

Abstract
Proton exchange membrane water electrolysis (PEMWE) is a promising technology to produce hydrogen directly from renewable electricity sources due to its high power density and potential for dynamic operation. Widespread application of PEMWE is, however, currently limited due to high cost and low efficiency, for which high loading of expensive iridium catalyst and high OER overpotential, respectively, are important reasons. In this study, we synthesize highly dispersed IrRu nanoparticles (NPs) supported on antimony-doped tin oxide (ATO) to maximize catalyst utilization. Furthermore, we study the effect of adding various amounts of Ni to the synthesis, both in terms of catalyst structure and OER activity. Through characterization using various X-ray techniques, we determine that the presence of Ni during synthesis yields significant changes in the structure of the IrRu NPs. With no Ni present, metallic IrRu NPs were synthesized with Ir-like structure, while the presence of Ni leads to the formation of IrRu oxide particles with rutile/hollandite structure. There are also clear indications that the presence of Ni yields smaller particles, which can result in better catalyst dispersion. The effect of these differences on OER activity was also studied through rotating disc electrode measurements. The IrRu-supported catalyst synthesized with Ni exhibited OER activity of up to 360 mA mgPGM–1 at 1.5 V vs RHE. This is ∼7 times higher OER activity than the best-performing IrOx benchmark reported in the literature and more than twice the activity of IrRu-supported catalyst synthesized without Ni. Finally, density functional theory (DFT) calculations were performed to further elucidate the origin of the observed activity enhancement, showing no improvement in intrinsic OER activity for hollandite Ir and Ru compared to the rutile structures. We, therefore, hypothesize that the increased activity measured for the IrRu supported catalyst synthesized with Ni present is instead due to increased electrochemical surface area.
Keywords: Proton exchange membrane water electrolysis, oxygen evolution reaction, platinum group metal minimization, alloyed iridium catalysts, supported iridium catalysts, amorphous iridium, hollandite iridium
Introduction
Hydrogen is a key energy carrier and an important chemical feedstock in today’s industry. Most hydrogen currently used is made from natural gas. However, if produced through water electrolysis that is driven by renewable electricity, direct carbon emissions can be avoided. This “green hydrogen” is a key element in the energy transition.1−3 Of the different water electrolysis systems, low-temperature water electrolysis based on proton exchange membranes (PEMWE) is the most promising technology for applications where high power densities and fast load flexibility are desired.4 However, the current requirement for extensive use of costly and scarce materials for PEMWE threatens the future widespread application of this technology. State-of-the-art PEMWE anodes utilize large amounts of Ir, which the European Commission has placed on its critical raw material (CRM) list over elements with particularly high supply risk to the EU.5 Ir is one of the scarcest materials on earth and is almost entirely extracted as a byproduct of Pt and Pd mining.6,7 The Ir production volume is therefore highly dependent on Pt demand, resulting in near-zero supply elasticity. As a result, future implementation of PEMWE technology at a scale of multiple GW annually would require nearly all the global Ir production, given the state-of-the-art Ir content of ∼2 mg per cm2 of geometric electrode area.7,8 Furthermore, Ir production is geographically unbalanced, with the main production originating from South Africa and Russia.9,10 In addition to the dependence on Ir for the anode catalyst, the use of other expensive materials such as Ti for bipolar plates and PFSA-type membranes contribute to driving up the cost of PEM systems.
A crucial target for enabling widespread application of PEMWE technology is lowering the Ir content in the anode catalyst layer.7,11,12 This requires a drastic increase in catalytic activity per unit mass of Ir used. There are two main approaches to achieve increased activity from the anode catalyst: (i) to develop catalysts with a higher electrochemically active surface area (ECSA), and (ii) to develop catalyst materials with higher intrinsic or specific activity (reaction rate per active surface area). In the first approach, small nanoparticles (NPs) are used as catalyst material, as they maximize the available surface with respect to catalyst mass. However, NPs tend to aggregate and thereby lose active surface area over time. Therefore, they are typically dispersed on conductive support materials, which provide electrical contact and stable anchoring under operation. Importantly, supported catalyst materials also typically exhibit better NP dispersion than their unsupported counterparts, which results in optimal utilization of the catalyst material and the need for lower catalyst loading. The second approach, on the other hand, consists of investigating modifications of the active phase of the catalyst that increase the intrinsic activity of the catalysts. The most common modification is alloying which can induce structural as well as electronic (ligand) effects.13,14
When aiming to increase the ECSA, identifying suitable support materials is a major challenge. The oxygen evolution reaction (OER), which is the anode process in PEMWEs, requires strongly oxidizing potentials. Thus, oxide support materials are obvious candidates.15 In order to create sufficient electrical conductivity, the oxide support material is typically doped.15 In particular, antimony-doped tin oxide (ATO) is often discussed as a promising support material candidate due to its high electrical conductivity.16−21 As leaching of the Sb dopant has been reported,22 the stability of ATO-based catalysts under OER working conditions needs to be assessed with care, especially at higher operating temperatures.
In the present work, we focus on combining both approaches, i.e., strategies for the optimization of the specific activity of highly dispersed NPs by supporting them on mixed-metal oxides. Adding Ru to an Ir catalyst can increase its catalytic activity; in previous work by some of the authors of the current study, it has been shown that the activity of Ir can be increased roughly 6 times by adding 60 at. % Ru.23 A composition optimization based on density functional theory (DFT) calculations proposed an IrRu mixed oxide with 30–45 at. % Ru as the most active catalyst for the OER.24 At the same time, studies have shown that there is typically a trade-off between activity and stability for Ir and Ru-based catalysts, where the most active catalysts are typically the least stable.25 Later work showed that this trend depends on measurement conditions and catalyst structure.26
Furthermore, it has been demonstrated that mixing of Ni into RuO2 and IrO2 results in improved OER performance.27−30 Theoretical calculations have shown that Ni doping results in an intrinsic improvement arising from a change in the energy levels of the reaction intermediates.27,31 Meanwhile, an experimental study of Ir–Ni mixed metal oxides also revealed a shift in crystal structure from the typically observed rutile IrOx to increasing amounts of edge-sharing octahedra when the Ni content was increased, suggesting that there could also be a structural contribution to the increased activity.29 Indeed, it has been shown that X-ray amorphous IrOx with a high degree of hollandite structure, which contains a larger fraction of edge-sharing octahedra, exhibits higher catalytic activity than its rutile counterpart.32,33 This effect was explained through increased intrinsic activity based on an alteration of the Ir/O active site electronic structure.32,33
In order to further study the influence of Ni on the structure and activity of Ir based OER catalysts, we utilize a solvothermal flow synthesis under supercritical conditions to produce Ir0.7Ru0.3 NPs on a commercial ATO support, with and without Ni added to the synthesis. The two different types of catalyst are named IrRu and IrRuNi, respectively, throughout this work. The solvothermal flow synthesis technique typically leads to the generation of catalyst NPs with high crystallinity and narrow size distribution34−36 and enables the addition of controlled amounts of Ni that is present during the catalyst NP nucleation. A wide range of characterization techniques are used to evaluate the effect of Ni on the IrRu morphology, crystal structure and OER activity. It turns out that the presence of Ni gives rise to an IrRu phase with amorphous structure (in X-ray diffraction (XRD) measurements), exhibiting similarity to hollandite structure in total X-ray scattering pair distribution function (PDF) experiments. Catalysts where Ni was present during synthesis exhibit a very high OER activity of up to ∼360 mA mgPGM–1 at 1.5 V vs RHE, which is more than 7 times higher than literature reports21,37 and benchmark measurements using unsupported IrOx. DFT modeling comparing hollandite with rutile structure for Ir and Ru suggests that the high activity observed is likely not due to increased intrinsic activity, but rather due to other factors such as higher ECSA. This work therefore gives rise to further information on X-ray amorphous Ir phases and the role of Ni in Ir-based OER catalysts and provides a path to significantly improved utilization of Ir and Ru in PEMWE anodes, which is key to enable widespread implementation this technology.
Results and Discussion
Synthesis and Structural Analysis with X-ray Diffraction
IrRu and IrRuNi NPs were synthesized directly on ATO support with the solvothermal flow approach described in the methodology. To discount any structural changes in the ATO support due to the synthesis conditions, bare ATO support was first flowed through the solvothermal reactor under synthesis conditions and analyzed. Ir, Ru and Ni precursor salts and ATO support were then added together to the reactor to synthesize supported IrRu and IrRuNi catalysts. The structure of the samples was analyzed by XRD and Rietveld refinement, with the data shown in Figure 1. A more comprehensive list of all the calculated XRD patterns for the various possible phases is shown in Figure S1. For the bare ATO support, the peaks in the XRD diffractogram are described well as a cassiterite, rutile crystal structure of SnO2 that takes the space group P42/mnm, see Figure 2A, Figure S2 and Table S2. Rietveld refinement of the data from the ATO particles indicates a crystallite size of ∼15 nm, which is comparable to the as-received powder that has a particle size of ∼12 nm. Regarding the supported catalyst particles, some of the structures that these catalysts could be expected to take are depicted in Figure 2B-E and their calculated powder patterns are shown in Figure S1. Metallic Ir adopts an fcc structure while metallic Ru adopts an hcp structure. Since the XRD powder patterns are very different for metallic Ir and Ru (see Figure S1B and C), it is possible to distinguish if an alloy is formed or if the two phases are crystalline and segregated. Furthermore, as described above, literature reports show that the IrOx-based catalysts can either adopt a rutile or hollandite phase, and that hollandite is more active than rutile IrOx.29,32 According to the calculated XRD patterns, the rutile IrOx will be nearly indistinguishable from the rutile ATO support (see Figure S1A and F). This is not the case for hollandite IrOx due to the presence of an extra peak at 17.6°2θ (Figure S1G) and a different intensity pattern for Bragg reflections between 20 and 40°2θ.
