

Abstract
Viral myocarditis is an important cause of human morbidity and mortality for which reliable and effective therapy is lacking. Using reovirus strain 8B infection of neonatal mice, a well-characterized experimental model of direct virus-induced myocarditis, we now demonstrate that myocardial injury results from apoptosis. Proteases play a critical role as effectors of apoptosis. The activity of the cysteine protease calpain increases in reovirus-infected myocardiocytes and can be inhibited by the dipeptide alpha-ketoamide calpain inhibitor Z-Leu-aminobutyric acid-CONH(CH2)3-morpholine (CX295). Treatment of reovirus-infected neonatal mice with CX295 protects them against reovirus myocarditis as documented by (i) a dramatic reduction in histopathologic evidence of myocardial injury, (ii) complete inhibition of apoptotic myocardial cell death as identified by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling, (iii) a reduction in serum creatine phosphokinase, and (iv) improved weight gain. These findings are the first evidence for the importance of a calpain-associated pathway of apoptotic cell death in viral disease. Inhibition of apoptotic signaling pathways may be an effective strategy for the treatment of viral disease in general and viral myocarditis in particular.
The mechanisms by which viruses produce cytopathic effects in their host cells are not well understood. Such knowledge is essential to an understanding of viral pathogenesis and development of novel antiviral therapies. Apoptosis is a mechanism of active cell death distinct from necrosis, characterized by DNA fragmentation, cell shrinkage, and membrane blebbing without rupture (26). Apoptosis plays a critical role in many physiologic (28, 74), as well as infectious and noninfectious, pathologic conditions (72). Viruses may either promote or inhibit apoptosis as a strategy to maximize pathogenicity in their hosts (40, 54, 67). Several viruses, including adenovirus, poxviruses, herpesviruses, and human papillomavirus, proliferate and evade host immune responses by interfering with programmed cell death (1, 19, 31, 68). Many other viruses, such as human immunodeficiency virus, human T-cell leukemia virus, influenza virus, measles virus, rubella virus, poliovirus, human herpesvirus 6, Sindbis virus, and reoviruses, cause cytopathic effect by induction of apoptosis in their target cells (11, 14, 21–23, 34, 40, 42, 50, 70).
We have used reovirus-induced apoptosis as an experimental model system to study the viral and cellular mechanisms involved in apoptotic cell death (39). Reoviruses are nonenveloped viruses that contain a genome of segmented, double-stranded RNA. Infection of cultured fibroblasts and epithelial cells with reoviruses induces apoptosis. Reoviral strains differ in the efficiencies with which they induce this cellular response, and these differences are determined by the viral S1 gene (44, 69). Apoptosis also occurs following reovirus infection in vivo and colocalizes with areas of pathologic injury (38, 39). This finding suggests that apoptosis is an important mechanism of tissue damage in reoviral infection.
Reovirus strain 8B is a reassortant reovirus that efficiently produces myocarditis in infected neonatal mice (55, 58). Damage has been shown to be a direct effect of viral infection of myocardiocytes (60). This damage differs from that of several other models of viral myocarditis (such as coxsackievirus and murine cytomegalovirus) in which secondary inflammatory responses, or lymphocyte recognition of viral or self-antigens on myocardial cells, may be the predominant cause of cardiac damage (12, 17, 20, 30, 46). SCID mice infected with reovirus 8B develop myocarditis, and passive transfer of reovirus-specific immune cells is protective, rather than harmful, to 8B-infected mice (58, 60). This finding indicates that immune mechanisms contribute to amelioration rather than induction of reovirus-induced viral injury (60). However, the mechanism by which direct myocardial injury occurs is not well characterized. Since tissue damage occurs by apoptosis in other in vivo models of reoviral infection (38), and apoptosis has been suggested in some models of viral myocarditis (6, 25), we wished to determine if reoviral myocarditis occurs as a result of apoptotic cell injury and, if so, whether manipulation of known signaling pathways preceding apoptosis is protective.
Protease cascades appear to play critical roles as effectors of apoptosis, as with the cysteine proteases caspases and calpain (10, 32, 41, 62, 79). Caspases are the most extensively investigated members of this class of protease and have been implicated in a wide variety of apoptotic models. However, the role of calpain in apoptosis has been recognized recently. Calpain is a calcium-dependent neutral cysteine protease that is ubiquitous in the cytosols of many cell types (35, 63). Calpains have recently been implicated in several models of apoptosis, including dexamethasone-induced thymocyte apoptosis (65), neuronal cell apoptosis (36), neutrophil apoptosis (64), ischemia-induced rat liver apoptosis (27, 61), myonuclear apoptosis in limb-girdle dystrophy (3), and chemical hypoxia-induced apoptosis of rat myocytes (8). We have recently shown that reovirus-induced apoptosis in vitro is preceded by increased cellular calpain activity and is inhibited by two classes of calpain inhibitors (13).
We now show that reovirus 8B-induced myocarditis occurs by apoptosis. Calpain activity increases in cardiomyocytes following infection with reovirus 8B, and calpain inhibition reduces myocardial injury and morbidity in infected mice. This is evidence that interference with apoptotic signaling pathways may prove of benefit as a therapeutic strategy in the treatment of viral infection in general and viral myocarditis in particular.
MATERIALS AND METHODS
Virus.
Reovirus 8B is an efficiently myocarditic reovirus that has been previously characterized (58). 8B stocks were subjected to plaque assay three times and passaged twice in mouse L cells prior to use.
Mice.
Swiss-Webster (Taconic) mouse litters were housed in individual filter-topped cages in an American Association for Laboratory Animal Care-accredited animal facility. All animal procedures were performed under protocols approved by the appropriate institutional committees.
Mouse inoculations.
Two-day-old Swiss-Webster (Taconic) mice were intramuscularly inoculated with 1,000 PFU of 8B reovirus in the left hind limb (20-μl volume). Mock-infected mice received gel saline vehicle inoculation (equal volume) (137 mM NaCl, 0.2 mM CaCl2, 0.8 mM MgCl2, 19 mM H3BO3, 0.1 mM Na2B4O7, 0.3% gelatin).
Histologic analysis.
At 7 days postinfection, mice were sacrificed and hearts were immediately immersed in 10% buffered formalin solution. After being mounted as transverse sections, hearts were embedded in paraffin and sectioned to 6 μm in thickness. For quantification of degree of myocardial injury, hematoxylin- and eosin-stained midcardiac sections (at least six per heart) were examined at a ×125 magnification by light microscopy and scored blindly. Scoring was performed using a previously validated system (58), with scores ranging from 0 to 4 (0, no lesions; 1, one or a few small lesions; 2, many small or a few large lesions; 3, multiple small and large lesions; and 4, massive lesions). Twenty-three to 24 mice were scored from each group.
DNA fragmentation.
The presence of internucleosomal DNA cleavage in myocardial tissue was investigated by phenol-chloroform extraction of DNAs from 8B-infected and mock-infected hearts and precipitation in 95% ethyl alcohol. The DNA was then end labeled with 5 μCi of [32P]dGTP using 10 U of terminal transferase (M187; Promega Corporation), resolved by electrophoresis on a 2% agarose gel, fixed in 5% acetic acid–5% methanol, dried, and scanned on a Instant Imager (Packard Instrument Company).
TUNEL.
