.1% formic acid at 0.1 μg/μL and 0.1 μg was introduced using a two-step linear gradient with 4–25 % buffer B (0.1% (v/v) formic acid and 5% DMSO in acetonitrile) for 20 min followed by 25–40 % buffer B for 5 min. MS/MS were acquired in data-dependent mode, with full MS scans acquired in the Orbitrap mass analyzer with a resolution of 60,000 and m/z scan range of 340–1,600. The top 20 most abundant ions with intensity threshold above 500 counts and charge states 2 and above were selected for fragmentation using collision-induced dissociation (CID) with an isolation window of 2 m/z, normalized collision energy of 35%, activation Q of 0.25, and activation time of 5 ms. The CID fragments were analyzed in the ion trap with rapid scan rate. Dynamic exclusion was enabled with repeat count of 1 and exclusion duration of 30 s. The AGC target was set to 1,000,000 and 50,000 for full FTMS scans and ITMSn scans, respectively. The maximum injection time was set to 250 ms and 100 ms for full FTMS scans and ITMSn scans, respectively.
Standard nonspecifically proteolyzed peptide samples analyzed on a Fusion Lumos mass spectrometer (for mass spectrometer instrument method optimization):
Pepsin-digested bovine brain peptides were resuspended in 0.1% formic acid at 0.1 μg/μL and 0.2 μg were injected, and analyzed on the Fusion Lumos mass spectrometer as described above for pMHC-I peptides.
Raji total proteome samples analyzed on a Fusion Lumos mass spectrometer:
Tryptic peptides resulting from digested Raji lysate were resuspended in 0.1% formic acid at 1 μg /μL and 1 μg was introduced into the Fusion Lumos mass spectrometer using a two-step linear gradient with 4–25 % buffer B (0.1% (v/v) formic acid in acetonitrile) for 135 min followed by 25–45 % buffer B for 15 min. Data was acquired in top speed data dependent mode with a duty cycle time of 3 s. Full MS scans were acquired in the Orbitrap mass analyzer with a resolution of 120 000 (FWHM) and m/z scan range of 340–1540. Selected precursor ions were subjected to fragmentation using higher-energy collisional dissociation (HCD) with quadrupole isolation, isolation window of 1.6 m/z, and normalized collision energy of 30%. HCD fragments were analyzed in the Orbitrap mass analyzer with a resolution of 15,000 (FWHM). Fragmented ions were dynamically excluded from further selection for a period of 15 seconds. The AGC target was set to 400,000 and 50,000 for full FTMS scans and FTMS2 scans, respectively. The maximum injection time was set to 50 ms for full FTMS scans and dynamic for FTMS2 scans.
Database search
pMHC data
The MSConvert program (v3.0.45) was used to generate peak lists from RAW data files25 which were subsequently queried against a “target-decoy” sequence proteome database consisting of the Uniprot human database (downloaded September 18, 2019), common contaminants and the corresponding reversed sequences using the SEQUEST algorithm (SEQUEST v.28 (rev. 12))26–28. The precursor mass range was set to 600–4000 Da, the mass error tolerance was set to 10 ppm, and the fragment mass error tolerance to 0.02 Da. Enzyme specificity was set to none and oxidation of methionines was considered as variable modifications (+15.995). High-confidence peptide identifications were selected at a 1% false discovery rate with a linear discriminator analysis27. Abundance quantification was based on precursor peak areas.
Raji whole proteome tryptic digest data
Raji tryptic digest data were searched using SEQUEST HT on the Proteome Discoverer (2.2.0.388) platform against a database containing known human proteins (downloaded September 18, 2019 from Uniprot) from which HLA-related proteins were substituted with the Raji cell line’s known HLA alleles, downloaded from the IPD-IMGT/HLA Database. Common contaminant proteins were added to this database prior to decoy protein creation and searching. The precursor mass range was set to 350–10000 Da, the mass error tolerance was set to 10 ppm, and the fragment mass error tolerance to 0.02 Da. Enzyme specificity was set to trypsin, carbamidomethylation of cysteines (57.021) was set as variable modifications, oxidation of methionines and acetylation of protein N-terminus (+42.011) was considered as variable modifications. Percolator was used to filter peptides and proteins to a false discovery rate of 1%. Abundance quantification was based on precursor ion intensities.
Bovine brain tryptic digest data
Bovine brain tryptic digest data were searched using SEQUEST HT on the Proteome Discoverer (2.2.0.388) platform, against a database combining Bovine proteins (downloaded from uniprot.org, January 21, 2016), common contaminants and reversed decoys generated from the above. The precursor mass range was set to 350–3000 Da, the mass error tolerance was set to 10 ppm, and the fragment mass error tolerance to 0.02 Da for Fusion data and 0.6 Da for Elite data. Enzyme specificity was set to trypsin, carbamidomethylation of cysteines (57.021) was set as static modifications, oxidation of methionines and acetylation of protein N-terminus (+42.011) was considered as variable modifications. Percolator was used to filter peptides to a false discovery rate of 1%.
Results and Discussion
Protein A cartridge binding capacity
We and others routinely use the W6/32 antibody to enrich for MHC class I peptides and the L243 antibody to enrich for MHC class II peptides8,11,17,29,30. For the manual sample preparation procedure we used in prior publications, 560 μg of antibody were immobilized on 400 μL of rProtein A Sepharose Fast Flow beads8. This was sufficient to capture MHC complexes from 1.25×108 Raji cells. It would be advantageous to reduce the amount of antibody, protein A resin, and input cell material since each can be expensive or time-consuming to generate. Since AssayMAP cartridges used here have a fixed protein A resin bed volume of just 5 μL, we expected we could use substantially less antibody per immunoprecipitation reaction. We found that approximately 250 μg of both W6/32 and L243 antibodies was sufficient to saturate the protein A resin. This amount is similar to the AssayMAP antibody purification protocol guide’s stated binding capacity of protein A cartridges31. We further found that 175 and 145 μg of W6/32 and L243 (respectively) remained bound after being washed with TBS (Supplemental Figure 2). We therefore pre-loaded 250 ug of antibody for all subsequent experiments described below.
Immunoprecipitation of MHC molecules: binding rate and elution volume optimization
We previously established that cell lysate incubated with antibody bound protein A-sepharose beads for 5 hours at 4°C was sufficient for effective MHC recovery8. Longer incubation durations could be expected to increase non-specific protein binding, while shorter incubations could lower overall assay sensitivity if fewer MHC molecules are captured from lysate. Other researchers reported immunoprecipitation durations ranging from 1 hour to overnight at 4°C with different binding systems including in-solution binding and affinity column binding 14–16,18. Towards optimizing the balance between assay speed, specificity and sensitivity, we sought to evaluate how lysate flow rates influenced MHC binding to antibody-bound protein-A cartridges.
It was important to first consider that the AssayMAP Bravo was operated in a room temperature environment, and that lysate remains on a 4°C cooling plate. This configuration should minimize sample proteolysis and other undesired reactions within the lysate but not on antibody-bound AssayMAP cartridges which are maintained at room temperature throughout the binding procedure. Consequently, we chose to limit the antibody binding step to <2 hours. With a target lysate volume of 0.8 mL per 1×108 cells and applying the default flow rate of 10 μL/min across the antibody-bound AssayMAP cartridge, 80 minutes would be needed. To evaluate how eventual peptide identifications might depend on the initial antibody binding flow conditions, we doubled the flow rate to 20 μL/min, and compared the number of unique peptide identifications we made relative to the default. As shown in Figure 1A–C, the number of unique peptides we identified decreased by an average of 28% with the faster flow rate. This decrease was fairly consistent across the range of peptide lengths (Figure 1A) and abundance (Figure 1D,E), although very abundant peptides were consistently identified regardless of flow rate (Figure 1D,E).
