

Abstract
Synaptic potentiation underlies various forms of behavior and depends on modulation by multiple activity-dependent transcription factors to coordinate the expression of genes necessary for sustaining synaptic transmission. Our current study identified the tumor suppressor p53 as a novel transcription factor involved in this process. We first revealed that p53 can be elevated upon chemically induced long-term potentiation (cLTP) in cultured primary neurons. By knocking down p53 in neurons, we further showed that p53 is required for cLTP-induced elevation of surface GluA1 and GluA2 subunits of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR). Because LTP is one of the principal plasticity mechanisms underlying behaviors, we employed forebrain-specific knockdown of p53 to evaluate the role of p53 in behavior. Our results showed that, while knocking down p53 in mice does not alter locomotion or anxiety-like behavior, it significantly promotes repetitive behavior and reduces sociability in mice of both sexes. In addition, knocking down p53 also impairs hippocampal LTP and hippocampus-dependent learning and memory. Most importantly, these learning-associated defects are more pronounced in male mice than in female mice, suggesting a sex-specific role of p53 in these behaviors. Using RNA sequencing (RNAseq) to identify p53-associated genes in the hippocampus, we showed that knocking down p53 up- or down-regulates multiple genes with known functions in synaptic plasticity and neurodevelopment. Altogether, our study suggests p53 as an activity-dependent transcription factor that mediates the surface expression of AMPAR, permits hippocampal synaptic plasticity, represses autism-like behavior, and promotes hippocampus-dependent learning and memory.
INTRODUCTION
In response to sensory stimuli, activity-dependent gene transcription is required for the consolidation of long-lasting changes in circuit function. The transcription can be mediated by multiple so-called activity-dependent transcription factors in neurons, such as cAMP response element-binding protein (CREB) (1), myocyte enhancer factor 2 (MEF2) (2), and serum response factor (SRF) (3). Upon activity stimulation, changes in the expression of their target genes shape the number, strength, and connectivity of old and also new synapses, ultimately leading to changes in behavior. This is particularly critical in the hippocampus, where communication between hippocampal sub-regions determines the formation and consolidation of learning and memory. Disruption of activity-dependent transcription has been frequently observed in neurodevelopmental and psychiatric disorders including autism spectrum disorders (4). Discovering novel and as yet uncharacterized regulation underlying activity-dependent transcription may help us piece together a better understanding of the molecular defects behind those diseases.
To approach this question, we study the tumor suppressor p53, a widely known transcription factor that regulates the expression of cellular stress response genes and that exerts anti-proliferative effects in dividing cells (5). In terminally differentiated cells, p53 also mediates the expression of genes responsible for DNA damage repair and apoptosis (6, 7). Previous studies have confirmed the expression of p53 in a variety of neuronal cells in the brain, particularly during early development (8, 9). Also, our earlier work suggested that p53 is involved in group 1 metabotropic glutamate receptor (Gp1-mGluR)-dependent homeostatic refinement of neural network activity (10, 11) and synaptic scaling (12) in fragile X syndrome mice. However, although the dysregulation of p53 has been observed in, and linked to, neurodevelopmental disorders, most studies of p53 in other pathological conditions still centers on p53’s fundamental functions in cellular growth control and cell death pathways (6, 7, 13, 14). It remains largely unclear whether p53 can exert other physiological functions in the nervous system, particularly toward behavior. Given the highly regulatory nature of the expression of p53, it is possible that certain types of neuronal activity may alter the expression of p53 and its associated genes in ways that subsequently contribute to behavior.
To test this possibility, we obtained data to show that the expression of p53 is elevated following a chemically induced long-term potentiation (cLTP) and this elevation is required for cLTP-induced elevation of the surface AMPAR and for the strengthening of excitatory synapses. Along with this function in synaptic plasticity, we also showed that p53 is crucial for repressing autism-like behavior and promoting hippocampus-dependent learning and memory, particularly in male mice. Using RNA sequencing to search for p53-associated genes in the hippocampus, we revealed multiple genes with known roles in neurodevelopmental or synaptic plasticity. Together, our findings suggest p53 as an activity-dependent protein and an activity-dependent transcription factor. Our findings also suggest that p53 contributes to behavior through a mechanism different from those used by p53 to regulate cell cycle or apoptosis. Overall, our study provides critical information toward the understanding of novel activity-dependent transcription and explains a previously unclear connection between p53 and autism spectrum disorders.
MATERIALS AND METHODS
Ethics statement
All experiments using animals followed the guidelines of Animal Care and Use provided by the Illinois Institutional Animal Care and Use Committee (IACUC) and the guidelines of the Euthanasia of Animals provided by the American Veterinary Medical Association (AVMA) to minimize animal suffering and the number of animals used. This study was performed under an approved IACUC animal protocol of University of Illinois at Urbana-Champaign (#20049 and #23016 to N.-P. Tsai.).
Animals
The breeders of wild-type (stock No. 000664), p53f/f (stock No. 008462) and Emx1-Cre (stock No. 022762) mice on a C57BL/6J background were obtained from The Jackson Laboratory. Mice were group-housed in cages two to five in a 12h/12h light/dark cycle in a temperature-controlled room with ad libitum access to water and food. All animal procedures were performed in accordance with our institutional animal care committee’s regulations. Transgenic mice were identified by PCR with genomic DNA prepared from toe clips. For genotyping, the primers used to detect p53 loxP allele are: 5’-GGTTAAACCCAGCTTGACCA-3′, and 5’-GGAGGCAGAGACAGTTGGAG-3′. The primers used to differentiate wild-type and Emx1 alleles are 5’-CGG TCT GGC AGT AAA AAC TAT C-3’ (Emx1-Cre), 5’-GTG AAA CAG CAT TGC TGT CAC TT-3’ (Emx1-Cre); 5’-AAG GTG TGG TTC CAG AAT CG-3’ (wild-type), and 5’-CTC TCC ACC AGA AGG CTG AG-3’ (wild-type).
Reagents and antibodies
Dimethyl sulfoxide (DMSO, BP231) was from ThermoFisher Scientific. Cycloheximide (CHX, 01810), actinomycin D (AcD, A1410) and strychnine (S8753) were from Sigma. Tetrodotoxin (TTX, 14964) was from Cayman Chemical. Bicuculine (2503) was from Tocris. Glycine (CG01) was from Bioland Scientific. The shRNA against Necdin and control scrambled shRNA (VB900053–2767fdt and VB010000–0023jze) was from VectorBuilder. Antibodies used in this study and their dilutions are summarized in Supplementary Information.
Primary neuronal cultures and transfection
Cortical regions including hippocampus were isolated from mice at postnatal day 0 (P0) or P1 and dissected on ice in Hank’s Balanced Salt Solution (HBSS; 21–021-CV, Corning) containing 20% fetal bovine serum, digested with trypsin (T479, Sigma) in the absence of serum, and plated on poly-D-lysine (sc-136156, Santa Cruz Biotechnology)-coated glass coverslips in 24-well plates for miniature excitatory synaptic currents (mEPSCs) and immunocytochemistry or poly-D-lysine-coated 10 cm plate for western blotting. The cultures were maintained in NeuralA basal medium (10888022, ThermoFisher Scientific) supplemented with B27 supplement (17504001, Invitrogen), GlutaMax (2 mM, 35050061, Invitrogen), and cytosine β-D-arabinofuranoside (AraC, 1 μM, C1768, Sigma). Each experiment was performed using sister cultures made from the same litter. Cultures were grown at 37°C with 5% carbon dioxide atmosphere. The culture medium was changed 50% on days in vitro (DIV) 2 and every 3–4 days thereafter until the experiments. Cells were transfected using Lipofectamine 3000 (L3000015, ThermoFisher Scientific) for 30–60 min followed by replacing the culture medium with conditioned medium to minimize toxicity produced by Lipofectamine.
Chemical LTP stimulation.
Chemical LTP (cLTP) was induced as described previously (15, 16). Briefly, cultured cortical neurons or hippocampal neurons were maintained for 20 min in artificial cerebrospinal fluid (aCSF, 125 mM NaCl, 25 mM HEPES, 2.5mM KCl, 1.5 mM CaCl2, 1 mM MgCl, 33 mM Glucose; pH 7.4) supplied with 0.5 μM TTX, 20 μM bicuculline and 1 μM strychnine before 5-min cLTP induction in the same solution as above with 200 μM glycine but without MgCl. After finished cLTP application, neurons recovered in normal aCSF for 15 or 30 min before lysis or mEPSC recording.
Electrophysiology
Whole-cell patch-clamp recordings were made on cultured primary hippocampal neurons at room temperature (23–25°C) in a submersion chamber continuously perfused with aCSF. For mEPSCs, bicuculline (20 μM), TTX (0.5 μM), and strychnine (1 μM) were added to the aCSF to block GABAA receptors, sodium channels, and glycin receptors, respectively. Whole cell recording pipettes (~4–6 MΩ) were filled with intracellular solution containing (in mM): 130 K-gluconate, 6 KCl, 3 NaCl, 10 HEPES, 0.2 EGTA, 4 Mg-ATP, 0.4 Na-GTP, 14 Tris-phosphocreatine, 2 QX-314 (pH 7.25, 285 mOsm). Neurons at DIV 12–16 were used for electrophysiological analyses. Membrane potential was clamped at −60 mV. Neurons were not included in analyses if the resting membrane potential was > −45 mV, access resistance was > 25 MΩ, or if access resistance changed by > 20%. All recordings were performed with Clampex 10.6 and Multiclamp 700B amplifier interfaced with Digidata 1550B data acquisition system (Molecular Devices). Recordings were filtered at 1 kHz and digitized at 10 kHz.
Mouse behavioral testing
Both male and female mice at 6–8 weeks of age from at least five litters were used in each experiment. Some litters were pooled before the experiment, therefore litter effects were not assessed. For each behavioral assay, the mice were transferred to the test room for habituation at least 30 min before all behavioral tests. The ambient noise level was approximately 65 dB. Unless otherwise indicated, the tests were performed during the light phase in a dimly lit room (<50 lux) with indirect lighting on the testing area to minimize fear and anxiety-like behavior (17–19). All behavioral equipment was cleaned with 70% ethanol before and after each animal test. Detailed procedures are provided in Supplementary Information.