Evaluation of fragmented DNA was performed by terminal deoxynucleotidyltransferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL), as previously described (38). Paraffin-embedded cardiac midsections were prepared by removing paraffin with xylene and then rehydrating them in 100, 95, and then 70% ethanol solutions. After digestion in proteinase K solution (Boehringer Mannheim) for 30 min at 37°C, slides were pretreated in 0.3% H2O2 in phosphate-buffered saline for 15 min at room temperature and then washed. The TdT labeling reaction was carried out under coverslips in a humidified chamber for 1 h at 37°C with TdT and digoxigenin 11-dUTP. (Boehringer Mannheim). The reaction was stopped with SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) buffer. After being blocked in 2% bovine serum albumin for 10 min, sections were probed with Vectastain ABC (avidin DH and biotinylated enzyme; Vector Laboratories) for 1 h at room temperature, and then visualized with a diaminobenzadine peroxidase substrate kit (Vector Laboratories). Negative and positive controls were used with all reactions.
Viral-antigen stain.
Cardiac midsections were prepared as noted above. Following the hydrogen peroxide incubation, slides were blocked in 2% normal goat serum for 30 min at room temperature. Sections were then incubated in rabbit polyclonal anti-reovirus type 3 Dearing antiserum as the primary antibody (gift of Terence Dermody, Vanderbilt University) at a dilution of 1:1,000 for 1 h at 37°C. Biotinylated goat anti-rabbit antibody was used as the secondary antiserum (1:200 dilution in 2% normal goat serum) for 30 min at 37°C. Sections were probed and visualized as noted above.
Calpain activity in myocytes.
The determination of the presence of calpain-specific spectrin (fodrin) breakdown products (150- and 145-kDa doublet) by immunoblotting was used as an assay of calpain activity (36). Mouse primary cardiac myocyte cultures were prepared as previously described (5). Cells were plated at 1.6 × 106 cells/well in 24-well plates and incubated for 48 h. Cells were then infected with reovirus strain 8B (multiplicity of infection [MOI], 20, in Dulbecco modified Eagle medium [DMEM]) or mock infected (DMEM) and then incubated at 37°C. Mock-infected cells were harvested at 48 h. 8B-infected cells were harvested at 24, 48, and 72 h postinfection. Cell lysates were prepared by sonication in lysis buffer (15 mM Tris [pH 7.4], 10 mM EDTA, 0.1% NP-40, 20% glycerol, 50 mM β-mercaptoethanol, 50 μg of pepstatin per ml, 100 μg of leupeptin per ml, 1 mM phenylmethylsulfonyl fluoride), and the cytoplasmic fractions were run on a 7.5% polyacrylamide gel. Protein loading for these gels (25 μg/well) was normalized by protein concentration analysis of cell lysates. Following transfer (15 V overnight), the nitrocellulose membrane was blocked in 5% nonfat dried milk–Tris normal saline for 2 h, probed with anti-fodrin mouse monoclonal antibody (ICN) at a dilution of 1:1,000 for 1.5 h, and then washed. Membranes were then incubated in anti-mouse immunoglobulin G horseradish peroxidase-linked whole antibody (Amersham) at a dilution of 1:1,000, as the secondary antibody. After the membranes were washed, ECL Plus (Amersham) was used for detection.
In additional experiments, primary cardiac myocytes were infected with 8B reovirus (using the method described above) with and without pretreatment in CX295 (100 μM). Cell lysates were prepared and analyzed for calpain activity by immunoblotting as described above.
Specificity of the calpain inhibitor CX295.
Z-Leu-aminobutyric acid- CONH(CH2)3-morpholine (CX295) is a dipeptide alpha-ketoamide compound which inhibits calpain at the active site (kindly provided by Gary Rogers at Cortex Pharmaceuticals, Inc.). To determine the efficacy of CX295 as a calpain inhibitor, 10 μg of purified μ-calpain (porcine RBC; Calbiochem) was added to the preferred fluorogenic calpain substrate sucrose-Leu-Tyr-amino-methyl-coumarin (SLY-AMC) in the presence and absence of CX295 (100 μM), as well as in the presence of the pan-caspase inhibitor Z-d-DCB (100 μM). The calpain assay was performed as previously described (16). Proteolytic hydrolysis of the substrate by purified calpain liberates the highly fluorescent AMC moiety. Fluorescence at a 380-nm excitation and 460-nm emission was quantified with a Hitachi F2000 spectrophotometer. An AMC standard curve was determined for each experiment. Calpain activity was expressed in picomoles of AMC released per minute of incubation time per microgram of purified calpain.
To determine the specificity of CX295 as a calpain inhibitor, 10 ng of purified caspase 3 (Upstate) was added to the preferred fluorogenic caspase-3 substrate DEVD-AMC in the presence and absence of the pan-caspase inhibitor Z-d-DCB (100 μM), as well as CX295 (100 μM). In addition, 57 ng of purified caspase-1 (provided by Nancy Thornberry, Merck) was added to the preferred fluorogenic caspase-1 substrate YVAD-AMC in the presence and absence of Z-d-DCB, as well as CX295. The caspase activity assay was performed as previously described (16). Caspase activity was expressed as picomoles of AMC released per minute of incubation time per nanogram of purified caspase. Experiments were all performed in triplicate.
Calpain inhibition in vivo.
For calpain inhibition experiments, animals received daily intraperitoneal injections of either active CX295 (70 mg/kg of body weight in a 50-μl volume) or its inactive saline diluent. The first dose was given 30 min prior to infection with 8B virus. A total of six doses were given, at 24-h intervals. Mice were sacrificed at 7 days postinfection.
Viral titer determination.
Injected hind limbs and whole hearts were placed in 1 ml of gel saline and immediately frozen at −70°C. After three freeze (−70°C)-thaw (37°C) cycles, the tissues were sonicated approximately 15 to 30 s by using a microtip probe (Heat Systems model XL2020) until a homogenous solution was obtained. The virus suspensions were serially diluted in 10-fold steps in gel saline and placed in duplicate on L-cell monolayers for plaque assay, as previously described (13). Virus titers were expressed as log10 PFU per milliliter.
Serum CPK.
Following decapitation of mice, whole blood was collected from individual mice into plasma separator tubes with lithium heparin to prevent coagulation (Microtainer; Becton Dickinson). Samples were collected from 8B-infected mice treated with CX295 (n = 20), 8B-infected mice treated with the inactive diluent (n = 20), and uninfected age-matched controls (n = 9). Serum creatine phosphokinase (CPK) measurements were performed by the University of Colorado Health Sciences Center Clinical Laboratory and were expressed as units per liter.
Growth.
Mice were infected with 10 PFU of 8B reovirus. Infected drug-treated (n = 15) and infected control (n = 15) mice were weighed daily on days 0 to 14 postinfection. Additional experiments using a higher dose of virus (1,000 PFU) were also completed, with daily weighing on days 0 to 7 postinfection. In these experiments, weights were also compared to those of normal age-matched uninfected mice.
Statistics.
The results of all experiments are reported as means ± standard errors of the means. Means were compared using parametric two-tailed t tests (Graph Pad; Prism), and differences were considered significant if P values were <0.05.
RESULTS
(i) Reovirus 8B induces myocardial injury by apoptosis.
Mice were inoculated intramuscularly into the hind limb with either 1,000 PFU of strain 8B reovirus or gel saline (mock infection). At 7 days postinfection, mice were sacrificed and transverse cardiac sections were prepared for histologic evaluation. In the 8B-infected hearts, there was marked myocardial disruption and diffuse edema (Fig. 1B). Numerous pyknotic nuclei and apoptotic bodies were present (Fig. 1B; see also Fig. 5E). Despite the degree of myocardial injury, only rare mononuclear cells (predominantly macrophages) and neutrophils were present. Myocardial disruption and edema were not seen in mock-infected hearts (Fig. 1A). Extensive areas of apoptotic TUNEL-positive nuclei were noted in the 8B-infected hearts (Fig. 1D), which correlated with the areas of histologic abnormality described above. Mock-infected hearts were TUNEL negative (Fig. 1C). Large regions of reovirus antigen-positive tissue were noted in the 8B-infected hearts (Fig. 1F) and occurred in the same distribution as the areas of histologic injury and TUNEL-positive cells.