Figure 1. MHC II binding rate comparison.

MHC II peptide immunoprecipitation and purification from 1×108 Raji cells was performed as described in Methods but with two immunoprecipitation binding rates (10 μL/min and 20 μL/min) (N=2 preparative replicates*) (see Supplemental Table 1 for detailed parameters). (A) Peptide length distribution for each binding rate. Error bounds are from the replicate experiments (maximum and minimum values). (B, C) The overlap between unique peptides identified from 10 μL/min and 20 μL/min. (D, E) Peptide abundance rank distribution for each replicate experiment. The peptides identified from 10 μL/min experiment were ranked by their peak areas (peptide abundance rank) and plotted against their corresponding log10 peak areas (light blue dots). The peptides identified in the 10 μL/min experiment but not in the 20 μL/min experiment were highlighted with smaller purple dots. (F,G) pMHC II predicted binding affinity distributions noting which pMHC I identified from 10 μL/min experiment (blue) were also identified from 20 μL/min experiment (dark green). %EL rank is the percentile of the predicted binding affinity compared to the distribution of binding affinities calculated from a set of random natural peptides32. NetMHCpan 4.1’s authors recommended that pMHC I found among the top 0.5% of binding predictions be considered strong binders and pMHC I found above 0.5% but below 2% considered weak binders. Peptides with %EL ranks greater than 2% are predicted not to bind. (H) MHC binding prediction of peptides identified from 10 μL/min and 20 μL/min experiments, and uniquely identified in each condition. Peptides were mapped to the single allele to which they were assigned the most stringent rank score. The total number of peptides considered per experiment is labeled above each bar. For economic considerations, we performed these optimization procedures only with MHC II immunoprecipitations rather than both MHC-I and MHC-II.
*In this manuscript “technical replicates” were defined as repeated injections of the same peptide mixture; “preparative replicates” were defined as parallel immunoprecipitations from a single lysate, and “biological replicates” were defined as independent samples (different animals, different cell cultures) reflecting the same biological state.
We further considered the predicted binding affinities of identified peptides using NetMHCIIpan 4.032 and found the peptides identified from both flow rates were distributed across the binding affinity range with no obvious bias (Figure 1F,G). Of all pMHC II identified from both flow rates, 85% of the peptides were predicted to bind MHC II, and 66% were predicted bind MHC II with high affinity (Figure 1F,G). These percentages are slightly higher than the pMHC II identified from 10 μL/min analysis, where 80% of the peptides were predicted to bind MHC II, and 59% were predicted to bind MHC II with high affinity (Figure 1F,G).
After assigning peptides to a single MHC-II allele based on predicted binding affinity, we next compared their frequencies across the three Venn diagram regions shown in Figure 2B–C (Figure 1H). We found that the faster 20 μL/min flow rate yielded proportionately more peptides predicted to bind DRB1*10:01 than the 10 μL/min analysis (38% compared to 27%). Peptides uniquely identified in the 20 μL/min experiment versus the 10 μL/min one exacerbated this DRB1*10:01 bias (56% compared to 19%). Conversely, we found that peptides predicted to bind the DRB1*03:01 allele were biased in the opposite direction (13% DRB1*03:01 binding found only in the 20 μL/min experiment compared to 46% DRB1*03:01 found only in the 10 μL/min experiment). The proportion peptides predicted to not bind any MHC-II alleles was slightly less in the 20 μL/min experiment than the 10 μL/min experiment (19.2% and 15.8%) (Figure 1H).
Figure 2. pMHC prepared with/without size filtration.

Four lysate aliquots equivalent to 1×108 Raji cells were subject to MHC immunoprecipitation (two aliquots for MHC I and two aliquots for MHC II). pMHC I or pMHC II were eluted from antibody-bound protein A cartridges, then either passed through 10 kDa spin column followed by desalting with RP-S cartridges, or desalted with RP-S cartridges without size filtration (see Supplemental Table 1 for details). Each resulting peptide sample was analyzed with an Orbitrap Elite mass spectrometer as described in the Methods. (A) Chromatograms of four LC-MS/MS runs varying the immunoprecipitation conditions or post-immunoprecipitation size filtration. The number of reported unique peptides from each sample was calculated from all three MS runs, although just one representative chromatogram (i.e., collected with the CID method including charge state +1) is shown per sample. Numeric labels above chromatographic peaks reflect the m/z values of the most abundant species associated with each given peak. PEG contamination peaks’ m/z values are shown in red. (B,C) Peptide length distributions comparing MHC I (B) and MHC II (C) prepared with or without size filtration.
To help understand the allelic differences we observed among MHC II binding predictions, we next examined the Raji cells’ MHC II allele expression levels by proteome analysis. We found that the protein abundance distribution of DQA1*05:01-DQB1*05:01,DQA1*01:01-DQB1*02:01,DRB1*10:01, and DRB1*03:01 mirrored the predicted binding affinity frequencies shown in Figure 2H (Supplemental Figure 3, Supplemental Table 3). However, this could not explain the shift in allele distributions we observed between the two flow rates. While we hypothesize that faster flow rates could enrich for peptides with greater binding affinities, more replicate experiments will be needed to investigate if the difference we observed is reproducible and significant.
In accordance with these data, we note that recent studies showed that different enrichment and desalting procedures alter allele-specific pMHC distributions measured by LC-MS.33,34 The authors concluded that if the sample amount is not limiting, multiple complementary procedures might be used for deeper immunopeptidome surveys. However, we propose that when sample amounts are limiting, a single strategy that yields the deepest coverage would be most appropriate. Considering these data, we did not believe the time savings of 40 minutes achieved with the faster 20ul/min flow rate justified the substantial sensitivity decrease we observed. Furthermore, without the ability to control the temperature of protein-bound AssayMAP cartridges during these wash steps, we did not believe testing slower flow rates was warranted.
Once MHC molecules are immunocaptured, their peptide cargo is released with 10% acetic acid. Towards identifying the minimum volume needed to efficiently disrupt the interaction of antibodies and MHC molecules we performed 4 sequential 50 μL elutions from immunoprecipitated MHC II molecules, and measured the peptides identified from each fraction (Table 2). We found that the first 50 μL elution recovered 98% of the total peptides identified in all the eluates and combining the first and the second elution (100 μL total) over 99% of the total peptides were recovered. Therefore, we chose 100 μL of 10% acetic acid with a flow rate of 5 μL/min as our final elution condition (Table 1).
Table 2.
Sequential elutions for MHC II immunoprecipitation
| Elution1 | PSMs2 | Unique peptides3 | |
|---|---|---|---|
| Rep1 | 1 | 13885 | 8359 |
| 2 | 2597 | 1641 | |
| 3 | 389 | 287 | |
| 4 | 263 | 183 | |
| Total | 17134 | 8572 | |
| Rep2 | 1 | 14078 | 8355 |
| 2 | 437 | 326 | |
| 3 | 118 | 105 | |
| 4 | 24 | 24 | |
| Total | 14657 | 8474 |
MHC II peptide immunoprecipitation and purification from 1×108 Raji cells was performed as described in Methods but with four sequential 50 μL elutions with 10% acetic acid (N=2 preparative replicates) (see Supplemental Table 1 for detailed parameters). Each elution was individually analyzed by LC-MS/MS.
