

Abstract
Hepatitis A virus (HAV) infects African green monkey kidney (AGMK) cells via the HAV cellular receptor-1 (havcr-1), a mucin-like type 1 integral-membrane glycoprotein of unknown natural function. The ectodomain of havcr-1 contains an N-terminal immunoglobulin-like cysteine-rich region (D1), which binds protective monoclonal antibody (MAb) 190/4, followed by an O-glycosylated mucin-like threonine-serine-proline-rich region that extends D1 well above the cell surface. To study the interaction of HAV with havcr-1, we constructed immunoadhesins fusing the hinge and Fc portion of human IgG1 to D1 (D1-Fc) or the ectodomain of the poliovirus receptor (PVR-Fc) and expressed them in CHO cells. These immunoadhesins were secreted to the cell culture medium and purified through protein A-agarose columns. In a solid-phase assay, HAV bound to D1-Fc in a concentration-dependent manner whereas background levels of HAV bound to PVR-Fc. Binding of HAV to D1-Fc was blocked by treatment with MAb 190/4 but not with control MAb M2, which binds to a tag epitope introduced between the D1 and Fc portions of the immunoadhesin. D1-Fc neutralized approximately 1 log unit of the HAV infectivity in AGMK cells, whereas PVR-Fc had no effect in the HAV titers. A similarly poor reduction in HAV titers was observed after treating the same stock of HAV with murine neutralizing MAbs K2-4F2, K3-4C8, and VHA 813. Neutralization of poliovirus by PVR-Fc but not by D1-Fc indicated that the virus-receptor interactions were specific. These results show that D1 is sufficient for binding and neutralization of HAV and provide further evidence that havcr-1 is a functional cellular receptor for HAV.
Hepatitis A virus (HAV), an atypical member of the Picornaviridae that causes acute hepatitis in humans (for a review, see reference 16), has a positive-sense RNA genome of approximately 7,500 bases encapsidated in a shell formed by 60 copies of at least three viral proteins (VP1, VP2, and VP3). HAV codes for a very small VP4, the fourth picornavirus structural protein, which has not been detected in mature virions. Most wild-type strains of HAV do not grow in cell culture; however, attenuated variants that grow efficiently in primate cell culture have been isolated on serial passaging of the virus (4, 5, 8, 10–12, 15, 30). HAV has also been adapted to grow in guinea pig, pig, and dolphin cell cultures (9), indicating that the cellular factors required for HAV replication are not restricted to primates.
Like other picornaviruses, the first step in the life cycle of HAV is its interaction with a cellular receptor that allows it to enter the cell. Using protective monoclonal antibody (MAb) 190/4 as a probe, Kaplan et al. (18) identified the HAV cellular receptor-1 (havcr-1) in African green monkey kidney cells as a receptor for HAV. Nucleotide sequence analysis revealed that havcr-1 is a class I integral membrane glycoprotein of unknown natural function. The extracellular domain of havcr-1 contains an N-terminal cysteine-rich region (D1), which has homology to members of the immunoglobulin superfamily, followed by a threonine-, serine-, and proline-rich (TSP-rich) region, which is characteristic of O-glycosylated mucin-like glycoproteins (27). D1, which is required for binding of HAV and MAb 190/4 (35), is most probably extended well above the cell surface by the TSP-rich region.
Immunoadhesins are antibody-like molecules resulting from the fusion of the hinge and Fc portion of an immunoglobulin and the ligand-binding region of a receptor or adhesion molecule (for a review, see reference 3). These chimeric immunoglobulins are frequently used as research tools because they are easy to construct, express, and purify through protein A or G columns. In addition, the structure and function of the fused receptors are usually maintained in the immunoadhesins as a result of the flexibility and separation provided by the hinge region. Further, due to their homomultimeric characteristics, immunoadhesins have higher ligand avidity than do the monomeric receptors from which they were derived. To study the interaction of HAV with havcr-1, we constructed immunoadhesins fusing the hinge and Fc region of human IgG1 to D1 (D1-Fc) or the ectodomain of the poliovirus receptor (PVR-Fc) and expressed them in CHO cells. These immunoadhesins were secreted to the cell culture medium and purified using protein A columns. Here we report that D1-Fc binds specifically and neutralizes HAV whereas PVR-Fc has no effect on the HAV titers. The data presented in this work indicate that D1 is sufficient for HAV receptor function and provide further evidence that havcr-1 is a functional receptor for HAV.
MATERIALS AND METHODS
Antisera.
Anti-HAV antiserum was produced in rabbits immunized with a commercially available HAV vaccine. After several boosts with the HAV vaccine, rabbit serum was collected and assayed for anti-HAV antibodies by an indirect immunofluorescence assay in HAV- and mock-infected cells (39). HAV-specific immunofluorescence was observed in HAV-infected African green monkey kidney (AGMK) cells treated with the rabbit anti-HAV antibodies up to a dilution of 5,000 to 10,000 but not in mock-infected cells.
Murine IgG1 MAbs P1B5 directed against human α3 integrin (Gibco BRL), 190/4 directed against havcr-1 (18), and M2 directed against the FLAG peptide DTKDDDDK (IBI, Inc.) were purified through affinity columns. Unlabeled, 125I-labeled, and peroxidase-labeled human anti-HAV polyclonal antisera were obtained from the HAVAB kit (Abbott Laboratories). Goat anti-human Fc antibodies, phosphatase-labeled goat anti-human IgG antibodies, phosphatase-labeled anti-mouse IgG antibodies, peroxidase-labeled goat anti-human IgG antibodies, and peroxidase-labeled rabbit anti-human Fc antibodies were used as suggested by the manufacturer (Kirkegaard & Perry Laboratories, Inc.).