Figure 1.
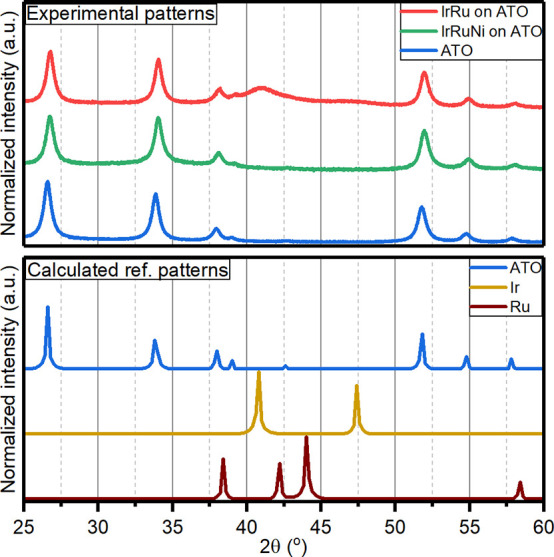
(Top) Measured XRD patterns of IrRu (red) and IrRuNi (green) catalysts, and ATO (blue). (Bottom) Calculated reference patterns for ATO (blue), Ir (dark yellow), and Ru (wine).
Figure 2.
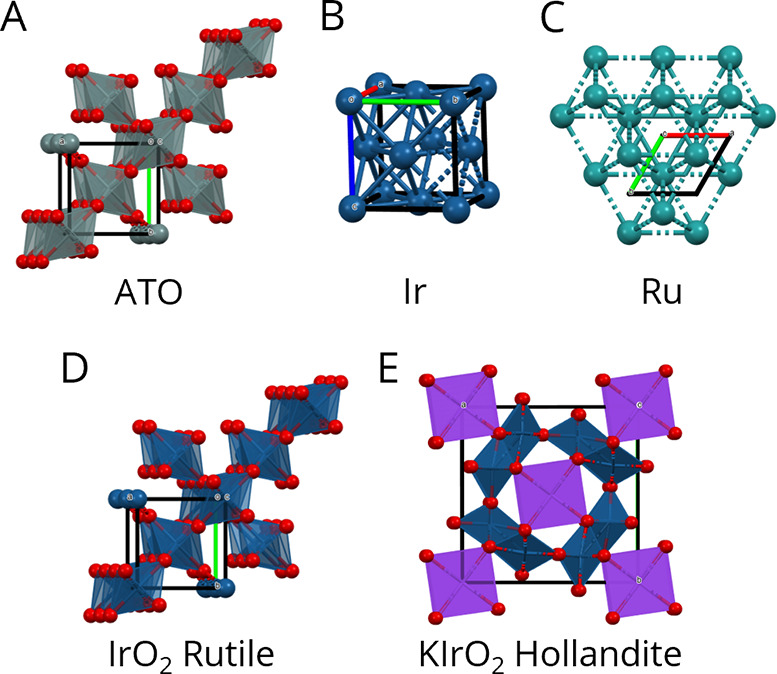
Possible structures produced during the synthesis of IrRu catalyst. Unit cells are shown within each structure. (A) Rutile ATO, space group P42/mnm.38 (B) Metallic iridium, space group Fm3̅m.39 (C) Metallic ruthenium, space group P63/mmc.40 (D) Rutile iridium oxide, space group P42/mnm.41 (E) Hollandite iridium oxide, space group I4/m.42,43 Figures were made with Mercury 2021.2.044 and Adobe Illustrator.
IrRu NPs supported on ATO show Bragg reflections consistent with rutile (space group P42/mnm) and metallic Ir NPs (space group Fm3̅m), see Figure 1. The particle size obtained from Rietveld refinement is 13 nm for ATO NPs and 3 nm for Ir NPs (Figure S3 and Table S3). While both Ir and Ru are found in the elemental analysis (see Table S1), there are no peaks consistent with metallic Ru nanoparticles, suggesting that Ru is forming an alloy with Ir. This can be further supported by the shift of the observed main peak for Ir, which was measured at 41.1°2θ compared to the calculated one at 40.8°2θ, implying a smaller unit cell. Analysis of the XRD data for IrRuNi reveals only Bragg peaks consistent with rutile ATO, with no evidence of metallic Ir (Figure 1). Rietveld refinement of the IrRuNi data gives 11–12 nm crystallite size of the ATO phase, slightly smaller compared to bare ATO and the IrRu catalyst (Figure S4 and Table S4).
Pair Distribution Function (PDF) Analysis
PDF analysis of total scattering measurements was used to further elucidate the catalyst structure, since no peaks consistent with crystalline IrRuNi were observed with XRD.43 Calculated PDF patterns of metallic Ir (dashed blue trace) and rutile IrO2 (dashed purple trace) are shown in Figure 3. These calculated peaks can be used as fingerprints of the different phases. Metallic Ir has a peak at 2.7 Å, which corresponds to the Ir–Ir distance, while no similar peak is observed for iridium oxide. The specific peaks for iridium oxide are located at 2.0, 3.1, and 3.5 Å, and correspond to Ir–O and Ir–O–Ir edge and corner distances, respectively. The calculated PDF for hollandite (Figure S7) has the same major peaks as those seen in the rutile phase except that the ratio of edge:corner sharing octahedra is larger.
Figure 3.
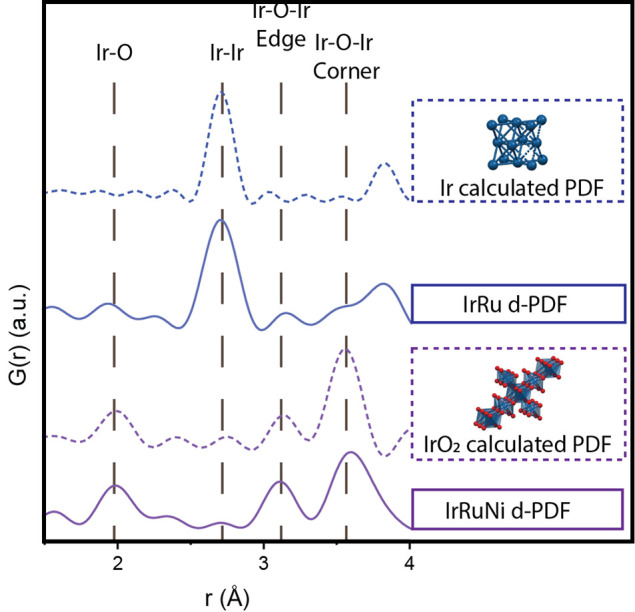
Calculated PDFs of Ir (dashed blue) and rutile IrO2 (dashed purple) and collected d-PDFs of IrRu (full blue) and IrRuNi (full purple) catalysts. The measured d-PDFs in the full range of r-values can be seen in Figure S8.
Collected PDFs were then analyzed using the calculated PDFs as reference. The contribution of the ATO support to the collected PDFs was removed by subtracting the signal from bare ATO, resulting in a difference-PDF (d-PDF).45 Collected d-PDFs for the IrRu (solid blue) and IrRuNi (solid purple) catalysts are shown in Figure 3. The IrRu d-PDF is dominated by peaks resembling metallic iridium. Some peaks consistent with rutile IrO2 are also present; it is, however, difficult to determine if these are undersubtracted rutile ATO peaks or some actual IrO2. The entire d-PDFs for IrRu and IrRuNi can be seen in Figure S5 and S6. In all cases, there is some leftover signal from rutile peaks after subtraction of ATO, which is probably due to small structural changes in ATO during the reaction that are not visible in the XRD. XRD data and Rietveld refinement indicate that the ATO changes to have a smaller crystallite size in the presence of reactants under synthesis conditions, meaning there is likely some minor change in the structure of ATO during the reaction with and without Ir and Ru. The d-PDF for IrRuNi only exhibits peaks consistent with rutile iridium oxide, and none that are consistent with metallic Ir. Moreover, there also appears to be a larger ratio of edge sharing octahedra in the IrRuNi d-PDF, as can be seen by the increase in intensity of the peak at 3.1 Å compared to the 3.5 Å peak, than in the calculated rutile iridium oxide PDF. This indicates the presence of either hollandite structure or of more edge-sharing octahedra in the particles. When comparing the d-PDF of IrRu to IrRuNi, it can also be seen that the former exhibits larger crystalline domains (peaks at higher r-values) than the latter (Figure S8).
