

Abstract
Introduction
Acute lung injury (ALI) as one kind of acute pulmonary inflammatory disorder, manifests primarily as damage to alveolar epithelial cells and microvascular endothelial cells. Activation of the complement system is a common pathological mechanism in ALI induced by diverse factors, with the complement alternative pathway assuming a pivotal role. Baicalin, a flavonoid derived from the root of Scutellaria baicalensis Georgi, exhibits noteworthy biological activities. The present study attempted the interventional effects and underlying mechanisms of baicalin in microangiopathy in ALI induced by complement alternative pathway activation.
Methods
Activation of the complement alternative pathway by cobra venom factor (CVF). HMEC cells were pretreated with baicalin and then exposed to complement activation products. The expression of inflammatory mediators was detected by ELISA, and the intranuclear transcriptional activity of NF-κB was assessed by a dual fluorescent kinase reporter gene assay kit. Before establishing the ALI mouse model, baicalin or PDTC was gavaged for 7 d. CVF was injected into the tail vein to establish the ALI model. The levels of inflammatory mediators in BALF and serum were determined by ELISA. HE staining and immunohistochemistry evaluated pathological changes, complement activation product deposition, and NF-κB p65 phosphorylation in lung tissue.
Results
Baicalin reduced complement alternative activation product-induced expression of HMEC cells adhesion molecules (ICAM-1, VCAM-1, E-selectin) and cytokines (IL-6, TNF-α) as well as upregulation of NF-κB intranuclear transcriptional activity. Baicalin intervention reduced the number of inflammatory cells and protein content in the BALF and decreased the levels of IL-6, TNF-α, and ICAM-1 in serum and IL-6, TNF-α, ICAM-1, and P-selectin in BLAF. In addition, baicalin attenuated inflammatory cell infiltration in the lung of ALI mice and reduced the deposition of complement activation products (C5a, C5b-9) and phosphorylation of NF-κB p65 in lung tissue.
Conclusion
Baicalin relieves complement alternative pathway activation-induced lung inflammation by inhibition of NF-κB pathway, delaying the progression of ALI.
Keywords: Baicalin, Acute lung injury, Complement alternative pathway, Microvascular endothelial cells, Inflammation, NF-κB
Introduction
Acute lung injury (ALI) and its severe manifestation, acute respiratory distress syndrome (ARDS), pose significant challenges to human health. These conditions are characterized by acute inflammation and microvascular endothelial cell damage during the early stages [1, 2]. ALI/ARDS encompasses a multifaceted etiology, marked by rapid onset and a mortality rate of up to 40% [3]. Presently, comprehensive therapeutic approaches for ALI include mechanical ventilation, extracorporeal membrane oxygenation, stem cell therapy, and pharmacotherapy [1]. However, clinical practice continues to grapple with the dearth of efficacious therapeutic drugs and modalities.
Both clinical and experimental investigations have underscored the pivotal role of the complement system in ALI pathogenesis [4–6]. In the initial phases of ALI, the complement system takes center stage, with abnormal or excessive activation serving as an initiating and amplifying factor, culminating in inflammation and tissue injury [7]. The complement system, an integral facet of innate immunity, encompasses activation pathways comprising classical, alternative, and lectin pathways [8]. ALI triggered by diverse factors invariably involves the activation of the complement system. Notably, the inflammatory response and microvascular endothelial cells damage stemming from the activation of the complement alternative pathway play a paramount role in ALI pathogenesis [9, 10]. Consequently, inhibition of inflammation and tissue damage resulting from complement or alternative pathway activation has emerged as a potential therapeutic strategy for ALI. Based on this strategy, we constructed a mouse model of CVF-activated complement alternative pathway-induced ALI [11].
CVF is a protein extracted from cobra venom and is capable of selectively activating the complement alternative pathway. Its capacity to activate the complement alternative pathway closely mirrors in vivo complement activation under pathological conditions [12]. CVF readily forms CVF▪Bb with factor B in serum, effectively activating the complement alternative pathway and generating substantial quantities of C3a, C5a, and membrane-attacking complex (MAC) [12]. CVF has gained widespread utility as an experimental tool for elucidating the role of the complement system in the pathogenesis of various diseases including acute lung inflammation [11, 13]. Within this context, we employed our constructed mouse model of CVF-activated complement alternative pathway-induced ALI to identify bioactive agents, leading to the discovery of the protective effects of baicalin.
Baicalin, a primary bioactive compound isolated from Scutellaria baicalensis, exhibits a diverse array of biological activities [14]. Baicalin has been shown to exhibit a variety of pharmacological effects, including anti-inflammatory [15], anti-apoptotic [16], antioxidant [17], antidiabetic [18], anticancer [19], and antibacterial [20]. Previous studies have shown that baicalin effectively suppressed inflammation in different models of lung injury mediated by downregulating NF-κB, TLR4, NLRP3, and MAPK (p38, JNK, and ERK) signals [21–29]. Nevertheless, the role and mechanism of baicalin in ALI induced by complement alternative pathway activation has not been revealed and report. Meanwhile, our pre-screening revealed that baicalin has an intervention effect on lung inflammation induced by complement alternative pathway activation in mice. Hence, this study aimed to comprehensively explore the interventional effects and molecular mechanisms of action of baicalin on complementary alternative pathway activation-induced ALI in mice, thereby deepening our understanding of the pharmacological and anti-inflammatory effects of baicalin in ALI treatment.
Materials and methods
Cell culture and animal feeding
Human microvascular endothelial cells (HMEC), generously provided by Dr. Yang Jin at the Kunming Institute of Zoology, Chinese Academy of Sciences, were maintained in RPMI 1640 medium (Gibco, Waltham, USA). The medium was supplemented with 10% fetal bovine serum (FBS, Gibco, Waltham, USA) and 1% penicillin/streptomycin, and the cells were cultured under conditions of 37 °C, 5% CO2, and 100% humidity [30].
Kunming mice, weighing between 18 and 22 g and of male sex, obtained from SPF Certification (No. 2012-0011), were procured from the Experimental Animal Center of the Third Military Medical University of the Chinese People’s Liberation Army. Prior to the commencement of experiments, the animals were provided with standard rodent chow and water and were housed in a controlled environment with a temperature regulation system, following a 12-hour light and 12-hour dark cycle, for a period of 5 days. All animal experiments complied with the ARRIVE guidelines and were conducted with strict adherence to protocols approved by the Laboratory Animal Ethics Committee of Guizhou Medical University (No. 201345). Mice were handled according to internationally accepted principles for the care of laboratory animals. Mice were anaesthetised by intraperitoneal injection of sodium pentobarbital (50 mg/kg), and at the end of the experiment, the mice were euthanized by cervical dislocation, which resulted in immediate unconsciousness and cessation of breathing.