Murine anti-HAV neutralizing MAbs K2-4F2 and K3-4C8 (24) were purchased from Commonwealth Serum Laboratories, Melbourne, Australia, and MAb 813 (7) was purchased from Accurate Chemical Scientific Corp.
Cells and viruses.
The continuous clone GL37 of AGMK cells (36), termed AGMK GL37 cells, was grown in Eagle's minimal essential medium (EMEM) plus 10% fetal bovine serum (FBS) at 37°C in a CO2 incubator. Chinese hamster ovary (CHO) cells deficient in the enzyme dihydrofolate reductase (dhfr−) were obtained from the American Type Culture Collection and expanded in growth medium consisting of Iscove's medium containing 10% FBS (GibcoBRL) and supplemented with 100 μM hypoxanthine and 16 μM thymidine (Sigma Chemical Co.). Selection of CHO cells transfected with a hamster DHFR minigene was done in Iscove's medium containing 10% dialyzed FBS but without any supplement (selection medium).
The human tissue culture-adapted HM175 strain of HAV, which was derived from infectious cDNA (6) and passaged approximately 100 times in BS-C-1 cells, was grown in AGMK GL37 cells and termed HAV PI. The tissue culture-adapted KRM003 strain of HAV (36) was grown in AGMK GL37 cells and purified through sucrose gradients. To do so, HAV was pelleted through a 40% sucrose cushion, resuspended in NET (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 100 mM NaCl), treated with 0.1% sarcosyl (Sigma Chemical Co.) and 50 μg of proteinase K (Boehringer Mannheim) per ml at 37°C for 1 h, and sedimented through a 5 to 30% sucrose gradient in a Beckman SW40 rotor at 4°C for 90 min. Gradients were collected in 20 fractions of 0.5 ml each and assayed for HAV antigen by enzyme-linked immunosorbent assay (ELISA). To do so, Maxisorb 96-well plates (Nunc Inc.) were coated with a 1:5,000 dilution of human anti-HAV positive control antisera from the HAVAB kit. The plates were washed extensively and blocked with 100 μl of phosphate-buffered saline (PBS)–0.05% Tween 20 (Sigma Chemical Co.)–0.1% gelatin (Sigma) per well, and then 10 μl of each sucrose gradient fraction was added in duplicate wells. After being incubated for 2 h at 37°C, the plates were washed extensively, stained with a 1:10 dilution of peroxidase-labeled human anti-HAV antiserum from the HAVAB kit in PBS–0.05% Tween 20–0.1% gelatin, washed extensively, and developed with 3,3′,5,5′-tetramethylbenzidine substrate containing hydrogen peroxide (TMB one-component substrate) as recommended by the manufacturer (Kirkegaard & Perry Laboratories, Inc.). The colorimetric reaction was stopped with 1% H2SO4, and absorbance was read in a 96-well ELISA reader (Bio-Rad Laboratories) at 450 nm. Gradient fractions containing the 150S viral peak were pooled and stored at −70°C.
HAV titer determination.
The titer of HAV was determined on 96-well plates containing confluent monolayers of AGMK GL37 cells. A total of 8 to 16 replicate wells were inoculated with 100 μl of 10-fold dilutions of HAV prepared in EMEM–10% FBS. The plates were incubated at 35°C for 2 weeks in a CO2 incubator, and cell monolayers were fixed with 90% methanol and stained with a 1:2,500 dilution of rabbit anti-HAV antibodies and a 1:25,000 dilution of peroxidase-labeled goat anti-rabbit antibodies. TMB one-component substrate (100 μl/well) was added, the plates were incubated at room temperature for 15 to 30 min, and the reaction was stopped with 1% H2SO4 (100 μl/well). Wells that developed at least 2.5 times the absorbance of the uninfected control wells were considered positive, and viral titers were determined using the Reed and Muench method (31).
Plasmids.
pDH1P, which is similar to pMG1 (26) except that the PstI site at the 3′ end of the insert was eliminated, codes for a Chinese hamster ovary cell dhfr 2.5-kb minigene that contains the six exons of the gene but lacks introns 2 to 5. pDH1P was cotransfected into CHO dhfr− cells with plasmids coding for soluble chimeric receptors. pEF-ICAM5(1–2)-Fc, a generous gift of Jose Casasnovas, Karolinska Institute, Stockholm, Sweden, codes for an intercellular adhesion molecule-1 (ICAM-1) immunoadhesin obtained by fusing the cDNA fragment of the first and second immunoglobulin-like domains of human ICAM-1 to a synthetic splicing donor (GGTGAGT) and a genomic fragment of the hinge and Fc portion of human human IgG1. The resulting ICAM-1 immunoadhesin in pEF-ICAM5(1–2)Fc is similar to ICI–2D/IgG (25) and is driven by the human elongation factor 1α promoter.
