

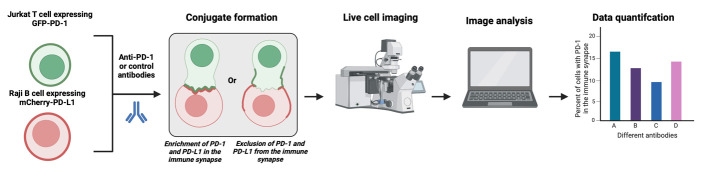
Abstract
PD-1 is an immune checkpoint on T cells. Antibodies to PD-1 or its ligand PD-L1 are gaining popularity as a leading immunotherapy approach. In the US, 40% of all cancer patients will be treated with anti-PD-1 or anti-PD-L1 antibodies but, unfortunately, only 30% will respond, and many will develop immune-related adverse events. There are nine FDA-approved anti-PD-1/PD-L1 antibodies, and approximately 100 are in different stages of clinical development. It is a clinical challenge to choose the correct antibody for a given patient, and this is critical in advanced malignancies, which often do not permit a second-line intervention. To resolve that, an in vitro assay to compare the performance of the different anti-PD-1/PD-L1 antibodies is not only a critical tool for research purposes but also a possible tool for personalized medicine. There are some assays describing the binding affinity and function of anti-PD-1/PD-L1 antibodies. However, a significant limitation of existing assays is that they need to consider the location of PD-1 in the immune synapse, the interface between the T cell and tumor cells, and, therefore, ignore a critical component in its biology. To address this, we developed and validated an imaging-based assay to quantify and compare the ability of different anti-PD-1/PD-L1 antibodies to remove PD-1 from the immune synapse. We correlated that with the same antibodies' ability to increase cytokine secretion from the targeted cells. The strong correlation between PD-1 location and its function in vitro and in vivo within the antibody treatment setting validates this assay's usability, which is easily recordable and straightforward.
Key features
• Live-cell imaging quantifies and compares how anti-PD-1 and anti-PD-L1 antibodies disrupt PD-1 localization, causing the removal of PD-1 during immune synapse formation.
• Hao et al. [1] validated the protocol, and the findings were extended to a live confocal microscopy method.
• It requires a Zeiss LSM 900 confocal microscope and appropriate imaging software and is optimized for the latest version of Zen Blue.
• Anti-PD-1 antibodies are commonly used in cancer therapies, and this protocol optimizes the analysis of their effectiveness.
Keywords: PD-1, PD-L1, B cells, Confocal microscopy, Immunotherapy, T cells, Cancer, Immune checkpoints, Immune synapse
Graphical overview
Background
PD-1, or programmed cell death protein 1, is a protein found on the surface of T cells. PD-1 acts as a brake on T cells, preventing them from overreacting and attacking healthy tissues. This is important for maintaining immune tolerance and preventing autoimmune diseases. However, cancer cells can sometimes exploit the PD-1 pathway to evade immune attacks. They can do this by expressing high levels of PD-L1 (programmed cell death ligand 1), the ligand for PD-1 that inhibits T-cell activity, and in some cases also PD-L2, a higher-affinity ligand for PD-1 [2].
This is where PD-1 inhibitors come in. These drugs block the interaction between PD-1 and PD-L1, allowing T cells to recognize and destroy cancer cells [3,4]. PD-1 and PD-L1 inhibitors have revolutionized cancer treatment, and they are now used to treat a variety of cancers including melanoma, lung cancer, and head and neck cancer. Unlike traditional chemotherapy, which often has short-lived effects, anti-PD-1/PD-L1 antibodies can induce long-lasting tumor shrinkage and even complete remission in some patients. Compared to chemotherapy, anti-PD-1/PD-L1 antibodies have a more favorable side effect profile. While they can cause immune-related adverse events, these are generally less severe and more manageable than the toxicities associated with chemotherapy. Therefore, anti-PD-1/PD-L1 antibodies have become the standard of care for many cancer types, and their use is rapidly expanding to new applications. This provides more treatment options for patients with advanced or aggressive cancers who may not have had many choices before. Unfortunately, not all patients respond to anti-PD-1 therapy [5]. The reasons for this are complex and not fully understood. Still, it highlights the need for further research to identify biomarkers and in vitro assays that can predict who will benefit most from this treatment.