FIG. 1.

Consecutive cardiac midsections from mock-infected (A, C, and E) and reovirus 8B-infected (B, D, and F) neonatal mice 7 days after left hind limb inoculation with 1,000 PFU of strain 8B reovirus or mock inoculation. Hematoxylin- and eosin-stained tissue reveals marked disruption of myocardial architecture in the 8B-infected heart (B) compared to that in the mock-infected heart (A). Despite the degree of injury, there is minimal inflammatory cell infiltrate. The degree of cellularity seen in both mock-infected and infected hearts is normal for neonatal mice. In situ detection of DNA nick ends by TUNEL revealed positively staining nuclei in the same region of an injured 8B-infected heart (D), which are absent in a mock-infected animal (C). Immunohistochemistry with anti-type 3 Dearing reovirus antibody reveals the presence of viral antigen in the areas of myocardial injury in the 8B-infected mouse (F), absent in the mock-infected animal (E). Original magnification, ×25.
FIG. 5.

Cardiac midsections from reovirus 8B-infected neonatal mice treated with the calpain inhibitor CX295 (B, D, F, and H) compared to those from inactive diluent control mice (A, C, E, and G) 7 days following intramuscular inoculation with 1,000 PFU of reovirus 8B. Hematoxylin- and eosin-stained sections at an original magnification of ×25 reveal extensive focal areas of myocardial injury (arrows) in the control animal (A), which is absent in the CX295-treated animal (B), despite identical viral infections. Views at an original magnification of ×50 demonstrate minimal inflammatory cell infiltrate in the affected area (C), but myocardial architecture is dramatically disrupted, compared to that of a CX295-treated mouse (D). At an original magnification of ×100, nuclei with apoptotic morphology are easily seen in the control animal (E), as are cells with condensed and pyknotic nuclei (long arrow) as well as apoptotic bodies (shorter arrows). These characteristics are absent in the drug-treated mouse (F). TUNEL analysis of the control animal reveals extensive areas of positively staining cells in the same regions of injury (G) but no TUNEL-positive areas in the drug-treated mouse (H).
In order to provide further confirmation that the morphological changes seen in virus-infected hearts were indeed due to apoptosis, DNAs were extracted from 8B-infected and mock-infected hearts, end labeled, and analyzed by agarose gel electrophoreses. DNAs from infected hearts, but not mock-infected controls, showed fragmentation into oligonucleosomal-length ladders, characteristic of apoptosis (Fig. 2). Taken together, these findings indicate that reovirus-induced myocardial injury is due to apoptosis.
FIG. 2.
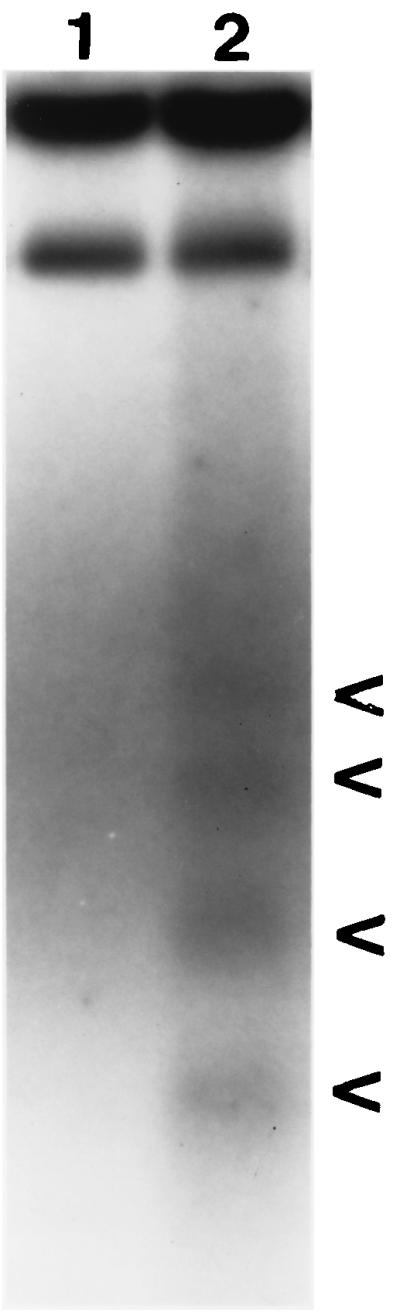
DNA laddering in 8B-infected-neonatal-mouse hearts. DNA was extracted from reovirus 8B-infected and mock-infected hearts and detected by end-labeling analysis. DNA from the heart of a representative 8B-infected mouse (lane 2) is fragmented into oligonucleosomal-length pieces. These fragments result from internucleosomal DNA cleavage, a hallmark of apoptosis. (Arrowheads indicate DNA fragments, ranging in length from 200 to 1,000 bp). Fragmentation is absent in the mock-infected animal (lane 1).
(ii) Calpain is activated in 8B-infected murine cardiac myocytes.
We have previously shown that reovirus infection in L929 cells results in increased calpain activity, which precedes apoptosis (13). We wished to determine whether calpain activity was also increased in myocardial cells following reovirus infection. Proteolysis of spectrin (fodrin), a preferred calpain substrate, was examined as an indication of calpain activation. Intact spectrin (280 kDa) is degraded by calpain, resulting in a characteristic spectrin breakdown product doublet seen at 150 and 145 kDa. Mouse primary cardiac myocyte cultures were infected with reovirus strain 8B (MOI, 20) or mock infected. Lysates were prepared after harvesting of cells at 24, 48, and 72 h postinfection. Western blot analysis of these cytoplasmic fractions revealed increased calpain activity following 8B infection compared to that of mock-infected cytoplasmic fractions. This increase was first detectable at 24 h, reached maximal intensity by 48 h, and was still present at 72 h postinfection (Fig. 3A). Densitometric analysis of calpain-specific breakdown products revealed a peak fourfold increase in calpain activity following virus infection (Fig. 3B). Caspase degradation of spectrin results in breakdown products at 120 kDa, which were not observed in this experiment. These data demonstrate that an increase in calpain activity occurs following infection of myocardiocytes with reovirus strain 8B, in a time course that parallels the onset of apoptosis.
FIG. 3.

Calpain activity in a reovirus-infected primary cardiac myocyte culture. (A) Activity was measured by monitoring proteolysis of spectrin (280 kDa), a preferred calpain substrate, to its calpain-specific breakdown products (150- to 145-kDa doublet) by immunoblot assay. Lysates of 8B-infected cardiomyocytes (MOI, 20) were prepared at 24, 48, and 72 h postinfection and compared to lysates of mock-infected cells at 48 h postinfection (experiments with all conditions were performed in duplicate). Compared to that of mock-infected mice, 8B-infected mice had increased calpain activity (150- to 145-kDa doublet) beginning at 24 h postinfection, which was markedly increased at 48 h postinfection and still present, but decreasing, at 72 h postinfection. No change was detectable in the intact (280-kDa) spectrin band following infection. (B) Densitometric analysis of calpain-specific spectrin breakdown products (150- to 145-kDa doublet) reveals a peak fourfold increase in calpain activity following infection with 8B reovirus.
(iii) CX295 is an effective calpain inhibitor and does not appreciably inhibit caspases.