Plasmids coding for fusion proteins of the cysteine-rich region of havcr-1 (D1) and the ectodomain of the PVR were constructed by replacing the ICAM-1 sequences in pEF-ICAM5(1–2) with PCR fragments coding for the D1 and the PVR ectodomain. Recombinant DNA manipulations were done by standard methods (32). The nucleotide sequences of all constructs were by automatic sequencing using an ABI Prism model 377 automatic sequencer and the ABI Prism Dye terminator cycle-sequencing ready reaction kit (Perkin-Elmer Cetus, Inc.). Both strands of the PCR products were sequenced using positive- and negative-sense synthetic oligonucleotides spaced 300 to 400 bases apart. All plasmids were grown in E. coli DH5α and purified by chromatography using plasmid preparation kits as recommended by the manufacturer (Qiagen, Inc.). Plasmid constructs were done as follows.
(i) pEF-HAVcr-1D1flag.
A PCR fragment coding for amino acids 1 to 138 of havcr-1 was amplified from pDR2GL37/5 (18). Synthetic oligonucleotide D1/Sal I (5′-CGGATACCCGTCGACATAATGCATCTTCAAGTGGTCATC-3′), which contains a SalI site before the initiation codon of havcr-1 followed by nucleotides 198 to 216 of the havcr-1 cDNA, was used as the sense primer. Synthetic oligonucleotide D1/flag/SpeI (5′-GGACTAGTACTCACCCTTGTCATCGTCGTCCTTGTAGTCGTCTGTTCGAACAGTTCTGACAATTGGAGTGACTCTTGGGGGCCCAAT-3′), containing an SpeI site followed by the antisense of an artificial splicing donor signal (GTGAGT), 24 nucleotides coding for the FLAG peptide DTKDDDDK, and nucleotides 615 to 565 of the havcr-1 cDNA, was used as the antisense primer. The single band amplified in the PCR was purified by electrophoresis with a TAE–1% low-melting-temperature agarose gel, cut with SalI and SpeI, ligated into SalI-SpeI-cut pEF-FcICAM5(1–2) vector, and used to transform E. coli.
(ii) pEF-PVR.
A PCR fragment coding for the full ectodomain of PVR was amplified by PCR using synthetic oligonucleotides PVR/Sal I (5′-CGGATACCCGTCGACATAATGGCCCGAGCCATGGCCGCC-3′) and PVR/SpeI (5′-G GACTAGTACTCACCTAAGGTCCCGGGAGGTCCCTCTTTGACCTGGA CGGTCAGTTCTGC-3′) and cloned into SalI-SpeI-cut and phospatase-treated pEF-FcICAM5(1–2) vector as described above. Similar constructs coding for chimeric molecules consisting of the extracellular domain of PVR and the hinge and Fc portion of human IgG (20) and mouse IgG2a (1) have been described.
Transfection and selection of CHO cells expressing high levels of soluble receptors.
CHO dhfr− cells were cotransfected with 0.45 μg of pDHIP and 3.5 μg of pEF-HAVcr-1D1flag or pEF-PVR using the Fugene6 reagent (Roche Laboratories) as specified by the manufacturer. At 6 h posttransfection, Iscove's medium containing 10% FBS supplemented with hypoxanthine and thymidine was added, and the cells were grown for 48 h at 37°C in a CO2 incubator. The cells were split 1:10 and grown in Iscove's medium containing dialyzed FBS without supplements (selection medium). After 14 days of selection, the cells were cloned by end-point dilution in 96-well plates. The culture medium of 24 single-cell clones was assayed for the expression of soluble receptors by a capture ELISA using 96-well plates coated with goat anti-human Fc antibody and staining with peroxidase-labeled rabbit anti-human Fc conjugate. Clones that secreted the soluble receptors to the cell culture medium were grown in the presence of 5 nM methotrexate. As soon as the cells became accustomed to growing at a given concentration of methotrexate and forming a confluent monolayer in 3 days after being split 1:5, the concentration of methotrexate was increased fourfold. Cells cotransfected with pDHIP and pEF-HAVcr-1D1flag were termed CHO D1-Fc and reached a maximum level of expression of D1-Fc at 0.32 μM methotrexate. Cells cotransfected with pDHIP and pEF-PVR were termed CHO PVR-Fc and reached a maximum level of expression of PVR-Fc at 1.28 μM methotrexate.
Purification of soluble receptors.
CHO/D1-Fc and CHO/PVR-Fc cells were grown in 15-cm-diameter dishes with selection medium containing 0.32 and 1.28 μM methotrexate, respectively. After the growth medium was replaced with 10 ml of serum-free OptiMEM medium, cell monolayers were incubated overnight at 37°C in a CO2 incubator and the medium containing the soluble receptors was harvested. An additional 10 ml of OptiMEM medium was added to each plate, and the medium was harvested 24 h later. The yields of the first and second harvests were pooled and frozen at −20°C. Immunoadhesins were purified from 300 ml of harvested medium by affinity chromatography using Affi-Gel protein A-agarose columns as recommended by the manufacturer (Bio-Rad Laboratories). Fractions containing the immunoadhesins were identified by dot-blot analysis staining with phosphatase-labeled anti-human Fc antibodies.
Protein analysis.
Soluble receptors were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Purified D1-Fc and PVR-Fc (1 μg each) were boiled in 2× SDS–β-mercatoethanol loading buffer and subjected to SDS-PAGE (21) through 4 to 20% Tris-glycine polyacrylamide gels (Novex; Invitrogen) and Coomassie blue staining. Alternatively, before being fixed, proteins were transferred to polyvinylidene difluoride membranes (Immobilon-P; Millipore, Inc.), and stained with a 1:1,000 dilution of goat anti-human Fc antibody and a 1:5,000 dilution of phosphatase-labeled rabbit anti-goat antibody. The substrate 5-bromo-4-chloro-3-indolylphosphate-nitroblue tetrazolium (Kirkegaard & Perry Laboratories) was used as recommended by the manufacturer.