There are currently nine FDA-approved anti-PD-1/PD-L1 antibodies [6], each with its specific indications and potential side effects: pembrolizumab (Keytruda), nivolumab (Opdivo), cemiplimab (Libtayo), atezolizumab (Tecentriq), avelumab (Bavencio), durvalumab (Imfinzi), dostarlimab (Jemperli), toripalimab (Loqtorzi), and tislelizumab (Tevimbra). It is important to note that this is a partial list, and the number of anti-PD-1 and PD-L1 antibodies in clinical trials continually evolves. Still, over 100 different anti-PD-1 and PD-L1 antibodies are currently in various stages of clinical development, according to the ClinicalTrials.gov database [7]. This number encompasses multiple anti-PD-1/PD-L1 agents, including monoclonal antibodies, bispecific antibodies, and antibody-drug conjugates (ADCs).
With so many different anti-PD-1/PD-L1 antibodies in development, there are several reasons why in vitro assays are crucial for studying them. In vitro assays offer a safe and relatively inexpensive way to test the properties of these antibodies before moving to complex and expensive animal or human studies. In vitro assays provide a controlled environment to study antibodies' specific mechanisms of action [8]. They can help understand how these antibodies interact with PD-1/PD-L1, T, and cancer cells, ultimately leading to better drugs and treatment strategies. In vitro assays can also identify biomarkers that predict which patients are more likely to respond to specific anti-PD-1/PD-L1 therapy [9]. This personalized approach can help optimize treatment for individual patients and avoid unnecessary side effects for those unlikely to benefit. In vitro assays help compare different formulations or combinations of antibodies, allowing the optimization of their efficacy and safety before clinical trials.
Promega developed an assay to measure the ability of anti-PD-1/PD-L1 antibodies to block immune checkpoint signals [10]. The assay consists of two genetically engineered cell lines, PD-1 effector cells and PD-L1 aAPC/CHO-K1 cells. When cocultured, the PD-1/PD-L1 interaction inhibits T-cell receptor (TCR)-mediated luminescence. When the PD-1/PD-L1 interaction is disrupted, TCR activation induces luminescence via activation of the NFAT pathway, which can be detected by adding bio-glo reagent and quantitation with a luminometer. Another report described a flow cytometry assay to evaluate the T-cell binding status of the anti-PD-1 antibodies using a single drop of peripheral blood [11]. Another group established a PD-1/PD-L1 blockade assay based on surface plasmon resonance (SPR) biosensors [12]. This assay immobilizes human PD-1 on a sensor chip, where the binding kinetics of PD-L1 to PD-1 and the blockade rates of PD-1 inhibitors are determined. Compared to other techniques, such as PD-1/PD-L1 pair ELISA, this method offers real-time and label-free detection with advantages including shorter experimental runs and more minor sample quantity requirements. We and others have published in vitro assays used to study the effects of anti-PD-1 antibodies on T-cell proliferation, cytotoxicity (to assess the killing ability of T cells activated by PD-1 blockade against cancer cell lines), and cytokine secretion [13], providing insights into the immune response triggered by PD-1. While in vitro assays are valuable tools, they have limitations. They cannot fully replicate the complex interactions in the human body, and validating findings with in vivo studies and clinical trials is crucial.
The formation of a synapse between a T cell and a tumor cell is a crucial initiator of the immune response, with interactions between the major histocompatibility complex (MHC) and TCR being essential for T-cell activation and killing [14]. Synapse formation causes the clustering of proteins in different regions of the contact area, called supramolecular activation complexes (SMACs). SMACs can be further separated into different subregions: the central SMAC (cSMAC), the peripheral SMAC (pSMAC), and the distal-SMAC (dSMAC), where the cSMAC contains the proteins and checkpoint receptors of interest for this assay [15]. PD-1 and PD-L1 are a part of the group of molecules that centralize to the cSMAC during the formation of the synapse [16]. Interactions between PD-1 and PD-L1 activate the phosphatase SHP2, causing suppression of T-cell activity and overall immune response by preventing stable T-cell interactions from forming. PD-L1 and PD-1 can also develop microclusters within the cSMAC, contributing to their suppressive function [17]. The localization of PD-1 in the synapse when bound or unbound by antibodies allows it to be a prime target for studying the efficacy of different antibodies. Specifically, antibody binding mislocalizes PD-1 and prevents interaction with PD-L1 [1]. The exclusion of antibody-bound PD-1 can be considered size-based, as the immune synapse is observed to exclude molecules above a specific size limit [18]. Removing PD-1 from the synapse prevents PD-1/PD-L1 clustering within the cSMAC and interactions with PD-L1 for inhibitory function. As such, antibodies with different characteristics may affect the localization of PD-1 in the immune synapse to different degrees.