Reagents
CVF was prepared in-house according to established laboratory procedures [31]. The myeloperoxidase (MPO) kit was procured from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). BCA protein assay kit was procured from Beyotime Biotechnology Co., Ltd. (Shanghai, China). Pyrrolidine dithiocarbamate ammonium (PDTC) was procured from Chengdu Elfa Biotechnology Co., Ltd. (Chengdu, China). Baicalin was procured from Beijing Solebaum Technology Co. Ltd. (Beijing, China). Carboxymethylcellulose sodium (CMC-Na) was procured from Shanghai Aladdin Biochemical Technology Corp. (Shanghai, China). Enzyme-linked immunosorbent assay (ELISA) kits for quantification of mouse IL-6, TNF-α, P-selectin, E-selectin, VCAM-1, and ICAM-1 were procured from Boster Biological Technology Co., Ltd. (Wuhan, China). The complement C5a polyclonal antibody was procured from Thermo Fisher Scientifi (Cat. No. PA578891, Thermo Fisher Scientifi, Waltham, USA). Rabbit polyclonal C5b-9 was procured from Abcam (Cat. No. ab55811, Abcam, Cambridge, England). p-RELA/NFκB p65 antibody (49.Ser 311) was procured from Santa Cruz Biotechnology (Cat. No. sc-135769, Santa Cruz, Dallas, USA).
In vitro study
Preparation of complement alternative pathway activation products
The CVF incubation product, denoted as CVF-activated complement (CAC) [11], was generated by combining normal human serum (NHS) with CVF at a concentration of 6.5 × 104 U/L in equal proportions. The mixture was subsequently incubated in a water bath at 37 °C for a duration of 30 min. Inactivated normal human serum (INHS) was used as a comparative control, following the method outlined above, to prepare an incubation product.
Adhesion molecule assay
HMEC were initially seeded in cell culture plates at a density of 1.0 × 104 cells and incubated for 24 h. Subsequently, the cells were rinsed twice with warm serum-free RPMI 1640 medium. A mixture of 50 µL of medium and 10 µL of varying concentrations of baicalin (final concentration 10− 10, 10− 11, and 10− 12 M) was added to pre-treat the cells for 2 h. Following this pre-treatment, 40 µL of CAC incubation products were gently mixed and added to the cells, followed by an incubation period of 6 h. Following the incubation, the supernatant was carefully collected and centrifuged. The resulting supernatant was used for quantification of ICAM-1, VCAM-1, and E-selectin, following the instructions provided with the ELISA kit.
For reference, the NHS + CVF incubation products were used as the model group (Model), whereas the INHS + CVF incubation products served as the normal control group (Control).
Cytokine assay
HMEC were seeded at a density of 2.0 × 104 cells in 96-well culture plates, each containing a total volume of 200 µL. After an incubation period of 24 h, the cells were washed twice with warmed serum-free RPMI 1640 medium. Subsequently, 100 µL of the medium and 20 µL of baicalin at varying concentrations (final concentration 10− 10, 10− 11, and 10− 12 M) were added to pre-treat the cells for 2 h. Following this pre-treatment, the incubation products were combined with 80 µL of CAC, resulting in a total volume of 200 µL. The cells were then incubated for an additional 48 h. After incubation, the supernatant of the culture medium was collected and centrifuged. The subsequent steps involved the determination of IL-6 and TNF-α levels, following the protocols provided with the ELISA kit.
For reference, the model group (Model) utilized NHS + CVF incubation products, while the normal control group (Control) employed INHS + CVF incubation products.
NF-κB transcriptional activity assay
Cells were seeded into black 96-well plates at a density of 1 × 105 cells/well and allowed to grow overnight. The cells were then washed thrice with warm serum-free RPMI 1640 medium. Subsequently, they were supplemented with 90 µL of RPMI 1640 containing 10% fetal bovine serum and subjected to transfection following the instructions outlined in the Liposome Transfection Kit (Cat. No. C0526; Beyotime Biotechnology Co., Ltd., Shanghai, China). Each well received 10 µL of the transfection mixture, comprising 88 ng of NF-κB expression plasmid, 45 ng of an internal reference plasmid, and 0.2 µL of liposome reagent, and was incubated for 16 h. Following transfection, the supernatant was aspirated and 90 µL of RPMI 1640 medium, along with 10 µL of baicalin at different concentrations or warm PBS, was added and incubated for 2 h. Subsequently, 30 µL of the cell supernatant was removed, replaced with 30 µL of CAC stimulation, and incubated for an additional 4 h [30]. A negative control was established using mixed incubates of CVF and INHS.
Fluorescence intensity was assessed using the Dual-Luciferase Reporter Assay System (Cat. No. E1910, Promega, USA), and the relative nuclear transcription activity (Ra) was calculated using the following formula:
Ra = (R1treat/R2 treat) / (R1control/R2 control) × 100,
where R1 is the value of firefly luciferase, and R2 is the value of Renilla luciferase.
In vivo study
Induction and treatment protocols for ALI in mice
Referring to the published literature, an ALI mouse model was established, and the appropriate dose of baicalin and PDTC administered was selected [21–30].Seventy-two KM mice were randomly allocated into six groups, each comprising 12 mice: control group (Control), CVF group (Model), baicalin 200 mg/kg + CVF group (high-dose baicalin), baicalin 100 mg/kg + CVF group (medium-dose baicalin), baicalin 50 mg/kg + CVF group (low-dose baicalin), and PDTC 100 mg/kg + CVF group (PDTC, used as a positive control). To initiate the ALI model in mice and specifically activate the complement alternative pathway [11, 30], CVF (35 µg/kg body weight, dissolved in sterile PBS with pH 7.4) was administered via tail vein injection. Control mice received sterile PBS injections. Preceding to model induction, the animals received oral pretreatment with baicalin and PDTC, both of which were prepared in 0.5% carboxymethylcellulose sodium solution for 7 days. An equal volume of 0.5% carboxymethylcellulose sodium (CMC-Na) was administered to the vehicle-treated control group. On the seventh day, CVF (35 mg/kg) was administered via tail vein injection, with an interval of 1 h post-administration (30 min after administration in the PDTC group). The normal control group received an equivalent volume of PBS via tail vein injection. An hour following the CVF or PBS challenge, the mice were anesthetized by intraperitoneal (i.p.) injection of pentobarbital sodium (50 mg/kg), and blood was collected from the retro-orbital venous plexus. Subsequently, the mice were euthanized by cervical dislocation, which resulted in immediate unconsciousness and cessation of breathing. Finally, bronchoalveolar lavage fluid (BALF), and lung tissues were collected from each group for further analysis following established protocols (Fig. 1) [11].
Fig. 1.