Detection of epitope 190/4 in the immunoadhesins.
A capture ELISA was used to study the expression of the 190/4 epitope in D1-Fc. MaxiSorb 96-well plates (Nunc Laboratories) were coated with 50 μl of 1-μg/ml MAb 190/4 or negative control MAb P1B5 in 50 mM sodium carbonate (pH 9.6) per well overnight at 4°C. The plates were blocked with 5% bovine serum albumin in PBS for 1 h at room temperature. Twofold dilutions of CHO D1-Fc and CHO PVR-Fc cell culture medium were added in duplicate wells, and the plates were incubated for 1 h at room temperature. The plates were washed extensively with PBS–0.05% Tween 20 and stained with a 1:50,000 dilution of peroxidase-labeled goat anti-human antibody for 1 h at room temperature. After the plates were washed extensively, 100 μl of TMB one-component substrate was added per well, the plates were incubated for 30 min at room temperature, and the absorbance at 450 nm was determined as described above.
Binding assays.
Binding of HAV to soluble receptors was done in 96-well plates. MaxiSorb 96-well plates were coated with 10 μg of protein A-purified soluble receptors or normal human immunoglobulins per ml (prepared from the negative control antiserum of the HAVAB kit) in 50 mM sodium carbonate (pH 9.6) overnight at 4°C. After the plates were washed extensively, 10-fold dilutions of sucrose gradient-purified HAV were added, and the plates were incubated for 2 h at room temperature and washed extensively. The plates were treated with a 1:10 dilution of 125I-labeled human anti-HAV antiserum for 2 h at 4°C, washed extensively, and exposed to X-ray film (XAR-2; Kodak) at −70°C in the presence of intensifying screens.
For binding assays using protein A-treated beads, different amounts of purified D1-Fc or PVR-Fc were bound to 25 μl of protein A-Trisacryl beads (Pierce Laboratories) in 500 μl of PBS containing 2% ovalbumin (Sigma Chemical Co.). After 2 h at 4°C, 50 μl of sucrose-purified HAV (5 × 107 50% tissue culture infective doses [TCID50]) in 500 μl of PBS–2% ovalbumin was added and the mixture was incubated with rotation overnight at 4°C. After the mixture was washed three times with PBS at 4°C, bound HAV was eluted with 100 μl of 6 M LiCl for 30 min at room temperature. After the beads were pelleted by centrifugation, the supernatant containing the eluted virus was transferred to a new tube, diluted 40-fold with EMEM, and subjected to titer determination on AGMK GL37 cells.
To determine whether MAb 190/4 could inhibit the binding of HAV to D1-Fc, 3 μg of D1-Fc was incubated at 4°C with 0, 0.5, 5, or 50 μg of MAb 190/4 or control MAb M2 in 500 μl of PBS–2% ovalbumin. Protein A-Trisacryl beads (25 μl) were added, and the mixture was incubated for 2 h at 4°C. Binding, elution, and HAV titer determination were done as described above.
Neutralization assays.
To neutralize HAV with the immunoadhesins, different amounts of purified D1-Fc and PVR-Fc (100, 10, and 1 μg) were incubated overnight at 4°C with 105 TCID50 of HAV PI stock in 200 μl of EMEM–10% FBS. Confluent monolayers of AGMK GL37 cells in 96-well plates were inoculated with 10-fold dilutions of the neutralization reaction products and incubated for 4 h at 37°C. After the plates were washed three times, EMEM–10% FBS was added, and the plates were incubated at 35°C for 10 days in a CO2 incubator. The cells were fixed with 90% methanol, and HAV titers were determined by ELISA as described above. It should be pointed out that all dilutions and incubations were done in the presence of 10 μg of the respective immunoadhesins per ml.
To neutralize HAV with MAbs, 105 TCID50 of HAV PI stock in a final volume of 200 μl of EMEM–10% FBS was treated with 50 μg of protein A-purified MAb K2-4F2, K3-4C8, or VHA 813 overnight at 4°C. Confluent monolayers of AGMK GL37 cells in 96-well plates were inoculated with 10-fold dilutions of the neutralization reaction products and incubated for 4 h at 37°C in a CO2 incubator. After the plates were washed three times, EMEM–10% FBS was added, and the plates were incubated at 35°C for 10 days. The cells were fixed with 90% methanol, and HAV titers were determined by ELISA as described above.
RESULTS
Expression of soluble receptors in CHO cells.