To obtain a better idea of structure and size of the catalyst NPs, the d-PDFs were fitted with Ir and IrO2 structural models. For IrRu samples, the d-PDF was fitted with a metallic Ir phase. The refinement described all major peaks and gave a 2–3 nm crystallite size (Table S6 and Figure S10). This is a similar crystallite size to that obtained from Rietveld refinement of XRD data.
For IrRuNi samples, the d-PDF was fit with rutile IrO2 over r-values from 1.5 to 30 Å, resulting in crystallite size of 1 nm. The main peak not described by this model is that corresponding to edge-sharing octahedra (Table S7 and Figure S11). Therefore, a refinement was also done with hollandite IrO2 (Table S8 and Figure S12), which in turn underestimates the corner sharing octahedra intensity. The actual structure of the IrO2 is therefore likely a mixture of the two phases, rutile and hollandite. Still, there were peaks at high r-values that were not described by this model, so another refinement was performed from 7 to 60 Å using a cassiterite phase (ATO). The data at higher r-values were well described by an ATO phase of 9–10 nm in diameter, and probably originated from the undersubtraction of the ATO phase due to the complications described above. This means that some of the rutile peaks at low r-values have a contribution from the ATO as well but the intensity of rutile peaks at low r-values cannot totally be explained by ATO alone.
The difference in crystallite size for IrRu and IrRuNi is corroborated by TEM images, as can be seen in Figure S13. The IrRu active NPs can be clearly distinguished from the ATO support, while the IrRuNi NPs, on the other hand, are barely distinguishable in TEM images, suggesting smaller particle size. These observations fit well with the information obtained from modeling of the PDF data.
X-ray Absorption Spectroscopy (XAS)
To gain further information on the oxidation state and local coordination of Ir and Ru, X-ray absorption spectra of the IrRu and IrRuNi sample were collected at the Ir LIII-edge and the Ru K-edge, respectively. X-ray absorption spectroscopy has the advantage that the iridium and ruthenium coordination can be studied without the ATO background contribution. The X-ray absorption near edge structure (XANES) part of the spectra reveals that the IrRu sample is less oxidized than the IrRuNi sample, both at the Ir and Ru edges (Figure 4A and B). The Fourier transformed magnitudes of the extended X-ray absorption fine structure (EXAFS) data shown in Figure 5C and D confirm a mixed metallic and oxide structure for the IrRu sample, while in the case of IrRuNi no metallic contribution is observed. This agrees well with the findings from the d-PDF analysis of the IrRu and IrRuNi samples.
Figure 4.
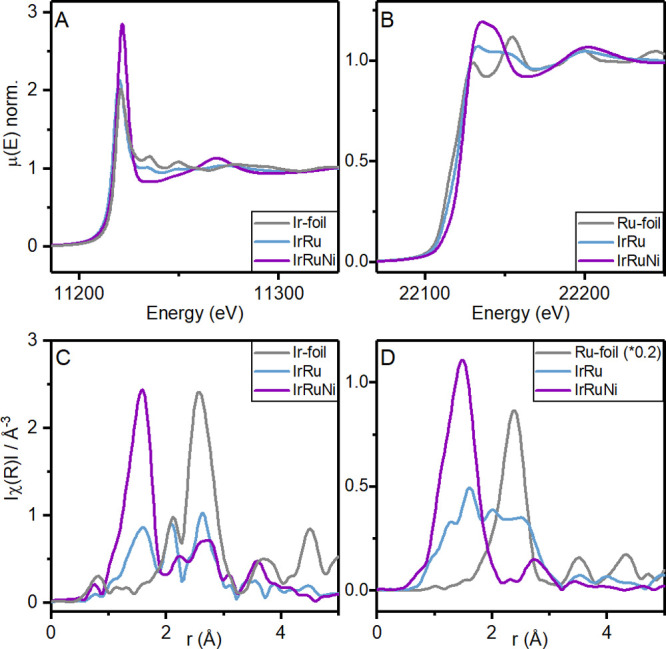
X-ray absorption near-edge (XANES) spectra of IrRu and IrRuNi samples measured (A) at the Ir LIII-edge, as well as an Ir metal foil reference, and (B) at the Ru K-edge, as well as a Ru metal foil reference. Fourier transform (FT) magnitudes of the k2-weighted extended X-ray absorption fine structure (EXAFS) data of IrRu and IrRuNi samples with respective metal foils, measured (C) at the Ir LIII-edge and (D) at the Ru K-edge.
Figure 5.
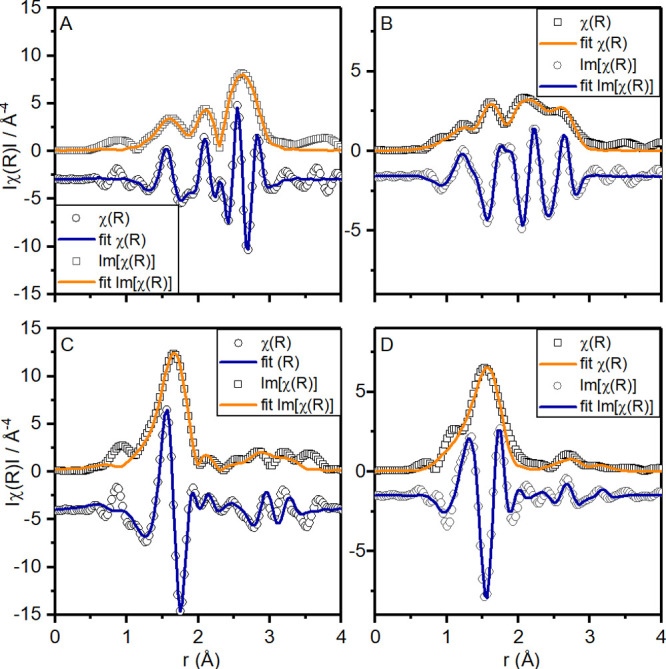
Fit of the FT magnitudes of the k2-weighted EXAFS of IrRu on the (A) Ir LIII-edge and (B) Ru K-edge as well as of IrRuNi on the (C) Ir LIII-edge and (D) Ru K-edge. The data were fit with k3 weights, in a k-range of 2.5–14 Å–1 and an R-range of 1.2–3 Å.
To extract detailed information on the local coordination environment of Ir and Ru in the structure, structural models were fitted to the EXAFS data (see Figure 5). The mixed oxide and metal coordination of the IrRu sample was fitted with a model that contained both scattering paths to nearest metal neighbors in an fcc structure as well as metal–oxygen coordination to represent the oxide contribution (see Table S9). The oxide part was modeled with two separate M-O distances to represent a distorted octahedron. The metal contribution included two metal–metal scattering paths, both to neighboring iridium and ruthenium atoms.
Based on the coordination numbers extracted from the fits of this model to the Ru K-edge EXAFS data, an oxide contribution of ca. 50% can be estimated if perfect 6-fold metal–oxygen coordination and 12-fold metal–metal coordination is assumed. Based on the same assumption, the oxide coordination of Ir is estimated at ca. 30%, revealing the slightly less oxidized nature of Ir compared to Ru in the structure. To describe the metal–metal contribution to the EXAFS data, both Ir and Ru had to be included in the model, which indicates an Ir–Ru alloy in agreement with the structural model found for the PDF data.
For the IrRuNi sample, on the other hand, no metallic contribution was found. In accordance with the PDF analysis, both the Ru K-edge and Ir LIII-edge EXAFS data could be fit with a hollandite structure model (see Table S10). The model included two metal–oxygen scattering paths for the distorted octahedral coordination as well as the metal–metal scattering to the nearest metal neighbors residing in edge-sharing octahedra.
Oxygen Evolution Reaction (OER) Activity Measurements
As the main goal of this work was to promote a higher OER activity of the IrRu catalysts for PEM electrolysis, further investigations were performed to study whether the addition of Ni in the IrRu catalyst synthesis was reflected in improved OER activity. The activity of the different catalysts was determined through chronoamperometry measurements using a rotating disc electrode (RDE) setup. The mass activity at 1.5 V vs RHE normalized to the platinum-group metal (PGM) content of the IrRu catalyst with different amounts of Ni (from 0 wt % to 20 wt %) is reported in Figure 6. The activity at a range of potentials between 1.4 and 1.6 V vs RHE can be seen in Figure S14. Cyclic voltammetry and electrochemical impedance spectroscopy for representative samples can be seen in Figure S15 and S16, respectively. The mass activity for a typical IrRu catalyst without Ni was measured as 140 mA mgPGM–1. This can be compared to 10–50 mA mgIr–1, which is a typical activity reported for commercial unsupported IrOx.21,37 The ∼3 times or more increase in mass activity of the IrRu catalyst compared to unsupported IrOx clearly illustrates the beneficial effects of dispersing the metal catalyst on ATO support and adding Ru.