Flowchart of the animal experimentation protocol. Experimental design to assess the effect of baicalin on ALI mice (n = 12/group)
Inflammatory cell counting and protein concentration determination in BALF
The BALF was subjected to centrifugation at 2000 rpm and 4 °C for a duration of 10 min, resulting in the pelleting of cells. The BALF supernatant was meticulously mixed and the total protein concentration was assessed using a BCA assay kit.
Simultaneously, cell pellets were reconstituted in ice-cold PBS. Following the exclusion of non-viable cells via trypan blue staining, the overall count of inflammatory cells in the BALF was determined using a hemocytometer.
MPO assay and ELISA of BALF and serum
The superior and middle lobes of the right lung were harvested and subjected to homogenization to assess MPO activity, which serves as an indicator of neutrophil accumulation [32]. This analysis was conducted according to the protocol outlined in the respective test kit.
In addition, serum levels of IL-6, TNF-α and ICAM-1, and BAFL levels of IL-6, TNF-α, ICAM-1 and P-selectin were detected using mouse ELISA kits.
Lung histopathology and immunohistochemical
Reference to previous methods [11, 30], the lower lobe of the right lung was fixed in 4% paraformaldehyde, dehydrated, embedded, and cut into approximately 5 μm-thick sections for histopathologic detection with hematoxylin and eosin (H&E) staining, and the protein examination of C5a, C5b-9 and phosphorylated NF-κB p65 with immunohistochemical method.
Twelve paraffin sections were taken from the tissue samples for H&E staining in each group. The morphological changes in the lung tissues of each group of mice were observed and photographed under a light microscope. The lung injury score was performed according to the recommendations of the official American Thoracic Society workshop report (Table 1) [33]. The inflammation of lung tissue was evaluated according to the degree of infiltration of inflammatory cells. The scoring contents included the following criteria: edema, inflammation, alveolar dilation, proliferation and necrosis of alveolar epithelial cells, and thickness of the alveolar wall. The score range was 0–4 points (0, absence or very mild; 1, 2, 3 and 4 represented mild, moderate, severe and maximal, respectively). Each slice was scored using three fields of view and averaged.
Table 1.
The scoring criteria for lung injury
| Scoring | Inflammation (per field) | Edema | Lveolar dilation/Thickness of the alveolar wall/Proliferation and necrosis of alveolar epithelial cells |
|---|---|---|---|
| 0 | absence | absence | absence |
| 1 | 10 inflammatory cells | 10% alveolar involvement | mild |
| 2 | 10–50 inflammatory cells | 10–50% alveolar involvement | moderat |
| 3 | 50–70 inflammatory cells | 50–70% alveolar involvement | severe |
| 4 | Outweigh 70 inflammatory cells | 70–100% alveolar involvement | maximal |
Each group took 11–12 paraffin sections from the tissue samples for immunohistochemistry. C5a (dilution 1:200, PA578891, Thermo, USA), C5b-9 (dilution 1:200, ab55811, Abcam, UK) and phosphorylated NF-κB p65 (dilution 1:100, sc-135769, Santa Cruz, USA) primary antibodies were used to detect lung tissue C5a, C5b-9 deposits and levels of phosphorylated NF-κB p65. A positive reaction was identified by brownish-yellow or brown particles in the nucleus or cytoplasm, respectively. The C5a, C5b-9, and NF-κB p65 phosphorylation was measured semi-quantitatively by detecting the total positive area under the microscope with Image-Pro Plus image analysis software.
Statistical analysis
Statistical analyses of the data were conducted using the SPSS software (version 19.0). Data are presented as mean ± standard deviation (SD). Differences between groups were assessed using analysis of variance (one-way ANOVA), followed by the least significant difference (LSD) test for post-hoc comparisons of means. Significance was defined as P < 0.05 or P < 0.01.
Results
Baicalin reduces the release of adhesion molecules in HMEC induced by CAC
The expression of ICAM-1, VCAM-1, and E-selectin was elevated following the stimulation of HMEC cells with CAC in comparison to the control group. Conversely, the expression of ICAM-1, VCAM-1, and E-selectin displayed a noteworthy and dose-dependent reduction upon pretreatment with baicalin at concentrations of 10− 10, 10− 11, and 10− 12 M (Fig. 2A, B, and C).
Fig. 2.

Effects of different concentrations of baicalin on the adhesion molecule expression of HMEC induced by CAC. HMEC were pretreated with different concentrations (10− 10, 10− 11, and 10− 12 M) for 2 h and then incubated with 30 µL of the CAC product for 6 h. The levels of ICAM-1 (A), VCAM-1 (B), and E-selectin (C) in the culture medium were determined using enzyme-linked immunosorbent assay (ELISA). Values represent the mean ± SD (n = 8). **P < 0.01 compared with control group; ##P < 0.01 compared with model group
Baicalin inhibits the inflammatory mediator expressions of HMEC induced by CAC
Following the stimulation of HMEC with CAC, the expression of IL-6 and TNF-α was elevated in comparison to the control group. However, pretreatment with baicalin at varying doses (10− 10, 10− 11, and 10− 12 M) resulted in a notable and dose-dependent reduction in the expression levels of IL-6 and TNF-α (Fig. 3A, B).
Fig. 3.

Effects of different concentrations of baicalin on the inflammatory mediator expression of HMEC induced by CAC. HMEC were pre-treated with different concentrations (10− 10,10− 11, and 10− 12 M) of baicalin for 2 h and then incubated with 30 µL of CAC for 48 h. IL-6 (A) and TNF-α (B levels in the medium were determined by ELISA. Values represent the mean ± SD (n = 8). **P < 0.01 compared with control group; #P < 0.05 and ##P < 0.01 compared with model group
Effects of baicalin on the transcriptional activity of NF-κB after exposure of HMEC to CAC
The nuclear transcriptional activity of NF-κB was significantly upregulated following CAC stimulation of HMEC. However, pretreatment with baicalin at various concentrations (10− 10, 10− 11, and 10− 12 M) exhibited a noteworthy dose-dependent inhibitory effect on the augmentation of NF-κB intranuclear transcriptional activity induced by CAC (Fig. 4).
Fig. 4.