To gain a better understanding of the mechanism of cell entry by HAV, we expressed soluble forms of havcr-1 and studied their interaction with HAV. In preliminary studies, we could not detect significant levels of binding and neutralization of HAV by soluble monomeric forms of the cysteine-rich region of havcr-1 (D1) expressed in insect, HeLa, and COS-7 cells (E. Silberstein, D. Feigelstock, and G. G. Kaplan, unpublished results). Since picornavirus cellular receptors expressed as dimeric immunoadhesins were demonstrated to be more active than their monomeric soluble forms (25), we decided to express a D1 immunoadhesin to test its HAV receptor activity. For this purpose, we constructed pEF-HAVcr-1D1flag, a plasmid coding for D1 fused to the hinge and Fc regions of human IgG1. A FLAG tag epitope was inserted between D1 and the IgG1 fragment to monitor the expression of the chimeric receptor with anti-FLAG MAb M2, and the resulting immunoadhesin was termed D1-Fc (Fig. 1A). CHO dhfr− cells were cotransfected with pEF-HAVcr-1D1flag and pDH1P, a plasmid coding for a hamster DHFR minigene. Stable transfectants were selected in Iscove's medium containing dialyzed bovine serum (selection medium). As a control, we constructed pEF-PVR, a plasmid coding for the three extracellular immunoglobulin-like domains of the PVR fused to the hinge and Fc portion of human IgG1. The resulting chimeric receptor, termed PVR-Fc (Fig. 1A), shared the hinge and Fc region with D1-Fc and was expressed in CHO cells as described above. CHO cell transfectants were single cell cloned by end-point dilution in 96-well plates. Since we expected that the immunoadhesins would be secreted from the cell, we used ELISA with anti-human Fc antibodies to test the cell culture medium of single-cell clones for the expression of chimeric receptors. Approximately 50% of the analyzed clones secreted the chimeric receptors to the cell culture medium. Two clones that produced the highest levels of D1-Fc and PVR-Fc were treated with increasing amounts of methotrexate until they secreted 1 to 10 μg of the soluble receptors per ml to the cell culture medium. These CHO cell clones, termed CHO D1-Fc and CHO PVR-Fc, were maintained in culture with the maximum achieved levels of methotrexate and expanded to produce milligram amounts of soluble receptors. D1-Fc and PVR-Fc were purified from the cell culture medium by affinity chromatography using protein A-agarose columns. Both chimeric receptors bound to the columns, which indicated that they formed the expected homodimers with active Fc portions capable of binding to protein A. D1-Fc and PVR-Fc were eluted from the columns at pH 3.0 and analyzed by SDS-PAGE followed by Coomassie blue staining (Fig. 1B). The predominant form of D1-Fc migrated as a 50-kDa band, which agrees with the expected size of the fully glycosylated form of the chimeric receptor. Two minor bands, one migrating just below the 50-kDa D1-Fc band and another migrating as a 35-kDa protein that corresponds to the expected molecular mass of the D1-Fc protein backbone, most probably represent glycosylated forms of D1-Fc. PVR-Fc migrated as a single 90-kDa band corresponding to the expected size of a fully glycosylated form of the chimeric receptor (20). Western blot analysis (Fig. 2) showed that the three bands of D1-Fc reacted with both anti-FLAG MAb M2 (lane 1) and anti-human Fc (lane 3) antibodies. The results in Fig. 2 further suggested that these three bands are glycosylated forms of D1-Fc; however, it is also possible that the 50- and 35-kDa bands are degradation products or other modifications of D1-Fc. As expected, the 90-kDa PVR-Fc band reacted with the anti-human Fc antibodies (lane 4) but not with MAb M2 (lane 2), which indicated that the 90-kDa band corresponded to the PVR-Fc immunoadhesin.
FIG. 1.

Expression of D1-Fc and PVR-Fc immunoadhesins in CHO cells. (A) Schematic representation of the D1-Fc and PVR-Fc immunoadhesins. To construct D1-Fc, the cysteine-rich region of havcr-1 (D1) was tagged at its N terminus with peptide DTKDDDDK (FLAG) and fused to the hinge and Fc regions of human IgG1 (IgG1 Fc). To construct PVR-Fc, the ectodomain of PVR containing the three immunoglobulin-like domains (V1, C1, and C2) was fused to the hinge and Fc regions of human IgG1. Two identical fusion proteins are linked by disulfide bonds (dashed lines), forming homodimers, which are secreted to the cell culture medium as soluble immunoadhesins. (B) D1-Fc and PVR-Fc were purified through protein A columns. The eluted immunoadhesins were analyzed by denaturing SDS-PAGE in a 4 to 20% polyacrylamide gel and stained with Coomassie blue. The arrow points to the fully glycosylated 50-kDa form of D1-Fc. The positions of prestained molecular mass markers and their sizes in kilodaltons are shown on the right.
FIG. 2.

Western blot analysis of purified D1-Fc and PVR-Fc immunoadhesins. Protein A-purified D1-Fc (lanes 1 and 3) and PVR-Fc (lanes 2 and 4) were analyzed by denaturing SDS-PAGE in a 4 to 20% polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, and probed with anti-FLAG MAb M2 (lanes 1 and 2) or anti-human Fc antibodies (lanes 3 and 4). The positions of prestained molecular mass markers and their sizes in kilodaltons are shown on the left.
D1-Fc binds to protective MAb 190/4.
It was important to confirm that the D1 portion of D1-Fc was antigenically identical to the cysteine-rich region of havcr-1 expressed at the cell surface of the AGMK GL37 cells. Therefore, we analyzed whether MAb 190/4, which binds to a conformational epitope in havcr-1 (18), would also react with D1-Fc. A capture ELISA was used to detect the binding of D1-Fc and PVR-Fc to 96-well plates coated with MAb 190/4 or negative control MAb P1B5. Twofold dilutions of cell culture medium from CHO D1-Fc or CHO PVR-Fc cells were added to the wells, and the plates were incubated for 1 h at room temperature. Bound chimeric receptors were detected with peroxidase-labeled goat anti-human antibodies and TMB one-component substrate. The colorimetric reaction was stopped with 1% H2SO4, and absorbance was read at 450 nm. Figure 3 shows that D1-Fc bound to MAb 190/4 in a concentration-dependent manner but not to MAb P1B5, which implied that D1-Fc expressed the protective epitope 190/4. As expected, control PVR-Fc did not bind to either MAb 190/4 or MAb P1B5, indicating that D1 bound specifically to MAb 190/4. These ELISA results showed that the D1 portions of havcr-1 and D1-Fc have the same antigenicity and suggested that they also share their structure.