Based on our findings that (i) 8B-induced myocarditis is due to apoptosis, (ii) calpain is activated in 8B-infected myocardiocytes, and (iii) inhibition of calpain activation inhibits reovirus-induced apoptosis in vitro, we wished to determine if calpain inhibition could prevent 8B-induced myocardial injury in vivo. CX295 is a dipeptide alpha-ketoamide compound that inhibits calpain at the active site and is nontoxic and effective in vivo (4, 48). To confirm the specificity of the compound, the activity of purified calpain was monitored in the presence and absence of CX295. The inactive diluent of CX295 and the pan-caspase inhibitor Z-d-DCB were used as controls. Purified calpain activity (measured as cleavage of SLY-AMC) was markedly reduced by CX295 (Fig. 4A). Calpain activity was decreased from 1,255 ± 22 to 206 ± 4 pmol/min/μg in the presence of CX295 (P < 0.0001). Neither the diluent nor Z-d-DCB controls had appreciable effect on calpain activity.
FIG. 4.




Efficacy and specificity of the calpain inhibitor CX295. The effects of the calpain inhibitor CX295 on purified calpain (A), caspase 3 (B), and caspase 1 (C) activity were evaluated using a fluorogenic substrate assay, in which SLY-AMC, DEVD-AMC, and YVAD-AMC, respectively, served as substrates. Activities are expressed as purified calpain or caspase activity per minute, per micromole or picomole of substrate. One hundred micro-molar CX295 significantly inhibited calpain activity (the asterisk indicates a P of <0.0001) (A) but had minimal effect on caspase 3 or caspase 1 activity. Z-d-DCB, a pan-caspase inhibitor, was used as a control. Z-d-DCB had no effect on calpain activity but nearly completely inhibited caspase 3 and 1 activities (B and C). CX295 also effectively inhibited calpain activity in 8B-infected cardiomyocytes in vitro (D). Calpain activity (measured by densitometric analysis of the 150- to 145-kDa calpain-specific fodrin degradation product) increased by 2.4-fold in 8B-infected cardiomyocytes compared to that in mock-infected cardiomyocytes. Calpain activity was significantly reduced in CX295-treated, 8B-infected cells compared to that in infected, untreated cells (P = 0.04). (The 150- to 145-kDa doublet appears as a single large band due to the gel conditions.)
To further confirm that CX295 is a specific calpain inhibitor and does not inhibit caspases, the activities of purified caspase 1 and caspase 3 were monitored in the presence and absence of CX295, as well as in the presence of the known pan-caspase inhibitor Z-d-DCB (positive control). CX295 had a minimal inhibitory effect on either caspase 1 or caspase 3 activity (measured as cleavage of YVAD-AMC and DEVD-AMC, respectively), whereas Z-d-DCB inhibited both caspases nearly completely (Fig. 4B and C). Purified caspase 1 activity decreased from 6,829 ± 39 to 6,496 ± 36 pmol/min/ng in the presence of CX295, compared to 0 pmol/min/ng in the presence of Z-d-DCB. Purified caspase 3 activity decreased from 1,752 ± 6 to 1,533 ± 9 pmol/min/ng in the presence of CX95, compared to 118 ± 4 pmol/min/ng in the presence of Z-d-DCB.
CX295 also effectively inhibited calpain activity in primary cardiomyocyte culture. Calpain activity was quantified by immunoblotting in reovirus-infected myocytes, in the presence and absence of CX295. Calpain activity (measured by densitometric analysis of the 150- and 145-kDa calpain-specific fodrin breakdown product) increased by 2.4-fold in 8B-infected cardiomyocytes compared to that in mock-infected cardiomyocytes. Calpain activity was significantly reduced in CX295-treated, 8B-infected cells compared to that in infected, untreated cells (P = 0.04) (Fig. 4D and E).
These experiments confirm that CX295 is both an effective and a specific calpain inhibitor.
(iv) Calpain inhibitor CX295 inhibits 8B-induced myocardial injury.
Two-day-old Swiss-Webster mice were infected with 1,000 PFU of 8B virus and received six daily intraperitoneal injections of either active CX295 (70 mg/kg) or its inactive saline diluent as a control (see Materials and Methods). Mice were sacrificed on day 7 postinfection, and myocardial sections were prepared.
Transverse cardiac sections from 8B-infected mice treated with CX295 or its inactive diluent (control) were stained with hematoxylin and eosin and viewed by light microscopy (Fig. 5A to F). Cardiac tissue from control mice (Fig. 5A) showed extensive focal areas of myocardial damage, which were absent in the CX295-treated animals, despite identical viral infection (Fig. 5B). Marked disruption of the normal myocardial architecture was evident in hearts of control mice (Fig. 5C), compared to that of drug-treated animals (Fig. 5D). Nuclei with apoptotic morphology were easily seen within these areas in the control mice (Fig. 5E), including in cells with condensed and pyknotic nuclei, as well as apoptotic bodies. These characteristics were absent in the drug-treated animals (Fig. 5F). Staining by TUNEL of comparable sections from drug-treated and diluent-treated (control) infected mice was examined. Nuclei that stained positive by TUNEL were virtually absent in the drug-treated mice (Fig. 5H), unlike with control infected animals (Fig. 5G).
For quantification of the degree of myocardial injury, hematoxylin- and eosin-stained midcardiac sections (at least six sections per heart) were scored using a previously validated scoring system (58). Twenty infected, CX295-treated animals and 23 control (infected, diluent-treated) mice were evaluated. There was a highly significant reduction in the myocardial injury scores of animals treated with CX295. The mean score for control animals was 3.0 ± 0.1 (range, 2 to 4), compared to 0.6 ± 0.1 (range 0 to 1.5) for CX295-treated animals (P < 0.0001) (Fig. 6).
FIG. 6.

Reduction in myocardial injury score. Myocardial injury of 8B-infected animals was quantified by blindly scoring hematoxylin- and eosin-stained midcardiac sections of drug-treated and control (inactive-diluent-treated) animals upon light microscopy. At least six sections per heart were scored from 20 to 23 animals in each group. A highly significant reduction in myocardial injury score was noted for drug-treated animals. The mean lesion score was 3.0 ± 0.1 (range 2 to 4) for controls, compared to 0.6 ± 0.1 (range 0 to 1.5) for CX295-treated animals. ∗, P < 0.0001. See Materials and Methods for explanations of mean lesion scores.
CPK is an intracellular enzyme present in cardiac and skeletal muscle that is released upon tissue injury. It can be measured in the serum and used as a quantitative marker of skeletal and cardiac muscle damage (2). Blood was collected from infected mice treated with CX295 and inactive diluent-treated controls at 7 days postinfection, as well as uninfected age-matched mice. Serum CPK levels were significantly elevated in 8B-inoculated mice compared to those in uninfected mice, indicative of 8B-induced muscle injury. There was a statistically significant reduction in serum CPK level toward a normal level in CX295-treated mice compared to the level in control mice (Fig. 7). Uninfected age-matched mice had a mean CPK level of 4,521 ± 431 U/liter. 8B-infected mice had a mean CPK level of 5,658 ± 359 U/liter, representing an increase of 1,137 U/liter above normal. Treatment of infected animals with CX295 reduced the mean CPK value to 4,634 ± 350 U/liter, not significantly elevated compared to normal levels but significantly reduced compared to levels in infected, nontreated animals (P < 0.05).
FIG. 7.

Reduction in serum CPK. CPK was measured as a marker of myocardial damage in 8B-infected animals treated with CX295, and levels were compared to those for control (inactive-diluent-treated) infected animals, as well as age-matched uninfected controls. There was a significant reduction in serum CPK toward normal levels in CX295-treated infected mice compared to levels in infected control animals (P < 0.05).