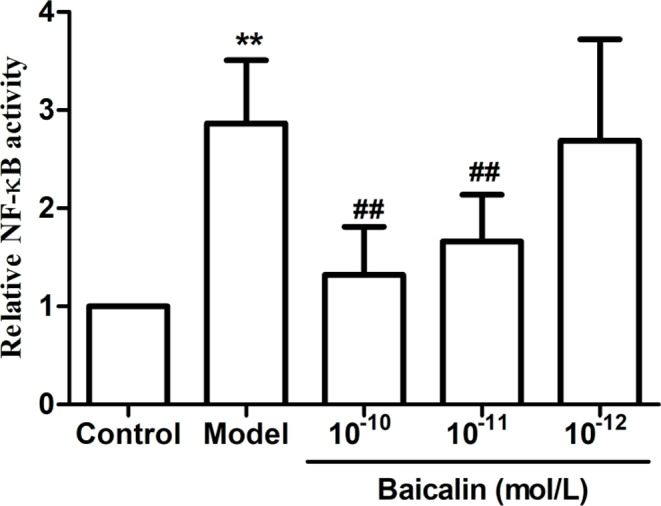
Effect of baicalin with different concentrations on transcriptional activity of NF-κB after exposure of HMEC to CAC. HMEC were inoculated into 96-well plates overnight and then transfected with NF-κB expression plasmid and liposome reagents. The cells were then pretreated with different concentrations (10− 10, 10− 11, and 10− 12 M) for 2 h and incubated with 30 µL of CAC product for 4 h. Luciferase activity was measured using a dual luciferase reporter gene assay system, and values represent the mean ± SD (n = 8). **P < 0.01 compared with control group; ##P < 0.01 compared with model group
Baicalin reduces inflammatory cell extravasation, elevated BALF protein levels and lung MPO activity in ALI mice
The concentration of proteins in BALF serves as a common indicator of pulmonary vascular permeability, which is a crucial feature of ALI. Comparatively, the Model group displayed significantly higher BALF protein concentrations than did the control group. Notably, pretreatment with baicalin at doses of 200 mg/kg and 100 mg/kg, along with PDTC, led to a significant reduction in protein concentrations in BALF when compared to the model group. However, no statistically significant difference was observed with 50 mg/kg baicalin pretreatment (Fig. 5A).
Fig. 5.

Baicalin inhibited protein concentrations and cells counts in BALF and suppressed neutrophil infiltration in lung tissue. Mice were orally administered different concentrations (200, 100 and 50 mg/kg/day) of baicalin or PDTC (100 mg/kg/day) or vehicle for 7 days. On the seventh day, CVF (35 µg/kg) was injected via the tail vein for 1 h. Lung tissue and BALF were then collected. total protein concentration (A) and cells count (B) in BALF were assayed. The upper and middle lobes of the right lung were used to detect MPO activity in the lung (C). Results are expressed as mean ± SD (n = 12).**P < 0.01 was compared with the control group; #P < 0.05 and ##P < 0.01 were compared with the model group
Furthermore, we assessed the total leukocyte count and lung MPO activity, an indicator of neutrophil infiltration, in BALF [32]. The BALF of the model group exhibited a substantial increase in total cells counts (Fig. 5B) and pulmonary MPO activity (Fig. 5C) compared to the control group, indicating the presence of inflammatory cells and neutrophil recruitment in the lung. Notably, pretreatment with baicalin (at doses of 200, 100, and 50 mg/kg) or PDTC led to a significant reduction in both total cells counts (Fig. 5B) and lung MPO activity (Fig. 5C) in BALF after CVF challenge compared with the Model group. These findings suggest that baicalin can mitigate pulmonary edema and inflammation in a dose-dependent manner in mice with complement alternative pathway activation-mediated ALI.
Effects of baicalin on inflammatory mediators and adhesion molecules in BALF and serum of ALI mice
We measured IL-6, TNF-α, and ICAM-1 levels in the serum and IL-6, TNF-α, ICAM-1, and P-selectin in the BALF using ELISA to evaluate the anti-inflammatory effects of baicalin in CVF-induced ALI. The levels of pro-inflammatory cytokines (TNF-α, IL-6) and adhesion molecule (ICAM-1) in the serum of mice in the model group were significantly elevated compared with those in the control group. Pretreatment with baicalin or PDTC resulted in a substantial reduction in the levels of pro-inflammatory cytokines (TNF-α, IL-6) and adhesion molecule (ICAM-1) in the serum of mice following CVF stimulation, compared to the Model group (Fig. 6A, B, and C).
Fig. 6.

Baicalin reduces the levels of inflammatory factors and adhesion molecules in serum and BALF of ALI mice. Mice were orally administered different concentrations (200, 100 and 50 mg/kg/day) of baicalin or PDTC (100 mg/kg/day) or vehicle for 7 days. Serum and BALF were collected 1 h after intravenous injection of CVF (35 µg/kg) and subjected to ELISA. A-C: serum levels of TNF-α, IL-6, and ICAM-1; D-G: BALF levels of TNF-α, IL-6, ICAM-1, and P-selectin. Results are expressed as mean ± SD (n = 12). **P < 0.01was compared with the control group; #P < 0.05 and ##P < 0.01 were compared with the model group
The levels of pro-inflammatory cytokines (TNF-α, IL-6) and adhesion molecule (ICAM-1, P-selectin) in the BALF of mice in the model group were significantly elevated compared with those in the control group. Pretreatment with baicalin or PDTC resulted in a substantial reduction in the levels of pro-inflammatory cytokines (TNF-α, IL-6) and adhesion molecule (ICAM-1, P-selectin) in the BALF of mice following CVF stimulation, compared to the Model group (Fig. 6D, E, F, and G).
Baicalin attenuates lung tissue damage in ALI mice
We examined the impact of baicalin on lung histopathological changes in mice subjected to challenges involving complement alternative pathway activation. The results obtained from H&E staining revealed that lung injury induced by complement alternative pathway activation in mice was characterized by several features, including alveolar septal thickening, proliferation and necrosis of alveolar epithelial cells, and inflammatory cell infiltration. Remarkably, pretreatment with baicalin and PDTC substantially attenuated lung injury induced by complement alternative pathway activation (Fig. 7).
Fig. 7.

Baicalin ameliorates complement alternative pathway activation-induced acute lung injury in mice. Mice were orally administered different concentrations (200, 100, and 50 mg/kg/day) of baicalin, PDTC (100 mg/kg/day), or vehicle for 7 days. CVF (35 µg/ kg) was then administered via the tail vein for 1 h. Lung histopathology of mice (H&E staining): lung H&E staining (A) and the lung injury score (B). The original magnifications are × 100. Results are expressed as the mean ± SD (n = 12). **P < 0.01 was compared with the control group; ##P < 0.01 were compared with the model group
Baicalin inhibits CVF-induced deposition of complement activation products in lung tissue
CVF is known for its specific induction of hyperactivation in the complement alternative pathway, resulting in the generation of various complement activation products, including C5a and C5b-9. In our investigation, we assessed the deposition of C5a and C5b-9 in lung tissue to evaluate the activation of the complement alternative pathway. Notably, there was a significant increase in the deposition of C5a and C5b-9 in the lung of mice treated with CVF. However, both the high and medium doses of baicalin, as well as PDTC pretreatment, led to a reduction in C5a and C5b-9 deposition in the lung tissue when compared to mice in the model group (Fig. 8).