FIG. 3.

Binding of protective MAb 190/4 to D1-Fc. Binding of D1-Fc to MAb 190/4 was determined by capture ELISA. Twofold dilutions of cell culture medium containing D1-Fc (■ and □) or PVR-Fc (● and ○) were bound to 96-well plates coated with MAb 190/4 (■ and ●) or negative control MAb P1B5 (□ and ○). Bound immunoadhesins were stained with peroxidase-labeled anti-human Fc antibodies. TMB was used as substrate, and the absorbance (O.D.) was read at 450 nm (y axis) and plotted against the dilution (x axis). Data are means of results from duplicate wells; duplicate values varied by less than 10%. The results are those of one experiment which was repeated at least twice with approximately 5 to 10% experimental error.
The Cys-rich region of havcr-1 (D1) is sufficient for binding of HAV.
Since we confirmed that D1-Fc expressed protective epitope 190/4, it was of interest to analyze whether this soluble receptor could also bind HAV. To do so, twofold dilutions of sucrose-purified HAV were added to 96-well plates coated with D1-Fc and the virus bound to the wells was detected with 125I-labeled human anti-HAV antibodies followed by autoradiography of the 96-well plate. Because the 125I-labeled human anti-HAV antibodies reacted with PVR-Fc (data not shown), protein A-purified normal human immunoglobulins were used as the negative control to coat the plates. Similarly, protein A-purified human anti-HAV immunoglobulins were used as the positive control for the assay. Figure 4 shows that HAV bound to D1-Fc in a concentration-dependent manner but did not bind to the normal human immunoglobulins (hu normal Ig). As expected, HAV bound strongly to the human anti-HAV immunoglobulins (hu anti-HAV Ig). Taken together, these capture ELISA results suggested that D1-Fc had HAV-binding activity. To further substantiate this, we set up a binding assay using protein A-coated beads as the solid phase, which allowed us to include PVR-Fc as the negative control for the experiment. Different amounts of D1-Fc and PVR-Fc were bound to protein A-Trisacryl beads for 2 h at 4°C, and purified HAV was added. After an overnight incubation at 4°C, the beads were washed extensively and the bound HAV was released from the beads with 6 M LiCl. The released virus was subjected to titer determination on 96-well plates containing AGMK GL37 cell monolayers. The results of this binding assay (Fig. 5) showed that 18, 9, and 4.5 μg of D1-Fc bound approximately 1.5, 1, and 0.5 log unit more HAV, respectively, than did similar amounts of PVR-Fc. Moreover, HAV bound to D1-Fc in a concentration-dependent manner whereas similar background levels of HAV bound to all the concentrations of PVR-Fc used in the assay. Variables such as the affinity of the virus for the receptor, the effect of extensive washing on the dissociation of the virus-receptor complexes, and the effectiveness of the virus release by 6 M LiCl affected the amount of HAV detected in our binding assay. We have not quantified the residual HAV at each step of the binding assay, but it is highly unlikely to be 100% of the input virus. A theoretically good efficiency of 50% at each of the three steps of the assay will result in a recovery of 12.5% of the input virus. Therefore, our recovery of 13% using D1-Fc compared to 1% using PVR-Fc (Fig. 5, 18 μg of soluble receptors) seems reasonable and shows a 10-fold increase of signal over the background level. Due to the nature of the assay, it is possible that the remaining 87% of the input HAV or part of it bound specifically to the D1-Fc agarose beads but dissociated from the immunoadhesin during the extensive washing or was not released by the 6 M LiCl treatment. It should be pointed out that the consistently high background levels of nonspecific binding of virus to the solid phase are probably due to the well-known “sticky” nature of HAV. The results presented in Fig. 5 further support the notion that D1-Fc has HAV-binding activity.
FIG. 4.

Binding of HAV to 96-well plates coated with D1-Fc. Twofold dilutions of purified HAV were bound to 96-well plates coated with 10 μg of purified D1-Fc or human normal immunoglobulins (hu normal Ig) per ml. A well coated with 10 μg of human anti-HAV immunoglobulins (hu anti-HAV Ig) per ml was used as a positive control for virus binding. Bound HAV was detected by 125I-labeled human anti-HAV antibodies, extensive washing, and direct autoradiography of the 96-well plate.
FIG. 5.

Binding of HAV to immunoadhesins attached to protein A-treated beads. Different amounts of purified D1-Fc or PVR-Fc were bound to 25 μl of protein A-Trisacryl beads for 2 h at 4°C. Sucrose-purified HAV (5 × 107 TCID50) was added, and the mixture was incubated with rotation overnight at 4°C. After the mixture was washed three times with PBS at 4°C, bound HAV was eluted with 100 μl of 6 M LiCl2 for 30 min at room temperature, diluted 40-fold with EMEM, and subjected to titer determination on AGMK GL37 cell monolayers. Values are the log10 of the HAV titers determined by the Reed and Muench method (32), and the standard deviations are shown as error bars.