Previously, we reported an in vitro assay to study PD-1 signaling in primary human T cells [13], based on the phospho-flow cytometry method to determine how PD-1 ligation alters the levels of CD3ζ phosphorylation on Tyr142, which can be easily applied to other proximal signaling proteins. We also reported a plate-bound assay to examine the long-term consequences of PD-1 ligation, such as cytokine production and T-cell proliferation. We also described an in vitro superantigen-based assay to evaluate T-cell responses to therapeutic agents targeting the PD-1/PD-L1 axis and immune synapse formation in the presence of PD-1 engagement [13]. Based on this, we developed an imaging-based assay, described here. This protocol utilizes PD-1 GFP-expressing Jurkat T cells and PD-L1-/PD-L2 mCherry-expressing Raji B cells, yielding a model to study the function and location of anti-PD-1/PD-L1/PD-L2 antibodies in real time. This study design requires a Zeiss LSM 900 confocal microscope and an updated version of Zen Blue software, at least version 3.3. Compared with other methods of quantifying PD-1 antibody strength, such as flow cytometry, this method allows for both visual and numerical analysis of synapse formation between B and T cells, simultaneously providing qualitative and quantitative data. Furthermore, it enables live-cell imaging, captures temporal data, and gives insights into the dynamics of these interactions over time. This protocol was validated by Hao et al., 2024.
Materials and reagents
Biological materials
Jurkat T cells (ATCC, catalog number: TIB-152)
Raji B cells (ATCC, catalog number: CCL-86)
Staphylococcus enterotoxin E (SEE) ET404 1 mg (Toxin Technology, catalog number: NC1467973)
Reagents
RPMI 1640 (Thermo Fisher Scientific, GibcoTM, catalog number: 31800)
Opti-MEM (Thermo Fisher Scientific, catalog number: 11058021)
Heat-inactivated fetal bovine serum (FBS) (Thermo Fisher Scientific, GibcoTM, catalog number: 10082147)
Penicillin/streptomycin (P/S) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140)
Human CD3/CD28/CD2 T-cell activator (StemCell Technologies, ImmunoCultTM, catalog number: 10970) (optional; see protocol)
10× Poly-L-Lysine (PLL) solution (0.1% w/v in H2O) (Sigma-Aldrich, Millipore, catalog number: P8920)
Trypan Blue stain (0.4%) (Thermo Fisher Scientific, InvitrogenTM, catalog number: T10282)
Molecular biology grade water (Corning, catalog number: 46-000-CI)
pHR-PD-1-GFP vector and pHR-PD-L1-mCherry vector (AddGene, catalog number: 180819) [19]
Solutions
1× Poly-L-Lysine solution (see Recipes)
RPMI (w/ FBS) media (see Recipes)
Staphylococcus enterotoxin E (SEE) working stock (see Recipes)
Recipes
-
1× Poly-L-Lysine solution
Reagent Final concentration Amount 10× PLL [0.1 % (w/v) in H2O] 1× 100 µL ddH2O n/a 900 µL Total n/a 1,000 µL -
RPMI (w/ FBS) media
Reagent Final concentration Amount RPMI 1640 n/a 500 mL FBS 10% 55 mL P/S 1% 5.5 mL Total n/a 560.5 mL -
Staphylococcus enterotoxin E (SEE) working stock (stored at -20 °C)
Reagent Final concentration Amount Staphylococcus enterotoxin E 1 mg/mL 20 µg ddH2O n/a 20 µL Total n/a 20 µL
Laboratory supplies
15 mL and 50 mL conical tube (Falcon, catalog numbers: 38009, 38010)
12-well cell culture plate (Thermo Fisher Scientific, catalog number: 150200)
Untreated cell culture flask with vent cap (Thermo Fisher Scientific, CorningTM, catalog number: 431463)
MatTek Plates (50 mm dish, No. 0 coverslip, 14 mm glass diameter, uncoated) (MatTek, catalog number: P50G-0-14-F)
Single-channel pipette, Pipetman 0.2–2 µL, 1–10 µL, 2–20 µL, 20–200 µL, 100–1,000 µL (Gilson, catalog numbers: F144054M, F144055M, F144058M, F144059M)
Filtered pipette tips (USA Scientific)
1.5 mL microcentrifuge tubes (Fisher Scientific, catalog number: 05-408-129)
Immersol autofluorescence-free immersion oil 518 F (Zeiss, catalog number: 444960-0000-000)
Aluminum foil (Amazon brand)
Cell counting chamber slides (CountessTM, catalog number: C10228)
KOVATM GlassticTM slide 10 with grids (Fisher Scientific, catalog number: 22-270141)
Cell culture multiwell plate 24 wells (Grenier Bio-One, catalog number: 662160)
Equipment
Confocal microscope (Zeiss, model: LSM 900)
Heracell 150i CO2 incubator (37 °C, 5% CO2) (Thermo Fisher Scientific, catalog number: 50116048)
Digital miniblock heater (VWR, catalog number: 460-0334)
Automated cell counter (InvitrogenTM, CountessTM, catalog number: AMQAX2000)
FisherbrandTM manual differential counter (Fisher Scientific, catalog number: 13-684-141)
5425 centrifuge (Eppendorf, catalog number: 5405000646)
5810R centrifuge (Eppendorf, catalog number: 022625501)
Software and datasets
Image J (Version 1.54)
Zen (Version 3.3 Blue Edition)
Procedure
-
Preparation of T and B cells
Note: For optimal imaging, the assay should be done with a fluorescent PD-1-expressing T-cell line (such as Jurkat T cells expressing pHR-PD-1-GFP) and a fluorescent PD-L1-expressing antigen-presenting cell line (such as Raji B cells expressing pHR-PD-L1-mCherry). However, primary T cells can also be used following transfection/viral transduction with a fluorescent PD-1 expression vector in combination with Raji cells [20]. Fluorescent T and B cell lines can be generated as described in Lerrer et al. [21].