Numbers of peptide-spectrum matches (PSMs) identified from each elution, or total across all four elutions.
Numbers of unique peptide sequences identified from each elution, or total across all four elutions.
pMHC purification from immunoprecipitation eluate
The 10% acetic acid used to elute peptides from captured MHC molecules does not discriminate between peptides, intact proteins or protein complexes. The latter two can be substantial sources of contamination in LC-MS/MS analyses, and include antibodies and MHC components. Commonly, acid-eluted peptides are enriched by size filtration with a 10 kDa membrane followed by C18 cleanup4–6. Alternatively, pMHC enrichment and desalting using C18 material alone 11,15,17,35 and HPLC separation34 have been reported.
To evaluate the utility size filtration could have with our approach, we passed 10% acetic acid MHC-I and MHC-II eluates through pre-conditioned 10 kDa spin columns, followed by desalting with the AssayMAP’s RP-S cartridge, or applied the 10% acetic acid eluate directly to RP-S cartridges in a single enrichment and desalting step. We observed that LC-MS chromatograms generated from size-filtered eluate was dominated by polyethylene glycol (PEG)-related contamination peaks (44 Da apart as shown in Figure 2A, m/z values highlighted in red). These contaminants were absent from analyses without size filtration. Furthermore, we found that excluding the size filtration step yielded nearly 50% more unique peptides without changing the general pMHC-I and pMHC-II length ranges (Figure 2B,C). We attribute this improvement to fewer processing steps which led to greater sample recovery, and to avoiding contaminating compounds leached from the size filters which can compete with peptides throughout the LC-MS process. Besides the desalting method with RP-S cartridges discussed in this manuscript, there are other separation and desalting strategies that we would include in future optimization efforts.
We were concerned that the immunoprecipitation eluate, largely composed of 10% acetic acid, could adversely affect peptide retention by or elution from the RP-S. To evaluate this in an economical fashion, we generated a peptide standard consisting of trypsin-digested bovine brain (Methods) and compared their LC-MS profiles with and without loading in 10% acetic acid (Supplemental Figure 4). We found that 10% acetic acid substantially reduced early-eluting hydrophilic peptides. However, this defect was effectively rescued by adding water to the peptide solution prior to their purification on RP-S cartridges, thereby diluting the acetic acid to 5% (Supplemental Figure 4). Likewise, adding 1% (v/v) of 2M sodium hydroxide raised the peptide solution’s pH to 3, and improved hydrophilic peptide recovery from the RP-S cartridges. The resulting chromatograms and numbers of identified peptides were similar to our standard peptide loading condition of 0.1% formic acid36 (Supplemental Figure 4). Since larger sample volumes require longer loading times, we proceeded with sodium hydroxide neutralization in our final protocol (Table 1) for greater time efficiency.
As with pMHC elution from protein A-bound antibodies (Table 2), we suspected that excess time-consuming elution volumes could be reduced without diminishing analysis sensitivity. To test this, we loaded MHC II peptides immunoprecipitated from 1×108 Raji cells on to RPS cartridges. We then carried out five sequential 20 μL elution steps with 50% acetonitrile/0.1% formic acid, and measured the peptides from each by LC-MS/MS (Table 3, Supplemental Figure 5). We found that 96–98% of all peptides identified across all elution steps were found in the first 20 μL elution. Over 99% of peptides were found from 40 μL of cumulative elution. Based on the LC-MS/MS runs corresponding with each elution step, we observed neither overwhelming amounts of intact proteins nor increased column pressure throughout all five runs (Supplemental Figure 5). This suggests the RPS cartridges can effectively replace membrane-based size filtration. Accordingly, we used 40 μL of 50% acetonitrile as the final elution parameter for recovering RP-S-bound peptides during the combined enrichment and desalting step (Table 1).
Table 3.
Sequential pMHC II elutions using RP-S cartridges
| Elution1 | PSMs | Unique peptides | |
|---|---|---|---|
| Rep1 | 1 | 14760 | 8768 |
| 2 | 1903 | 984 | |
| 3 | 81 | 65 | |
| 4 | 7 | 7 | |
| 5 | 8 | 2 | |
| Total | 16759 | 8968 | |
| Rep2 | 1 | 14401 | 8650 |
| 2 | 2713 | 1477 | |
| 3 | 517 | 279 | |
| 4 | 116 | 116 | |
| 5 | 29 | 21 | |
| Total | 17776 | 8902 |
Five sequential 20 μL elution steps were performed on RP-S cartridges using pMHC II immunoprecipitated from 1×108 Raji Cells (N=2) (see Supplemental Table 1 for details).
Serial immunoprecipitations with two antibodies can increase contaminants and decrease pMHC yield
Often, peptide ligands which bind multiple MHC alleles or MHC classes (e.g., MHC I and MHC II) are desired from a single specimen6. Immunoprecipitating MHCs with different antibodies can be performed in parallel -- which requires available lysate to be divided into two pools -- or in series -- which requires that the flow-through from one immunoprecipitation serve as the input to a second. The latter makes more efficient use of available lysate but takes more time and risks contamination of the second immunoprecipitation by the first. We compared these options using the optimized parameters described above, and evaluated evidence of cross-contamination from the corresponding LC-MS search results. As shown in Figure 3A, pMHC I identifications resulting from serial immunoprecipitation (the L243 MHC II antibody followed by the MHC I W6/32 antibody) yielded 2.5 times more 12–16-mer peptides relative to MHC I IP performed on the initial lysate (parallel). This suggests 17% of the putative pMHC I peptides from the second immunoprecipitation could be attributed to pMHC II contamination from the first. Similarly, pMHC II identifications resulting from the reverse serial immunoprecipitation (the W6/32 MHC I antibody followed by the L243 MHC II antibody) yielded close to 5.6 times more 9-mer peptides relative to the MHC II IP alone (Figure 3B), indicative of at least 5% of putative pMHC II were contaminated by the first pMHC I immunoprecipitation.
Figure 3. pMHC identified from and serial immunoprecipitation can harbor increased contaminants and have reduced sensitivity relative to parallel immunoprecipitations.

MHC I and MHC II were immunoprecipitated from lysate equivalent to 1×108 Raji cells in parallel (P), using W6/32 and L243 antibodies, respectively (see Supplemental Table 1 for detailed parameters). Resulting pMHC I and pMHC II were analyzed by LC-MS/MS as described above. Flowthroughs from each immunoprecipitation were collected, and added to protein A cartridges conjugated with the alternate antibody (L243 and W6/32, respectively). The resulting serial (S) immunoprecipitations were collected and processed as described above. (A) pMHC I length distributions resulting from parallel and serial immunoprecipitation (N=2 preparative replicates). (B) pMHC II length distributions resulting from parallel and serial immunoprecipitation (N=2 preparative replicates. Error bounds describe the maximum and minimum values observed across replicate experiments. (C)(D) MHC-I and MHC-II binding prediction of peptides identified in different experiments using NetMHCpan 4.1 (C) and NetMHCIIpan 4.0 (D), respectively. The allele with the best rank score for each peptide sequence is plotted. NetMHCpan 4.1 recommended that peptides with %EL rank lower than 2% are predicted to be MHC I binders and NetMHCIIpan 4.0 recommended that peptides with %EL rank lower than 10% are predicted to be MHC II binders. (E) Overlap between predicted MHC I binders identified from MHC I parallel immunoprecipitation and MHC II series immunoprecipitation. (F) Overlap between predicted MHC-II binders identified from MHC II parallel immunoprecipitation and MHC I series immunoprecipitation. (C-F) report data from one preparative replicate. Similar charts from the other replicate are shown in Supplemental Figure 6.