Figure 6.
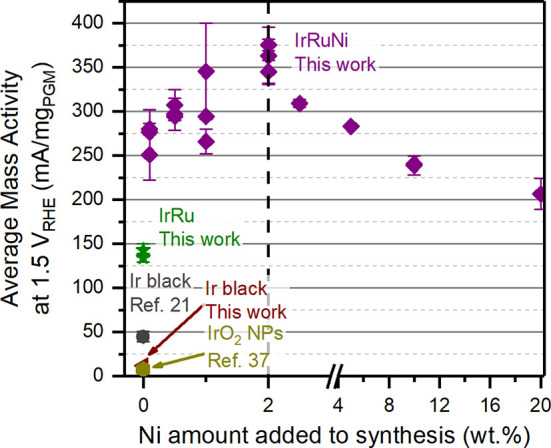
OER activity per mass of PGM (Ir and Ru) measured by chronoamperometry at 1.5 V vs RHE, plotted against the wt % of Ni present during catalyst synthesis. Data from IrRuNi (purple) and IrRu (green) from this work are compared to an Ir black benchmark from this work (red), as well as literature data from Ir black21 and IrO2 NPs.37 Each data point represents a unique catalyst synthesis, while error bars represent ±σ from at least two individual measurements. Measurements were performed in N2-saturated 0.1 M HClO4 using an RDE setup.
When adding varying amounts of Ni during synthesis, a clear trend can be observed between OER activity and concentration of Ni precursor in the reaction mixture. With a Ni content of 0.1 wt %, the OER activity at 1.5 V vs RHE increases to 270 mA mgPGM–1, revealing the strong effect of having even minute amounts of Ni present during synthesis. The activity further increases to 360 mA mgPGM–1 when 2 wt % Ni is added to the reaction mixture. This is 2.6 times higher than the IrRu catalyst activity and roughly 7 times higher than the IrOx benchmark. As the Ni concentration is further increased beyond 2 wt % target Ni loading, the mass activity starts to drop. The mass activity of the 20 wt % Ni sample is 200 mA mgPGM–1. It is likely that the decrease in activity plateaus with even higher Ni content, which would be consistent with previous studies showing that Ni-rich mixed metal oxides with Ir and Ru exhibit increased activity compared to pure IrOx and RuOx,28,29 albeit with low stability of the Ni phase.29 To investigate stability of the Ni present in some of the catalyst materials, acid wash was performed on two relevant catalysts (cf. Table S11). No significant changes in Ni content was observed, indicating apparent chemical stability for the catalyst Ni content, within the uncertainty of EDX quantification, under the acidic conditions in which the mass activity measurements were performed.
The presence of a clear optimum at 2 wt % Ni added to the synthesis, with the OER mass activity decreasing with increasing amounts of Ni present, indicates that Ni alone is not the main active element. If this was the case, a continuous increase in mass activity would be expected with increasing Ni content. Instead, one or both of the following hypotheses are likely causing the higher activity for the IrRuNi catalyst: (i) The presence of Ni during synthesis of the IrRuNi catalyst gives rise to oxide iridium–ruthenium NPs with partial hollandite-like structure, while the lack of Ni in the IrRu synthesis yields metallic NPs. The latter will get oxidized during OER operating conditions, where the Ir oxidation state can vary between various oxides and hydrous oxides, depending on the exact electrochemical conditions and history.25 Previous literature results have indicated that hollandite structured IrOx exhibits elevated OER activity,32,33 possibly explaining the IrRuNi performance. (ii) The smaller crystallite size determined by PDF of IrRuNi catalyst NPs compared to IrRu indicates a smaller NP size for the former, which is also corroborated by TEM. This likely yields larger ECSA per mass of active metal for IrRuNi, resulting in higher OER mass activity. Furthermore, it has been suggested that the more open structure of hollandite crystallites with tunnel-like motifs improves mass transport, providing a surface area effect as well.33 To evaluate the potential surface area effect, estimations of the double layer capacitance of the various catalysts were performed (see Figure S17). A clear correlation can be observed between added Ni amount, double-layer capacitance and resulting mass activity, indicating that increased surface area is at least part of the explanation.
Density Functional Theory (DFT) Calculations
To elucidate whether the different performances of the IrRuNi and IrRu catalysts can also be attributed to a higher intrinsic activity caused by the partial hollandite-like structure of IrRuNi, DFT calculations were performed comparing the OER activity of rutile and hollandite RuO2 and IrO2. The conventional pathway for the OER, which consists of four proton-coupled electron transfer steps, is considered:
| 1 |
| 2 |
| 3 |
| 4 |
where * denotes a vacant adsorption site. For the *OH and *OOH intermediates we also consider the possibility of the hydrogen being transferred to a neighboring bridging oxygen, as is thought to be the case on RuO2 (see Figure S18).46 The active sites on the rutile (110) surface are the coordinatively unsaturated (cus) sites, which are arranged in rows, separated by rows of bridging oxygens, as shown in Figure 7A. The (100) surface of the hollandite structure, shown in Figure 7B, is similar in structure and has corresponding rows of cus sites; however, the distance between neighboring rows is longer.
Figure 7.

Computational models of (A) the rutile 110 surface in top and side view and (B) the hollandite 100 surface in top and side view. Metal atoms (Ru or Ir) are gray-blue, and oxygen is red. The cus site considered as the active site is marked by *, and neighboring cus sites are shown without preadsorbed *O for clarity. (C) Overpotential for rutile and hollandite oxides of Ru (red) and Ir (blue). Black lines indicate the volcano defined by the scaling relations for the conventional reaction pathway.
The adsorption energies of the reaction intermediates were calculated and used to determine the lowest energy reaction path and the corresponding overpotential (see Figure S18). This is plotted against ΔG2 in Figure 7C for rutile and hollandite RuO2 and IrO2. The figure shows that the calculated overpotential for the hollandite surface is significantly higher than that of the rutile surface for Ru, while it is comparable for Ir. This indicates that the experimentally observed mass activity increase for IrRuNi compared to IrRu is not a result of an intrinsic increase in activity. Instead, we hypothesize that the reason for the higher mass activity for IrRuNi is a combination of smaller particle size that likely results in higher ECSA, and/or a more open structure.
Conclusions
In this investigation, the presence of Ni during solvothermal flow synthesis of IrRu-based catalysts supported on ATO is shown to yield drastic changes in the structure of the synthesized catalyst NPs. While Ni-free synthesis yielded metallic IrRu catalyst NPs, the presence of Ni led to the synthesis of oxide catalyst NPs with a mixture of hollandite and rutile structures. Further characterization also indicates that IrRu catalyst NPs are larger than IrRuNi catalyst NPs. We further show that IrRuNi catalysts exhibit significantly improved OER mass activity. In fact, IrRuNi exhibits more than twice the activity over the entire voltage window from 1.4 to 1.6 V vs RHE compared to IrRu, and more than 7 times higher than state-of-the-art unsupported IrOx catalysts. Surface area estimations indicate that the mass activity correlates with specific surface area, suggesting that increased surface area is at least responsible for part of the activity improvements. To investigate if the presence of a hollandite phase in the IrRuNi catalyst could also contribute to the higher mass activity, DFT calculations were performed on the rutile and hollandite structure for both Ir and Ru. The results show a larger calculated overpotential for hollandite Ru and a comparable overpotential for hollandite Ir, indicating that the intrinsic activity is, in fact, not improved. We therefore hypothesize that the increased mass activity observed experimentally for IrRuNi arises from a higher electrochemically active surface area due to smaller catalyst NPs, yielding improved utilization of Ir and Ru. These results provide a path for achieving drastically reduced Ir loading in PEMWE anodes, which is a key development to mitigate the impact of limited Ir demand and high costs on the large-scale implementation of PEMWE technology.
Materials and Methods
Catalyst Synthesis
The supported catalyst nanoparticles were synthesized in a custom-built solvothermal flow reactor that is described in detail in previous publications.34−36 The ATO powder (Sb2O5:SnO2 15:85 wt %, 99.5% purity, Alfa Aesar) was suspended in deionized water together with IrCl3·xH2O (reagent grade, Sigma-Aldrich) and RuCl3·xH2O (99.9% PGM basis, Alfa Aesar) in the stoichiometric amounts necessary to ensure a metal loading of 40 wt % and an Ir:Ru molar ratio of 70:30. For the Ni-containing catalyst, Ni(NO3)2·6H2O (98%, Alfa Aesar) was added to achieve the desired IrRu:Ni ratio in the precursor solution. The support and precursor concentrations were 0.75 mg mL–1 ATO, 1.8 mM Ir and 0.8 mM Ru. The precursors were put into receptacles at the top of the reactor and magnetically stirred with a stir bar. The reactor was preheated so that the reactant mixing point was at a temperature of 270 °C and the reactor was pressurized to 300 bar. After the reactor reached operating temperature and pressure, the reactant mixture was pumped at a flow rate of 60 mL/min, and the water was pumped at a flow rate of 41 mL/min. It took approximately 10 min for the reactant mixture to move all the way through the reactor and the product was collected in the syntheses here for approximately 10 min.