Fig. 8.

Baicalin inhibits C5a and C5b-9 deposition in lung tissue of ALI mice. The C5a (A) and C5b-9 (B) depositions were determined by immunohistochemistry, and the positive cells are marked by tan or brown. The original magnifications are × 100. C5a (C) and C5b-9 (D) scores are shown as mean ± SD (n = 11–12). **P < 0.01 was compared with the control group; #P < 0.05 and ##P < 0.01 were compared with the model group
Baicalin inhibits NF-κB p65 phosphorylation level in lung tissues of ALI mice
Prior research has established that activation of the complement alternative pathway by CVF leads to the phosphorylation of NF-κB p65 in mouse lung tissue [11, 30]. Consistent with these findings, our immunohistochemical analysis of lung tissue (Fig. 9) revealed an increase in NF-κB p65 phosphorylation in the lung of mice in the model group compared to the normal group. However, the levels of NF-κB p65 phosphorylation in the lung of mice pretreated with baicalin at doses of 200 mg/kg and 100 mg/kg were significantly lower than those in the CVF-stimulated group. Additionally, as a positive control, PDTC (100 mg/kg) significantly reduced CVF-induced NF-κB p65 phosphorylation.
Fig. 9.

Effect of baicalin on phosphorylation of NF-κB p65 in lung tissue of ALI mice. A: Immunohistochemical detection of NF-κB p65 phosphorylation with positive cells appearing tan or brown. B: Semi-quantitative scoring of results by mean optical density values per high-magnification field of view. The original magnifications are × 100. Results are expressed as the mean ± SD (n = 12). **P < 0.01 was compared with the control group; ##P < 0.01 were compared with the model group
Discussion
The hallmark of ALI pathobiology is unchecked inflammation-driven alveolar-capillary barrier dysfunction and diffuse alveolar damage; suppressing lung inflammation can considerably reduce lung damage in several ALI models [34–36]. Early in the pathogenesis of ALI, the complement system is first activated, and its activation triggers an inflammatory response [4]. Notably, all three complement activation pathways (classical pathway, alternative pathway, and lectin pathway [37]) require the involvement of an alternative pathway to induce tissue injury in vivo, including acute lung inflammation [38]. In the present study, baicalin had a significant intervention effect on the inflammatory response of human microvascular endothelial cells induced by the activation of the complement alternative pathway and on the acute inflammatory response of mouse lungs.
Previous studies have shown that baicalin effectively suppressed inflammation in different models of lung injury mediated by downregulating NF−κB signal [21–29]. Shen et al. found that baicalin relieves LPS-induced lung inflammation via the NF-κB and MAPK pathways [21]. Peng et al. found that baicalin alleviated APEC-induced acute lung injury in chicken by inhibiting NF-κB pathway activation [22]. Long et al. also demonstrated that baicalin alleviates LPS-induced ALI in mice via inhibiting TLR4/NF-κB pathway [28]. Our previous study showed that inhibition of NF−κB p65 phosphorylation attenuates lung inflammation induced by activation of the complement alternative pathway [11, 30]. Consistent with other findings, our results showed that baicalin significantly reduced the activation of NF-κB p65. Therefore, the mechanism by which baicalin attenuates the inflammatory response induced by the activation of the complement alternative pathway may be related to the inhibition of NF-κB signaling activation.
A prominent feature of ALI is the increased permeability of the alveolar-capillary barrier, allowing the influx of proteins and inflammatory fluid rich in cells into the alveolar lumen, ultimately impairing gas exchange [1, 39]. Endothelial permeability is a key aspect of ALI pathogenesis, and vascular endothelial cells express various complement proteins, regulatory factors, and their receptors [37, 40]. During acute lung inflammation, vascular endothelial cells are the primary targets of complement system activation, leading to endothelial injury, increased capillary permeability, and influx of protein-rich fluid into the alveolar space, fostering the formation of pulmonary edema [38]. In the early stages of ALI, the complement system becomes hyperactive, resulting in the deposition of complement activation products on vascular endothelial cells. This, in turn, induces cellular activation, leading to the expression of adhesion molecules (ICAM-1, VCAM-1, P-selectin, and E-selectin) and release of cytokines (TNF-α and IL-6) [11, 41]. Importantly, all these proteins are regulated by the transcription factor NF-κB [42].Given the pivotal role of microvascular endothelial cells in ALI pathogenesis, we initially investigated the influence of baicalin on the inflammatory responses of these cells. We employed an in vitro model of HMEC cells, in which complement alternative pathway activation induced an inflammatory response (Figs. 2 and 3). Our findings indicate that baicalin significantly suppresses the expression of adhesion molecules (Fig. 2), inflammatory factor (Fig. 3), and upregulates NF-κB nuclear transcriptional activity (Fig. 4) in HMEC cells induced by complement alternative pathway activation. These results suggest that baicalin effectively intervenes in the inflammatory response of HMEC triggered by complement alternative pathway activation, primarily through the inhibition of NF-κB activation.
Activation of the complement system is a common feature across various ALI models, and both experimental and clinical evidence underscores the crucial role of complement activation in ALI development [43, 44]. In the early stages of ALI, excessive complement activation results in the generation of various products such as C5a and the membrane attack complex C5b-9. These products can either stimulate neutrophils to adhere to the lung capillary endothelium or directly activate endothelial cells, enhancing the upregulation of adhesion molecules including ICAM-1, and P-selectin which contributes to vascular endothelial cell injury [45, 46]. In our study, we induced ALI in mice by activating the complement alternative pathway with CVF to explore the impact of baicalin on lung inflammation resulting from complement alternative pathway activation. Our results showed that baicalin significantly inhibited the production of IL-6, TNF-α, and ICAM-1 in the serum of CVF-induced ALI mice, as well as the production of IL-6, TNF-α, ICAM-1, and P-selectin in BLAF (Fig. 6). Furthermore, baicalin reduced cell number and protein content in BALF and reduced MPO activity in the lung tissue (Fig. 5). Baicalin also effectively ameliorated pathological changes in the lung of ALI mice (Fig. 7).
NF-κB plays a pivotal role as a master regulator of inflammation, exerting control over every aspect from initiation to resolution of the inflammatory response [47–49]. The transcription factor NF-κB plays a crucial role in the acute phase of inflammation by acting upstream of the synthesis of proinflammatory mediators [50]. Under normal conditions, NF-κB resides in the cytoplasm and binds IκBα. However, when cells encounter extracellular pro-inflammatory cytokines, NF-κB is liberated from the cytoplasm and translocates to the nucleus, leading to the expression of inflammatory genes [11, 30]. Importantly, ALI/ARDS is characterized by endothelial cell dysfunction, which is largely regulated by NF-κB [51, 52]. The severity and lethality of ALI/ARDS are notably associated with an NF-κB-mediated “cytokine storm” [53, 54]. Upon activation of complement activation products, vascular endothelial cells undergo NF-κB activation, causing translocation of NF-κB p65 to the nucleus. This event triggers the transcription of adhesion proteins and cytokines, initiating a potent pro-inflammatory cascade that ultimately results in lung injury [55]. Our study revealed that baicalin significantly inhibits NF-κB p65 phosphorylation in mice lung with ALI induced by activation of the complement alternative pathway. The results from animal experiments further corroborate that baicalin’s protective effect against inflammation induced by complement alternative pathway activation in mouse lung is achieved through the inhibition of NF-κB signaling activation.