HAV binds specifically to D1-Fc.
We previously showed that protective MAb 190/4 binds to havcr-1 and blocks the binding of HAV to this receptor (18). Therefore, we tested whether MAb 190/4 could block the binding of HAV to the D1 portion of D1-Fc. Anti-FLAG MAb M2, which binds to the FLAG octapeptide tag introduced between D1 and the hinge and Fc regions of D1-Fc (Fig. 1A), was used as the control MAb for the blocking experiment. D1-Fc bound to protein A-coated beads was first treated with different amounts of MAbs 190/4 and M2, then incubated with purified HAV, and finally processed as indicated above for the binding assay. PVR-Fc bound to protein A-coated beads was used to determine the amount of HAV bound nonspecifically to the solid phase. Bound HAV was eluted with 6 M LiCl and subjected to titer determination on 96-well plates containing AGMK GL37 cells (Fig. 6). Treatment with 50 and 5 μg of MAb 190/4 blocked the binding of HAV to D1-Fc and resulted in background levels similar to those observed with PVR-Fc (0 μg of blocking MAb). Treatment with 0.5 μg of MAb 190/4 did not block the binding of HAV to D1-Fc, which showed that MAb 190/4 blocked the binding of HAV in a concentration-dependent manner. Treatment with similar amounts of MAb M2 (50, 5, and 0.5 μg) did not block the binding of HAV to D1-Fc, which clearly indicated that HAV bound specifically to D1-Fc by interacting with its D1 portion.
FIG. 6.

Inhibition of binding of HAV to D1-Fc by protective MAb 190/4. Equal amounts (3 μg) of D1-Fc and PVR-Fc were treated with 0, 0.5, 5, or 50 μg of MAb 190/4 or control MAb M2 at 4°C. Protein A-Trisacryl beads (25 μl) were added, and the mixture was incubated for 2 h at 4°C. Sucrose-purified HAV (5 × 107 TCID50) was added, and the mixture was incubated with rotation overnight at 4°C. HAV was eluted and subjected to titer determination as described in the legend to Fig. 5. Values are the log10 of the HAV titers determined by the Reed and Muench method (32), and the standard deviations are shown as error bars.
Neutralization of HAV by D1-Fc.
To assess whether D1-Fc could neutralize HAV, 105 TCID50 of HAV PI was incubated with different amounts of purified D1-Fc or control PVR-Fc overnight at 4°C. Tenfold dilutions of the neutralization reaction products were subjected to titer determination on 96-well plates containing AGMK GL37 cell monolayers. After adsorbing the virus at 37°C for 4 h, monolayers were washed extensively, medium containing 10 μg of the soluble receptor used in the neutralization reaction per ml was added, and the mixture was incubated for 2 weeks at 35°C (Fig. 7). Treatment with 100 and 10 μg of D1-Fc reduced the HAV titers by approximately 1.5 and 0.5 log unit, respectively, compared to control samples treated with the same amounts of PVR-Fc. Treatment with 1 μg of D1-Fc had almost no effect on the HAV titer compared to treatment with a similar amount PVR-Fc. Taken together, these data clearly showed that D1-Fc neutralized HAV in a concentration-dependent manner. Since the low level of HAV neutralization induced by D1-Fc was intriguing, we compared it to the level of neutralization induced by murine MAbs under similar experimental conditions. HAV PI (105 TCID50) was treated with 50 μg of neutralizing MAbs VHA 813, K3-4C8, and K2-4C8 or negative control anti-FLAG MAb M2, which does not react with HAV (Fig. 8). Treatment with MAbs VHA 813, K2-4C8, and K3-4C8 reduced the HAV titers by approximately 0.75, 0.45, and 0.4 log unit compared to treatment with MAb M2. These poor neutralization levels are consistent with the significant nonneutralizable fraction of virus found in crude preparations of HAV (22, 28, 34). These neutralization results indicated that D1-Fc neutralized HAV to a similar extent to that for the murine neutralizing MAbs.
FIG. 7.

Neutralization of HAV by D1-Fc. Different amounts of purified D1-Fc and PVR-Fc (100, 10, and 1 μg) were incubated overnight at 4°C with 105 TCID50 of HAV PI stock. Tenfold dilutions of the neutralization reactions were subjected to titer determination on 96-well plates containing confluent monolayers of AGMK GL37 cells. After 4 h of adsorption at 37°C, the plates were washed three times and incubated for 10 days at 35°C under CO2. All dilutions and incubations were done in the presence of 10 μg of the respective immunoadhesin per ml. HAV titers were determined by ELISA. Values are the log10 of the HAV titers determined by the Reed and Muench method (32), and the standard deviations are shown as error bars.
FIG. 8.