To further examine the extent of cross-contamination between serial immunoprecipitations, we compared predicted MHC binding affinities of the identified peptides across all four elution configurations. As shown in Figure 3C, 8% of the pMHC II from serial immunoprecipitation were consistent with binding affinity to known MHC I alleles, while only 1.5% of pMHC II from parallel immunoprecipitation showed binding affinity to these alleles. Furthermore, of the predicted MHC I binders resulting from serial MHC II immunoprecipitation, 86% were also identified from parallel MHC I immunoprecipitation (Figure 3E). Similarly, 15% of the pMHC from serial MHC I immunoprecipitation were predicted to bind the Raji cells’ MHC II alleles, while only 3% of pMHC from parallel MHC I immunoprecipitation were predicted to bind these alleles (Figure 3D). Of the predicted MHC II binders from serial MHC I immunoprecipitation, 89% were also identified from parallel MHC II immunoprecipitation (Figure 3F). Thus considering both predicted binding affinity and repeated peptide identification, serial immunoprecipitations bore substantially more peptides predicted to bind the MHC alleles expected from the first immunoprecipitation.
Considering the above evidence, we attribute the contamination we observed to our choice not to covalently crosslink antibodies to the protein A cartridge: antibodies which spontaneously dissociated from the cartridge and into the flow-through could be available to bind the protein A resin in the second immunoprecipitation stage. These data suggest in our experiment configuration, parallel immunoprecipitations may be necessary to achieve accurate MHC assignment across multiple immunopeptide assays. Alternatively, strategies such as antibody crosslinking or an additional antibody clearing step using protein A prior to the second immunoprecipitation could improve specificity. However, we also observed a lower yield for serial immunoprecipitation than the corresponding parallel immunoprecipitation. As shown in Figure 3A, the number of 9-mers identified from serial MHC I immunoprecipitation was 21% less than from parallel immunoprecipitation. Similarly, as shown in Figure 3B, the number of 13–25mers identified from serial MHC II immunoprecipitation was 14% less than from parallel immunoprecipitation. We attribute this decrease to sample loss occurring from more extensive sample handling. Thus, we believe additional antibody clearing steps will decrease pMHC yield from serial immunoprecipitations, and recommend the parallel immunoprecipitation approach if sample amounts allow.
Evaluation of input quantity versus immunopeptidome depth
Clinical samples are often precious and in limited amounts. The amount of sample that is needed for isolating enough MHC peptides for a sufficiently deep immunopeptide analysis can be more than three orders of magnitude greater than the amount that is needed for whole proteome analysis. Therefore, assay sensitivity is a key factor to evaluate the success of MHC peptide preparation. By concentrating analytes on 5 μL bed volume cartridges with almost no dead volume, the AssayMAP system should increase binding efficiency and minimize sample loss relative to batch-format procedures using beads in test tubes or plates.
Using the number of unique pMHC identified as a sensitivity metric, we evaluated how a range of input cell numbers (Figure 4) and lysate concentrations (Supplemental Figure 7) impacted our assay. As expected, the number of identified pMHC I monotonically increased with the amount of input, ranging from over 1,300 pMHC I when 2.5×106 cells were used to over 6,000 pMHC I from 1×108 cells (Figure 4A). We found an overall high degree of concordance between pMHC I identified from low (2.5×106) versus high (1×108) cell numbers, supporting the validity of the low cell number experiment. As shown in Figure 4C, over 80% of the 1,310 pMHC I identified from 2.5×106 cells were also identified from 1×108 cells; furthermore, most of these were present in high abundance in both samples (Figure 4E). We found substantial agreement in MHC I binding affinity between pMHC I identified from 2.5×106 and from 1×108 cells (Figure 4G). Of the pMHC I identified from both 2.5×106 and 1×108 cell experiments, 71% were predicted to bind MHC I (Figure 4G). However, greater predicted binding affinities were found from the larger cell number experiment (91% vs 81%) (Figure 4I).
Figure 4. Evaluation of input quantity versus sensitivity.

Triplicate Raji cell cultures were harvested in amounts of 5×105, 2.5×106, 5×106, 2.5×107, and 1×108 cells. Each cell pellet was lysed and subjected to MHC I and MHC II immunoprecipitation (see Supplemental Table 1 for detailed parameters) and LC-MS/MS analysis as described in Methods. (A) Numbers of unique pMHC-I and pMHC-II identified from different cell numbers using the present AssayMAP protocol (Table 1). Each number is the averaged value from three biological replicates. Error bars denote standard deviation of the triplicate experiments. (B) Numbers of unique pMHC-I and pMHC-II identified from our prior manual protocol (see Methods; N=1). (C) Overlap between MHC I peptides identified from 1×108 cells and 2.5×106 cells. (D) Overlap between MHC II peptides identified from 1×108 cells and 5×105 cells. (E) pMHC I abundance rank distributions noting which pMHC I identified from 1×108 cells (blue) were also identified from 2.5×106 cells (tan). (F) pMHC II abundance rank distributions noting which pMHC II identified from 1×108 cells (purple) were also identified from 5×105 cells (green). (G) pMHC I predicted binding affinity distributions noting which pMHC I identified from 1×108 cells (blue) were also identified from 2.5×106 cells (tan). (H) pMHC II predicted binding affinity distributions noting which pMHC II identified from 1×108 cells (purple) were also identified from 5×105 cells (green). (I) MHC I allele binding prediction for peptides identified with different cell numbers. (J) MHC II allele binding prediction for peptides identified with different cell numbers. The allele with the best rank score for each peptide sequence is plotted. Binding affinity predictions are as described in Figure 1F–G.
It is noteworthy that 50% to almost 300% more peptides were identified from MHC II than MHC I immunoprecipitations, which also increased monotonically with increasing input amounts (Figure 4B, Supplemental Figure 7B). We detected an average of 700 and 1,141 pMHC II peptides from just 5 × 105 cells and lysate equivalents, in comparison to 60 and 294 for MHC I. Similar to pMHC I, 57% of pMHC II were in common between low (5×105) and high (1×108) cell numbers and over 90% of these peptides were predicted to bind MHC II (Figure 4D,F,H). We also found the predicted allelic distributions were very similar for peptides identified from either 5×105 or 1×108 cells (Figure 4J). However, as with MHC I, the percentage of predicted MHC II binders identified from 1×108 cells was higher than from 1×105 cells (84% vs 62%) (Figure 4J).