PDTC serves as an effective inhibitor of NF-κB by impeding IκB phosphorylation, preventing NF-κB translocation into the nucleus, and reducing downstream cytokine expression [56, 57]. In our study, baicalin exhibited a pattern of action similar to that of the NF-κB inhibitor PDTC. Both baicalin and PDTC significantly restrained the release of inflammatory mediators and levels of NF-κB p65 phosphorylation induced by complement alternative pathway activation in the lung of mice. These results further validated that the protective effect of baicalin against lung inflammation induced by complement alternative pathway activation in mice operates through the inhibition of NF-κB signaling activation.
In conclusion, this study showed that baicalin could attenuate the inflammatory response of HMEC cells induced by the activation of the complement alternative pathway and had a significant intervention effect on the lung inflammation caused by the activation of the complement alternative pathway in ALI mice. Its core mechanism of action was related to inhibiting the activation of the NF-κB signaling pathway (Fig. 10).
Fig. 10.

Schematic drawing of pharmacological effects and mechanisms of baicalin. Baicalin relieves complement alternative pathway activation-induced lung inflammation by inhibition of NF-κB pathway, delays the progression of ALI. FD: Factor D; FB: Factor B
Conclusion
In summary, our findings strongly indicate that baicalin is a potent inhibitor of acute lung inflammation triggered by the activation of the complement alternative pathway. Its mechanism of action primarily involves the inhibition of the NF-κB pathway. Baicalin is expected to be a promising candidate for the prevention of ALI, as well as for prevention complement-related inflammatory conditions and diseases.
Acknowledgements
Not applicable.
Abbreviations
- ALI
Acute injury lung
- BALF
Bronchoalveolar lavage fluid
- CVF
Cobra venom factor
- CAC
CVF-activated complement
- IL-6
Interleukin-6
- ICAM-1
Intracellular adhesion molecule
- H&E
Hematoxylin and eosin
- NF-κB
Nuclear factor kappa-B
- PDTC
Pyrrolidinedithiocarbamic acid
- TNF-α
Tumor necrosis factor-α
- FD
Factor D
- FB
Factor B
Author contributions
Jiao Li conducted most of the experiments; Min Li, Qing-Yu Lu, and Li Guo provided statistical support; Qiao-Zhou Liu contributed to some experiments. Qian-Yun Sun contributed to manuscript revision; Jiao Li, Qi-Yun Zhang, Min Li, and Qian-Yun Sun designed the study and analyzed the data. Qi-Yun Zhang drafted the initial version of the paper, and all authors reviewed the subsequent versions and read and approved the final manuscript.
Funding
This work was supported by funding from the National Natural Science Foundation of China (No. U1812403) and Guizhou Provincial Science and Technology Foundation (QKHRC2016-4018, QKHPTRC 2016–5625, and QKHPTRC 2019–5702), and Scientific and Technological Research Projects on Chinese Medicine and Ethnomedicine of the Administration of Traditional Chinese Medicine of Guizhou Province (QZYY-2023-153).
Data availability
The datasets generated and/or analyzed during the current study are not publicly available, because a follow-up study on baicalin is underway. Further inquiries can be available from the corresponding author.
Declarations
Ethics approval and consent to participate
All procedures in this study adhered to the Guidelines for Care and Use of Laboratory Animals of Guizhou Medical University. The experimental animal protocol received ethical approval from the Laboratory Animal Ethics Committee of Guizhou Medical University (No. 201345). The study is reported in accordance with ARRIVE guidelines. All methods were carried out in accordance with relevant guidelines and regulations in the declarations section.
Consent for publication
No applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jiao Li and Qi-Yun Zhang contributed equally to this work.
Contributor Information
Min Li, Email: limin1@gz5055.com.
Qian-Yun Sun, Email: sunqy@hotmail.com.
References
- 1.Mokrá D. Acute lung injury - from pathophysiology to treatment. Physiol Res. 2020;69(Suppl 3):S353–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. Lancet (London England). 2021;398(10300):622–37. 10.1016/S0140-6736(21)00439-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Máca J, Jor O, Holub M, Sklienka P, Burša F, Burda M, Janout V, Ševčík P. Past and Present ARDS Mortality Rates: a systematic review. Respir Care. 2017;62(1):113–22. 10.4187/respcare.04716 [DOI] [PubMed] [Google Scholar]
- 4.Yang Z, Nicholson SE, Cancio TS, Cancio LC, Li Y. Complement as a vital nexus of the pathobiological connectome for acute respiratory distress syndrome: an emerging therapeutic target. Front Immunol. 2023;14:1100461. 10.3389/fimmu.2023.1100461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammerschmidt DE, Weaver LJ, Hudson LD, Craddock PR, Jacob HS. Association of complement activation and elevated plasma-C5a with adult respiratory distress syndrome. Pathophysiological relevance and possible prognostic value. Lancet (London England). 1980;1(8175):947–9. 10.1016/S0140-6736(80)91403-8 [DOI] [PubMed] [Google Scholar]
- 6.Ali YM, Lynch NJ, Shaaban AA, Rizk DE, Abdel-Rahman SH, Khatri P, Yabuki M, Yaseen S, Dudler T, Demopulos G, et al. Inhibition of the lectin pathway of complement activation reduces LPS-induced acute respiratory distress syndrome in mice. Front Immunol. 2023;14:1192767. 10.3389/fimmu.2023.1192767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Yang Z, Chavko M, Liu B, Aderemi OA, Simovic MO, Dubick MA, Cancio LC. Complement inhibition ameliorates blast-induced acute lung injury in rats: potential role of complement in intracellular HMGB1-mediated inflammation. PLoS ONE. 2018;13(8):e0202594. 10.1371/journal.pone.0202594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement System Part I - Molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262. 10.3389/fimmu.2015.00262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flierl MA, Rittirsch D, Sarma JV, Huber-Lang M, Ward PA. Adrenergic regulation of complement-induced acute lung injury. Adv Exp Med Biol. 2008;632:93–103. [PubMed] [Google Scholar]