RNA sequencing
RNA sequencing (RNAseq) was conducted using the services provided by GENEWIZ LLC (South Plainfield, NJ). Hippocampi from 5 male or female, p53 WT or p53 cKD, mice were used. Following the sequencing, sequence reads were trimmed to remove possible adapter sequences and nucleotides with poor quality using Trimmomatic. The trimmed reads were mapped to the Mus musculus genome using the STAR aligner. Unique gene hit counts were calculated by using featureCounts from the Subread package. After extraction of gene hit counts, using DESeq2, a comparison of gene expression between the p53 WT and p53 cKD was performed. The Wald test was used to generate p-values and log2 fold changes for subsequently generating the volcano plot.
Bioinformatics analyses
To obtain differentially expressed genes between p53 WT and p53 cKD mice in males and females, we performed regression-based methods, specifically a linear regression model. The expression values of each gene were considered as covariates, while the response variables were whether the mice are p53 WT or p53 cKD. In short, we tested the significance of the regression coefficient from the expression values to the response variable (WT vs. KD). To ensure accuracy, we adjusted for sex differences and utilized advanced regression methods to calculate the p-values of the regression coefficient, as detailed below.
To account for sex differences, we used a separate linear regression model to calculate the coefficient of the sex covariate for each gene. Specifically, we use to denote the gene expression matrix of samples of gene expression values, where each row denotes the expression of a mice, and we use to denote the sex. For each gene , we first calculated , and then calculated a residue matrix where . To calculate the regression coefficient, we utilized a recently developed advanced regression model that connects regression to linear mixed model and ridge regression. This approach allows us to calculate p-values even in high-dimensional settings (20).
When examining the correlation between differentially expressed genes in p53 cKD mice and the 102 known autism-linked genes (21), we first identified the 102 genes in our datasets and found 100 of them. We then proceeded to calculate the correlation by utilizing the Pearson correlation function from the scikit learn python package, which provides both the correlation value and the corresponding p-value. Furthermore, we utilized the sex-adjusted gene expression values, obtained as described earlier, to calculate the correlation instead of using the original values.
Experimental Design and Statistical Analysis
Student’s t-test was used when two conditions or groups were compared. ANOVA with post hoc Tukey HSD test was used when making multiple comparisons between treatments versus genotypes. Tests were performed two-tailed. The expected sample sizes for primary cultures and animal studies were estimated based on our previous studies (22). Samples were randomized but no formal randomization criteria was used. No samples were excluded from analyses. Because of the design of our research, no blinding was performed. The data presented in this study have been tested for normality using Kolmogorov-Smirnov Test. Specific sample numbers, including the numbers of cells or repeats, are indicated in the figure legends. Data analyses were performed using OriginPro 2021b (OriginLab). Differences are considered significant at the level of p < 0.05. p values are directly labeled in figures while the results of group comparisons in ANOVA tests are summarized in Supplemental Information.
RESULTS
p53 expression is up-regulated following cLTP
To begin determining whether and how the tumor suppressor p53 is involved in the synaptic plasticity process, we employed an established protocol (22) to chemically induced N-methyl-D-aspartate (NMDA) receptor–dependent long-term potentiation (cLTP) in cortical neuron cultures at days-in-vitro (DIV) 14–16, during which 200 μM of glycine was applied to cultures for 5 minutes (Fig. 1A1). We chose to use cortical neuron cultures in order to obtain sufficient amount of cells for biochemical assays. Following the induction of cLTP and allowing for recovery for 30 minutes, we observed a significant elevation of p53 in comparison to the control condition (CTL) (Fig. 1A2,3) without significant changes in GAPDH (Supplementary Fig. 1). The signal of p53 was validated using total brain lysates from p53 knockout mice (Supplementary Fig. 2). This cLTP-induced expression of p53 can also be observed when cultures were prepared separately from either male or female mice (Supplementary Fig. 3) or when cultures were made with only hippocampal regions (Supplementary Fig. 4). Also noted are the two bands of p53 signal on the blot, which represent the two major protein products from various isoforms that were frequently observed in different organs (23). In addition to the changed expression, p53 activity can also be regulated by post-translational modifications, such as phosphorylation at serine resides 15 (Ser15) and 33 (Ser33) (24, 25). As shown in Supplementary Figure 5, however, we did not observe significant changes in phosphorylation on either of the residues following cLTP induction. While there might be changes in other uncharacterized modifications on p53 following cLTP induction, based on our observation in Figure 1A, we decided to focus on the changes in the expression of p53. To validate that the elevation of p53 was indeed dependent on activation of the NMDA receptor, we pretreated the cultures with or without an NMDA receptor antagonist, 2-Amino-5-phosphonovalerate (APV, 100 μM), and confirmed that APV efficiently blunts the elevation of p53 (Fig. 1B1,2). Because the levels of p53 are well known to be regulated by an E3 ubiquitin ligase named murine double minute-2 (Mdm2) (26), we measured the levels of Mdm2 (Fig. 1A2,3) and p53 ubiquitination (Supplementary Fig. 6) following the induction of cLTP but observed no significant changes. When measuring the levels of another substrate of Mdm2 in neurons, postsynaptic density protein 95 (PSD-95) (2), we also failed to observe any significant changes (Fig. 1A2,3). These data suggest that the elevated p53 following cLTP likely occurs through an Mdm2-independent, potentially also degradation-independent, pathway.
Figure 1. p53 protein expression is induced by cLTP.
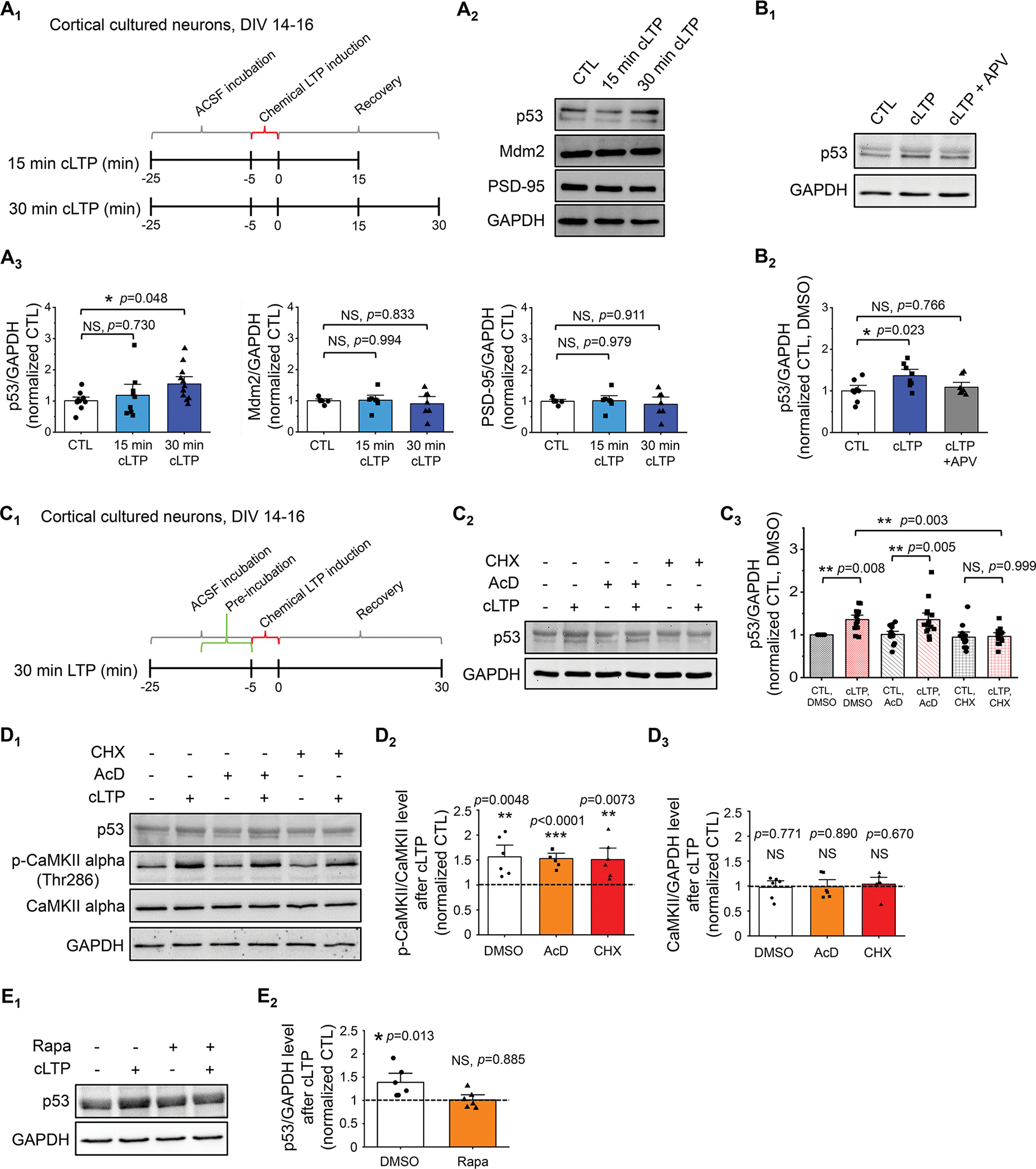
(A) Schematic representation of the experimental design for 5-min glycin-induced cLTP (A1), representative western blots of p53 and representative western blots of p53, Mdm2, PSD-95, and GAPDH after cLTP induction in WT cortical culture neurons at DIV 14–16 (A2), and quantification (A3) are shown (n = 9–12 individual cultures per condition for p53/GAPDH and n = 6–7 individual cultures per condition for Mdm2/GAPDH and PSD-95/GAPDH). For quantification, data from 15 min and 30 min cLTP were normalized to data from control condition (CTL). (B) Quantification and representative western blots of p53 following cLTP induction in the presence or absence of 10-min pre-treatment of 100 μM APV, an NMDA receptor blocker (n = 8 individual cultures per condition). (C) Schematic representation of the experimental design for cLTP with pretreatment of drugs (C1), representative western blots of p53 following cLTP induction in the presence or absence of 10 min pre-treatment of 0.1% DMSO (Control, CTL), 60 μM cycloheximide (CHX), or 20 μM actinomycin-D (AcD) (C2) and quantification (C3) are shown (n = 12–15 individual cultures per condition). (D) Quantification of p53 and representative western blots of p53, GAPDH, CaMKII alpha, and phospho-CaMKII alpha-Thr286 after cLTP induction in the presence or absence of 10 min pre-treatment of 0.1% DMSO, 20 μM actinomycin-D (AcD), or 60 μM cycloheximide (CHX) (n = 6 individual cultures per condition). (E) Quantification of p53 and representative western blots of p53 and GAPDH after cLTP induction in the presence or absence of 10 min pre-treatment of 0.1% DMSO or 1 μM rapamycin (Rapa) (n = 6 individual cultures per condition). Data were analyzed by one-way ANOVA with Tukey test (A, B), two-way ANOVA with Tukey test (C), or Student’s t-test (D, E) and presented as mean ± SEM with *p < 0.05, **p < 0.01 and NS: non-significant.