Based on these data, we conclude that starting with as few as 2.5×106 cells for MHC I and 5×105 cells for MHC II, we can confidently identify many of the abundant pMHC that can be measured from larger cell amounts. For example, 66% of the top 20% of pMHC I identified from 1×108 cells were identified from 2.5×106 cells, and 26% of the top 20% of pMHC II identified from 1×108 cells were also identified from 5×105 cells (Supplemental Table 11,12).
It has been previously reported that pMHC I and II repertoires are enriched in gene functional categories such as protein translation and intramembrane components.37,38 We found that for both MHC I and MHC II, the low cell number experiments largely recapitulated the gene categories that were prevalent in the large cell number experiments (Supplemental Figure 8). Thus, relatively small cell numbers could yield accurate, though more limited functional assessments of cells based on immunopeptide repertoires.
We found that MHC immunoprecipitations from cells diluted over two orders of magnitude yielded similar numbers of pMHC identifications as those made from lysate diluted to a similar extent. We had anticipated that pMHC yields would be substantially lower when handling small cell numbers, but this difference seemed to be modest (slope for cell versus lysis dilutions = 1.09 and 0.92 for MHC I and II, respectively, with R2 values of 0.8661 and 0.9909. This indicates that the pMHC isolation procedure is both sensitive and reproducible across a wide range of cell concentrations (Supplemental Figure 9).
The larger number of pMHC II identified here, coupled with the plateau of these identifications made from lysate equivalent to 5 × 107 or more cells (Supplemental Figure 7B) could suggest Raji cells express more MHC II than MHC I molecules. In support of this, proteomic analysis of Raji cells showed that MHC II was roughly two to three times as abundant as MHC I (label free quantification, N=2, see Supplemental Table 3). We believe, therefore, that the antibody-bound protein A cartridges became saturated with MHC II molecules at lower amounts of diluted lysate than with MHC I molecules. While different antibody binding efficiencies and peptide repertoire diversity could also contribute to this difference, we expect that different cells and tissues not tested here could vary considerably in the numbers of pMHC I and II recovered from them. Thus, the values reported here from Raji cells should best be considered in the context of this experiment and not benchmarks against which other cell or tissue preparations should be compared.
Data collected with our previous manual preparation protocol yielded 4,200 and 8,400 unique pMHC I and pMHC II from 3×108 Raji cells (Figure 4B, see Methods for sample preparation details). The automated pipeline described here identified similar numbers of peptides with 1/10th of the number of input cells. This is great improvement in terms of assay sensitivity. We note that since the AssayMAP Bravo can dispense a maximum of 1mL of input lysate onto each cartridge under the configuration we used here, we did not attempt to load more lysate than shown in Figure 4 and Supplemental Figure 7. Additional volume could be applied manually in a sequential fashion to load, for example, 2×108 cells’ worth of lysate at our working concentration (1 ×108 cells per mL). However, since we found that lysate equivalent to 1×108 and 2.5×107 Raji cells was sufficient to nearly saturate one W6/32 antibody and one L243 antibody bound protein A cartridge, respectively (Supplemental Figure 7), we recommend dividing greater volumes across multiple cartridges to achieve higher loading capacity. Alternatively, one could use larger capacity cartridges – we understand that AssayMAP cartridges with five-times greater capacity were recently made commercially available.
Reproducibility evaluation
By dramatically reducing the need for human intervention, by employing precise flow rate control, and by using very small volumes, we expect that the automated liquid handling capability described here should significantly reduce operator error, thereby improving reproducibility of our immunopeptide assay. We measured the reproducibility of this protocol by processing two aliquots (1×108 cells equivalent each) of the same lysate pool for MHC II (L243 immunoprecipitation (preparative replicates). We compared these data to a third MHC II immunoprecipitation (1×108 cells equivalent lysate) which was analyzed with two LC-MS injections on the same day (technical replicates). We found that 62% of the peptides were identified in both preparative replicates (Figure 5A), whereas 70% were identified in both technical replicates (Figure 5B), similar to previously reported replicate overlap by Nicastri et al.34 For both evaluations, peak area quantification were nearly identical (Pearson correlation coefficient = 0.889 vs. 0.884, respectively (Figure 5C,D)), similar to previously reported reproducibility by Chong et al17.
Figure 5. Reproducibility of preparative replicates and LC-MS/MS technical replicates.

(A) Overlap of identified peptides between preparative replicates (two MHC II immunopreciptations from a single lysate aliquot; prep-1 and prep-2). (B) Overlap of identified peptides between technical replicates (two back-to-back LCMS analyses of a single pMHC II immunoprecipitation; tech-1 and tech-2). (C) Correlation of peak area quantification between preparative replicates. (D) Correlation of peak area quantification between technical replicates.
We then evaluated the reproducibility of the triplicate cell dilution experiments described in Figure 4. As shown in Figure 6, the number of pMHC I identified in all three replicates was around 60% of that identified in any single experiment, for input amounts of 2.5×107 cells or greater. Similarly, the number of pMHC II identified in all three replicates was around 60%−70% of that identified in one replicate experiment. The percentages of peptides identified in all three replicates from 5×105 cells were substantially lower than this (25% for MHC I and 36% for MHC II). We found that for cell numbers 2.5×106 or greater, 10%−20% of pMHC I and pMHCII in any replicate experiment were never identified in another replicate. Not surprisingly, this percentage was higher in replicate analyses of 5×105 cells (26% for MHC I and 35% for MHC II). From these data, we concluded that as few as 2.5×106 cells can yield similar reproducibility as much higher cell numbers. Furthermore, the degree of reproducibility we observed from biological replicate analyses of different cell pellets was consistent with the preparative and technical reproducibility we described in Figure 5. Thus, our sample handling steps prior to immunoprecipitation were robust and reproducible.
Figure 6. Reproducibility of biological replicates.

Peptides identified in only one of three replicate experiments, two of three replicate experiments, and all three of replicate experiments were counted per set of MHC IP (MHC I or II) and number of input cells. These counts were expressed as the percentage of unique peptides found within each individual immunoprecipitation experiment. Each percentage shown here is the averaged value from three biological replicates. Error bars denote standard deviation of the triplicated experiments.
Mass spectrometry analysis method
Since the abundance profiles of peptides eluted from MHC molecules are expected to be quite unlike those of conventional tryptic whole cell lysate digests, we suspected that several interrelated mass spectrometer data acquisition parameters would need to be optimized for this distinct peptide class. Accordingly, we evaluated the automatic gain control (AGC) target and maximum ion injection time settings – both of which regulate the number of charged particles within the orbitrap that can contribute to a mass spectrum. We first loaded 0.2 μg of a standard peptide mixture resulting from nonspecific proteolysis (see Methods) to simulate low-abundant pMHC samples. Fixing the AGC at 50k and varying the maximum injection time parameter, we recorded the number of confident identifications made, the distribution of ion injection times measured over the span of each LC-MS run (Figure 7A). We found a surprisingly large degree of variation across both dimensions depended on the maximum ion injection time setting. The default “Automatic” mode in the instrumentation software resulted in nearly all spectra being acquired at a rate of 22 ms per spectrum (Figure 7B). While this translated to the greatest number of MS/MS spectra being acquired, this setting yielded the least number of confident peptide-spectrum matches (PSMs) and unique peptide identifications (Figure 7A). Similarly, the “dynamic” setting caused the 22 ms injection time threshold to be applied to more than 50% of the PSMs, with the remaining spectra allowed to be acquired for up to 300 ms (Figure 7C). This additional time per MS/MS spectrum decreased the total number of acquired MS/MS spectra by 5%, but resulted in 24% and 18% more confident PSMs and unique peptides, respectively. This indicated that longer injection times tended to yield higher-quality MS/MS spectra from which more confident identifications could be made. To further test this, we extended the maximum injection times to 50 ms, 100 ms, and 200 ms. Remarkably, over 70%, 50%, 33% of confidently assigned PSMs reached the maximum injection time ceiling, (Figure 7D–F). Even at a long 500 ms maximum injection time setting, 15% of the PSMs reached this ceiling (Figure 7G). These data showed that most maximum injection time limits were insufficient to meet the desired 50K AGC target for these kinds of very low abundance peptide mixtures. However, we note that 100 ms appeared to be an optimal parameter at this AGC setting, based the number of confident identifications we could make (Figure 7A).