Day 1
Note: If activation via CD3/CD28/CD2 ImmunoCult T-cell activator for T cells is needed, the steps delineated for day 1 and below for day 2 must be done separately. Otherwise, the steps for day 1 and day 2 can be combined into a single day. See Figure 1.
-
Maintain T and B cells in T75 flasks with 12–15 mL of RPMI (w/ FBS) media (see Recipes) at the appropriate optimal growing concentrations.
~2 × 105 to 2 × 106 cells/mL are optimal growing concentrations for Jurkat.
~3 × 105 to 2 × 106 cells/mL are optimal growing concentrations for Raji.
Collect around 5 mL from flasks and ensure that any clumps are broken up with pipetting.
-
Count viable T cells manually or using an automated cell counter with Trypan Blue.
Take 10 µL of cells and 10 µL of Trypan Blue.
Mix with a pipette.
-
If counting using an automated cell counter:
i. Add 10 µL of the mixture to a cell counting chamber slide.
ii. Insert the slide into an automated cell counter, with the side containing the cell/Trypan mixture inserted first.
iii. Focus the machine, press scan, and record cell viability as reported.
-
If counting manually:
i. Take 10 µL of the mixture and add it to a single chamber of the KOVATM GlassticTM slide.
ii. Place the slide under a microscope and count the number of viable cells per grid using an appropriate magnification to visualize cells in each box.
1) Viable cells will appear shiny or bright, whereas nonviable or dead cells will be permeable to the Trypan Blue dye and appear darker.
iii. Calculate the number of viable cells by converting 10 viable cells/box = 2 million viable cells/mL.
1) Count multiple boxes, convert, and then average the resulting viable cell numbers together to get the most accurate estimate of total cell viability.
-
Calculate the appropriate volume needed for 106 viable cells (or more, if desired) and centrifuge the cells at 500× g for 5 min in a microcentrifuge tube.
If needed, centrifuge in a 15 mL or 50 mL tube at 500× g, and then resuspend and transfer to a microcentrifuge tube.
Remove the supernatant and leave around 50 µL of media.
Add fresh RPMI (w/ FBS) media to a final concentration of 106 cells per milliliter (950 µL of media if 106 cells were used in step A5).
Move isolated 106 cells (total volume 1 mL) to one well of a 24-well cell culture plate for overnight culture.
Add 12.5 µL of CD3/CD28/CD2 ImmunoCult T-cell activator for T cells per milliliter and incubate overnight (not necessary for PD-1-expressing Jurkat cells) at 37 °C in a tissue culture incubator.
Day 2
Note: Both day 1 and day 2 steps can be done on the same day if overnight T-cell activation is not necessary, e.g., when using PD-1-expressing Jurkat cells. See Figure 1.
-
Repeat steps 2–6 from Day 1 for B cells or perform in parallel for B and T cells if overnight T-cell activation was not performed.
Collect around 5 mL from flasks and ensure that any clumps are broken up with pipetting.
Count viable cells manually or using an automated cell counter with Trypan Blue, as described in the steps for day 1.
-
Calculate the appropriate volume needed for 106 viable cells (or more, if desired) and centrifuge them in a microcentrifuge tube at 500× g for 5 min.
If needed, centrifuge in a 15 mL or 50 mL tube at 500× g, and then resuspend and transfer to a microcentrifuge tube.
Remove the supernatant and leave around 50 µL of media.
Add Opti-MEM media to a final concentration of 106 cells per milliliter (950 µL of media if 106 cells were used in step 1d).
-
If T-cell activation is required, change the media with the T-cell activator solution to fresh media.