We next asked whether elevated p53 resulted from elevated expression by employing pretreatment of either the transcription inhibitor actinomycin-D (AcD, 20 μM) or the translation inhibitor cycloheximide (CHX, 60 μM) 10 mins prior to cLTP induction (Fig. 1C1). As shown (Fig. 1C2,3), pretreatment of CHX, but not AcD, inhibited cLTP-induced elevation of p53. The concern of AcD or CHX interfering with the induction of cLTP was ruled out because a significant elevation of Ca²⁺/calmodulin-dependent protein kinase II (CaMKII) phosphorylation at threonine 286 (Thr286) induced upon cLTP was unaffected by either AcD or CHX pretreatment (Fig. 1D). These data suggest that the elevation of p53 following cLTP occurs through elevated expression via a posttranscriptional mechanism. Because cLTP is known to trigger mammalian target of rapamycin (mTOR)-dependent protein synthesis (27), we employed an mTOR inhibitor, rapamycin (1 μM), and confirmed that cLTP-induced elevation of p53 is blunted (Fig. 1E). Altogether, our results suggest that cLTP elevates the expression of p53 through an mTOR-dependent mechanism.
Knocking down p53 impairs cLTP-dependent modulation of AMPA receptors
To study the functional roles of p53 during cLTP, we generated a conditional knockdown of p53 in mice by crossing p53 floxed mice (p53f/f) with Emx1-Cre mice to obtain p53f/+-Emx1-Cre+ (p53 cKD) and p53f/+-Emx1-Cre- (p53 WT) mice. Emx1-Cre can selectively reduce p53 in forebrain excitatory neurons, starting as early as embryonic day 10.5 (28). We used heterozygous p53 mice (p53f/+) to minimize the possibility of strong compensatory effects and apoptosis caused by the complete knockout of p53 (29). The knockdown efficiency of p53 in p53 cKD hippocampi at postnatal day (P) 14 was roughly 40% in hippocampus (Fig. 2A). We focus on hippocampus because the LTP in hippocampus is considered the most relevant brain plasticity mechanism associated with learning and memory. Upon obtaining these mice, we asked how p53 might participate in LTP. Because an induction of cLTP is known to potentiate the functions of AMPA receptors particularly in hippocampus and because our previous work has linked p53 to post-transcriptional regulation of AMPA receptors (12), we determined whether p53 regulates the basal levels of AMPA receptors. As shown (Fig. 2B), we found no significant difference in the levels of either GluA1 or GluA2 subunits of AMPA receptors when comparing the hippocampi of p53 WT and p53 cKD mice. We next asked whether p53 is required for any functional changes in AMPA receptors. To evaluate this, we prepared primary hippocampal neuron cultures from p53 WT and p53 cKD mice. Because functional AMPA receptors depend on functional synapses, we first determined whether knocking down p53 affects relative synapse numbers in neurons. We performed immunocytochemistry using antibodies against the presynaptic marker synapsin-I and the postsynaptic marker PSD-95 and quantified colocalization of pre- and postsynaptic puncta, as we have done recently (22). Following this experiment, we observed no significant changes in the puncta number and area of PSD-95, synapsin-I or their colocalization in p53 WT versus p53 cKD cultures at DIV 14–16 (Fig. 2C and Supplementary Fig. 7), suggesting no basal defects in synapse development after knocking down p53.
Figure 2. Knocking down p53 impairs cLTP-induced surface expression of AMPAR and strengthening of synapses.
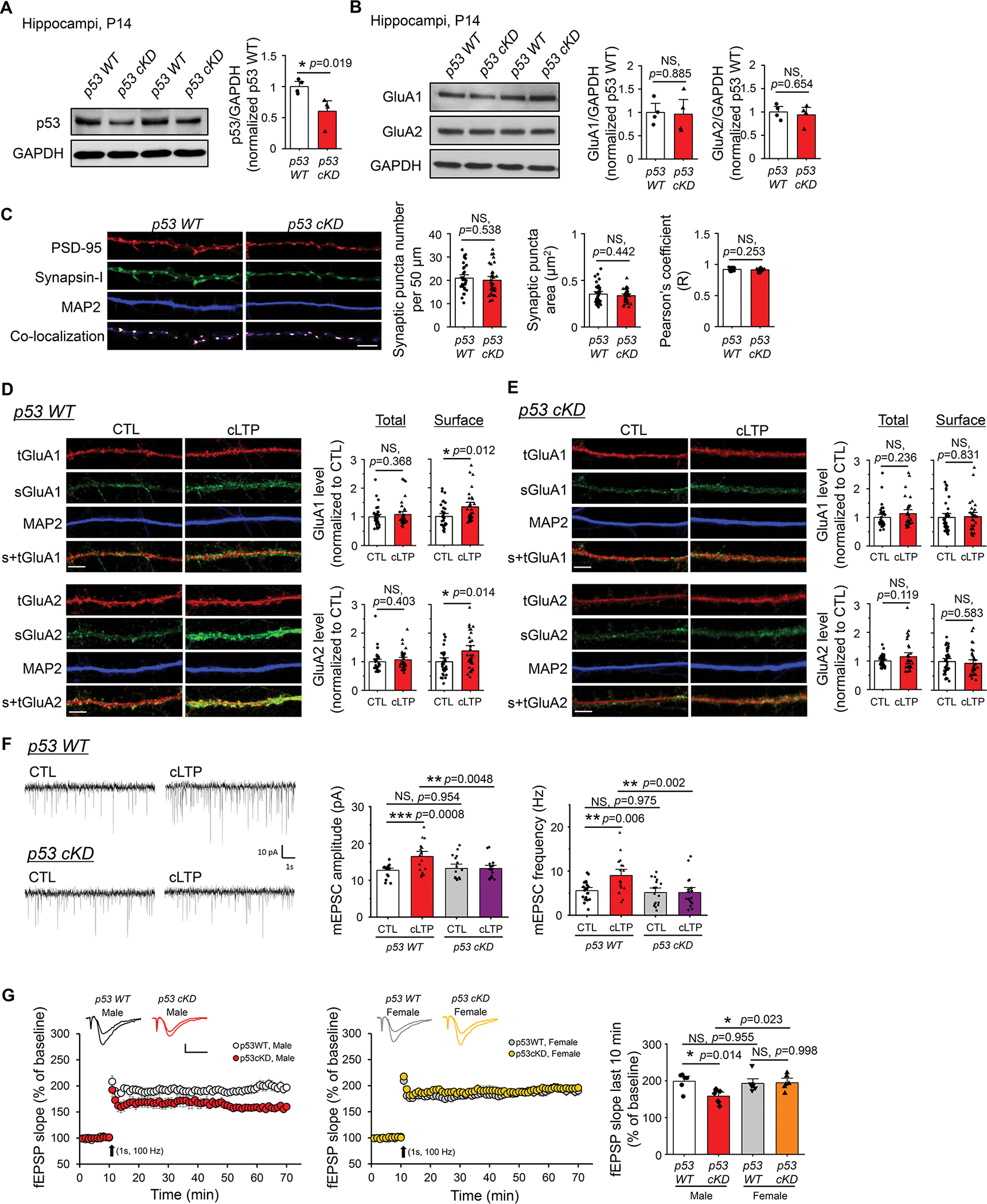
(A) Quantification of p53 and representative western blots of p53 and GAPDH from p53 WT and p53 cKD hippocampi at P14 (n = 4 mice per genotype). For quantification, data from p53 cKD hippocampi were normalized to data from p53 WT hippocampi. (B) Quantification of GluA1 and GluA2 and representative western blots of GluA1, GluA2, and GAPDH from p53 WT and p53 cKD hippocampi at P14 (n = 4 mice per genotype). For quantification, data from p53 cKD hippocampi were normalized to data from p53 WT hippocampi. (C) Quantification of colocalized synaptic puncta number, synaptic puncta area, and Pearson’s correlation coefficient and representative dendrites from dissociated p53 WT or p53 cKD hippocampal neurons at DIV 14. Immunocytochemistry showing post-synaptic marker PSD-95 (red), pre-synaptic marker synapsin-I (green), dendritic marker MAP2 (blue), and co-localization of PSD-95 and synapsin-I (n = 34 for p53 WT neurons and n =33 for p53 cKD neurons). Scale bar: 5 μm. (D, E) Immunocytochemistry showing total (t) and surface (s) GluA1 and GluA2, and dendritic marker MAP2 from p53 WT (D) or p53 cKD (E) hippocampal neurons induced with or without cLTP at DIV 16. Representative images of dendrites and quantification for GluA1 (top) and GluA2 (bottom) are shown (n = 31–37 neurons for GluA1 and n = 34–37 neurons for GluA2). For quantification, data from cLTP induction were normalized to data from CTL condition. Scale bar: 5 μm. (F) Patch-clamp recording of cultured p53 WT or p53 cKD hippocampal neurons induced with or without cLTP at DIV 12–16. Representative mEPSC traces (left) and quantification of mEPSC amplitude and frequency (right) are shown (n = 17–20 neurons). (G) A train of high-frequency stimulation (HFS; 100 Hz for 1 s) induced LTP at Schaffer collateral synapses in male p53 WT (n = 6 slices from 4 mice), male p53 cKD (n = 7 slices from 5 mice), female p53 WT (n = 7 slices from 5 mice), and female p53 cKD (n = 6 slices from 4 mice). Representative fEPSP traces were recorded before and 60 min after LTP induction (Scale bars: 0.4 mV, 10 s). Summary bar graphs showing the fEPSP slopes measured 50–60 min after a HFS at Schaffer collateral synapses were on the right. Data were analyzed by Student’s t-test (A-E) or two-way ANOVA with Tukey test (F, G) and presented as mean ± SEM with *p < 0.05, **p < 0.01, *** p < 0.001 and NS: non-significant.