- 10.Ehrnthaller C, Ignatius A, Gebhard F, Huber-Lang M. New insights of an old defense system: structure, function, and clinical relevance of the complement system. Mol Med (Cambridge Mass). 2011;17(3–4):317–29. 10.2119/molmed.2010.00149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo J, Li M, Yang Y, Zhang L, Zhang LW, Sun QY. Pretreatment with atorvastatin ameliorates cobra venom factor-induced acute lung inflammation in mice. BMC Pulm Med. 2020;20(1):263. 10.1186/s12890-020-01307-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogel CW, Fritzinger DC. Cobra venom factor: structure, function, and humanization for therapeutic complement depletion. Toxicon: Official J Int Soc Toxinology. 2010;56(7):1198–222. 10.1016/j.toxicon.2010.04.007 [DOI] [PubMed] [Google Scholar]
- 13.Hagio T, Nakao S, Matsuoka H, Matsumoto S, Kawabata K, Ohno H. Inhibition of neutrophil elastase activity attenuates complement-mediated lung injury in the hamster. Eur J Pharmacol. 2001;426(1–2):131–8. 10.1016/S0014-2999(01)01191-8 [DOI] [PubMed] [Google Scholar]
- 14.Wang ZL, Wang S, Kuang Y, Hu ZM, Qiao X, Ye M. A comprehensive review on phytochemistry, pharmacology, and flavonoid biosynthesis of Scutellaria baicalensis. Pharm Biol. 2018;56(1):465–84. 10.1080/13880209.2018.1492620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan G, Chen L, Wang H, Wu S, Li S, Wang X. Baicalin inhibits LPS-induced inflammation in RAW264.7 cells through miR-181b/HMGB1/TRL4/NF-κB pathway. Am J Translational Res. 2021;13(9):10127–41. [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng WX, Wang F, Cao XL, Pan HY, Liu XY, Hu XM, Sun YY. Baicalin protects PC-12 cells from oxidative stress induced by hydrogen peroxide via anti-apoptotic effects. Brain Injury. 2014;28(2):227–34. 10.3109/02699052.2013.860469 [DOI] [PubMed] [Google Scholar]
- 17.Kong F, Luan Y, Zhang ZH, Cheng GH, Qi TG, Sun C. Baicalin protects the myocardium from reperfusion-induced damage in isolated rat hearts via the antioxidant and paracrine effect. Experimental Therapeutic Med. 2014;7(1):254–9. 10.3892/etm.2013.1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang P, Yu M, Shi M, Bo P, Gu X, Zhang Z. Baicalin and its aglycone: a novel approach for treatment of metabolic disorders. Pharmacol Rep. 2020;72(1):13–23. 10.1007/s43440-019-00024-x [DOI] [PubMed] [Google Scholar]
- 19.Gao Y, Snyder SA, Smith JN, Chen YC. Anticancer properties of baicalein: a review. Med Chem Research: Int J Rapid Commun Des Mech Action Biologically Act Agents. 2016;25(8):1515–23. 10.1007/s00044-016-1607-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui XD, Zhang JK, Sun YW, Yan FB, Zhao JF, He DD, Pan YS, Yuan L, Zhai YJ, Hu GZ. Synergistic antibacterial activity of baicalin and EDTA in combination with colistin against colistin-resistant Salmonella. Poult Sci. 2023;102(2):102346. 10.1016/j.psj.2022.102346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen B, Zhang H, Zhu Z, Ling Z, Zeng F, Wang Y, Wang J. Baicalin relieves LPS-Induced lung inflammation via the NF-κB and MAPK pathways. Molecules 2023, 28(4). [DOI] [PMC free article] [PubMed]
- 22.Peng LY, Yuan M, Song K, Yu JL, Li JH, Huang JN, Yi PF, Fu BD, Shen HQ. Baicalin alleviated APEC-induced acute lung injury in chicken by inhibiting NF-κB pathway activation. Int Immunopharmacol. 2019;72:467–72. 10.1016/j.intimp.2019.04.046 [DOI] [PubMed] [Google Scholar]
- 23.Changle Z, Cuiling F, Feng F, Xiaoqin Y, Guishu W, Liangtian S, Jiakun Z. Baicalin inhibits inflammation of lipopolysaccharide-induced acute lung injury toll like receptor-4/myeloid differentiation primary response 88/nuclear factor-kappa B signaling pathway. J Traditional Chin Med = Chung i tsa Chih Ying Wen pan. 2022;42(2):200–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou M, Yang L, Niu L, Zhao Y, Sun Y, Fu Y, Peng X. Baicalin ameliorates Mycoplasma gallisepticum-induced lung inflammation in chicken by inhibiting TLR6-mediated NF-κB signalling. Br Poult Sci. 2021;62(2):199–210. 10.1080/00071668.2020.1847251 [DOI] [PubMed] [Google Scholar]
- 25.Zhang H, Li X, Wang J, Cheng Q, Shang Y, Wang G. Baicalin relieves Mycoplasma pneumoniae infection–induced lung injury through regulating microRNA–221 to inhibit the TLR4/NF–κB signaling pathway. Mol Med Rep 2021, 24(2). [DOI] [PMC free article] [PubMed]
- 26.Bai C, Li T, Sun Q, Xin Q, Xu T, Yu J, Wang Y, Wei L. Protective effect of baicalin against severe burn–induced remote acute lung injury in rats. Mol Med Rep. 2018;17(2):2689–94. [DOI] [PubMed] [Google Scholar]
- 27.Ding XM, Pan L, Wang Y, Xu QZ. Baicalin exerts protective effects against lipopolysaccharide-induced acute lung injury by regulating the crosstalk between the CX3CL1-CX3CR1 axis and NF-κB pathway in CX3CL1-knockout mice. Int J Mol Med. 2016;37(3):703–15. 10.3892/ijmm.2016.2456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Long Y, Xiang Y, Liu S, Zhang Y, Wan J, Yang Q, Cui M, Ci Z, Li N, Peng W. Baicalin Liposome Alleviates Lipopolysaccharide-Induced Acute Lung Injury in Mice via Inhibiting TLR4/JNK/ERK/NF-κB Pathway. Mediators of inflammation 2020, 2020:8414062. [DOI] [PMC free article] [PubMed]
- 29.Meng X, Hu L, Li W. Baicalin ameliorates lipopolysaccharide-induced acute lung injury in mice by suppressing oxidative stress and inflammation via the activation of the Nrf2-mediated HO-1 signaling pathway. Naunyn Schmiedebergs Arch Pharmacol. 2019;392(11):1421–33. 10.1007/s00210-019-01680-9 [DOI] [PubMed] [Google Scholar]
- 30.Guo J, Liu QZ, Zhu FJ, Li M, Li J, Guo L, Sun QY, Yang QX. Acteoside attenuates acute lung injury following administration of cobra venom factor to mice. Heliyon. 2022;8(11):e11622. 10.1016/j.heliyon.2022.e11622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun QY, Chen G, Guo H, Chen S, Wang WY, Xiong YL. Prolonged cardiac xenograft survival in guinea pig-to-rat model by a highly active cobra venom factor. Toxicon: Official J Int Soc Toxinology. 2003;42(3):257–62. 10.1016/S0041-0101(03)00140-5 [DOI] [PubMed] [Google Scholar]