To determine if reductions in myocardial injury were also associated with reduction in viral titer at primary (hind limb) and secondary (heart) sites of replication, the titers of virus in tissues were determined by plaque assay of tissue homogenates. There was a 0.5-log10-PFU/ml reduction in viral titers in the hind limbs of CX295-treated animals compared to those for controls (7.2 ± 0.1 to 6.7 ± 0.2 log10 PFU/ml; P = 0.003). Hearts of CX295-treated animals had a 0.7-log10-PFU/ml reduction in viral titer (6.1 ± 0.2 to 5.4 ± 0.2 log10 PFU/ml; P < 0.01) (Fig. 8). Although these decrements were statistically significant, they were modest in degree, and substantial viral replication occurred in both the drug-treated and the control animals (1,000- to 10,000-fold).
FIG. 8.

Tissue-specific viral titers. Titers were measured by plaque assay at 7 days postinfection from homogenates of limbs (site of primary replication) and hearts (site of secondary replication) of 8B-infected mice. A slight reduction in peak viral titers was seen in the CX295-treated group. Both CX295-treated and control animals showed a >3-log10-unit increase in virus over the input inoculum (103 PFU/mouse).
We wished to determine whether the reduction in myocardial damage caused by CX295 treatment reduced morbidity in mice. We therefore measured growth (weight gain) in infected, drug-treated mice and compared to that in infected, untreated controls. CX295-treated mice had improved growth compared to that of control mice (4.6 ± 0.4 versus 3.6 ± 0.1 g at 7 days postinfection; P = 0.008), and growth was not significantly different from that of uninfected age-matched animals (4.6 ± 0.4 versus 4.8 ± 0.1 g; P = 0.6, not significant) (Fig. 9).
FIG. 9.

Growth of infected mice. Growth, as measured by weight gain, was assessed in 8B-infected mice treated with CX295 and inactive diluent (control). Improved growth was noted for infected, CX295-treated animals at 7 days postinfection compared to the growth of infected controls (∗, P = 0.008). Weights of infected, treated animals were not significantly different from those of uninfected, age-matched controls.
DISCUSSION
Viral myocarditis remains a serious disease without reliable or effective treatment. The events following viral attachment and replication in myocardial tissue that lead to myocarditis are not clearly understood. A variety of mechanisms from various models have been suggested, including direct viral injury and persistence (9, 24), autoimmune phenomena (17, 45, 51), cytokine fluxes (18, 33, 52, 59), inflammation (29, 53), and apoptosis (12, 77, 78). A clearer understanding of pathogenic mechanisms is crucial for the development of effective therapeutic strategies, since currently employed antiviral agents have not made a significant impact on outcomes from this clinical syndrome. Reoviral myocarditis is an ideal model with which to study these events, since myocardial injury is a direct effect of virus infection and does not involve immune-mediated effects.
Reovirus 8B induces myocarditis by apoptosis.
Reovirus 8B induces myocarditis in mice by direct viral injury to myocytes, and we now show that this occurs by induction of apoptosis. This conclusion is supported by the presence of distinctive morphologic criteria upon microscopic examination of infected heart tissues; TUNEL-positive nuclei were found exclusively in regions of viral infection and myocardial injury. The presence of apoptosis was confirmed by the presence of the characteristic intranucleosomal cleavage pattern of extracted DNA. It has been shown previously that multiple viral genes (M1, L1 and L2, and S1) encoding core and attachment proteins are determinants of reovirus-induced acute myocarditis (56). Interactions between these proteins determine myocarditic potential. Several of these genes have been associated with reovirus RNA synthesis and reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures, which are determinants of reovirus myocarditic potential. In addition, the S1 gene, which codes for the viral attachment protein ς1, is the primary determinant of apoptotic potential among strains of reovirus (69). It is likely, therefore, that the extent of reovirus-induced myocardial injury is determined by a combination of host responses, encompassing both the interferon and the apoptosis pathways. Indeed, just as inhibition of the interferon pathway was sufficient to enhance reovirus-induced myocarditis (59), we show here that inhibition of the apoptotic pathway is sufficient to abrogate reovirus-induced myocarditis. Thus, apoptosis is an integral component of reovirus-induced myocardial injury.
Calpain activity is increased in reovirus-infected myocardiocytes, and calpain inhibition is protective against reovirus-induced myocarditis.
Calpain is a calcium-activated cysteine protease that has proven importance with regard to the initiation of apoptosis in the reoviral model, as well as several other unrelated models of apoptosis (see the introduction). We first demonstrated that calpain activity is increased in cardiac myocytes in vitro following 8B infection, in a time course paralleling induction of apoptosis. We then demonstrated that calpain inhibition resulted in a reduction of calpain activity in infected cells and dramatic reductions in reovirus-induced injury, as well as apoptosis. Clinically significant reduction in myocardial injury was documented by reduced serum CPK levels and improved growth in treated mice.
It is likely that CX295 acted primarily by interfering with crucial signal transduction cascades involving calpain, required for induction of apoptotic cell death. Additionally, it is possible that CX295 provided some portion of its effect by inhibiting viral growth at either the primary (hind limb) or secondary (heart) site of replication, since viral titers were slightly lower in drug-treated animals than in controls. Slight reductions in viral titer do not seem a likely explanation for the majority of drug effect, since in prior experiments involving reovirus-induced myocarditis, yields of virus at early and late times postinfection did not correlate with the degree of myocardial injury (57). In addition, efficiently myocarditic and poorly myocarditic reovirus strains replicate to similar titers in the heart; thus, differences in myocarditic potential do not simply reflect viral growth in the heart (56).
One must be cautious in attributing a role for calpain in disease pathogenesis based solely on data derived from calpain inhibition. Currently available calpain inhibitors suitable for in vivo use have weak, but measurable, inhibitory activities against other cysteine proteases. However, the inhibitor employed in our experiments, CX295, is 500- to 900-fold more active against calpains than cathepsins (the Ki for calpain is 0.027 − 0.042 μM, versus a Ki for cathepsin B of 24 μM) and failed to inhibit caspase activity in vitro, as described in this paper. We believe that inhibition of calpain, rather than of other cysteine proteases, is the essential element of CX295's protective effect against 8B-induced myocardial apoptosis and injury.
It is not clear what constitutes the upstream and downstream components of a signaling cascade within which calpain might fit, either during reovirus infection or in other systems where calpain is involved. The mechanisms by which reovirus triggers increased cellular calpain activity are not known but may include initiation of calcium fluxes following viral attachment, as demonstrated with rotavirus, a closely related virus (15); upregulation of growth factors which facilitate calpain activation (37, 66); or upregulation of endogenous calpain activator proteins which have been characterized for several cell types (49). Calpain may play a physiologic role in the regulation of a variety of cellular transcription factors and cell cycle-regulating factors implicated in apoptosis, including Jun, Fos, p53, cyclin D, and NF-κB (3, 7, 73). We have recently shown that activation of NF-κB is required for reovirus-induced apoptosis (J. L. Connolly, S. E. Rodgers, B. Pike, P. Clarke, K. L. Tyler, and T. S. Dermody, Abstr. 17th Ann. Meet. Am. Soc. Virology, abstr. 17-2, 1998), suggesting the possibility that calpain inhibition acts to modulate NF-κB-induced signal transduction. Calpains may also modulate cell death by cleaving Bax, a proapoptotic protein located in the cytosol (75). In addition, the caspase and calpain proteolytic cascades may interact. Caspases may play a role in the regulation of calpain by cleavage of calpastatin, the endogenous inhibitor of calpain (43, 71, 76). Reflexively, calpain may be involved in the proteolytic activation of some caspases (47).