We next measure functional changes in AMPA receptors. To this end, we measured the surface levels of AMPA receptors following cLTP. This is because the surface levels of AMPA receptors are known to be elevated after glycine stimulation (30) and such elevation is crucial to sustain LTP (31, 32). We again employed primary hippocampal neuron cultures from p53 WT and p53 cKD mice and performed immunocytochemistry at DIV 14 to compare the surface levels of GluA1 or GluA2 at the baseline. As shown (Supplementary Fig. 8), we observed a reduction of both surface GluA1 and surface GluA2 in p53 cKD neurons, suggesting a role for p53 in regulating basal surface expression of AMPA receptors. We next determine how GluA1 and GluA2 are affected following induction of cLTP. As shown (Fig. 2D, E), while cLTP did not change the levels of total GluA1 or GluA2 in either p53 WT or p53 cKD neurons, cLTP significantly elevated the levels of surface GluA1 and GluA2 in p53 WT, but not in p53 cKD neurons. These observations indicate that p53 is required for the potentiation of AMPA receptors following cLTP. Because AMPA receptors are the major group of glutamate receptors that mediate the majority of fast synaptic transmission, we asked whether the defect on the surface of AMPA receptors in p53 cKD can be reflected on functional synaptic transmission. To test this, we measured the miniature excitatory postsynaptic currents (mEPSCs) in primary hippocampal neuron cultures made from p53 WT or p53 cKD mice at DIV 12–16. Interestingly, despite a reduction of surface GluA1 and GluA2 in p53 cKD neurons at the baseline (Supplementary Fig. 8), we did not observe a significant difference in the amplitude of mEPSCs between p53 WT and p53 cKD neurons in the control condition (Fig. 2F). This suggests a possibility that the size of presynaptic vesicles may be elevated in p53 cKD neurons and this idea would need to be tested by future experiments. Most importantly, however, as our immunocytochemistry data in Figs. 2D and 2E suggested, increased mEPSC amplitude and frequency following cLTP induction were observed in p53 WT neurons but not in p53 cKD neurons (Fig. 2F, G). When we induced LTP by a single train of high-frequency stimulation (HFS; 100 Hz for 1s) and recorded the field excitatory post-synaptic potentials (fEPSPs) to access hippocampal LTP at Schaffer collateral synapses using acute hippocampal slices from p53 WT or p53 cKD mice at 3–4 weeks of age, we surprisingly observed a significantly impaired LTP in hippocampal slices from male, but not female, p53 cKD mice. Altogether, our data indicate that p53 is required for cLTP-induced elevation of surface AMPA receptors and the strengthening of excitatory synapses in cultured hippocampal neurons as well as high frequency stimulation-induced LTP in hippocampal slices from male mice.
Knocking down p53 does not alter locomotor activity or anxiety-like behavior
Long-term potentiation (LTP) represents a cellular mechanism for learning and memory and many other cognitive behaviors. To determine the effects of knocking down p53 on behavioral measures of cognition, we employed p53 WT or p53 cKD mice at 6–8 weeks of age for a series of behavioral experiments. Because we observed a defect of hippocampal LTP only in male p53 cKD mice (Fig. 2G), we employed mice of both sexes with the intent to evaluate sex-dependent changes in behavior. We first performed an open field test to examine locomotor activity and anxiety-like behavior because many behavioral assays require proper motor skills and could be affected by anxiety. As shown, p53 WT and p53 cKD mice of both sexes displayed similar levels of average speed, immobile time, time spent in a center zone, number of entries in a center zone, and defecation (Fig. 3A). To further probe anxiety behavior, we employed the elevated plus maze test. As shown (Fig. 3B), the average velocity and total distance traveled through the maze as well as the time and entries in open or closed arms were not significantly different between p53 WT and p53 cKD mice, except for elevated time in open arms observed in female p53 cKD mice. Taken all criteria together, our data suggest that p53 cKD mice exhibit grossly unchanged locomotor activity and levels of anxiety when compared to p53 WT mice.
Figure 3. Knocking down p53 does not alter locomotor activity or anxiety-like behavior in mice.
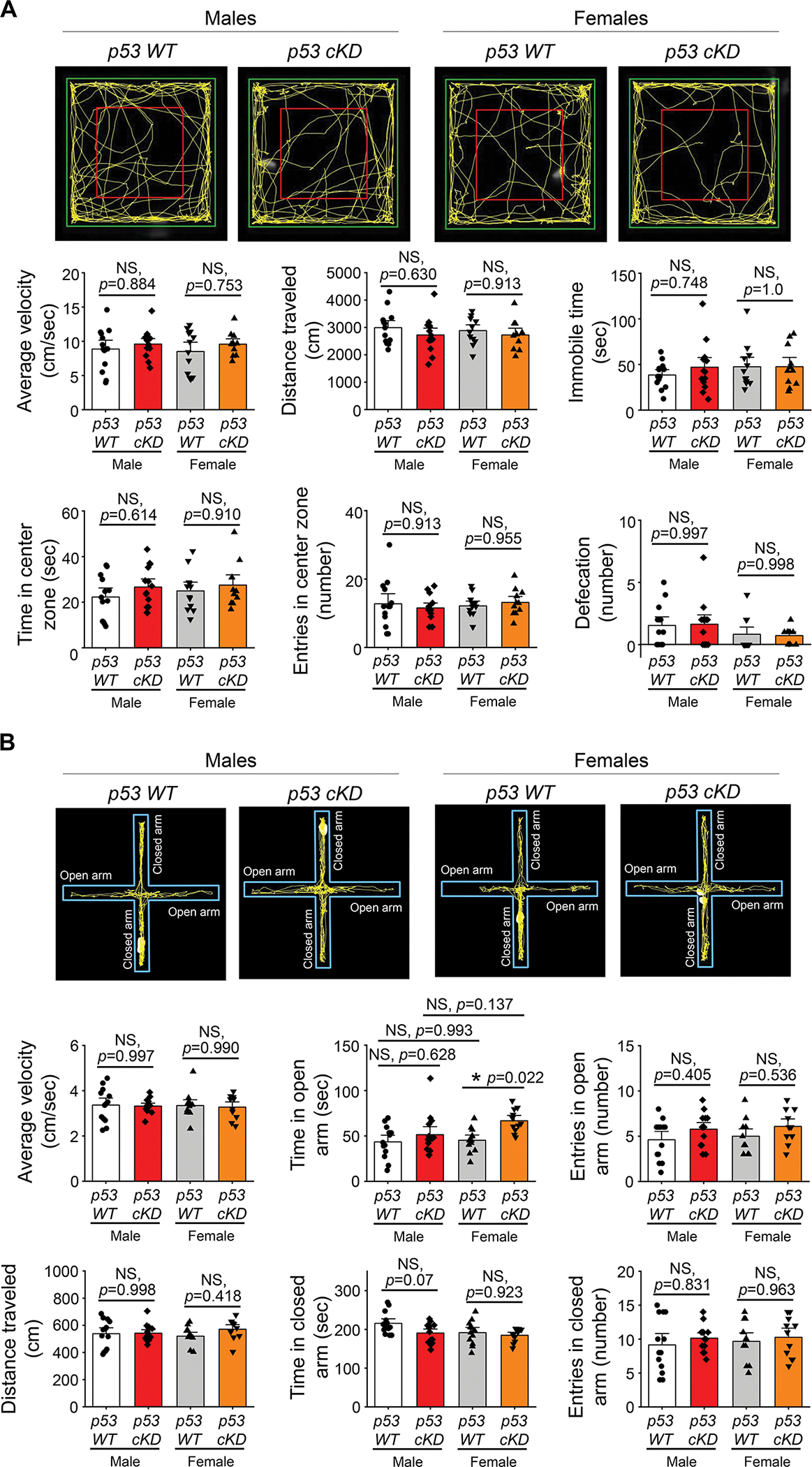
(A) Open field test from male or female p53 WT and p53 cKD mice (n = 13 for male p53 WT, n = 14 for male p53 cKD, n = 12 for female p53 WT, and n = 11 for female p53 cKD mice). Representative image showing the explorative activity during 5 min open field test period (top). Quantification of average velocity, total distance traveled, immobile time, time spent in center zone, number of entries in center zone, and defecation number is shown on the bottom. (B) Elevated plus maze from male or female p53 WT and p53 cKD mice (n = 13 for male p53 WT, n = 14 for male p53 cKD, n = 12 for female p53 WT, and n = 11 for female p53 cKD mice). Representative traces showing the explorative activity mice for 5 min in the maze are on the top. Quantification of average velocity, time spent in open arm, number of entries in open arm, total distance traveled, time spent in closed arm, and number of entries in closed arm in the maze is on the bottom. Data were analyzed by two-way ANOVA with Tukey test and presented as mean ± SEM with *p < 0.05, and NS: non-significant.
p53 cKD mice display increased repetitive behavior and reduced sociability
A defect in LTP is commonly observed in autism animal models, and many autism-linked genes are found to be critical to LTP (33–35). Because recent studies have suggested the potential roles of p53 in regulating the expression of autism-linked genes (36, 37), we asked whether knocking down p53 could reduce or elevate autism-like behaviors. To this end, we first employed a marble burying test to evaluate repetitive behaviors. As shown (Fig. 4A), both male and female p53 cKD mice buried significantly more marbles than p53 WT mice. Although increased marble burying behavior can also indicate elevated anxiety (38), since we did not observe changes in anxiety in p53 cKD mice (Fig. 3), our data suggest that knocking down p53 elevates repetitive behavior in mice.
Figure 4. Knocking down p53 increases repetitive behavior and reduces sociability in mice.