- 32.Till GO, Morganroth ML, Kunkel R, Ward PA. Activation of C5 by cobra venom factor is required in neutrophil-mediated lung injury in the rat. Am J Pathol. 1987;129(1):44–53. [PMC free article] [PubMed] [Google Scholar]
- 33.Kulkarni HS, Lee JS, Bastarache JA, Kuebler WM, Downey GP, Albaiceta GM, Altemeier WA, Artigas A, Bates JHT, Calfee CS, et al. Update on the features and measurements of experimental Acute Lung Injury in animals: an official American thoracic Society Workshop Report. Am J Respir Cell Mol Biol. 2022;66(2):e1–14. 10.1165/rcmb.2021-0531ST [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li D, Ren W, Jiang Z, Zhu L. Regulation of the NLRP3 inflammasome and macrophage pyroptosis by the p38 MAPK signaling pathway in a mouse model of acute lung injury. Mol Med Rep. 2018;18(5):4399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong J, Liao W, Tan LH, Yong A, Peh WY, Wong WSF. Gene silencing of receptor-interacting protein 2 protects against cigarette smoke-induced acute lung injury. Pharmacol Res. 2019;139:560–8. 10.1016/j.phrs.2018.10.016 [DOI] [PubMed] [Google Scholar]
- 36.Li L, Chen J, Lin L, Pan G, Zhang S, Chen H, Zhang M, Xuan Y, Wang Y, You Z. Quzhou Fructus Aurantii Extract suppresses inflammation via regulation of MAPK, NF-κB, and AMPK signaling pathway. Sci Rep. 2020;10(1):1593. 10.1038/s41598-020-58566-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Acosta J, Qin X, Halperin J. Complement and complement regulatory proteins as potential molecular targets for vascular diseases. Curr Pharm Design. 2004;10(2):203–11. 10.2174/1381612043453441 [DOI] [PubMed] [Google Scholar]
- 38.Mowery NT, Terzian WTH, Nelson AC. Acute lung injury. Curr Probl Surg. 2020;57(5):100777. 10.1016/j.cpsurg.2020.100777 [DOI] [PubMed] [Google Scholar]
- 39.Long ME, Mallampalli RK, Horowitz JC. Pathogenesis of pneumonia and acute lung injury. Clinical science (London, England: 1979) 2022, 136(10):747–769. [DOI] [PMC free article] [PubMed]
- 40.Langeggen H, Pausa M, Johnson E, Casarsa C, Tedesco F. The endothelium is an extrahepatic site of synthesis of the seventh component of the complement system. Clin Exp Immunol. 2000;121(1):69–76. 10.1046/j.1365-2249.2000.01238.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karpman D, Ståhl AL, Arvidsson I, Johansson K, Loos S, Tati R, Békássy Z, Kristoffersson AC, Mossberg M, Kahn R. Complement interactions with blood cells, endothelial cells and Microvesicles in thrombotic and inflammatory conditions. Adv Exp Med Biol. 2015;865:19–42. 10.1007/978-3-319-18603-0_2 [DOI] [PubMed] [Google Scholar]
- 42.Millar MW, Fazal F, Rahman A. Therapeutic targeting of NF-κB in Acute Lung Injury: a double-edged Sword. Cells 2022, 11(20). [DOI] [PMC free article] [PubMed]
- 43.Wang R, Xiao H, Guo R, Li Y, Shen B. The role of C5a in acute lung injury induced by highly pathogenic viral infections. Emerg Microbes Infections. 2015;4(5):e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu X, Yao D, Bao L, Liu D, Xu X, An Y, Zhang X, Cao B. Ficolin a derived from local macrophages and neutrophils protects against lipopolysaccharide-induced acute lung injury by activating complement. Immunol Cell Biol. 2020;98(7):595–606. 10.1111/imcb.12344 [DOI] [PubMed] [Google Scholar]
- 45.Ward PA. Role of complement, chemokines, and regulatory cytokines in acute lung injury. Ann N Y Acad Sci. 1996;796:104–12. 10.1111/j.1749-6632.1996.tb32572.x [DOI] [PubMed] [Google Scholar]
- 46.Bosmann M, Ward PA. Role of C3, C5 and anaphylatoxin receptors in acute lung injury and in sepsis. Adv Exp Med Biol. 2012;946:147–59. 10.1007/978-1-4614-0106-3_9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell. 2017;168(1–2):37–57. 10.1016/j.cell.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tiruppathi C, Soni D, Wang DM, Xue J, Singh V, Thippegowda PB, Cheppudira BP, Mishra RK, Debroy A, Qian Z, et al. The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nat Immunol. 2014;15(3):239–47. 10.1038/ni.2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430(7000):694–9. 10.1038/nature02794 [DOI] [PubMed] [Google Scholar]
- 50.Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Therapy. 2017;2:17023. 10.1038/sigtrans.2017.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fan J, Ye RD, Malik AB. Transcriptional mechanisms of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2001;281(5):L1037–1050. 10.1152/ajplung.2001.281.5.L1037 [DOI] [PubMed] [Google Scholar]
- 52.Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol. 2006;290(4):L622–45. 10.1152/ajplung.00477.2005 [DOI] [PubMed] [Google Scholar]
- 53.Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. 2017;39(5):517–28. 10.1007/s00281-017-0639-8 [DOI] [PubMed] [Google Scholar]
- 54.Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383(23):2255–73. 10.1056/NEJMra2026131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng H, Liang W, He W, Huang C, Chen Q, Yi H, Long L, Deng Y, Zeng M. Ghrelin attenuates sepsis-induced acute lung injury by inhibiting the NF-κB, iNOS, and akt signaling in alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2019;317(3):L381–91. 10.1152/ajplung.00253.2018 [DOI] [PubMed] [Google Scholar]
- 56.Schreck R, Meier B, Männel DN, Dröge W, Baeuerle PA. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J Exp Med. 1992;175(5):1181–94. 10.1084/jem.175.5.1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang JY, Liu DJ, Liu MX. The protective effect of NF-κB signaling pathway inhibitor PDTC on mice with chronic atrophic gastritis. Scand J Gastroenterol. 2021;56(10):1131–9. 10.1080/00365521.2021.1953130 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analyzed during the current study are not publicly available, because a follow-up study on baicalin is underway. Further inquiries can be available from the corresponding author.