Figure 7. Instrument method optimization with different maximum injection time modes and automatic gain control.

(A) Number of unique standard nonspecific peptide, PSMs and MS/MS identified from different maximum injection time modes. *MIT: maximum injection time. Error bounds were determined from two replicate runs (maximum and minimum values). Color intensity relates to the magnitude of each value, as shown in the scale bar. (B-G) Injection time distributions for confidently assigned PSMs under different maximum injection modes. Note that the injection time bin widths varied along with the range of injection times investigated for each experiment. (H) Number of unique pMHC, PSMs and MS/MS identified from different AGC values. pMHC I or pMHC II were pooled from ten parallel immunoprecipitations (1×108 cells per immunoprecipitation). 1/30th of the peptide material was analyzed with LC-MS/MS per run, equivalent to the amount analyzed per LC-MS/MS run from a single immunoprecipitation with 1×108 Raji cells (see Supplemental Table 1 for detailed parameters). Two replicate runs were performed for each method and error bounds were calculated from the replicate runs (maximum and minimum values).
Having selected 100 ms as an optimal maximum injection time, we next considered whether decreasing the AGC threshold could be more appropriately matched to this setting, yielding more spectra and increased identification rates. To optimize this parameter for pMHC more closely, we performed further tests using pools of pMHC I and II purified from several parallel immunoprecipitations (see Supplemental Table 1 for detailed parameters). We found that while the number of MS/MS scans collected increased with decreasing AGC values as expected, the number of confident identifications did not improve (Figure 7H). Therefore, we selected an AGC value of 50K and maximum injection time of 100 ms as the optimal method for MHC peptide identification. However, we note that if fewer cells are used for a single immunoprecipitation experiment, increasing dynamic repeat count or MS/MS microscan count parameters may improve the overall peptide identification results – data acquisition parameters that were not explicitly tested here. Besides the parameters optimized in this manuscript, there are more optimizations to be done regarding instrument method. We would like to further optimize the instrument method in the future.
Conclusion
Here, we describe steps towards optimizing a robust, high-throughput and automated pipeline for pMHC purification. As shown in Figure 8A, our protocol can process up to 96 cell lysate samples to mass-spectrometry-ready peptides within 5.5 hours. Importantly, this process only required 1 hour of manual intervention. This is a nearly 40-fold improvement in throughput over our previous manual preparation protocol which took two full days to process only up to 10 samples (Figure 8B). Data collected with our previous manual preparation protocol yielded just 4,000 unique pMHC I and 8,000 unique pMHC II from 3×108 Raji cells, while the automated pipeline allowed the identification of similar numbers of unique pMHCs from 10% of the cells (Figure 4B). In summary, we expect that the use of AssayMAP Bravo significantly reduces operational error, enhances reproducibility and sensitivity. The tailored MS instrument method also facilitates peptide sequencing and boosts the numbers of peptides identified. More importantly, we hope the throughput gains offered by this platform will facilitate a new era in clinical-scale immunopeptidome profiling greatly speeding immunotherapy and vaccination development. In addition, this platform could be applied to capturing any ligands bound to a protein that can be affinity-purified, or immunoprecipitation off proteins in general.
Figure 8. Overview of the pipeline and time distribution.

(A) Automated MHC peptide preparation pipeline and time spent for each step. The steps with green background need human intervention. (B) Comparison of the experimental times needed for different protocols.
Supplementary Material
Supplemental Figure 1. pMHC capture and purification with AssayMAP Bravo in four steps (PDF)
Supplemental Figure 2. Representative protein A cartridge binding capacity estimations for W6/32 and L243 antibodies.
Supplemental Figure 3. Protein abundance distribution for different Raji HLA alleles
Supplemental Figure 4. Increasing pH improves peptide recovery from RP-S resin
Supplemental Figure 5. Sequential pMHC II elutions using RP-S cartridges (PDF)
Supplemental Figure 6. Peptides identified from parallel and serial immunoprecipitation (preparative replicate 2)
Supplemental Figure 7. Evaluation of input quantity versus sensitivity using different amounts of cell lysates
Supplemental Figure 8. Gene biological function analysis of pMHC I and pMHC II identified from high and low cell numbers.
Supplemental Figure 9. Dilluted cells or diluted cell lysate yield similar pMHC recovery rates.
Supplemental Table 4-16. Lists of peptide-spectrum match (PSM) for all the MS runs (xlxs)
Supplemental Table 1. AssayMAP Bravo parameters and sample information (xlxs)
Supplemental Table 2. Decision tree of mass ranges used for precursor selection (PDF)
Supplemental Table 10. Lists of PSM for sensitivity evaluation experiment using different amounts of cell lysates
Supplemental Table 9. Lists of PSM for parallel versus in series immunoprecipitation experiment
Supplemental Table 8. Lists of PSM for RP-S cartridge elution experiment
Supplemental Table 7. Lists of PSM for RP-S cartridge sample loading experiment
Supplemental Table 6. Lists of PSM for size filtration experiment
Supplemental Table 5. Lists of PSM for immunoprecipitation elution experiment
Supplemental Table 4. Lists of PSM for immunoprecipitation flow rate experiment
Supplemental Table 3. Quantification of MHC I and MHC II from Raji whole proteome analysis (xlxs)
Supplemental Table 11. Lists of PSM for sensitivity evaluation experiment using different cell numbers (MHC I)
Supplemental Table 12. Lists of PSM for sensitivity evaluation experiment using different cell numbers (MHC II)
Supplemental Table 13. Lists of PSM for manual immunoprecipitation
Supplemental Table 14. Lists of PSM for reproducibility evaluation experiment
Supplemental Table 15. Lists of PSM for MS method optimization experiment (standard nonspecific peptides)
Supplemental Table 16. Lists of PSM for MS method optimization experiment (MHC peptides)
Acknowledgement
We thank colleagues Niclas Olsson and Kavya Swaminathan for discussions, Sarah Lin for help with data deposition, and Tom Chen previously from Agilent Technologies for initial help with AssayMAP Bravo method design. The table of contents graphic was partially created with BioRender.com with permission. We acknowledge the following sources of funding: Chan Zuckerberg Biohub (L.Z. & J.E.E.); NIH/R01-DE027750-01 (P.L.M.); the Molecular and Pharmacology Training Program (T32GM113854) from the Department of Chemical and Systems Biology, Stanford University (M.L.H.).