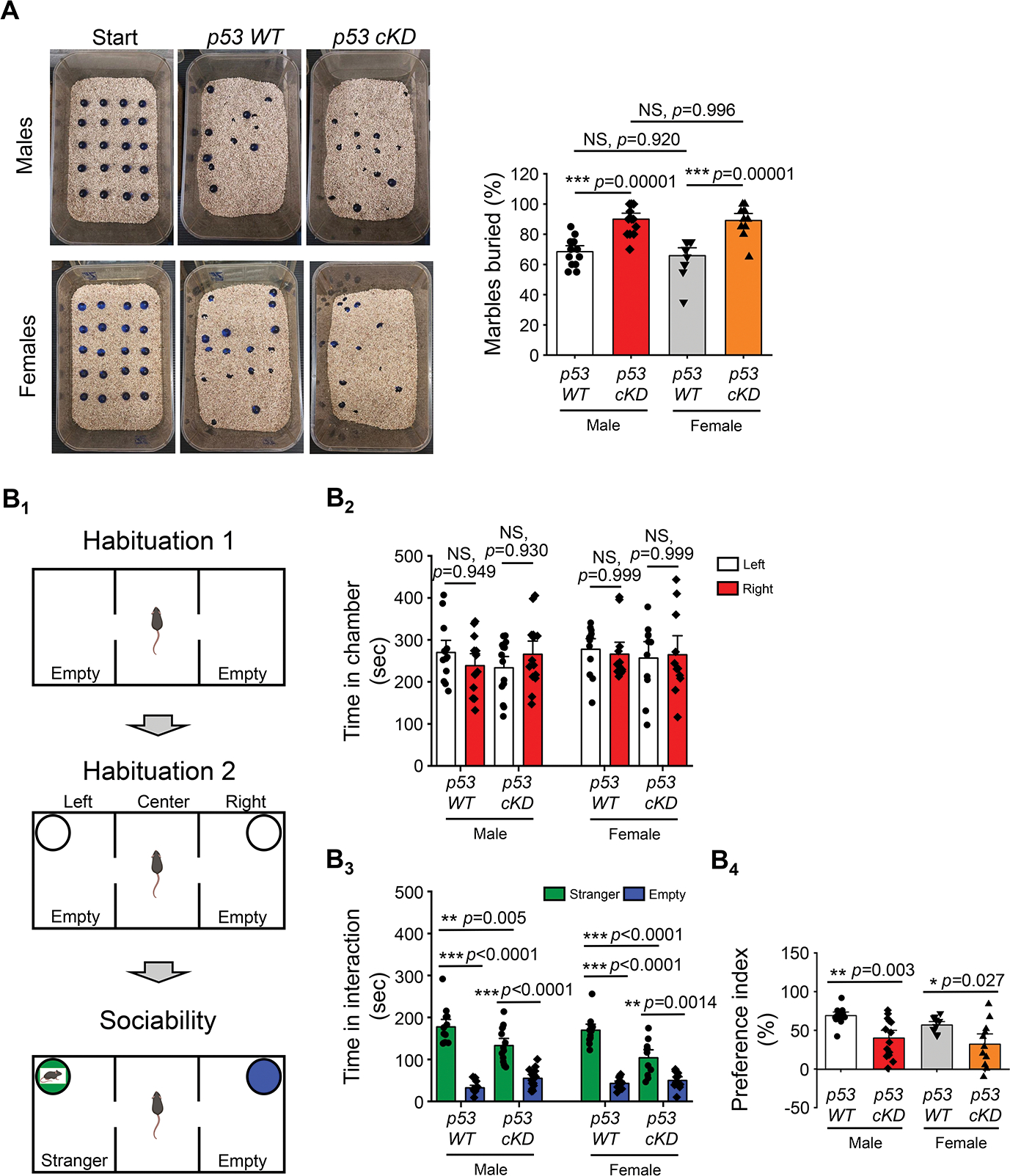
(A) Marble burying test from male or female p53 WT and p53 cKD mice. Representative images (left) and quantification of the marble burying activity (right) are shown (n = 13 for male p53 WT, n = 14 for male p53 cKD, n = 12 for female p53 WT, and n = 11 for female p53 cKD mice). (B) The three-chamber social interaction test from male or female p53 WT and p53 cKD mice (n = 13 for male p53 WT, n = 14 for male p53 cKD, n = 12 for female p53 WT, and n = 11 for female p53 cKD mice). Schematic representation depicting the three-phase sociability protocol for the three-chamber social interaction test (B1). Quantification of the time spent in left or right chamber at second habituation from male or female p53 WT and p53 cKD mice when an empty cylinder was presented (B2). Quantification showing the time spent interacting with the stranger mouse or empty cylinder from male or female p53 WT and p53 cKD mice (B3). Quantification of social preference index from male or female p53 WT and p53 cKD mice (B4). Data were analyzed by two-way ANOVA with Tukey test (A and B4) or three-way ANOVA with Tukey test (B2, B3) and presented as mean ± SEM with *p < 0.05, **p < 0.01, ***p < 0.001 and NS: non-significant.
We next employed a three-chamber social interaction test to measure the sociability behavior of the mice (Fig. 4B). After allowing the mice to habituate to the chamber, empty wired cylinders were placed in both right and left chambers for a second round of habituation and the time spent in the right and left compartments was measured. As shown (Fig. 4B2), neither p53 WT nor p53 cKD mice of both sexes showed a preference toward either of the compartments. To evaluate their sociability, p53 WT or p53 cKD mice were put in the chamber again after a stranger mouse of same sex was put into one of the cylinders with the other cylinder left empty. Following that, the time each mouse spent interacting with the stranger mouse or the empty cylinder was measured. As shown (Fig. 4B3), p53 WT and p53 cKD mice of both sexes spent significantly more time with the stranger mouse than with the empty cylinder. Interestingly, both male and female p53 cKD mice spent less time interacting with the stranger mouse when compared to p53 WT mice. Using a preference index to measure the preference toward the stranger mouse or the empty cylinder, we confirmed that both male and female p53 cKD mice showed a significantly lower preference toward the stranger mouse (Fig. 4B4). Because further group analyses indicated no significant differences between sexes in all the criteria (Supplementary Information), our data altogether suggest that reduction of p53 leads to an elevation in autism-like behaviors in both male and female mice.
Hippocampus-dependent learning and memory are impaired in male but not in female p53 cKD mice
Since synaptic plasticity, especially LTP, is widely believed to be the cellular mechanism underlying learning and memory (39, 40), we aimed to determine how p53 might participate in learning and memory behavior. To do so, we first performed the Barnes maze test to determine whether a reduction of p53 affects hippocampus-dependent spatial learning and memory (Fig. 5A1) (22, 41). Following the habituation and adaptation to the set-up, the mice were trained for four days to locate an escape hole in order to escape a brightly lighted circular field. During these training days, as shown (Fig. 5A2,3), male p53 cKD mice exhibited a significant increase in the number of exploring errors on the third and fourth days of training with no significant changes in primary escape latency. We then performed probe trials at day 5, during which the escape box under the target hole was removed, to assess spatial memory formation (Fig. 5A4). As shown (Fig. 5A5), the male p53 cKD mice exhibited slightly but not significantly reduced quadrant occupancy. When performing the same experiment using female mice, we did not observe significant differences in any of the parameters (Fig. 5A5).
Figure 5. Knocking down p53 impairs hippocampus-dependent learning and memory particularly in male mice.
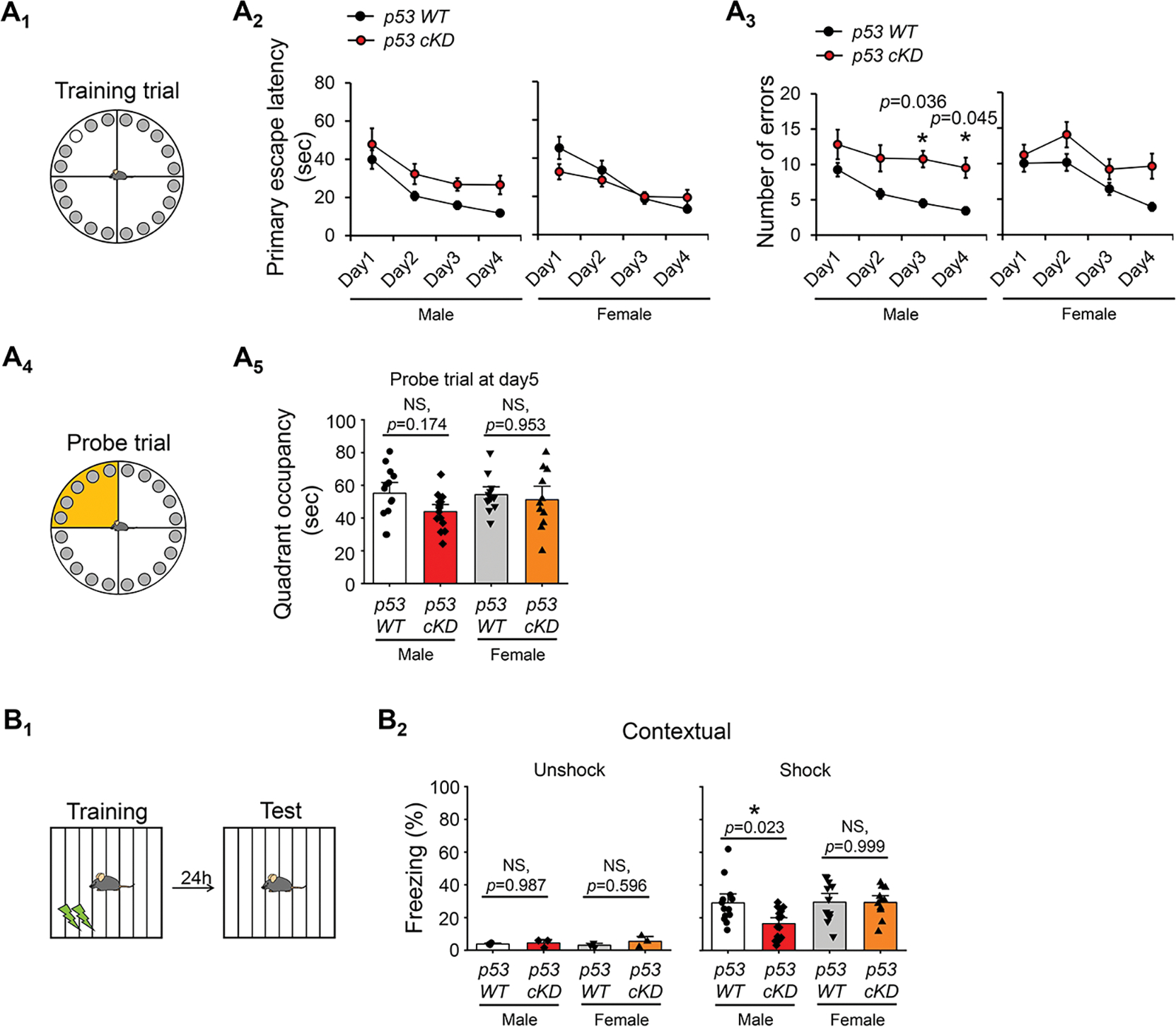
(A) Barnes maze test from male or female p53 WT and p53 cKD mice. Schematic representation of Barnes maze on training trials (A1, top) and on probe trials (A4, bottom). Mice were trained for 4 days with three training trials on each day to find the escape box (top, white circle). Learning curve during 4 training days of Barnes maze task showing the primary escape latency (A2, left) and number of errors before escaping (A3, right) from male or female p53 WT and p53 cKD mice (n = 13 for male p53 WT, n = 14 for male p53 cKD, n = 12 for female p53 WT, and n = 11 for p53 cKD female mice). Quadrant occupancy on probe trial at day5 (A5) was assessed by recording the time spent in the target area (yellow quadrant). (B) Contextual fear conditioning test from male or female p53 WT and p53 cKD mice. Schematic representation of fear conditioning test (B1). Mice were placed in the fear conditioning chamber for 180 s with a metal grid floor to deliver two electrical shocks at 120 and 150 s (0.5 mV, 2s). Twenty-four hours later, mice were placed in the same chamber for 180 s in the absence of the shock. Freezing behavior from male or female p53 WT and p53 cKD mice (n = 13 for male p53 WT, n = 14 for male p53 cKD, n = 12 for female p53 WT, and n = 11 for female p53 cKD mice) as well as mice from unshock control group (n = 3 mice per genotype) (B2) was shown. Data were analyzed by two-way ANOVA with Tukey test (A5, B2) or three-way ANOVA with Tukey test (A2, A3) and presented as mean ± SEM with *p < 0.05 and NS: non-significant.