Catalyst Composition Determination Using Energy Dispersive X-ray Spectroscopy (EDX)
The elemental composition of synthesized samples was analyzed using SEM-EDX to determine how much of the added Ni incorporated into the catalyst. A field emission scanning electron microscope FEG-SEM (XB-1540 EsB, Carl Zeiss NTS GmbH, Oberkochen, Germany) equipped with an energy dispersive X-ray spectroscopy system EDX (Oxford X-Max 50 mm2 SDD, Oxford Instruments, Oxfordshire, UK) and AZtec software was used. The powders were mounted on double adhesive carbon tape and flattened to minimize the effect of the microstructure on the EDX spectra. EDX was performed on dried catalyst powders at 20 kV high tension and approximately 5.5 mm WD. For each sample, EDX spectra were collected from 3 random areas at 250× magnification (455*340 μm2) with a lifetime of 60 s. Finally, the data were postprocessed in order to obtain the metal loadings on the support and the ratios between the different elements. The Sn and Sb data were converted to SnO2 and Sb2O5 assuming full oxidation, and the catalyst loadings were determined by dividing their mass fractions with the mass fractions of all catalyst elements and support oxides combined.
Transmission Electron Microscopy (TEM)
TEM micrographs of the ATO support and the supported NPs were acquired with Tecnai Spirit operated at 80 kV. The samples were dispersed in EtOH, drop-casted on a Cu grid (Quantifoil, 100 Lacey Carbon films) and dried under air at room temperature.
XRD Data Collection
Data was collected using a Malvern Panalytical Empyrean Cu source diffractometer. Rietveld refinement of the data was completed using HighScore Plus software version 3.0 (2012). Samples were measured on zero background plates in reflection mode using the reflection/transmission spinner sample stage. Data was measured in three scans from 15 to 145°2θ for a total measuring time of 1 h. Reference spectra were calculated using Mercury 2021.6.0.
Rietveld Refinement
Rietveld refinement was performed using the software Highscore plus version 3.0.5 using the Crystalographic Open Database (COD) for searching and matching functions. A shifted Chebychev II background was used with 5 or 6 coefficients. The scale factor, cell parameters and peak shape parameters U and W were refined. Crystallite size was determined by fitting the phase profile using the size only analysis pseudo voigt profile function and a Finger, Cox, Jephcoat asymmetry type.
PDF Data Collection
PDFs are obtained by collecting data to higher values of Q (nm–1) than XRD patterns and therefore require higher energy X-ray sources. Data was collected at MaxIV in Lund, Sweden at the DanMax beamline. Powder was packed in 1 mm diameter Kapton capillaries for data collection. The wavelength of the beam was 35.011 keV and the detector distance was 100 mm. Data was acquired using the RA-PDF setup (site Chupas 200347). Total scattering data is then integrated, and Fourier transformed to give something like a histogram of all atomic distances in a material or a PDF. Therefore, different peaks seen in PDF data correspond to different atomic distances. Data was integrated using PyFai48 and PDFs were generated using xPDFsuite.49 Parameters used were a QMaxInstrument of 20 Å–1, QMax of 17 Å–1, QMin of 1 Å–1 and rpoly of 0.9. Detector distance was determined from calibration with a Lab6 standard. The dampening parameters, Qdamp 0.01997 and Qbroad 0.01389, were determined from modeling a Si standard using PDFgui.50
d-PDF
A PDF of ATO was subtracted out of the data collected from supported IrRu and IrRuNi catalysts in Q space using xPDFsuite.49 ATO was entered as the background and f(Q) was used to determine how much of the ATO Bragg peaks could be subtracted.
PDF Modeling
As ATO and IrO2 take the same rutile structure with IrO2 having shorter M-M distances, the background subtraction was difficult in these samples. Therefore, the strategy was to analyze the data in two different ways to ensure consistency between the results. First, the samples were analyzed by only subtracting out the background and kapton, and then fitting the samples using PDFgui. Only scale, unit cell parameters, delta2, spdiameter and thermal parameters were refined. For the samples that have IrRu NPs, they could be fit with a cassiterite phase and an Ir metal phase. For the samples with Ni, which had amorphous Ir or IrRu content, these were fit with either rutile IrO2 in addition to ATO. The d-PDF curve was also analyzed by a model free analysis to determine what was not described by the model. Second, the ATO was subtracted in r-space at an r value of approximately 5.8 because there was a peak in ATO that was shifted to higher r values than in the IrO2. These were used for model free comparison, especially comparison of the edge and corner sharing peaks. The subtraction for all d-PDFs was done in q space using xPDFsuite.
XAS Analysis
Ex-situ XAS measurements were carried out at the SuperXAS - beamline of the Swiss Light Source (SLS) at PSI, Switzerland (2.9T superbend source, storage ring current of 400 mA) via a send in service. The samples were protected in Kapton-tape and mounted directly to a sample holder. The incident beam was collimated by mirror at 2.9 mrad (Rh-coated for Ir LIII) and monochromatized with a liquid nitrogen cooled channel-cut Si(111) monochromator in QEXAFS mode. Energy calibration was performed with a Ru reference foil to the Ru K-edge position or with a Pt reference foil to the Ir LIII-edge. Ion chambers with 2 bar of N2 were used for XAS detection in transmission mode at the Ir LIII-edge. Ru K-edge spectra were collected in quick fluorescence mode with a PIPS detector.
The data were processed using ProQEXAFS for calibration, interpolation, normalization, and averaging (300 s of measurement on each sample).
The averaged XAS spectra were analyzed by using the DEMETER software package. The raw spectra were energy aligned to a metal reference foil, background corrected, and normalized by the edge step. After the conversion of the energy units (eV) into photoelectron wavenumber k units (Å–1), the resulting χ(k) functions of the XAS spectra were weighted with k2 and then Fourier transformed to obtain pseudoradial structure functions. The fits to the EXAFS spectra were performed in ARTEMIS of the DEMETER software package based on IFFEFIT.51 XAS spectra of the pure metal foils were used as references to estimate the amplitude reduction factors (S02). The EXAFS data were fitted in R-space, with a fitting weight of k3. The k-range for the Fourier transform was from 2.5 to 14 Å–1 with a fit window in an R-range of 1.2–3.0 Å.
Electrode and Ink Preparation
Glassy carbon RDEs (5 mm diameter) were polished with 0.05 μm alumina suspension (Buehler MicroPolish) for 5 min and subsequently rinsed by immersion in Milli-Q water (twice) and isopropanol (twice) for 5 min each in an ultrasonic bath (VWR USC1700D).
The catalyst ink was prepared by first adding approximately 4.0 mg of catalyst powder (the exact amount was recorded for accurate loading estimation) to 6.2 g of a solvent solution of Milli-Q water and isopropanol at a volume ratio of 3:1. The catalyst powder was then dispersed in the ink using an ultrasonic horn (Hielscher UP200 St) for 10 min, before adding Nafion solution (DuPont D520, Ion Power GmbH) at a ratio of 5:1 compared to the supported catalyst content. The ink was sonicated once more for 10 min to ensure full dispersion of the catalyst powder.
The active electrodes were prepared by drop-casting 10 μL of the catalyst ink onto the cleaned glassy carbon RDEs. The mass of the added ink was recorded for exact loading estimations (typically 8.8–9.5 mg). The electrodes were dried under a flow of N2 saturated with 3:1 water/isopropanol to achieve reproducible coating of the catalyst layer on the RDE.
Electrochemical Characterization
Before each measurement, the jacketed electrochemical cells (BioLogic EL-ELECTRO-150DJ) were warmed up to 30 °C using a thermostatic bath (CETAC Technologies Inc. 2050). The cells flushed with Milli-Q water and filled with a 0.1 M solution of HClO4 (67–72% TraceSELECT, Honeywell Fluka). H2 (Linde/AGA Instrument H2 4.5–99.995% purity) was bubbled through the solution, and the reference electrodes (Hg/Hg2SO4; Radiometer Analytical XR200) were calibrated by measuring the equilibrium potential of a clean Pt wire versus the reference electrode. The gas bubbling was then switched to N2 for the OER activity measurements.
The RDEs were immersed in the electrolyte solution under potentiostatic control, keeping them connected with a Pt wire at +1 V vs RHE during the whole immersion step. Once the RDEs were in position, the Pt wire was disconnected and removed from the cell, and the rotation at 3600 rpm of the RDE was started.
The electrochemical measurement sequence for each sample followed the protocol from a previous publication:52
-
1.
Potentiostatic electrochemical impedance spectroscopy (PEIS) at 1 VRHE (from 100 kHz to 1 Hz, amplitude 5 mV, 8 points per decade, 2 measurements per point).
-
2.
Determination of the iR compensation (85% of the series resistance measured at 5 kHz).