Footnotes
The authors declare no competing financial interest
All mass spectrometry proteomics data have been deposited to the PRIDE Archive (http://www.ebi.ac.uk/pride/archive/) via the PRIDE partner repository with the data set identifier PXD022629.
Username: reviewer_pxd022629@ebi.ac.uk
Password: UorQs5f0
References
- (1).Abelin JG; Keskin DB; Sarkizova S; Hartigan CR; Zhang W; Sidney J; Stevens J; Lane W; Zhang GL; Eisenhaure TM; Clauser KR; Hacohen N; Rooney MS; Carr SA; Wu CJ Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-Allelic Cells Enables More Accurate Epitope Prediction. Immunity 2017, 46, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Elias JE Lymphoma Neoantigens. HemaSphere 2018, 2, 87–89. [Google Scholar]
- (3).von Boehmer H; Karjalainen K; Pelkonen J; Borgulya P; Rammensee HG The T-Cell Receptor for Antigen in T-Cell Development and Repertoire Selection. Immunol. Rev. 1988, 101, 21–37. [DOI] [PubMed] [Google Scholar]
- (4).Hunt DF; Henderson RA; Shabanowitz J; Sakaguchi K; Michel H; Sevilir N; Cox AL; Appella E; Engelhard VH Characterization of Peptides Bound to the Class I MHC Molecule HLA-A2.1 by Mass Spectrometry. Science 1992, 255, 1261–1263 [DOI] [PubMed] [Google Scholar]
- (5).Hunt DF; Michel H; Dickinson TA; Shabanowitz J; Cox AL; Sakaguchi K; Appella E; Grey HM; Sette A. Peptides Presented to the Immune System by the Murine Class II Major Histocompatibility Complex Molecule I-Ad. Science 1992, 256, 1817–1820. [DOI] [PubMed] [Google Scholar]
- (6).Khodadoust MS; Olsson N; Wagar LE; Haabeth OAW; Chen B; Swaminathan K; Rawson K; Liu CL; Steiner D; Lund P; Rao S; Zhang L; Marceau C; Stehr H; Newman AM; Czerwinski DK; Carlton VEH; Moorhead M; Faham M; Kohrt HE; Carette J; Green MR; Davis MM; Levy R; Elias JE; Alizadeh AA Antigen Presentation Profiling Reveals Recognition of Lymphoma Immunoglobulin Neoantigens. Nature 2017, 543, 723–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Olsson N; Schultz LM; Zhang L; Khodadoust MS; Narayan R; Czerwinski DK; Levy R; Elias JE T-Cell Immunopeptidomes Reveal Cell Subtype Surface Markers Derived From Intracellular Proteins. Proteomics 2018, 18, 1700410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Khodadoust MS; Olsson N; Chen B; Sworder B; Shree T; Liu CL; Zhang L; Czerwinski DK; Davis MM; Levy R; Elias JE; Alizadeh AA B-Cell Lymphomas Present Immunoglobulin Neoantigens. Blood 2019, 133, 878–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chen B; Khodadoust MS; Olsson N; Wagar LE; Fast E; Liu CL; Muftuoglu Y; Sworder BJ; Diehn M; Levy R; Davis MM; Elias JE; Altman RB; Alizadeh AA Predicting HLA Class II Antigen Presentation through Integrated Deep Learning. Nat. Biotechnol. 2019, 37, 1332–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kathuria KR; Chen B; Khodadoust MS; Olsson N; Davis MM; Elias JE; Levy R; Altman RB; Alizadeh AA Maria-I: A Deep-Learning Approach for Accurate Prediction of MHC Class I Tumor Neoantigen Presentation. Blood 2019, 134, 84. [Google Scholar]
- (11).Bassani-Sternberg M; Bräunlein E; Klar R; Engleitner T; Sinitcyn P; Audehm S; Straub M; Weber J; Slotta-Huspenina J; Specht K; Martignoni ME; Werner A; Hein R; H. Busch D; Peschel C; Rad R; Cox J; Mann M; Krackhardt AM Direct Identification of Clinically Relevant Neoepitopes Presented on Native Human Melanoma Tissue by Mass Spectrometry. Nat. Commun. 2016, 7 (1), 13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Stopfer LE; Mesfin JM; Joughin BA; Lauffenburger DA; White FM Multiplexed Relative and Absolute Quantitative Immunopeptidomics Reveals MHC I Repertoire Alterations Induced by CDK4/6 Inhibition. Nat. Commun. 2020, 11 (1), 2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kowalewski DJ, Biochemical SS Large-Scale Identification of MHC Class I Ligands. In Antigen Processing. Methods in Molecular Biology; van Endert P, Ed.; Humana Press: Totowa, NJ, 2013; pp 1689–1699. [DOI] [PubMed] [Google Scholar]
- (14).Penny SA; Malaker SA Isolation of Major Histocompatibility Complex (MHC)-Associated Peptides by Immunoaffinity Purification. In Immunoproteomics. Methods in Molecular Biology; Fulton K, T. S., Ed.; Humana: New York, NY, 2019; pp 235–243. [DOI] [PubMed] [Google Scholar]
- (15).Purcell AW; Ramarathinam SH; Ternette N. Mass Spectrometry–Based Identification of MHC-Bound Peptides for Immunopeptidomics. Nat. Protoc. 2019, 14, 1687–1707. [DOI] [PubMed] [Google Scholar]
- (16).Nelde A, Kowalewski DJ, Purification SS and Identification of Naturally Presented MHC Class I and II Ligands. In Antigen Processing. Methods in Molecular Biology; van E. P, Ed.; Humana: New York, NY, 2019; pp 123–136. [DOI] [PubMed] [Google Scholar]
- (17).Chong C; Marino F; Pak H; Racle J; Daniel RT; Müller M; Gfeller D; Coukos G; Bassani-Sternberg M. High-Throughput and Sensitive Immunopeptidomics Platform Reveals Profound Interferonγ-Mediated Remodeling of the Human Leukocyte Antigen (HLA) Ligandome. Mol. Cell. Proteomics 2018, 17, 533–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Marino F, Chong C, Michaux J, High-Throughput B-SM, Fast, and Sensitive Immunopeptidomics Sample Processing for Mass Spectrometry. In Immune Checkpoint Blockade. Methods in Molecular Biology; Pico de Coaña Y, Ed.; Humana Press: New York, NY, 2019; pp 67–79. [DOI] [PubMed] [Google Scholar]
- (19).Boegel S; Löwer M; Bukur T; Sahin U; Castle JC A Catalog of HLA Type, HLA Expression, and Neo-Epitope Candidates in Human Cancer Cell Lines. Oncoimmunology 2014, 3 (8), e954893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Devabhaktuni A; Elias JE Application of de Novo Sequencing to Large-Scale Complex Proteomics Data Sets. J. Proteome Res. 2016, 15, 732–742. [DOI] [PubMed] [Google Scholar]
- (21).Barnstable CJ; Bodmer WF; Brown G; Galfre G; Milstein C; Williams AF; Ziegler A. Production of Monoclonal Antibodies to Group A Erythrocytes, HLA and Other Human Cell Surface Antigens-New Tools for Genetic Analysis. Cell 1978, 14, 9–20. [DOI] [PubMed] [Google Scholar]
- (22).Lampson LA; Levy R. Two Populations of Ia-like Molecules on a Human B Cell Line. J. Immunol. 1980, 125, 293–299. [PubMed] [Google Scholar]
- (23).Agilent AssayMAP Bravo Cartridges for Automated Protein Sample Preparation Workflows. https://www.agilent.com/cs/library/selectionguide/public/5991-4863EN.pdf