To further evaluate learning and memory, we performed the contextual fear conditioning test to test associative learning and memory. During the training day, mice were placed in a training context and received brief aversive stimuli (footshock, 2×, 0.5 mA, 2 s duration, 30 s interval; (22, 41). On the test day (24 h after the training), mice were re-exposed to the training context and assessed for fear memory by measuring freezing behavior in the absence of footshock stimuli (Fig. 5B1). As shown (Fig. 5B2), male p53 cKD mice exhibited a significant decrease in freezing behavior compared to p53 WT mice. However, surprisingly, female p53 cKD mice did not exhibit any significant changes in freezing behavior compared to female p53 WT mice (Fig. 5B2). When we attempted to probe the expression of p53 during the task, we did not observe significant differences between mice receiving footshock stimuli or those without (Supplementary Fig. 9). These data suggest that while induction of LTP elevates p53 in cultured neurons, changed expression of p53 in vivo might be transient or only selective in certain cells following stimulation. This would require further investigation in the future. Altogether, based on our results in the Barnes maze test and the contextual fear conditioning test, we conclude that knocking down p53 negatively impacts hippocampus-dependent learning and memory behavior, especially in male mice.
Identifying p53-associated genes involved in synaptic plasticity and memory behavior
To search for p53-associated genes potentially involved in synaptic plasticity and learning and memory behavior, we performed transcriptomic analysis of hippocampi from male p53 WT and male p53 cKD mice at P14 with RNA sequencing (RNAseq, Fig. 6A). We started with male mice because of the more profound deficits that we observed in male p53 cKD mice (Figs. 2G and 5). The RNAseq analysis has detected a total of 17,779 genes in hippocampi from five p53 WT and five p53 cKD littermate mice (Supplementary Table 1). Surprisingly, only 11 genes exhibited at least a 50% increase (1.5-fold) and 63 genes exhibited at least a 33% reduction (0.67-fold) in p53 cKD hippocampi compared to p53 WT hippocampi (P < 0.05) (Supplementary Table 1). Because multiple genes—such as Necdin (Ndn), Follistatin (Fst), Forkhead transcription factor S1 (Foxs1), and Sushi repeat-containing protein (Srpx2) (42–45)—have known functions in synaptic plasticity and learning, we speculate that p53 regulates the expression of one or more of those genes to guide synaptic plasticity and behavior. To begin testing whether certain p53-associated genes are particularly crucial to synaptic plasticity, we decided to focus on Necdin, which shows the most consistent increase in expression in p53 cKD hippocampi (Fig. 6A). Necdin, a previously known p53 target gene (46), encodes a protein called Necdin that belongs to the melanoma antigen (MAGE) family proteins. Necdin has been linked to Prader-Willi syndrome (PWS), a neurodevelopmental and autism spectrum disorder (45, 47). Necdin has been implicated in neuronal excitability homeostasis and structural development of the nervous system (48–50). To explore the possibility that knocking down p53 disrupts synaptic plasticity through Necdin, we first confirmed a significant elevation of Necdin in hippocampi of both male and female p53 cKD mice at P14 (Fig. 6B). Because a reduction of p53 precludes cLTP-induced strengthening of excitatory synapses (Fig. 2), we asked whether inhibition of Necdin could rescue this deficit in p53 cKD neurons. To this end, we obtained an shRNA against Necdin and confirmed a knockdown efficiency of 30% in transfected p53 cKD hippocampal neurons (Fig. 6C), which brings Necdin to a level similar to that in p53 WT. Following the induction of cLTP in either p53 WT or p53 cKD neurons transfected with a scrambled shRNA or Necdin shRNA, we performed mEPSC recordings to measure the strength of individual synapses. While cLTP-induced elevations in mEPSC amplitude and frequency can still be seen in p53 WT neurons receiving either scrambled shRNA or Necdin shRNA (Fig. 6D), the cLTP effects were not restored in p53 cKD neurons receiving either of the shRNAs (Fig. 6E). These data suggest that Necdin is not required, or not the only contributing factor, to sustain p53-dependent elevation of synapse strengthening upon cLTP induction.
Figure 6. RNAseq reveals p53-associated genes potentially involved in synaptic plasticity, learning and memory.
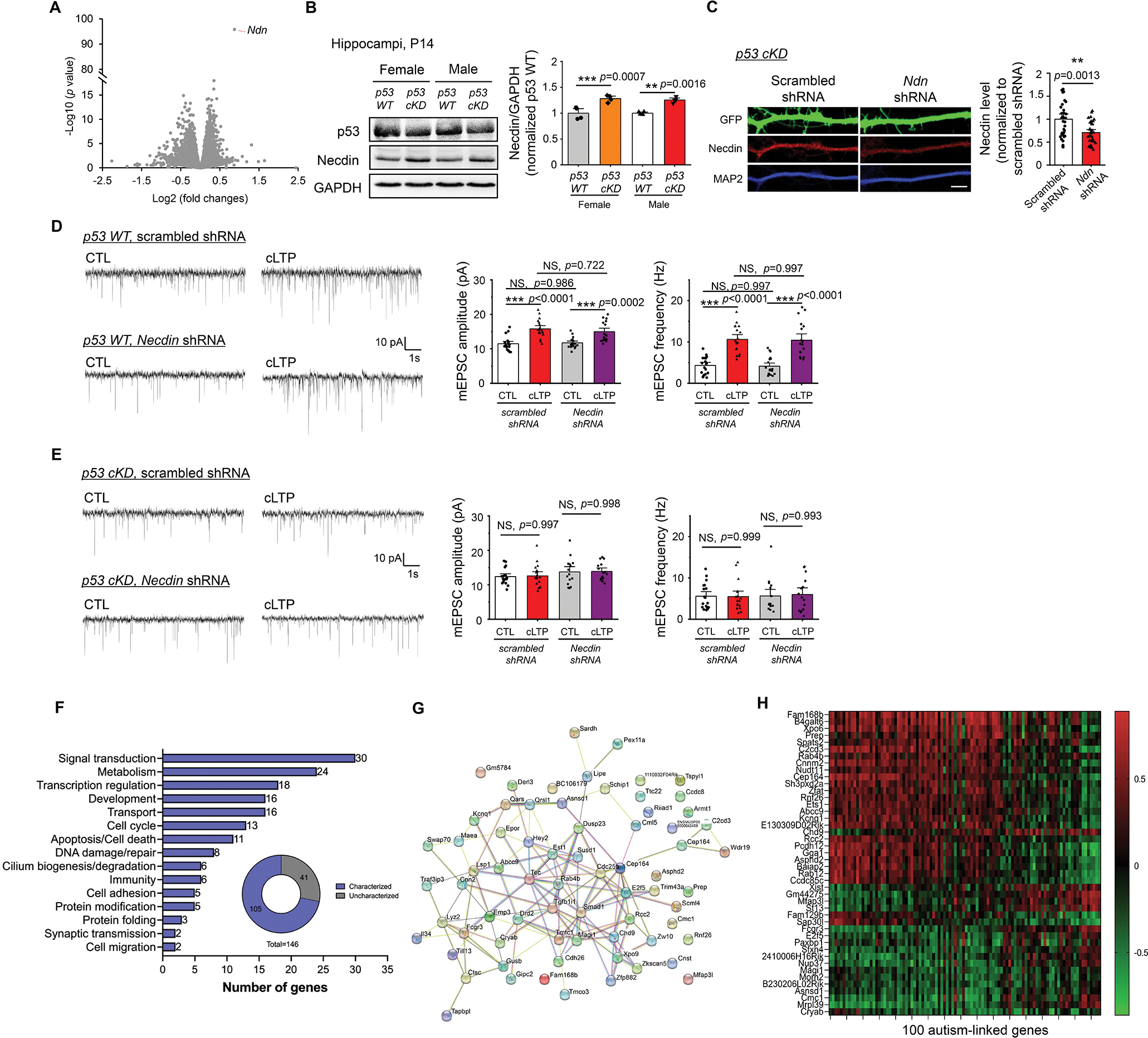
(A) A volcano plot following RNA sequencing analysis using hippocampi from male p53 WT and male p53 cKD mice at P14 (n = 5 mice per genotype). (B) Western blots of Necdin and GAPDH from male or female, p53 WT or p53 cKD, hippocampi at P14. Representative western blots (left) and quantification (right) are shown. (n = 4 mice per genotype). For quantification, data from p53 cKD hippocampi were normalized to data from p53 WT hippocampi. (C) Immunocytochemistry showing GFP, Necdin, and dendritic marker MAP2 from cultured p53 cKD hippocampal neurons transfected with a scrambled shRNA or Necdin shRNA. Representative dendritic images (left) and quantification for Necdin (right) are shown (n = 34 and 32 neurons for scrambled shRNA and Necdin shRNA, respectively). Scale bar: 5 μm. (D, E) Patch-clamp recording from cultured p53 WT (D) or p53 cKD (E) hippocampal neurons transfected with scrambled shRNA or Necdin shRNA and induced with or without cLTP at DIV 12–16. Representative mEPSC traces (left) and quantification of mEPSC amplitude and frequency (right) are shown (n = 15–19 neurons per condition). (F, G, H) Summary of major functional categories of 105 genes that are associated with p53 and differentially expressed in a sex-dependent manner (F), STRING analysis showing functional connection network of those genes identified (G), and a heatmap showing the levels of correlation between the top 44 genes that exhibit significant correlations with the known known autism-linked genes (H). Data were analyzed by Student’s t-test (A, B, C) or two-way ANOVA with Tukey test (D, E) and presented as mean ± SEM with **p < 0.01, *** p < 0.001 and NS: non-significant.