Collect cells from the well into a microcentrifuge tube.
-
Spin cells down at 500× g for 5 min.
If desired, this spin can be done at the same time as the spin for step 1.
Remove supernatant, leaving up to 50 µL above the cells, and resuspend in 1 mL Opti-MEM media.
Add 5 µL of Staphylococcus enterotoxin E (SEE) working stock (see Recipes) for each 1 mL of B cells and incubate at 37 °C in a tissue culture incubator for 30–60 min in the microcentrifuge tube.
-
Add the desired amount of anti-PD-1/PD-L1 antibodies to the appropriate tube and incubate at 37 °C in a tissue culture incubator for 30–60 min in the microcentrifuge tubes:
-
If an anti-PD-1 antibody is to be tested, add the antibody to the T-cell tube.
i. Add the anti-PD-1 antibody in parallel with the Staphylococcus enterotoxin E (SEE) so that both T and B cells will incubate for 30–60 min.
-
If an anti-PD-L1 antibody is to be tested, add the antibody to the B-cell tube.
i. Add the anti-PD-L1 antibody in parallel with the Staphylococcus enterotoxin E (SEE) so that both will incubate with the B cells for 30–60 min.
ii. Keep the T-cell tube incubating at 37 °C during this process.
-
During T-cell and B-cell incubation, coat MatTek plates with 200 µL of 1× Poly-L-Lysine (PLL) (see Recipes) solution for 30–45 min at room temperature or 37 °C in a tissue culture incubator.
-
Remove PLL from the plate via vacuum before plating cells.
Note: No wash is needed after removing the PLL.
-
Add 100 µL (105 cells) of treated B cells to a MatTek dish and let them settle for 5 min at room temperature.
Note: This step does not have to be done in a sterile hood.
-
Add 100 µL (105 cells) of T cells on top of the previously plated B cells.
Note: Ensure that T cells are added dropwise from the air, gently and evenly over the plate, to help maximize cell dispersion and minimize air bubble formation.
Let the combined solution settle for at least 10 min at room temperature before imaging.
Store microcentrifuge tubes with remaining treated B and T cells in a mini block heater at 37 °C, covered with tin foil to prevent photobleaching.
-
-
Imaging of T and B cells synapse formation
Note: Before proceeding with this protocol portion, ensure that the microscope is turned on and calibrated and that Zen Blue software is functional.
-
Load MatTek plate with cell solution (200 µL total) onto the microscope stage and center above 10× objective for initial observations of cell settling in the plate.
Note: Be gentle with the transfer and ensure that the cell solution is not disturbed from the center of the plate.
Locate the area of interest on the MatTek plate using the 10× objective and eyepiece to ensure that cells have fully settled in.
-
Remove the MatTek plate from the microscope stage and change to 63× objective with a small amount of oil on the lens.
Note: To prevent damage to the lens, ensure it is at the lowest Z-level before reloading the MatTek plate.
-
Adjust the objective height to ensure cells are in focus.
Note: The oil must touch the plate and the objective lens for clear images (Figure 2).
Select wavelengths in the fluorescence setting to be imaged.
Adjust gain, intensity, and wavelength absorbance until the background is sufficiently low, but PD-1 and PD-L1 fluorescent signals can be imaged.
Press Capture to take an image for further analysis when settings are acceptable.
-
The colocalization of both fluorescent signals will evidence synapse formation in a “straight line” between B and T cells (Figure 3).
The number of synapses formed for each antibody tested can be used to visually analyze synapse abundance and quality.
-
Count the number of synapses formed per X of pairs of T and B cells in contact with one another and compare the number of synapses counted between antibody treatment groups and the control group.
Note: Synapses should be counted only when GFP-PD-1 is enriched in the contact area.
-
-
Quantification of synapse intensity using microscopic colocalization analysis
Note: A proper version of Zen Blue is recommended for this step. Other software, such as AIM or Image J, can also be utilized, but these steps are outlined explicitly for Zen Blue. For best results, use a .czi file. See Figure 4.
After capturing the images, restart Zen Blue, select image processing, and open the images to be quantified in the image processing suite.
Select colocalization on the sidebar.
-
Markdown channels associated with PD-1 and PD-L1 fluorescence in the colocalization tab (e.g., GFP and mCherry).
Note: Channels will usually be automatically labeled according to how they were captured during the image acquisition step.
-
Utilize the region of interest (ROI) tool to mark areas where contact between T and B cells is observed.
Notes:
ROIs should not be limited to GFP-PD-1-enriched contact areas, especially in antibody treatment groups.