Potential therapeutic efficacy of calpain inhibitors.
In conclusion, our data suggest that reovirus-induced myocarditis occurs by direct viral induction of apoptotic cell death and that injury can be markedly reduced with the use of a calpain inhibitor. To our knowledge, this is the first successful demonstration of the use of calpain inhibition in vivo to ameliorate myocarditis in particular and virus-induced disease in general. However, future experiments are needed to determine whether calpain inhibition remains effective when it is administered after the onset of viral infection. Our results demonstrate the utility of apoptosis inhibition as a strategy for protection against viral infection.
ACKNOWLEDGMENTS
We thank Gary Rogers of Cortex Pharmaceutical for providing the calpain inhibitor CX295 and its inactive diluent. The University of Colorado Cancer Center provided core tissue culture and medium facilities.
This work was supported by Public Health Service grant 1RO1AG14071 from the National Institute of Aging, Merit and REAP grants from the Department of Veterans Affairs, a U.S. Army Medical Research and Material Command grant (USAMRMC 98293015) (K.L.T.), a Young Investigator Award from the National Kidney Foundation (C.L.E.), Public Health Service grant 1RO1HL57161, and North Carolina State University College of Veterinary Medicine grant 204743 (B.S.).
REFERENCES
- 1.Aubert M, Blaho J A. The herpes simplex virus type 1 regulatory protein ICP27 is required for the prevention of apoptosis in human cells. J Virol. 1999;73:2803–2813. doi: 10.1128/jvi.73.4.2803-2813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bachmeir K, Mair J, Offner F, Pummerer C, Neu N. Serum cardiac troponin T and creatine kinase-MB elevations in murine autoimmune myocarditis. Circulation. 1995;92:1927–1932. doi: 10.1161/01.cir.92.7.1927. [DOI] [PubMed] [Google Scholar]
- 3.Baghdiguian S, Martin M, Richard I, Pons F, Astier C, Bourg N, Hay R T, Chemaly R, Haleby G, Loiselet J, Anderson L V, Lopez de Muvigin A, Fardeu M, Mangeat P, Beckmann J S, Lefranc G. Calpain 3 deficiency is associated with myonuclear apoptosis and profound perturbation of the IκBα/NF-KB pathway in limb-girdle muscular dystrophy type 2A. Nat Med. 1999;5:503–511. doi: 10.1038/8385. [DOI] [PubMed] [Google Scholar]
- 4.Bartus R T, Baker K L, Heiser A D, Sawyer S D, Dean R L, Elliott P J, Straub J A. Postischemic administration of AK275, a calpain inhibitor, provides substantial protection against focal ischemic brain damage. J Cerebr Blood Flow Metab. 1994;14:537–544. doi: 10.1038/jcbfm.1994.67. [DOI] [PubMed] [Google Scholar]
- 5.Baty C J, Sherry B. Cytopathogenic effect in cardiac myocytes but not in cardiac fibroblasts is correlated with reovirus-induced myocarditis. J Virol. 1993;67:6295–6298. doi: 10.1128/jvi.67.10.6295-6298.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowles N E, Towbin J A. Molecular aspects of myocarditis. Curr Opin Cardiol. 1998;13:179–184. [PubMed] [Google Scholar]
- 7.Chen F, Lu Y, Kuhn D C, Maki M, Shi X, Demers L M. Calpain contributes to silica-induced IKB-degradation and nuclear factor κB activation. Arch Biochem Biophys. 1997;34:383–388. doi: 10.1006/abbi.1997.0132. [DOI] [PubMed] [Google Scholar]
- 8.Chen S J, Bradley M E, Lee T C. Chemical hypoxia triggers apoptosis of cultured neonatal rat cardiac myocytes: modulation by calcium-regulated proteases and protein kinases. Mol Cell Biochem. 1998;178:141–149. doi: 10.1023/a:1006893528428. [DOI] [PubMed] [Google Scholar]
- 9.Chow L H, Beisel K W, McManus B M. Enteroviral infection of mice with severe combined immunodeficiency. Evidence for direct viral pathogenesis of myocardial injury. Lab Investig. 1992;66:24–31. [PubMed] [Google Scholar]
- 10.Cohen G M. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colamussi M L, White M R, Crouch E, Hartshorn K L. Influenza A virus accelerates neutrophil apoptosis and markedly potentiates apoptotic effects of bacteria. Blood. 1999;93:2395–2403. [PubMed] [Google Scholar]
- 12.Colston J T, Chandrasakar B, Freeman G L. Expression of apoptosis-related proteins in experimental coxsackie myocarditis. Cardovasc Res. 1998;38:158–168. doi: 10.1016/s0008-6363(97)00323-4. [DOI] [PubMed] [Google Scholar]
- 13.DeBiasi R L, Squier M K T, Pike B, Wynes M, Dermody T S, Tyler K L. Reovirus-induced apoptosis is preceded by increased cellular calpain activity and is blocked by calpain inhibitors. J Virol. 1999;73:695–701. doi: 10.1128/jvi.73.1.695-701.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dockrell D H, Badley A D, Villacian J S, Heppelman C J, Algeciras A, Ziesvnar S, Yagita H, Lynch D H, Roche P C, Leibson P J, Paya C V. The expression of Fas ligand by macrophages and its upregulation by human immunodeficiency virus infection. J Clin Investig. 1998;101:2394–2405. doi: 10.1172/JCI1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong Y, Zeng C Q, Ball J M, Estes M K, Morris A P. The rotavirus enterotoxin NSP4 mobilizes calcium in human intestinal cells by stimulating phospholipase C-mediated inositol 1,4,5-triphosphate production. Proc Natl Acad Sci USA. 1997;94:3960–3965. doi: 10.1073/pnas.94.8.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edelstein C L, Ling H, Gengaro P E, Nemenoff R A, Bahr B A, Schrier R W. Effect of glycine on prelethal and postlethal increases in calpain activity in rat renal proximal tubules. Kidney Int. 1997;52:1271–1278. doi: 10.1038/ki.1997.452. [DOI] [PubMed] [Google Scholar]
- 17.Fairweather D, Lawson C M, Chapman A J, Brown C M, Booth T W, Papadimitriou J M, Snellam G R. Wild isolates of murine cytomegalovirus induce myocarditis and antibodies that cross-react with virus and cardiac myosin. Immunology. 1998;94:263–270. doi: 10.1046/j.1365-2567.1998.00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freeman G L, Colston J T, Zabalgoitia M, Chandrasekar B. Contractile depression and expression of proinflammatory cytokines and iNOS in viral myocarditis. Am J Physiol. 1998;274:H249–H258. doi: 10.1152/ajpheart.1998.274.1.H249. [DOI] [PubMed] [Google Scholar]
- 19.Galvan V, Brandimarti R, Roizman B. Herpes simplex virus 1 blocks caspase-3-independent and caspase-dependent pathways to cell death. J Virol. 1999;73:3219–3226. doi: 10.1128/jvi.73.4.3219-3226.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gauntt C J. Roles of the humoral response in coxsackievirus B-induced disease. Curr Top Microbiol Immunol. 1997;223:259–282. doi: 10.1007/978-3-642-60687-8_12. [DOI] [PubMed] [Google Scholar]