-
3.
Activation chronoamperometry at 1.6 VRHE for 5 min.
-
4.
Potentiostatic electrochemical impedance spectroscopy at 1 VRHE (from 100 kHz to 1 Hz, amplitude 5 mV, 8 points per decade, 2 measurements per point).
-
5.
Cyclic voltammetry from 1 VRHE to 1.6 VRHE at 10 mV/s for 5 cycles.
-
6.
Chronoamperometry measurements from 1 VRHE to 1.6 VRHE and back to 1 VRHE (with 2 min steps at 1, 1.1, 1.2, 1.3, 1.4, 1.45, 1.475, 1.5, 1.525, 1.55, 1.575, 1.6, 1.575, 1.55, 1.525, 1.5, 1.475, 1.45, 1.4, 1.3, 1.2, 1.1, 1).
-
7.
Potentiostatic electrochemical impedance spectroscopy at 1 VRHE (from 100 kHz to 1 Hz, amplitude 5 mV, 8 points per decade, 2 measurements per point).
-
8.
Cyclic voltammetry from 1 VRHE to 1.6 VRHE at 10 mV/s for 3 cycles.
The resulting data were postcorrected with the precise reference electrode potential determined before each measurement, as well as the complete ohmic drop calculated from the average of the 3 PEIS measurements. The chronoamperometry data were averaged over the last 10 s of each potential step to obtain the activity.
Density Functional Theory Calculations
DFT calculations were performed using the grid-based projector augmented wave (GPAW) software package.53,54 The Atomic Simulation Environment (ASE) was used to set up and analyze the calculations.55 The calculations were performed with a plane-wave basis with an energy cutoff of 500 eV and the exchange and correlation energy was described by the RPBE functional.56
The computationally optimized lattice constants for the rutile oxides were calculated to be a = 4.59 Å, c = 3.19 Å for IrO2 and a = 4.57 Å, c = 3.14 Å for RuO2 (experimentally a = 4.50 Å, c = 3.15 Å for IrO2 and a = 4.49 Å, c = 3.11 Å for RuO257). For the hollandite structures the computationally optimized lattice constants were found to be a = 10.13 Å, c = 3.18 Å for IrO2 and a= 10.02 Å, c = 3.16 Å for RuO2 (experimental values for KRu4O8 a = 9.91, c = 3.1158). The (110) surface of rutile IrO2 and RuO2 was modeled by a 3 × 1 slab with 4 layers, of which 2 were fixed at the bulk position (see Figure 7A). The hollandite (001) surface is used for comparison, represented by a 3 × 1 unit cell with 6 atomic layers (3 frozen) corresponding to 1.5 repeats of the unit cell (see Figure 7B). All cus sites on the surface except the active site are covered by *O, as is expected to be the case for catalysts on the strong-binding (left) side of the volcano close to the onset potential of OER. Both surfaces were sampled with 2 × 2 k-points and 20 Å of vacuum separating periodic images of the slab.
The adsorption free energies (ΔGads) of the OER reaction intermediates at a given potential (U) are calculated using the computational hydrogen electrode described in reference.59 Free energy corrections taken from this reference are added to the 0 K adsorption energies calculated by DFT (ΔEads) to account for changes in entropy (ΔS) and zero-point energy (ΔZPE).
where n is the number of electrons involved in the reaction. ΔGads is calculated for the two possible reaction pathways shown in Figure S15, and it is assumed that the reaction follows the pathway with the lowest free energy. The values of ΔG1–4 for this pathway is used to calculate the overpotential (η) for OER as
Acknowledgments
Part of this study was performed in the RECYCALYSE project which has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No. 861960. It was further supported by the Danish National Research Foundation in the Center for High Entropy Alloy Catalysis (CHEAC) DNRF 149. We acknowledge MAX IV Laboratory for time on DanMAX under Proposal 20220789. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. DanMAX is funded by the NUFI grant no. 4059-00009B. We would like to thank Mads Ry Jørgensen and Innokenty Kantor at DanMAX for their contributions in performing the PDF measurements. Thanks also to beamline scientist Dr. Adam H. Clarke at the SuperXAS beamline for performing ex situ XAS measurements at the Swiss Lightsource (SLS) at PSI, Villingen/Switzerland, through a send-in service. We also acknowledge access to the Microscopy Imaging Center (MIC) facilities of the University of Bern.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmaterialsau.4c00025.
EDX analysis results; calculated XRD spectra; XRD and Rietveld refinement; PDF data analysis; transmission electron microscopy; XAS data analysis; electrochemical measurements; acid wash experiments; DFT reaction pathways (PDF)
Author Contributions
# Erlend Bertheussen, Simon Pitscheider, and Susan R. Cooper contributed equally to this work. CRediT: Erlend Bertheussen formal analysis, investigation, methodology, validation, visualization, writing-original draft, writing-review & editing; Simon Pitscheider conceptualization, formal analysis, investigation, methodology, validation, visualization, writing-original draft, writing-review & editing; Susan R. Cooper conceptualization, formal analysis, investigation, methodology, software, validation, visualization, writing-original draft, writing-review & editing; Rebecca K. Pittkowski formal analysis, investigation, methodology, software, validation, visualization, writing-original draft, writing-review & editing; Katrine Svane formal analysis, investigation, methodology, software, validation, visualization, writing-original draft, writing-review & editing; Aline Bornet investigation, visualization, writing-review & editing; Erik M. Wisaeus formal analysis, investigation, validation; Kirsten M. Ø. Jensen funding acquisition, project administration, resources, supervision, writing-review & editing; Jan Rossmeisl funding acquisition, methodology, project administration, resources, supervision; Matthias Arenz conceptualization, funding acquisition, methodology, resources, supervision, writing-review & editing; Christian Kallesøe conceptualization, funding acquisition, project administration, resources, supervision, writing-review & editing; Christoffer Mølleskov Pedersen conceptualization, formal analysis, methodology, project administration, resources, software, supervision, validation, visualization, writing-review & editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Barbir F. PEM Electrolysis for Production of Hydrogen from Renewable Energy Sources. Sol. Energy 2005, 78 (5), 661–669. 10.1016/j.solener.2004.09.003. [DOI] [Google Scholar]
- Pellow M. A.; Emmott C. J. M.; Barnhart C. J.; Benson S. M. Hydrogen or Batteries for Grid Storage? A Net Energy Analysis. Energy Environ. Sci. 2015, 8 (7), 1938–1952. 10.1039/C4EE04041D. [DOI] [Google Scholar]
- Ayers K. E.; Capuano C.; Anderson E. B. Recent Advances in Cell Cost and Efficiency for PEM-Based Water Electrolysis. ECS Trans 2012, 41 (10), 15–22. 10.1149/1.3684798. [DOI] [Google Scholar]
- Carmo M.; Fritz D. L.; Mergel J.; Stolten D. A Comprehensive Review on PEM Water Electrolysis. Int. J. Hydrogen Energy 2013, 38 (12), 4901–4934. 10.1016/j.ijhydene.2013.01.151. [DOI] [Google Scholar]
- European Commission . Study on the Critical Raw Materials for the EU 2023 - Final Report.