This step can also be excluded if a whole image analysis is desired, in which case the rest of the protocol can be conducted similarly without an ROI.
-
To ensure reproducibility, utilize the Costes auto threshold option by clicking the Costes button. Alternatively, if desired, select the crosshair option to set proper quadrants for use in colocalization analysis. Quadrants are labeled on the cytofluorogram.
Quadrant 4 will contain pixels with low-intensity levels for both channels.
Quadrants 1 and 2 will have high-intensity pixels in one of the channels.
Quadrant 3 will contain pixels with high intensity in both channels.
Cytofluorogram threshold bar placements can be determined using single fluorescence control samples to determine the optimal boundaries to eliminate background signals.
-
Check the table option to view values for colocalization coefficients and compare between groups with higher colocalization coefficients indicative of more pixels with signals from both channels.
-
The unweighted colocalization coefficient values (labeled Colocalization Coefficient 1 and Colocalization Coefficient 2) for the region of interest can be utilized to analyze colocalization. Values for weighted colocalization coefficients will range from 0 to 1, with 0 indicating no colocalization and 1 indicating complete colocalization.
i. Unweighted colocalization coefficients are derived from Mander’s coefficients as described in Manders et al. [22].
ii. Weighted colocalization coefficient values (labeled as W Colocalization Coefficient 1 and W Colocalization Coefficient 2 on Zen Blue) consider pixel intensity in their calculation but otherwise use the same methodology as unweighted colocalization coefficient values.
iii. Either coefficient can be utilized at one’s discretion.
1) Colocalization Coefficient 1 (or W Colocalization Coefficient 1) displays the percentage of the cytofluorogram horizontal axis channel that overlaps with the vertical axis channel.
2) Colocalization Coefficient 2 (or W Colocalization Coefficient 2) displays the percentage of the cytofluorogram vertical axis channel that overlaps with the horizontal axis channel.
3) Record the colocalization coefficient (or W Colocalization Coefficient) that correlates with the channel for PD-1.
a) If PD-1 is the horizontal axis channel, use Colocalization Coefficient 1 (or W Colocalization Coefficient 1).
b) If PD-1 is the vertical axis channel, use Colocalization Coefficient 2 (or W Colocalization Coefficient 2)
-
Higher colocalization coefficient values mean increased colocalization of PD-1 and PD-L1 in the synapse, which indicates poorer antibody efficiency due to the lessened exclusion of PD-1 from the synapse.
Note: Colocalization coefficients can be plotted in GraphPad Prism to analyze quantified synapse intensity values between antibody treatment groups.
Multiple images for each antibody are used to ensure the colocalization coefficient derived is accurate.
-
-
Alternative ImageJ quantification of synapse intensity using microscopic colocalization analysis
Note: The JACoP plugin can be utilized as an alternative method for quantifying colocalization. Image processing can be optimally done using a Czi or TIFF file. If not already integrated, the JACoP plugin must be downloaded to the plugin folder in ImageJ. A region of interest cannot easily be specified using the JACoP plugin, so specific synapses must be preemptively isolated for analysis before using ImageJ. Please see the ImageJ website for more details regarding the usage of JACoP.
Following capturing images, open the image to be quantified using ImageJ.
Select Hyperstack for Stack viewing.
Select Default for Color options and check the Autoscale box.
-
Select Split channels to split into separate windows.
Note: This will separate the PD-L1 and PD-1 fluorescent channels, allowing the JACoP plugin to work correctly.
Select the Plugins tab and click JACoP.
-
On the JACoP menu, select M1 and M2 coefficients, which will give Mander’s coefficients as calculated in Manders et al. [22].
M1 will give the percentage of channel 1 that overlaps with channel 2.
M2 will give the percentage of channel 2 that overlaps with channel 1.
-
Record the Mander’s coefficient that correlates with the channel for PD-1.
For example, if PD-1 is labeled as channel 1, look at M1.
-
Click the threshold tab and adjust thresholds for each channel.
The area utilized for the colocalization analysis will appear in red when the threshold is adjusted.
-
Once thresholding is satisfactory, click analyze.
Results for M1 and M2 will appear in a separate window.
M1 and M2 can be interpreted in the same manner as described for Zen colocalization analysis.
Figure 1. Timeline of the cell preparation protocol.

Timelines for the protocol A) with T-cell activation or B) without T-cell activation are given. Steps shown on the same tick mark are to be completed in parallel. Steps should be completed in the order shown and according to the protocol written.
Figure 2. Oil is applied to the 63× objective lens, and contact is formed with the MatTek plate.