- 21.Girard S, Couderc T, Destombes J D, Thiesson J, Delpeyroux F, Blondel B. Poliovirus induces apoptosis in the mouse central nervous system. J Virol. 1999;73:6066–6072. doi: 10.1128/jvi.73.7.6066-6072.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ichimi R, Jin-o T, Ito M. Induction of apoptosis in cord blood lymphocytes by HHV-6. J Med Virol. 1999;58:63–68. doi: 10.1002/(sici)1096-9071(199905)58:1<63::aid-jmv10>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 23.Jaworski A, Crowe S M. Does HIV cause depletion of CD4+ T cells in vivo by the induction of apoptosis? Immunol Cell Biol. 1999;77:90–98. doi: 10.1046/j.1440-1711.1999.00798.x. [DOI] [PubMed] [Google Scholar]
- 24.Kanda T, Koike H, Arai M, Wilson J E, Carthy C M, Yang D, McManus B M, Nagai R, Kobayashi I. Increased severity of viral myocarditis in mice lacking lymphocyte maturation. Int J Cardiol. 1999;68:13–22. doi: 10.1016/s0167-5273(98)00250-2. [DOI] [PubMed] [Google Scholar]
- 25.Kawai C. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death: learning from the past for the future. Circulation. 1999;99:1091–1100. doi: 10.1161/01.cir.99.8.1091. [DOI] [PubMed] [Google Scholar]
- 26.Kerr J F R, Wylie A H, Currie A R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohli V, Madden J F, Bentley R C, Clavien P A. Calpain mediates ischemic injury of the liver through modulation of apoptosis and necrosis. Gastroenterology. 1999;116:168–178. doi: 10.1016/s0016-5085(99)70241-6. [DOI] [PubMed] [Google Scholar]
- 28.Krammer P H, Behrmann I, Daniel P, Dhein J, Debatin K M. Regulation of apoptosis in the immune system. Curr Opin Immunol. 1994;6:276–289. doi: 10.1016/0952-7915(94)90102-3. [DOI] [PubMed] [Google Scholar]
- 29.Lee J K, Zaidi S H, Liu P, Dawood F, Cheah A Y, Wen W H, Saiki Y, Rabinovitch M. A serine elastase inhibitor reduces inflammation and fibrosis and preserves cardiac function after experimentally-induced murine myocarditis. Nat Med. 1998;4:1383–1391. doi: 10.1038/3973. [DOI] [PubMed] [Google Scholar]
- 30.Liu P, Martino T, Opavsky M A, Penninger J. Viral myocarditis: balance between viral infection and immune response. Can J Cardiol. 1996;12:935–943. [PubMed] [Google Scholar]
- 31.Marshall W L, Yim C, Gustafson E, Graf T, Sage D R, Hanify K, Williams L, Fingeroth J, Finberg R W. Epstein-Barr virus encodes a novel homolog of the bcl-2 oncogene that inhibits apoptosis and associates with Bax and Bak. J Virol. 1999;73:5181–5185. doi: 10.1128/jvi.73.6.5181-5185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin S J, Green D R. Protease activation during apoptosis: death by a thousand cuts? Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 33.Matsumori A. Cytokines in myocarditis and cardiomyopathies. Curr Opin Cardiol. 1996;11:302–309. doi: 10.1097/00001573-199605000-00011. [DOI] [PubMed] [Google Scholar]
- 34.Megyeri K, Berencsi K, Halazonetis T D, Prendergast G C, Gri G, Plotkin S A, Rovera G, Gonczol E. Involvement of a p53-dependent pathway in rubella virus-induced apoptosis. Virology. 1999;259:74–84. doi: 10.1006/viro.1999.9757. [DOI] [PubMed] [Google Scholar]
- 35.Murachi T. Calpain and calpastatin. Trends Biochem Sci. 1983;8:167–169. [Google Scholar]
- 36.Nath R, Raser K J, Stafford D, Hajimohammadreza I, Posner A, Allen H, Talanian R V, Yuen P, Gilbertsen R B, Wang K K. Non-erythroid α-spectrin breakdown by calpain and interleukin 1 B-converting-enzyme-like protease(s) in apoptotic cells: contributory roles of both protease families in neuronal apoptosis. Biochem J. 1996;319:686–690. doi: 10.1042/bj3190683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neuberger T, Chakrabarti A K, Russell T, DeVreis G H, Hogan E L, Banik N L. Immunolocalization of cytoplamic and myelin mCalpain in transfected Schwann cells. I. Effect of treatment with growth factors. J Neurosci Res. 1997;47:521–530. [PubMed] [Google Scholar]
- 38.Oberhaus S M, Smith R L, Clayton G H, Dermody T S, Tyler K L. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J Virol. 1997;71:2100–2106. doi: 10.1128/jvi.71.3.2100-2106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oberhaus S M, Dermody T S, Tyler K L. Apoptosis and the cytopathic effects of reovirus. Curr Top Microbiol Immunol. 1998;233:23–49. doi: 10.1007/978-3-642-72095-6_2. [DOI] [PubMed] [Google Scholar]
- 40.O'Brien V. Viruses and apoptosis. J Gen Virol. 1998;79:1833–1834. doi: 10.1099/0022-1317-79-8-1833. [DOI] [PubMed] [Google Scholar]
- 41.Patel T, Gores G J, Kaufmann S H. The role of proteases during apoptosis. FASEB J. 1996;10:587–597. doi: 10.1096/fasebj.10.5.8621058. [DOI] [PubMed] [Google Scholar]
- 42.Pignata C, Fiore M, deFilippo S, Cavalcanti M, Gaetaniello L, Scotese I. Apoptosis as a mechanism of peripheral blood mononuclear cell death after measles and varicella-zoster virus infections in children. Pediatr Res. 1998;43:77–83. doi: 10.1203/00006450-199801000-00012. [DOI] [PubMed] [Google Scholar]
- 43.Porn-Ares M I, Samali A, Orenius S. Cleavage of the calpain inhibitor, calpastatin, during apoptosis. Cell Death Differ. 1998;5:1028–1033. doi: 10.1038/sj.cdd.4400424. [DOI] [PubMed] [Google Scholar]
- 44.Rodgers S E, Barton E S, Oberhaus S M, Pike B, Gibson C A, Tyler K L, Dermody T S. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J Virol. 1997;71:2540–2546. doi: 10.1128/jvi.71.3.2540-2546.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rose N R, Herskowitz A, Neumann D A. Autoimmunity in myocarditis: models and mechanisms. Clin Immunol Immunopathol. 1993;68:95–99. doi: 10.1006/clin.1993.1102. [DOI] [PubMed] [Google Scholar]
- 46.Rose N R, Hill S L. The pathogenesis of postinfectious myocarditis. Clin Immunol Immunopathol. 1996;80:S92–S99. doi: 10.1006/clin.1996.0146. [DOI] [PubMed] [Google Scholar]
- 47.Ruiz-Vela A, Gonzalez de Buitrago G, Martinez A-C. Implication of calpain in caspase activation during B cell clonal deletion. EMBO J. 1999;18:4988–4998. doi: 10.1093/emboj/18.18.4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saatman K E, Murai H, Bartus R T, Smith D H, Hayward N J, Perri B R, McIntosh T K. Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc Natl Acad Sci USA. 1996;93:3428–3433. doi: 10.1073/pnas.93.8.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salamino F, DeTullio R, Mengotti P, Viotti P L, Melloni E, Pontremoli S. Site-directed activation of calpain is promoted by a membrane-associated natural activator protein. Biochem J. 1993;290:191–197. doi: 10.1042/bj2900191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schultz-Cherry S, Hinshaw V S. Influenza virus neuraminidase activates latent transforming growth factor beta. J Virol. 1996;70:8624–8629. doi: 10.1128/jvi.70.12.8624-8629.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwimmbeck P L, Huber S A, Schultheiss H P. Roles of T cells in coxsackie-B induced disease. Curr Top Microbiol Immunol. 1997;223:283–303. doi: 10.1007/978-3-642-60687-8_13. [DOI] [PubMed] [Google Scholar]