- Vesborg P. C. K.; Jaramillo T. F. Addressing the Terawatt Challenge: Scalability in the Supply of Chemical Elements for Renewable Energy. RSC Adv. 2012, 2 (21), 7933. 10.1039/c2ra20839c. [DOI] [Google Scholar]
- Minke C.; Suermann M.; Bensmann B.; Hanke-Rauschenbach R. Is Iridium Demand a Potential Bottleneck in the Realization of Large-Scale PEM Water Electrolysis?. Int. J. Hydrogen Energy 2021, 46 (46), 23581–23590. 10.1016/j.ijhydene.2021.04.174. [DOI] [Google Scholar]
- Bernt M.; Siebel A.; Gasteiger H. A. Analysis of Voltage Losses in PEM Water Electrolyzers with Low Platinum Group Metal Loadings. J. Electrochem. Soc. 2018, 165 (5), F305–F314. 10.1149/2.0641805jes. [DOI] [Google Scholar]
- Sverdrup H. U.; Ragnarsdottir K. V. A System Dynamics Model for Platinum Group Metal Supply, Market Price, Depletion of Extractable Amounts, Ore Grade, Recycling and Stocks-in-Use. Resour Conserv Recycl 2016, 114, 130–152. 10.1016/j.resconrec.2016.07.011. [DOI] [Google Scholar]
- Rasmussen K. D.; Wenzel H.; Bangs C.; Petavratzi E.; Liu G. Platinum Demand and Potential Bottlenecks in the Global Green Transition: A Dynamic Material Flow Analysis. Environ. Sci. Technol. 2019, 53 (19), 11541–11551. 10.1021/acs.est.9b01912. [DOI] [PubMed] [Google Scholar]
- Babic U.; Suermann M.; Büchi F. N.; Gubler L.; Schmidt T. J. Critical Review—Identifying Critical Gaps for Polymer Electrolyte Water Electrolysis Development. J. Electrochem. Soc. 2017, 164 (4), F387–F399. 10.1149/2.1441704jes. [DOI] [Google Scholar]
- Bernt M.; Hartig-Weiß A.; Tovini M. F.; El-Sayed H. A.; Schramm C.; Schröter J.; Gebauer C.; Gasteiger H. A. Current Challenges in Catalyst Development for PEM Water Electrolyzers. Chem. Ing Tech 2020, 92 (1–2), 31–39. 10.1002/cite.201900101. [DOI] [Google Scholar]
- Ruban A.; Hammer B.; Stoltze P.; Skriver H. L.; Nørskov J. K. Surface Electronic Structure and Reactivity of Transition and Noble Metals. J. Mol. Catal. A Chem. 1997, 115 (3), 421–429. 10.1016/S1381-1169(96)00348-2. [DOI] [Google Scholar]
- Demirci U. B. Theoretical Means for Searching Bimetallic Alloys as Anode Electrocatalysts for Direct Liquid-Feed Fuel Cells. J. Power Sources 2007, 173 (1), 11–18. 10.1016/j.jpowsour.2007.04.069. [DOI] [Google Scholar]
- Puthiyapura V. K.; Mamlouk M.; Pasupathi S.; Pollet B. G.; Scott K. Physical and Electrochemical Evaluation of ATO Supported IrO2 Catalyst for Proton Exchange Membrane Water Electrolyser. J. Power Sources 2014, 269, 451–460. 10.1016/j.jpowsour.2014.06.078. [DOI] [Google Scholar]
- Marshall A. T.; Haverkamp R. G. Electrocatalytic Activity of IrO2-RuO2 Supported on Sb-Doped SnO2 Nanoparticles. Electrochim. Acta 2010, 55 (6), 1978–1984. 10.1016/j.electacta.2009.11.018. [DOI] [Google Scholar]
- Oh H.-S.; Nong H. N.; Strasser P. Preparation of Mesoporous Sb-, F-, and In-Doped SnO 2 Bulk Powder with High Surface Area for Use as Catalyst Supports in Electrolytic Cells. Adv. Funct Mater. 2015, 25 (7), 1074–1081. 10.1002/adfm.201401919. [DOI] [Google Scholar]
- Skoromets V.; Němec H.; Kopeček J.; Kužel P.; Peters K.; Fattakhova-Rohlfing D.; Vetushka A.; Müller M.; Ganzerová K.; Fejfar A. Conductivity Mechanisms in Sb-Doped SnO 2 Nanoparticle Assemblies: DC and Terahertz Regime. J. Phys. Chem. C 2015, 119 (33), 19485–19495. 10.1021/acs.jpcc.5b05091. [DOI] [Google Scholar]
- Ohno H.; Nohara S.; Kakinuma K.; Uchida M.; Uchida H. Effect of Electronic Conductivities of Iridium Oxide/Doped SnO2 Oxygen-Evolving Catalysts on the Polarization Properties in Proton Exchange Membrane Water Electrolysis. Catalysts 2019, 9 (1), 74. 10.3390/catal9010074. [DOI] [Google Scholar]
- Böhm D.; Beetz M.; Schuster M.; Peters K.; Hufnagel A. G.; Döblinger M.; Böller B.; Bein T.; Fattakhova-Rohlfing D. Efficient OER Catalyst with Low Ir Volume Density Obtained by Homogeneous Deposition of Iridium Oxide Nanoparticles on Macroporous Antimony-Doped Tin Oxide Support. Adv. Funct Mater. 2020, 30 (1), 1906670. 10.1002/adfm.201906670. [DOI] [Google Scholar]
- Hartig-Weiss A.; Miller M.; Beyer H.; Schmitt A.; Siebel A.; Freiberg A. T. S.; Gasteiger H. A.; El-Sayed H. A. Iridium Oxide Catalyst Supported on Antimony-Doped Tin Oxide for High Oxygen Evolution Reaction Activity in Acidic Media. ACS Appl. Nano Mater. 2020, 3 (3), 2185–2196. 10.1021/acsanm.9b02230. [DOI] [Google Scholar]
- Geiger S.; Kasian O.; Mingers A. M.; Mayrhofer K. J. J.; Cherevko S. Stability Limits of Tin-Based Electrocatalyst Supports. Sci. Rep 2017, 7 (1), 3–9. 10.1038/s41598-017-04079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornet A.; Pittkowski R.; Nielsen T. M.; Berner E.; Maletzko A.; Schröder J.; Quinson J.; Melke J.; Jensen K. M. Ø.; Arenz M. Influence of Temperature on the Performance of Carbon- and ATO-Supported Oxygen Evolution Reaction Catalysts in a Gas Diffusion Electrode Setup. ACS Catal. 2023, 13 (11), 7568–7577. 10.1021/acscatal.3c01193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svane K. L.; Rossmeisl J. Theoretical Optimization of Compositions of High-Entropy Oxides for the Oxygen Evolution Reaction. Angew. Chem., Int. Ed. 2022, 61 (19), e202201146 10.1002/anie.202201146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherevko S.; Geiger S.; Kasian O.; Kulyk N.; Grote J. P.; Savan A.; Shrestha B. R.; Merzlikin S.; Breitbach B.; Ludwig A.; Mayrhofer K. J. J. Oxygen and Hydrogen Evolution Reactions on Ru, RuO2, Ir, and IrO2 Thin Film Electrodes in Acidic and Alkaline Electrolytes: A Comparative Study on Activity and Stability. Catal. Today 2016, 262, 170–180. 10.1016/j.cattod.2015.08.014. [DOI] [Google Scholar]
- Roy C.; Rao R. R.; Stoerzinger K. A.; Hwang J.; Rossmeisl J.; Chorkendorff I.; Shao-Horn Y.; Stephens I. E. L. Trends in Activity and Dissolution on RuO 2 under Oxygen Evolution Conditions: Particles versus Well-Defined Extended Surfaces. ACS Energy Lett. 2018, 3 (9), 2045–2051. 10.1021/acsenergylett.8b01178. [DOI] [Google Scholar]
- Halck N. B.; Petrykin V.; Krtil P.; Rossmeisl J. Beyond the Volcano Limitations in Electrocatalysis - Oxygen Evolution Reaction. Phys. Chem. Chem. Phys. 2014, 16 (27), 13682–13688. 10.1039/C4CP00571F. [DOI] [PubMed] [Google Scholar]
- Moghaddam R. B.; Wang C.; Sorge J. B.; Brett M. J.; Bergens S. H. Easily Prepared, High Activity Ir-Ni Oxide Catalysts for Water Oxidation. Electrochem commun 2015, 60, 109–112. 10.1016/j.elecom.2015.08.015. [DOI] [Google Scholar]
- Reier T.; Pawolek Z.; Cherevko S.; Bruns M.; Jones T.; Teschner D.; Selve S.; Bergmann A.; Nong H. N.; Schlögl R.; Mayrhofer K. J. J.; Strasser P. Molecular Insight in Structure and Activity of Highly Efficient, Low-Ir Ir-Ni Oxide Catalysts for Electrochemical Water Splitting (OER). J. Am. Chem. Soc. 2015, 137 (40), 13031–13040. 10.1021/jacs.5b07788. [DOI] [PubMed] [Google Scholar]
- Spöri C.; Briois P.; Nong H. N.; Reier T.; Billard A.; Kühl S.; Teschner D.; Strasser P. Experimental Activity Descriptors for Iridium-Based Catalysts for the Electrochemical Oxygen Evolution Reaction (OER). ACS Catal. 2019, 9 (8), 6653–6663. 10.1021/acscatal.9b00648. [DOI] [Google Scholar]
- Buvat G.; Eslamibidgoli M. J.; Zhang T.; Prabhudev S.; Youssef A. H.; Ruediger A.; Garbarino S.; Botton G. A.; Zhang P.; Eikerling M.; Guay D. Understanding the Effect of Ni-Substitution on the Oxygen Evolution Reaction of (100) IrO 2 Surfaces. ACS Catal. 2022, 12 (17), 10961–10972. 10.1021/acscatal.2c02591. [DOI] [Google Scholar]
- Sun W.; Song Y.; Gong X.-Q.; Cao L.; Yang J. Hollandite Structure K x ≈0.25 IrO 2 Catalyst with Highly Efficient Oxygen Evolution Reaction. ACS Appl. Mater. Interfaces 2016, 8 (1), 820–826. 10.1021/acsami.5b10159. [DOI] [PubMed] [Google Scholar]
- Willinger E.; Massué C.; Schlögl R.; Willinger M. G. Identifying Key Structural Features of IrOx Water Splitting Catalysts. J. Am. Chem. Soc. 2017, 139 (34), 12093–12101. 10.1021/jacs.7b07079. [DOI] [PubMed] [Google Scholar]
- Kallesøe C.; Clausen H. F.; Christensen L. H.. Method of Preparing a Catalyst Structure. WO2015104025A1, 2015.