Areas of interest are highlighted using white circles. A) The microscope objective is at the lowest Z-position, with nothing placed on the lens. B) The objective is at the lowest Z-position with a small amount of Immersol autofluorescence immersion oil applied to the lens. C) The MatTek plate is centered above the lens without contact between the plate, oil, and lens. D) MatTek plate centered above the lens with contact between the plate, oil, and lens formed by adjusting using the focus knobs.
Figure 3. Conjugates formation between Jurkat T cells and Raji B cells.

Jurkat T cells expressing GFP-PD-1 were cocultured with Raji B cells expressing mCherry-PD-L1 and pulsed with SEE. The cells on the left panel (A) were imaged after 20 min of coculturing, while the cells on the right (B) were imaged after treatment with anti-PD-1 antibodies. Without anti-PD-1 antibodies, GFP-PD-1 is enriched in the synapse between the Raji and the Jurkat cells (yellow arrows). Scale bar in 20 μm.
Figure 4. Colocalization analysis using ZEN Blue software.

Areas of note are highlighted in white (colocalization tab, threshold adjustment bars, quadrant numbers, region of interest indicator, colocalization coefficients, region of interest tool, Costes auto threshold button, table option). For the example image, the colocalization coefficient to be recorded is 0.725, and the weighted colocalization coefficient to be recorded is 0.767, indicating that 72.5% of GFP-PD-1 signal colocalizes with mCherry-PD-L2 without taking into account pixel intensity, and 76% of GFP-PD-1 signal colocalizes with mCherry-PD-L2 when taking into account pixel intensity. A) Click on the colocalization option on the sidebar. B) Select the region of interest (ROI) using the shape tools. C) Appropriately position the ROI over the synapse and ensure it remains selected. D1) Either manually adjust the bars on the cytofluorogram, or D2) Click the Costes button to auto-set the threshold bars with the selected region of interest using the mouse. E) Click the table option to view the values for the localization coefficients. F) Unweighted and weighted colocalization coefficient values will be listed from left to right (Colocalization Coefficient 1, Colocalization Coefficient 2, W Colocalization Coefficient 1, W Colocalization Coefficient 2). G) Colocalization coefficient values for the region of interest (synapse) will be displayed in the table.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article(s):
Hao et al. (2024). Exclusion of PD-1 from the immune synapse: A novel strategy to modulate T cell function. Molecular Therapy: Oncology (Figures 1-3, S1).
General notes and troubleshooting
This protocol outlines the process for Zen Blue software analysis of confocal microscopy images, which is commonly utilized in conjunction with the Zeiss confocal microscope. Image analysis can also be conducted in other programs, such as ImageJ (the most recent version is preferred), but the steps taken to quantify colocalization will differ. The rest of the steps involving plating and imaging remain the same. Confocal microscopes, such as the Leica SP8, can also be utilized. In this case, the analysis would need to be done using the JACoP plugin for ImageJ.
The amount of SEE added to B cells for incubation can be increased as needed to help form immune synapses. SEE concentration can vary, with the minimum recommended volume added being 0.5 µL. It was found that 5 µL of SEE was necessary to optimize this protocol, but the amount can be altered as needed.
Utilizing the CD3/CD28/CD2 ImmunoCult T-cell activator does not harm imaging quality when using a cell line such as PD-1-expressing Jurkat T cells to enhance results further. Still, it is unnecessary, as adding the activator only sometimes leads to significant improvements in synapse formation and image.
Optimal synapse formation begins after cells settle for at least 10 min. Synapses are mostly readily noted by a bright line of PD-1 GFP signal in the contact area between Raji and Jurkat cells.
Following the 10-min settling time, there is an approximately 40–50 min window for capturing images. Synapses will begin to form after this settling time, but the optimal imaging window for synapses will be 20–60 min after coculture. Cells have been observed to dissociate 1 h after being cocultured together (Figure 5).
Figure 5. Image capture timeline and synapse formation.

Synapses can be observed to begin forming at 10 min after coculture. The optimal image window for more developed synapses is 20–60 min after coculture. Cells are observed to dissociate at times greater than 60 min after coculture or greater than 50 min after initial synapse formation.
Acknowledgments
Grants from the NIH AI125640, AI150597, and AI175498 supported this work.
Competing interests
The authors report no competing interests.
Ethical considerations
No animal or human subjects were used in this protocol.