- 52.Seko Y, Takahashi N, Yagita H, Okumura K, Yazaki Y. Expression of cytokine mRNA's in murine hearts with acute myocarditis caused by coxsackievirus b3. J Pathol. 1997;183:105–108. doi: 10.1002/(SICI)1096-9896(199709)183:1<105::AID-PATH1094>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 53.Seko Y, Takahashi N, Azuma M, Yagita H, Okumura K, Yazaki Y. Expression of costimulatory molecule CD40 in murine heart with acute myocarditis and reduction of inflammation by treatment with anti-CD40L/B7–1 monoclonal antibodies. Circ Res. 1998;83:463–469. doi: 10.1161/01.res.83.4.463. [DOI] [PubMed] [Google Scholar]
- 54.Shen Y, Shenk T E. Viruses and apoptosis. Curr Opin Genet Dev. 1995;5:105–111. doi: 10.1016/s0959-437x(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 55.Sherry B. Pathogenesis of reovirus myocarditis. In: Tyler K L, Oldstone M B A, editors. Reoviruses II: cytopathogenicity and pathogenesis. Berlin, Germany: Springer-Verlag; 1998. pp. 51–66. [Google Scholar]
- 56.Sherry B, Blum M A. Multiple viral core proteins are determinants of reovirus-induced acute myocarditis. J Virol. 1994;63:8461–8465. doi: 10.1128/jvi.68.12.8461-8465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sherry B, Baty C J, Blum M A. Reovirus-induced acute myocarditis in mice correlates with viral RNA synthesis rather than generation of infectious virus in cardiac myocytes. J Virol. 1996;70:6709–6715. doi: 10.1128/jvi.70.10.6709-6715.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sherry B, Schoen F J, Wenske E, Fields B N. Derivation and characterization of an efficiently myocarditic reovirus variant. J Virol. 1989;63:4840–4849. doi: 10.1128/jvi.63.11.4840-4849.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sherry B, Torres J, Blum M A. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J Virol. 1998;72:1314–1323. doi: 10.1128/jvi.72.2.1314-1323.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sherry B, Li X-Y, Tyler K L, Cullen J M, Virgin H W. Lymphocytes protect against and are not required for reovirus-induced myocarditis. J Virol. 1993;67:6119–6124. doi: 10.1128/jvi.67.10.6119-6124.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sindram D, Kohli V, Madden J F, Clavien P A. Calpain inhibition prevents sinusoidal endothelial cell apoptosis in the cold ischemic rat liver. Transplantation. 1999;68:136–140. doi: 10.1097/00007890-199907150-00025. [DOI] [PubMed] [Google Scholar]
- 62.Solary E, Eymin B, Droin N, Haugg M. Proteases, proteolysis, and apoptosis. Cell Biol Toxicol. 1998;14:11–32. doi: 10.1023/a:1007481921502. [DOI] [PubMed] [Google Scholar]
- 63.Sorimachi H, Ishiura S, Suzuki K. Structure and physiological function of calpains. Biochem J. 1997;328:721–732. doi: 10.1042/bj3280721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Squier M K, Sehnert A J, Sellins K S, Malkinson A M, Takanoand E, Cohen J J. Calpain and calpastatin regulate neutrophil apoptosis. J Cell Physiol. 1999;178:311–319. doi: 10.1002/(SICI)1097-4652(199903)178:3<311::AID-JCP5>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 65.Squier M K T, Cohen J J. Calpain, an upstream regulator of thymocyte apoptosis. J Immunol. 1997;158:3690–3697. [PubMed] [Google Scholar]
- 66.Strong J E, Tang D, Lee P W K. Evidence that the epidermal growth factor receptor on host cells confers reovirus infection efficiency. Virology. 1993;197:405–411. doi: 10.1006/viro.1993.1602. [DOI] [PubMed] [Google Scholar]
- 67.Teodoro J G, Branton P E. Regulation of apoptosis by viral gene products. J Virol. 1997;71:1739–1746. doi: 10.1128/jvi.71.3.1739-1746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thomas M, Banks L. Human papillomavirus (HPV) E6 interactions with Bak are conserved amongst E6 proteins from high and low risk HPV types. J Gen Virol. 1999;80:1513–1517. doi: 10.1099/0022-1317-80-6-1513. [DOI] [PubMed] [Google Scholar]
- 69.Tyler K L, Squier M K T, Rodgers S E, Schneider B E, Oberhaus S M, Grdina T A, Cohen J J, Dermody T S. Differences in the capacity of reovirus strains to induce apoptosis are determined by viral attachment protein ς1. J Virol. 1995;69:6972–6979. doi: 10.1128/jvi.69.11.6972-6979.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Valentin H, Azocar O, Horvat B, Willeims R, Garrone R, Evlasher A, Toribio M L, Rabourdin-Combe C. Measles virus infection induces terminal differentiation of human thymic epithelial cells. J Virol. 1999;73:2212–2221. doi: 10.1128/jvi.73.3.2212-2221.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang K K, Postmantur R, Nadimpalli R, Nath R, Mohan P, Nixon R A, Talanian R V, Keegan M, Herzog L, Allen H. Caspase-mediated fragmentation of calpain inhibitor protein calpastatin during apoptosis. Arch Biochem Biophys. 1998;356:187–196. doi: 10.1006/abbi.1998.0748. [DOI] [PubMed] [Google Scholar]
- 72.Wang K K W, Yuen P. Calpain inhibition: an overview of its therapeutic potential. Trends Pharmacol Sci. 1994;15:412–419. doi: 10.1016/0165-6147(94)90090-6. [DOI] [PubMed] [Google Scholar]
- 73.Watt F, Molloy P L. Specific cleavage of transcription factors by the thiol protease, m-calpain. Nucleic Acids Res. 1993;21:5092–5100. doi: 10.1093/nar/21.22.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weller M, Schulz J B, Wullner U, Loschmann P A, Klockgether T, Dichgans J. Developmental and genetic regulation of programmed neuronal death. J Neural Transm. 1997;50:115–123. doi: 10.1007/978-3-7091-6842-4_12. [DOI] [PubMed] [Google Scholar]
- 75.Wood D E, Thomas A, Devi L A, Berman Y, Beavis R C, Reed J C, Newcomb E W. Bax cleavage is mediated by calpain during drug-induced apoptosis. Oncogene. 1998;17:1069–1078. doi: 10.1038/sj.onc.1202034. [DOI] [PubMed] [Google Scholar]
- 76.Wood D E, Newcomb E W. Caspase-dependent activation of calpain during drug-induced apoptosis. J Biol Chem. 1999;274:8309–8315. doi: 10.1074/jbc.274.12.8309. [DOI] [PubMed] [Google Scholar]
- 77.Yang D, Yu J, Luo Z, Carthy C M, Wilson J E, Liu Z, McManus B M. Viral myocarditis: identification of five differentially expressed genes in coxsackievirus B3-infected mouse heart. Circ Res. 1999;84:704–712. doi: 10.1161/01.res.84.6.704. [DOI] [PubMed] [Google Scholar]
- 78.Yeh E T. Life and death in the cardiovascular system. Circulation. 1997;95:782–786. doi: 10.1161/01.cir.95.4.782. [DOI] [PubMed] [Google Scholar]
- 79.Zhivotovsky B, Burgess D H, Vanags D M, Orrenius S. Involvement of cellular proteolytic machinery in apoptosis. Biochem Biophys Res Commun. 1997;230:481–488. doi: 10.1006/bbrc.1996.6016. [DOI] [PubMed] [Google Scholar]