- Kallesøe C.; Clausen H. F.; Christensen L. H.; Lund-Olesen T.; Mamakhe A. H. M.; Iversen B. B.; Becker-Christensen J.; Aarup D. F.; Hales J.. Method of Preparing a Catalytic Structure. US2015202598A1, 2015.
- Silva H.; Hernandez-Fernandez P.; Baden A. K.; Hellstern H. L.; Kovyakh A.; Wisaeus E.; Smitshuysen T.; Chorkendorff I.; Christensen L. H.; Chakraborty D.; Kallesøe C. Supercritical Flow Synthesis of PtPdFe Alloyed Nanoparticles with Enhanced Low-Temperature Activity and Thermal Stability for Propene Oxidation under Lean Exhaust Gas Conditions. Catal. Sci. Technol. 2019, 9 (23), 6691–6699. 10.1039/C9CY00634F. [DOI] [Google Scholar]
- Lee Y.; Suntivich J.; May K. J.; Perry E. E.; Shao-Horn Y. Synthesis and Activities of Rutile IrO 2 and RuO 2 Nanoparticles for Oxygen Evolution in Acid and Alkaline Solutions. J. Phys. Chem. Lett. 2012, 3 (3), 399–404. 10.1021/jz2016507. [DOI] [PubMed] [Google Scholar]
- Baur W. H.; Khan A. A. Rutile-Type Compounds. IV. SiO2, GeO2 and a Comparison with Other Rutile-Type Structures. Acta Crystallogr. B 1971, 27 (11), 2133–2139. 10.1107/S0567740871005466. [DOI] [Google Scholar]
- Owen E. A.; Yates E. L. XLI. Precision Measurements of Crystal Parameters. London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science 1933, 15 (98), 472–488. 10.1080/14786443309462199. [DOI] [Google Scholar]
- Wyckoff R. W. G.Crystal Structures - Vol. 1, 2nd ed.; Interscience Publishers: New York, 1963. [Google Scholar]
- Bolzan A. A.; Fong C.; Kennedy B. J.; Howard C. J. Structural Studies of Rutile-Type Metal Dioxides. Acta Crystallogr. B 1997, 53 (3), 373–380. 10.1107/S0108768197001468. [DOI] [Google Scholar]
- Bestaoui N.; Deniard P.; Brec R. Structural Study of a Hollandite-Type KxIrO2. J. Solid State Chem. 1995, 118 (2), 372–377. 10.1006/jssc.1995.1356. [DOI] [Google Scholar]
- Talanov A.; Phelan W. A.; Kelly Z. A.; Siegler M. A.; McQueen T. M. Control of the Iridium Oxidation State in the Hollandite Iridate Solid Solution K 1- x Ir 4 O 8. Inorg. Chem. 2014, 53 (9), 4500–4507. 10.1021/ic5001667. [DOI] [PubMed] [Google Scholar]
- Macrae C. F.; Sovago I.; Cottrell S. J.; Galek P. T. A.; McCabe P.; Pidcock E.; Platings M.; Shields G. P.; Stevens J. S.; Towler M.; Wood P. A. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53 (1), 226–235. 10.1107/S1600576719014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl Christiansen T.; Kjær E. T. S.; Kovyakh A.; Röderen M. L.; Høj M.; Vosch T.; Jensen K. M. Ø. Structure Analysis of Supported Disordered Molybdenum Oxides Using Pair Distribution Function Analysis and Automated Cluster Modelling. J. Appl. Crystallogr. 2020, 53 (1), 148–158. 10.1107/S1600576719016832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divanis S.; Frandsen A. M.; Kutlusoy T.; Rossmeisl J. Lifting the Discrepancy between Experimental Results and the Theoretical Predictions for the Catalytic Activity of RuO 2 (110) towards Oxygen Evolution Reaction. Phys. Chem. Chem. Phys. 2021, 23 (35), 19141–19145. 10.1039/D1CP02999A. [DOI] [PubMed] [Google Scholar]
- Chupas P. J.; Qiu X.; Hanson J. C.; Lee P. L.; Grey C. P.; Billinge S. J. L. Rapid-Acquisition Pair Distribution Function (RA-PDF) Analysis. J. Appl. Crystallogr. 2003, 36 (6), 1342–1347. 10.1107/S0021889803017564. [DOI] [Google Scholar]
- Kieffer J.; Valls V.; Blanc N.; Hennig C. New Tools for Calibrating Diffraction Setups. J. Synchrotron Radiat 2020, 27 (2), 558–566. 10.1107/S1600577520000776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Juhás P.; Farrow C. L.; Billinge S. J. L.. XPDFsuite: An End-to-End Software Solution for High Throughput Pair Distribution Function Transformation, Visualization and Analysis. ArXiv, February 23, 2015. 10.48550/ArXiv.1402.3163 (accessed 2024–03–22). [DOI]
- Farrow C. L.; Juhas P.; Liu J. W.; Bryndin D.; Božin E. S.; Bloch J.; Proffen T.; Billinge S. J. L. PDFfit2 and PDFgui: Computer Programs for Studying Nanostructure in Crystals. J. Phys.: Condens. Matter 2007, 19 (33), 335219. 10.1088/0953-8984/19/33/335219. [DOI] [PubMed] [Google Scholar]
- Ravel B.; Newville M. ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis for X-Ray Absorption Spectroscopy Using IFEFFIT. J. Synchrotron Radiat 2005, 12 (4), 537–541. 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
- Schröder J.; Mints V. A.; Bornet A.; Berner E.; Fathi Tovini M.; Quinson J.; Wiberg G. K. H.; Bizzotto F.; El-Sayed H. A.; Arenz M. The Gas Diffusion Electrode Setup as Straightforward Testing Device for Proton Exchange Membrane Water Electrolyzer Catalysts. JACS Au 2021, 1 (3), 247–251. 10.1021/jacsau.1c00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen J. J.; Hansen L. B.; Jacobsen K. W. Real-Space Grid Implementation of the Projector Augmented Wave Method. Phys. Rev. B 2005, 71 (3), 035109. 10.1103/PhysRevB.71.035109. [DOI] [Google Scholar]
- Enkovaara J.; Rostgaard C.; Mortensen J. J.; Chen J.; Dułak M.; Ferrighi L.; Gavnholt J.; Glinsvad C.; Haikola V.; Hansen H. A.; Kristoffersen H. H.; Kuisma M.; Larsen A. H.; Lehtovaara L.; Ljungberg M.; Lopez-Acevedo O.; Moses P. G.; Ojanen J.; Olsen T.; Petzold V.; Romero N. A.; Stausholm-Møller J.; Strange M.; Tritsaris G. A.; Vanin M.; Walter M.; Hammer B.; Häkkinen H.; Madsen G. K. H.; Nieminen R. M.; Nørskov J. K.; Puska M.; Rantala T. T.; Schiøtz J.; Thygesen K. S.; Jacobsen K. W. Electronic Structure Calculations with GPAW: A Real-Space Implementation of the Projector Augmented-Wave Method. J. Phys.: Condens. Matter 2010, 22 (25), 253202. 10.1088/0953-8984/22/25/253202. [DOI] [PubMed] [Google Scholar]
- Hjorth Larsen A.; Jørgen Mortensen J.; Blomqvist J.; Castelli I. E.; Christensen R.; Dułak M.; Friis J.; Groves M. N.; Hammer B.; Hargus C.; Hermes E. D.; Jennings P. C.; Bjerre Jensen P.; Kermode J.; Kitchin J. R.; Leonhard Kolsbjerg E.; Kubal J.; Kaasbjerg K.; Lysgaard S.; Bergmann Maronsson J.; Maxson T.; Olsen T.; Pastewka L.; Peterson A.; Rostgaard C.; Schiøtz J.; Schütt O.; Strange M.; Thygesen K. S.; Vegge T.; Vilhelmsen L.; Walter M.; Zeng Z.; Jacobsen K. W. The Atomic Simulation Environment—a Python Library for Working with Atoms. J. Phys.: Condens. Matter 2017, 29 (27), 273002. 10.1088/1361-648X/aa680e. [DOI] [PubMed] [Google Scholar]
- Hammer B.; Hansen L. B.; Nørskov J. K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 1999, 59 (11), 7413–7421. 10.1103/PhysRevB.59.7413. [DOI] [Google Scholar]
- Rogers D. B.; Shannon R. D.; Sleight A. W.; Gillson J. L. Crystal Chemistry of Metal Dioxides with Rutile-Related Structures. Inorg. Chem. 1969, 8 (4), 841–849. 10.1021/ic50074a029. [DOI] [Google Scholar]
- Kobayashi W. Transport Properties of Quasi-One-Dimensional KRu4O8. Phys. Rev. B 2009, 79 (15), 155116. 10.1103/PhysRevB.79.155116. [DOI] [Google Scholar]
- Nørskov J. K.; Rossmeisl J.; Logadottir A.; Lindqvist L.; Kitchin J. R.; Bligaard T.; Jónsson H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108 (46), 17886–17892. 10.1021/jp047349j. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.