References
- 1. Hao L. Y., Lerrer S., Paiola M., Moore E. K., Gartshteyn Y., Song R., Goeckeritz M., Black M. J., Bukhari S., Hu X., et al.(2024). Exclusion of PD-1 from the immune synapse: A novel strategy to modulate T cell function. Mol Ther Oncol. 32(3): 200839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Solinas C., Aiello M., Rozali E., Lambertini M., Willard-Gallo K. and Migliori E.(2020). Programmed cell death-ligand 2: A neglected but important target in the immune response to cancer? Transl Oncol. 13(10): 100811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu R., Li H. F. and Li S.(2024). PD-1-mediated inhibition of T cell activation: Mechanisms and strategies for cancer combination immunotherapy. Cell Insight. 3(2): 100146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shiravand Y., Khodadadi F., Kashani S. M. A., Hosseini-Fard S. R., Hosseini S., Sadeghirad H., Ladwa R., K. O’Byrne and Kulasinghe A.(2022). Immune Checkpoint Inhibitors in Cancer Therapy. Current Oncology 29(5): 3044-3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen S., Zhang Z., Zheng X., Tao H., Zhang S., Ma J., Liu Z., Wang J., Qian Y., Cui P., et al.(2021). Response Efficacy of PD-1 and PD-L1 Inhibitors in Clinical Trials: A Systematic Review and Meta-Analysis. Front Oncol. 11: e562315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cancer Research Institute(2024). FDA approval timeline of active immunotherapies: CRI. https://www.cancerresearch.org/regulatory-approval-timeline-of-active-immunotherapies
- 7. National Library of Medicine(2000). Clinicaltrials.gov. https://clinicaltrials.gov/ [DOI] [PubMed]
- 8. Townsend D. R., Towers D. M., Lavinder J. J. and Ippolito G. C.(2024). Innovations and trends in antibody repertoire analysis. Curr Opin Biotechnol. 86: 103082. [DOI] [PubMed] [Google Scholar]
- 9. Cottrell T. R. and Taube J. M.(2018). PD-L1 and Emerging Biomarkers in Immune Checkpoint Blockade Therapy. Cancer J. 24(1): 41-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Promega(2019). PD-1/PD-L1 blockade bioassay. https://www.promega.com/products/reporter-bioassays/immune-checkpoint-bioassays/pd1_pdl1-blockade-bioassays/?catNum=J1250
- 11. Naito Y., Osa A., Masuhiro K., Hirai T., Koyama S. and Kumanogoh A.(2020). Monitoring PD-1-Blocking Antibodies Bound to T Cells Derived from a Drop of Peripheral Blood. J Visualized Exp. doi.org/10.3791/60608. [DOI] [PubMed] [Google Scholar]
- 12. Puopolo T., Li H., Gutkowski J., Cai A., Seeram N., Ma H. and Liu C.(2023). Establishment of Human PD-1/PD-L1 Blockade Assay Based on Surface Plasmon Resonance(SPR) Biosensor. Bio Protoc. 13(15): e4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tocheva A. S., Lerrer S. and Mor A.(2020). In Vitro Assays to Study PD‐1 Biology in Human T Cells. Curr Protoc Immunol. 130(1): e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dustin M. L.(2014). What Counts in the Immunological Synapse? Mol Cell. 54(2): 255-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alarcón B., Mestre D. and Martínez-Martín N.(2011). The immunological synapse: a cause or consequence of T-cell receptor triggering? Immunology. 133(4): 420-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zinselmeyer B. H., Heydari S., Sacristán C., Nayak D., Cammer M., Herz J., Cheng X., Davis S. J., Dustin M. L., McGavern D. B., et al.(2013). PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J Exp Med. 210(4): 757-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yokosuka T., Takamatsu M., Kobayashi-Imanishi W., Hashimoto-Tane A., Azuma M. and Saito T.(2012). Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 209(6): 1201-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cartwright A. N. R., Griggs J. and Davis D. M.(2014). The immune synapse clears and excludes molecules above a size threshold. Nat Commun. 5(1): e1038/ncomms6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu X., Masubuchi T., Cai Q., Zhao Y. and Hui E.(2021). Molecular features underlying differential SHP1/SHP2 binding of immune checkpoint receptors. eLife. 10: e74276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tocheva A. S., Peled M., Strazza M., Adam K. R., Lerrer S., Nayak S., Azoulay-Alfaguter I., Foster C. J., Philips E. A., Neel B. G., et al.(2020). Quantitative phosphoproteomic analysis reveals involvement of PD-1 in multiple T cell functions. J Biol Chem. 295(52): 18036-18050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lerrer S., Tocheva A. S., Bukhari S., Adam K. and Mor A.(2021). PD-1-stimulated T cell subsets are transcriptionally and functionally distinct. iScience. 24(9): 103020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Manders E. M. M., Verbeek F. J. and Aten J. A.(1993). Measurement of co‐localization of objects in dual‐colour confocal images. J Microsc. 169(3): 375-382. [DOI] [PubMed] [Google Scholar]