- (24).Rappsilber J; Ishihama Y; Mann M. Stop and Go Extraction Tips for Matrix-Assisted Laser Desorption/Ionization, Nanoelectrospray, and LC/MS Sample Pretreatment in Proteomics. Anal. Chem. 2003, 75, 663–670. [DOI] [PubMed] [Google Scholar]
- (25).Adusumilli R, Data MP Conversion with ProteoWizard MsConvert. In Proteomics. Methods in Molecular Biology; Comai L, Katz J, M. P., Ed.; Humana Press: New York, NY, 2017; pp 339–368. [DOI] [PubMed] [Google Scholar]
- (26).Elias JE; Gygi SP Target-Decoy Search Strategy for Increased Confidence in Large-Scale Protein Identifications by Mass Spectrometry. Nat. Methods 2007, 4, 207–214. [DOI] [PubMed] [Google Scholar]
- (27).Huttlin EL; Jedrychowski MP; Elias JE; Goswami T; Rad R; Beausoleil SA; Villén J; Haas W; Sowa ME; Gygi SP A Tissue-Specific Atlas of Mouse Protein Phosphorylation and Expression. Cell 2010, 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Eng JK; Mccormack AL; Yates JR An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [DOI] [PubMed] [Google Scholar]
- (29).Álvaro-Benito M; Morrison E; Abualrous ET; Kuropka B; Freund C. Quantification of HLA-DM-Dependent Major Histocompatibility Complex of Class II Immunopeptidomes by the Peptide Landscape Antigenic Epitope Alignment Utility. Frontiers in Immunology. 2018, 9, 872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Racle J; Michaux J; Rockinger GA; Arnaud M; Bobisse S; Chong C; Guillaume P; Coukos G; Harari A; Jandus C; Bassani-Sternberg M; Gfeller D. Robust Prediction of HLA Class II Epitopes by Deep Motif Deconvolution of Immunopeptidomes. Nat. Biotechnol. 2019, 37, 1283–1286. [DOI] [PubMed] [Google Scholar]
- (31).AssayMAP Antibody Purification Protocol Guide. https://www.agilent.com/cs/library/usermanuals/Public/G5496-90003_R00_AM-AbPurificationProtocol_S_EN.pdf
- (32).Reynisson B; Alvarez B; Paul S; Peters B; Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved Predictions of MHC Antigen Presentation by Concurrent Motif Deconvolution and Integration of MS MHC Eluted Ligand Data. Nucleic Acids Res. 2020, 48 (W1), W449–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Pandey K; Mifsud NA; Lim Kam Sian TCC; Ayala R; Ternette N; Ramarathinam SH; Purcell AW In-Depth Mining of the Immunopeptidome of an Acute Myeloid Leukemia Cell Line Using Complementary Ligand Enrichment and Data Acquisition Strategies. Mol. Immunol. 2020, 123, 7–17. [DOI] [PubMed] [Google Scholar]
- (34).Nicastri A; Liao H; Muller J; Purcell AW; Ternette N. The Choice of HLA-Associated Peptide Enrichment and Purification Strategy Affects Peptide Yields and Creates a Bias in Detected Sequence Repertoire. Proteomics 2020, 20 (12), 1900401. [DOI] [PubMed] [Google Scholar]
- (35).Lanoix J; Durette C; Courcelles M; Cossette É; Comtois-Marotte S; Hardy M-P; Côté C; Perreault C; Thibault P. Comparison of the MHC I Immunopeptidome Repertoire of B-Cell Lymphoblasts Using Two Isolation Methods. Proteomics 2018, 18, 1700251. [DOI] [PubMed] [Google Scholar]
- (36).Zhang Lichao;Elias JE Relative Protein Quantification Using Tandem Mass Tag Mass Spectrometry. In Proteomics: Methods and Protocols; Comai L, Katz J, Mallick P, Ed.; Humana Press: New York, NY, 2017; pp 185–198. [DOI] [PubMed] [Google Scholar]
- (37).Granados DP; Yahyaoui W; Laumont CM; Daouda T; Muratore-Schroeder TL; Côté C; Laverdure JP; Lemieux S; Thibault P; Perreault C. MHC I–Associated Peptides Preferentially Derive from Transcripts Bearing MiRNA Response Elements. Blood 2012, 119 (26), e181–e191. [DOI] [PubMed] [Google Scholar]
- (38).Clement CC; Becerra A; Yin L; Zolla V; Huang L; Merlin S; Follenzi A; Shaffer SA; Stern LJ; Santambrogio L. The Dendritic Cell Major Histocompatibility Complex II (MHC II) Peptidome Derives from a Variety of Processing Pathways and Includes Peptides with a Broad Spectrum of HLA-DM Sensitivity. J. Biol. Chem. 2016, 291 (11), 5576–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. pMHC capture and purification with AssayMAP Bravo in four steps (PDF)
Supplemental Figure 2. Representative protein A cartridge binding capacity estimations for W6/32 and L243 antibodies.
Supplemental Figure 3. Protein abundance distribution for different Raji HLA alleles
Supplemental Figure 4. Increasing pH improves peptide recovery from RP-S resin
Supplemental Figure 5. Sequential pMHC II elutions using RP-S cartridges (PDF)
Supplemental Figure 6. Peptides identified from parallel and serial immunoprecipitation (preparative replicate 2)
Supplemental Figure 7. Evaluation of input quantity versus sensitivity using different amounts of cell lysates
Supplemental Figure 8. Gene biological function analysis of pMHC I and pMHC II identified from high and low cell numbers.
Supplemental Figure 9. Dilluted cells or diluted cell lysate yield similar pMHC recovery rates.
Supplemental Table 4-16. Lists of peptide-spectrum match (PSM) for all the MS runs (xlxs)
Supplemental Table 1. AssayMAP Bravo parameters and sample information (xlxs)
Supplemental Table 2. Decision tree of mass ranges used for precursor selection (PDF)
Supplemental Table 10. Lists of PSM for sensitivity evaluation experiment using different amounts of cell lysates
Supplemental Table 9. Lists of PSM for parallel versus in series immunoprecipitation experiment
Supplemental Table 8. Lists of PSM for RP-S cartridge elution experiment
Supplemental Table 7. Lists of PSM for RP-S cartridge sample loading experiment
Supplemental Table 6. Lists of PSM for size filtration experiment
Supplemental Table 5. Lists of PSM for immunoprecipitation elution experiment
Supplemental Table 4. Lists of PSM for immunoprecipitation flow rate experiment
Supplemental Table 3. Quantification of MHC I and MHC II from Raji whole proteome analysis (xlxs)
Supplemental Table 11. Lists of PSM for sensitivity evaluation experiment using different cell numbers (MHC I)
Supplemental Table 12. Lists of PSM for sensitivity evaluation experiment using different cell numbers (MHC II)
Supplemental Table 13. Lists of PSM for manual immunoprecipitation
Supplemental Table 14. Lists of PSM for reproducibility evaluation experiment
Supplemental Table 15. Lists of PSM for MS method optimization experiment (standard nonspecific peptides)
Supplemental Table 16. Lists of PSM for MS method optimization experiment (MHC peptides)