To further explore the mechanisms underlying sex-dependent effects of p53 on behaviors, we performed RNAseq using hippocampi from female p53 WT and female p53 cKD littermate mice at P14 (Supplementary Table 2). Following that, we compared the results between genotypes (p53 WT and p53 cKD) and sexes (male and female), and obtained 146 genes associated with p53 that are differentially expressed in male versus female mice (Supplementary Table 3). Analysis of the function of these 146 genes using a gene ontology program (GO Term Mapper) showed 105 out of 146 genes with clearly defined molecular functions that can be grouped into several major functional categories including signal transduction, metabolism, transcription and development (Fig. 6F). We followed by using the Search Tool for Retrieval of INteracting Genes (STRING) to analyze functional association network and observed several functionally interactive genes with known roles in synaptic transmission or plasticity, including Tec tyrosine kinase (Tec) (51), Ras-Related GTP-Binding Protein 4b (RAB4B) (52), and Membrane-associated guanylate kinase, WW and PDZ domain-containing protein 1 (MAGI-1) (53) (Fig. 6G). We further examined the correlation between these 146 differentially expressed genes and the 100 known autism-linked genes based on a previous study (21). As shown (Fig. 6H and Supplementary Table 3), we found that 44 out of 146 genes exhibited significant correlations, either positively or negatively, with 20 or more of the 100 autism-linked genes. Altogether, our data suggest that p53 may be an upstream regulator for other critical genes associated with cognitive behaviors and autism, and such an effect may be modulated in a sex-dependent manner.
DISCUSSION
Our study suggests p53 is an activity-dependent transcription factor and further that p53 functions to promote synaptic plasticity by allowing for cLTP-induced elevation of surface AMPA receptors and hippocampal LTP. Using a conditional knockdown strategy to reduce p53 in forebrain excitability neurons, we further showed that p53 is crucial for repressing repetitive behavior and promoting sociability, learning, and memory. Our previous work has shown that impaired p53 ubiquitination caused by Mdm2 contributes to defects in mGluR-dependent neural plasticity and scaling in fragile X syndrome (10–12). These studies suggest that p53 is likely regulated tightly during development and crucial to brain development. Despite these previous findings, however, this current study shows for the first time that p53 is linked directly to autism-like behavior and hippocampus-dependent learning and memory. While it remains unclear how p53 regulates the surface expression of AMPA receptors, we speculate that certain p53-associated genes may regulate AMPA receptor trafficking, either directly or indirectly. When searching for p53-associated genes that may participate in AMPA receptor trafficking or synaptic plasticity, our data identified multiple genes that fit into the category. We first tested Necdin because of its known connection to autism and neurodevelopment (45, 54). However, restoring the levels of Necdin in p53 cKD neurons does not restore cLTP-induced strengthening of synapses. These data suggest a possibility that restoring one gene is not sufficient to restore a complex plasticity mechanism like LTP. Other plausible genes to consider include Follistatin (Fst) and Sushi repeat-containing protein (Srpx2). Fst has been shown to promote synaptic plasticity and learning, presumably through promoting neurogenesis (43). On the other hand, Srpx2 has been shown to mediate the stability of glutamatergic synapse during neurodevelopment, and disruption of Srpx2 has also been linked to intellectual disability and epilepsy (44, 55). The altered expression of these genes in p53 cKD supports the critical role of p53 in brain development. It also supports the assumption that p53 exerts its functions in synaptic plasticity and behavior through multiple, rather than just one, downstream genes, either directly or indirectly, and AMPA receptor trafficking might be only one of the cellular mechanisms underlying synaptic plasticity that are mediated by p53.
The elevation of p53 following induction of cLTP suggests p53 itself is an activity-dependent protein. Our results suggest that elevation of p53 results from elevated protein expression through an mTOR-dependent posttranscriptional mechanism. A previous study has suggested that the 5’ untranslated region (5’UTR) of p53 mRNA can be bound by ribosomal protein RPL26, leading to enhanced translation of p53 (56). Another study has shown that the 3’UTR of p53 mRNA can be bound by an RNA-binding protein HuR and such binding also leads to enhanced translation of p53 (57). On the contrary, the binding of p53 mRNA by another RNA-binding protein, RNA-binding protein 1c (RNP1c), leads to reduced translation of p53 (58). Interestingly, but not surprisingly, the interaction between p53 mRNA and either RPL26 or HuR can be enhanced, while the interaction between p53 mRNA and RNP1c can be reduced, when cells are under challenges and insults. This is consistent with the past several decades of knowledge about p53’s role in apoptosis especially in the context of tumor suppression. However, these past studies also led us to speculate that an induction of LTP, such as through activation of NMDA receptors, can be considered as a challenge to neurons. This assumption can be supported by the fact that activation of NMDA receptors can also trigger a cellular stress response (59). Furthermore, cellular stress-associated, mTOR-dependent, protein synthesis is known to require La-related protein 1 (LARP1) and RBM38 (60, 61). Because the translation of p53 is also known to be regulated by LARP1 and RBM38 (62, 63), the availability and activity of LARP1 and RBM38 toward p53 mRNA may be altered upon mTOR activation, leading to changes in p53 translation. Following these rationales, in future studies it would be interesting to delineate the molecular connection between mTOR activation and p53 translation and determine whether the elevation of p53 induced upon cLTP is associated with or regulated by cellular stress pathways.
Another interesting result from our study is that, while sex-dependent effects were not observed in cultures, the impact of knocking down p53 on hippocampal LTP and behavior is clearly more profound in male mice than in female mice. This observation suggests a possibility that p53 is involved in the sex-dependent regulation of circuit- or system-level activity. Although determining the mechanisms of these sex differences is beyond our current study, our RNAseq results (Fig. 6) suggest that p53 may achieve the sex-dependent effect directly or indirectly through multiple genes and/or mechanisms, and there are several examples that support this model. First, while the p53 gene is not on the X chromosome, many of p53’s target genes are, such as Srpx2 from our screening and the Xist RNA gene from a previous study (64). Different degrees of change in those X-linked genes by the knockdown of p53 in male versus female mice may explain the differences that we observed in behavior. Second, differential expression of p53 in certain cells or certain types of cells following activity stimulation in males versus in females could also be a possibility. A previous study has observed different expression patterns of p53 in neurons upon activity stimulation in male and female mice (65). If expression of p53 is differentially stimulated during the behavioral experiments in male and female mice, it could explain why knocking down p53 affects behavior in a sex-dependent manner. Third, a previous study has revealed basally different transcription activity of p53 in males versus females (66). While this observation was made specifically during the aging process, it could still suggest a possibility that p53-dependent transcription is basally different in male and female mice. If this is true, knocking down p53 would be expected to impact male and female mice differently. Lastly, the biological activity of p53 as well as the transcriptional targets of p53 can be affected by various hormones such as estrogen (67). Estrogen, through estrogen receptors and estrogen-response elements, differentially regulates gene expression in males and females. Knocking down p53 could omit hormonal effects on p53-dependent transcription differently in males and females, subsequently leading to sex-specific physiological responses. Altogether, p53 could exert its sex-specific function in behavior through one or more of these mechanisms, which merit further study in the future.
Supplementary Material
ACKNOWLEDGEMENTS
This work is supported by National Institute of Health R01NS105615, R01MH124827 and R21MH122840 to N-P.T.
Footnotes
CONFLICT OF INTEREST
The authors declare no competing interests.
DATA AVAILABILITY
The RNA sequencing results can be found at doi.org/10.6084/m9.figshare.23560812.v1.
REFERENCE
- 1.Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60(4):610–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsai NP, Wilkerson JR, Guo W, Maksimova MA, DeMartino GN, Cowan CW, et al. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell. 2012;151(7):1581–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramanan N, Shen Y, Sarsfield S, Lemberger T, Schütz G, Linden DJ, et al. SRF mediates activity-induced gene expression and synaptic plasticity but not neuronal viability. Nature neuroscience. 2005;8(6):759–67. [DOI] [PubMed] [Google Scholar]
- 4.Faust TE, Gunner G, Schafer DP. Mechanisms governing activity-dependent synaptic pruning in the developing mammalian CNS. Nature reviews Neuroscience. 2021;22(11):657–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017;170(6):1062–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camins A, Verdaguer E, Folch J, Beas-Zarate C, Canudas AM, Pallas M. Inhibition of ataxia telangiectasia-p53-E2F-1 pathway in neurons as a target for the prevention of neuronal apoptosis. Current drug metabolism. 2007;8(7):709–15. [DOI] [PubMed] [Google Scholar]
- 7.Folch J, Junyent F, Verdaguer E, Auladell C, Pizarro JG, Beas-Zarate C, et al. Role of cell cycle re-entry in neurons: a common apoptotic mechanism of neuronal cell death. Neurotoxicity research. 2012;22(3):195–207. [DOI] [PubMed] [Google Scholar]
- 8.Simon DJ, Belsky DM, Bowen ME, Ohn CYJ, O’Rourke MK, Shen R, et al. An anterograde pathway for sensory axon degeneration gated by a cytoplasmic action of the transcriptional regulator P53. Developmental cell. 2021;56(7):976–84.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slomnicki LP, Pietrzak M, Vashishta A, Jones J, Lynch N, Elliot S, et al. Requirement of Neuronal Ribosome Synthesis for Growth and Maintenance of the Dendritic Tree. J Biol Chem. 2016;291(11):5721–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu DC, Lee KY, Lizarazo S, Cook JK, Tsai NP. ER stress-induced modulation of neural activity and seizure susceptibility is impaired in a fragile X syndrome mouse model. Neurobiology of disease. 2021;158:105450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu DC, Soriano S, Yook Y, Lizarazo S, Eagleman DE, Tsai NP. Chronic Activation of Gp1 mGluRs Leads to Distinct Refinement of Neural Network Activity through Non-Canonical p53 and Akt Signaling. eNeuro. 2020;7(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee KY, Jewett KA, Chung HJ, Tsai NP. Loss of Fragile X Protein FMRP Impairs Homeostatic Synaptic Downscaling through Tumor Suppressor p53 and Ubiquitin E3 Ligase Nedd4–2. Human molecular genetics. 2018;27:2805–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy OA, Malagelada C, Greene LA. Cell death pathways in Parkinson’s disease: proximal triggers, distal effectors, and final steps. Apoptosis : an international journal on programmed cell death. 2009;14(4):478–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller FD, Pozniak CD, Walsh GS. Neuronal life and death: an essential role for the p53 family. Cell death and differentiation. 2000;7(10):880–8. [DOI] [PubMed] [Google Scholar]
- 15.Fortin DA, Davare MA, Srivastava T, Brady JD, Nygaard S, Derkach VA, et al. Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J Neurosci. 2010;30(35):11565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hussain NK, Diering GH, Sole J, Anggono V, Huganir RL. Sorting Nexin 27 regulates basal and activity-dependent trafficking of AMPARs. Proc Natl Acad Sci U S A. 2014;111(32):11840–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bouwknecht JA, Spiga F, Staub DR, Hale MW, Shekhar A, Lowry CA. Differential effects of exposure to low-light or high-light open-field on anxiety-related behaviors: relationship to c-Fos expression in serotonergic and non-serotonergic neurons in the dorsal raphe nucleus. Brain Res Bull. 2007;72(1):32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kastenberger I, Lutsch C, Herzog H, Schwarzer C. Influence of sex and genetic background on anxiety-related and stress-induced behaviour of prodynorphin-deficient mice. PLoS One. 2012;7(3):e34251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puschban Z, Sah A, Grutsch I, Singewald N, Dechant G. Reduced Anxiety-Like Behavior and Altered Hippocampal Morphology in Female p75NTR(exon IV−/−) Mice. Front Behav Neurosci. 2016;10:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, Aragam B, Xing EP. Trade-offs of Linear Mixed Models in Genome-Wide Association Studies. Journal of computational biology : a journal of computational molecular cell biology. 2022;29(3):233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 2020;180(3):568–84.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee KY, Zhu J, Cutia CA, Christian-Hinman CA, Rhodes JS, Tsai NP. Infantile spasms-linked Nedd4–2 mediates hippocampal plasticity and learning via cofilin signaling. EMBO reports. 2021;22(10):e52645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, et al. p53 isoforms can regulate p53 transcriptional activity. Genes & development. 2005;19(18):2122–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loughery J, Cox M, Smith LM, Meek DW. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic acids research. 2014;42(12):7666–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turenne GA, Price BD. Glycogen synthase kinase3 beta phosphorylates serine 33 of p53 and activates p53’s transcriptional activity. BMC cell biology. 2001;2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, et al. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40(3):595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(1):467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22(15):6309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fortin A, Cregan SP, MacLaurin JG, Kushwaha N, Hickman ES, Thompson CS, et al. APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. The Journal of cell biology. 2001;155(2):207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaafari N, Henley JM, Hanley JG. PICK1 mediates transient synaptic expression of GluA2-lacking AMPA receptors during glycine-induced AMPA receptor trafficking. J Neurosci. 2012;32(34):11618–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang M, Ramasamy VS, Kang HK, Jo J. Oleuropein promotes hippocampal LTP via intracellular calcium mobilization and Ca(2+)-permeable AMPA receptor surface recruitment. Neuropharmacology. 2020;176:108196. [DOI] [PubMed] [Google Scholar]
- 32.Yap KA, Shetty MS, Garcia-Alvarez G, Lu B, Alagappan D, Oh-Hora M, et al. STIM2 regulates AMPA receptor trafficking and plasticity at hippocampal synapses. Neurobiology of learning and memory. 2017;138:54–61. [DOI] [PubMed] [Google Scholar]
- 33.Kim YJ, Khoshkhoo S, Frankowski JC, Zhu B, Abbasi S, Lee S, et al. Chd2 Is Necessary for Neural Circuit Development and Long-Term Memory. Neuron. 2018;100(5):1180–93.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leach PT, Poplawski SG, Kenney JW, Hoffman B, Liebermann DA, Abel T, et al. Gadd45b knockout mice exhibit selective deficits in hippocampus-dependent long-term memory. Learning & memory (Cold Spring Harbor, NY; ). 2012;19(8):319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seese RR, Maske AR, Lynch G, Gall CM. Long-term memory deficits are associated with elevated synaptic ERK1/2 activation and reversed by mGluR5 antagonism in an animal model of autism. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2014;39(7):1664–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rahman MR, Petralia MC, Ciurleo R, Bramanti A, Fagone P, Shahjaman M, et al. Comprehensive Analysis of RNA-Seq Gene Expression Profiling of Brain Transcriptomes Reveals Novel Genes, Regulators, and Pathways in Autism Spectrum Disorder. Brain Sci. 2020;10(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hurley S, Mohan C, Suetterlin P, Ellingford R, Riegman KLH, Ellegood J, et al. Distinct, dosage-sensitive requirements for the autism-associated factor CHD8 during cortical development. Molecular autism. 2021;12(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas A, Burant A, Bui N, Graham D, Yuva-Paylor LA, Paylor R. Marble burying reflects a repetitive and perseverative behavior more than novelty-induced anxiety. Psychopharmacology. 2009;204(2):361–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nicoll RA. A Brief History of Long-Term Potentiation. Neuron. 2017;93(2):281–90. [DOI] [PubMed] [Google Scholar]
- 40.Sudhof TC, Malenka RC. Understanding synapses: past, present, and future. Neuron. 2008;60(3):469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ujjainwala AL, Courtney CD, Wojnowski NM, Rhodes JS, Christian CA. Differential impacts on multiple forms of spatial and contextual memory in diazepam binding inhibitor knockout mice. J Neurosci Res. 2019;97(6):683–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heglind M, Cederberg A, Aquino J, Lucas G, Ernfors P, Enerbäck S. Lack of the central nervous system- and neural crest-expressed forkhead gene Foxs1 affects motor function and body weight. Molecular and cellular biology. 2005;25(13):5616–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen YJ, Deng SM, Chen HW, Tsao CH, Chen WT, Cheng SJ, et al. Follistatin mediates learning and synaptic plasticity via regulation of Asic4 expression in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2021;118(39). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cong Q, Soteros BM, Wollet M, Kim JH, Sia GM. The endogenous neuronal complement inhibitor SRPX2 protects against complement-mediated synapse elimination during development. Nature neuroscience. 2020;23(9):1067–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jay P, Rougeulle C, Massacrier A, Moncla A, Mattei MG, Malzac P, et al. The human necdin gene, NDN, is maternally imprinted and located in the Prader-Willi syndrome chromosomal region. Nature genetics. 1997;17(3):357–61. [DOI] [PubMed] [Google Scholar]
- 46.Asai T, Liu Y, Di Giandomenico S, Bae N, Ndiaye-Lobry D, Deblasio A, et al. Necdin, a p53 target gene, regulates the quiescence and response to genotoxic stress of hematopoietic stem/progenitor cells. Blood. 2012;120(8):1601–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MacDonald HR, Wevrick R. The necdin gene is deleted in Prader-Willi syndrome and is imprinted in human and mouse. Hum Mol Genet. 1997;6(11):1873–8. [DOI] [PubMed] [Google Scholar]
- 48.Wu RN, Hung WC, Chen CT, Tsai LP, Lai WS, Min MY, et al. Firing activity of locus coeruleus noradrenergic neurons decreases in necdin-deficient mice, an animal model of Prader-Willi syndrome. Journal of neurodevelopmental disorders. 2020;12(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuwajima T, Hasegawa K, Yoshikawa K. Necdin promotes tangential migration of neocortical interneurons from basal forebrain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30(10):3709–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller NL, Wevrick R, Mellon PL. Necdin, a Prader-Willi syndrome candidate gene, regulates gonadotropin-releasing hormone neurons during development. Human molecular genetics. 2009;18(2):248–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karge A, Bonar NA, Wood S, Petersen CP. tec-1 kinase negatively regulates regenerative neurogenesis in planarians. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. eLife. 2012;18(9):1413–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ito H, Morishita R, Sudo K, Nishimura YV, Inaguma Y, Iwamoto I, et al. Biochemical and morphological characterization of MAGI-1 in neuronal tissue. Journal of neuroscience research. 2012;90(9):1776–81. [DOI] [PubMed] [Google Scholar]
- 54.Chibuk TK, Bischof JM, Wevrick R. A necdin/MAGE-like gene in the chromosome 15 autism susceptibility region: expression, imprinting, and mapping of the human and mouse orthologues. BMC genetics. 2001;2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roll P, Rudolf G, Pereira S, Royer B, Scheffer IE, Massacrier A, et al. SRPX2 mutations in disorders of language cortex and cognition. Proceedings of the National Academy of Sciences of the United States of America. 2006;15(7):1195–207. [DOI] [PubMed] [Google Scholar]
- 56.Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 2005;123(1):49–63. [DOI] [PubMed] [Google Scholar]
- 57.Mazan-Mamczarz K, Galbán S, López de Silanes I, Martindale JL, Atasoy U, Keene JD, et al. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J, Cho SJ, Shu L, Yan W, Guerrero T, Kent M, et al. Translational repression of p53 by RNPC1, a p53 target overexpressed in lymphomas. Genes & development. 2011;25(14):1528–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nosyreva E, Kavalali ET. Activity-dependent augmentation of spontaneous neurotransmission during endoplasmic reticulum stress. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30(21):7358–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lahr RM, Fonseca BD, Ciotti GE, Al-Ashtal HA, Jia JJ, Niklaus MR, et al. La-related protein 1 (LARP1) binds the mRNA cap, blocking eIF4F assembly on TOP mRNAs. eLife. 2017;6:e24146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ogami K, Oishi Y, Sakamoto K, Okumura M, Yamagishi R, Inoue T, et al. mTOR- and LARP1-dependent regulation of TOP mRNA poly(A) tail and ribosome loading. Cell reports. 2022;41(4):111548. [DOI] [PubMed] [Google Scholar]
- 62.Gentilella A, Morón-Duran FD, Fuentes P, Zweig-Rocha G, Riaño-Canalias F, Pelletier J, et al. Autogenous Control of 5′TOP mRNA Stability by 40S Ribosomes. Molecular cell. 2017;67(1):55–70.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun W, Laubach K, Lucchessi C, Zhang Y, Chen M, Zhang J, et al. Fine-tuning p53 activity by modulating the interaction between eukaryotic translation initiation factor eIF4E and RNA-binding protein RBM38. Genes & development. 2021;35(7–8):542–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Delbridge ARD, Kueh AJ, Ke F, Zamudio NM, El-Saafin F, Jansz N, et al. Loss of p53 Causes Stochastic Aberrant X-Chromosome Inactivation and Female-Specific Neural Tube Defects. Cell reports. 2019;27(2):442–54.e5. [DOI] [PubMed] [Google Scholar]
- 65.LaRese TP, Rheaume BA, Abraham R, Eipper BA, Mains RE. Sex-Specific Gene Expression in the Mouse Nucleus Accumbens Before and After Cocaine Exposure. Journal of the Endocrine Society. 2019;3(2):468–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(42):16633–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berger C, Qian Y, Chen X. The p53-estrogen receptor loop in cancer. Current molecular medicine. 2013;13(8):1229–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing results can be found at doi.org/10.6084/m9.figshare.23560812.v1.