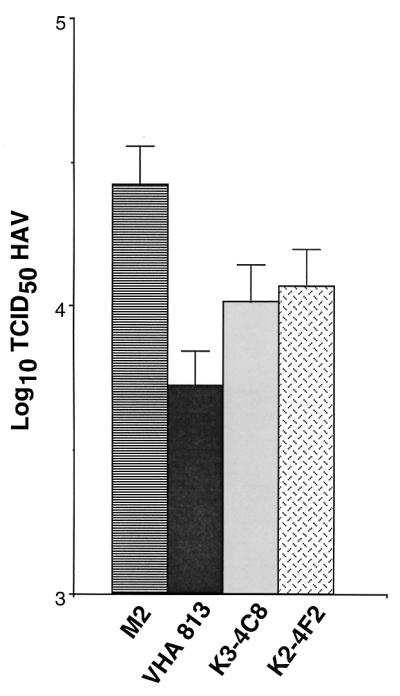
Neutralization of HAV by MAbs. HAV PI stock (105 TCID50) was treated with 50 μg of protein A-purified MAbs K2-4F2, K3-4C8, and VHA 813 for 1 h at 37°C. Tenfold dilutions of the neutralization reaction products were subjected to titer determination on 96-well plates containing confluent monolayers of AGMK GL37 cells. After 4 h of adsorption at 37°C, the plates were washed three times and incubated for 10 days at 35°C under CO2. HAV titers were determined by ELISA. Values are the log10 of the HAV titers calculated by the Reed and Muench method (32), and the standard deviations are shown as error bars.
D1-Fc neutralizes HAV but not poliovirus.
To ascertain whether the neutralization effect of D1-Fc was restricted to HAV, we tested the immunoadhesin-mediated neutralization of poliovirus, a related picornavirus. PV1/M (107 PFU) was treated with 100 μg of PVR-Fc or D1-Fc or mock treated for 1 h at 37°C. Tenfold dilutions of the neutralization reaction products were subjected to titer determination on 96-well plates containing confluent AGMK GL37 cell monolayers. The plates were incubated at 37°C for 72 h, and the cytopathic effect was assessed under a microscope. Treatment with PVR-Fc completely neutralized PV1/M (i.e., no cytopathic effect was observed at any dilution), whereas treatment with D1-Fc had no effect on the poliovirus titers (data not shown). This observation confirmed that D1-Fc neutralized HAV specifically and had no effect on poliovirus infectivity.
DISCUSSION
Picornaviruses share genomic, structural, and antigenic features; however, they bind to a wide variety of cellular receptors (13) that result in different mechanisms of viral cell entry. The better-understood picornavirus-receptor interactions so far are those of the major group of rhinoviruses with ICAM-1 and poliovirus with PVR. These closely related picornaviruses bind to the first domains of their immunoglobulin superfamily cellular receptors in a distinctive way (reference 38 and references therein): ICAM-1 interacts with residues on a single protomer of rhinovirus, whereas PVR interacts with residues from adjacent protomers of poliovirus. Some picornaviruses do not interact with the first domains of their receptors, and others require coreceptors or additional host factors to bind and enter the cell (29, 37). HAV is an atypical picornavirus with a mature virion that probably lacks VP4 and an antigenic structure that is quite different from those of the other members of the family (for a review, see reference 16). Very little is known about the cell entry mechanism of HAV, which, due to the peculiarities of this virus, could differ substantially from the mechanisms used by the other members of the family.
We have identified havcr-1 as an AGMK cell receptor for HAV and provided evidence of its functionality (18). The requirements for binding of HAV to havcr-1 are not well defined. We previously showed that the N-terminal immunoglobulin-like cysteine-rich region of havcr-1, referred to in this paper as D1, is required for binding of HAV (35) and protective MAb 190/4 (14). However, it was unknown whether D1 was sufficient for binding of HAV. The data presented in this paper demonstrate that HAV binds specifically to D1-Fc (Fig. 4 to 6) and indicate that D1 is indeed sufficient for binding of HAV, which is consistent with the results obtained with other picornavirus receptors (for a review, see reference 13). For instance, deletion analysis has shown that the V domain of PVR is necessary and sufficient for virus binding, uncoating, and infection (19, 33). However, constructs containing only the N-terminal virus-binding V domain of PVR do not function well as viral receptors. Domains 2 and 3 of PVR do not participate directly in virus binding, but they provide a “scaffold” for maintaining the conformation of the V domain that binds poliovirus (2). If this is also the case for havcr-1 (currently under investigation), it is possible that the low level of HAV neutralization induced by D1-Fc could be increased with soluble receptors containing D1 plus the TSP-rich region. Our comparison of the neutralization of HAV with soluble receptors and the MAbs (Fig. 8) suggested that, besides the structure of D1-Fc, other factors contribute to the low level of neutralization of HAV. Since soluble picornavirus receptors and MAbs behave similarly in neutralization assays and generate comparable rates of escape mutants (17), it is likely that factors such as viral aggregation and masking of epitopes (22, 28, 34) that affect antibody-mediated neutralization also contribute to the low rate of soluble receptor-mediated neutralization. It should be pointed out that this low level of neutralization is characteristic of HAV and contrasts with the high-level neutralization of poliovirus induced by PVR-Fc, which contains the three Ig-like domains of PVR.
Due to its mucin-like nature, it was hypothesized that havcr-1 functioned as an accessory factor that mediated the initial loose and nonspecific attachment of HAV to the cells and that other functional receptors were necessary for cell entry of HAV (23). However, several lines of evidence indicate that havcr-1 is indeed a specific and functional cellular receptor for HAV. First, we previously determined that D1 and not the mucin-like region of havcr-1 is necessary for binding of HAV (35), and in this paper we report that a purified soluble form of D1 is sufficient for binding of HAV. These results demonstrate that HAV interacts primarily with D1 and support the concept that the mucin-like region of havcr-1 is not necessary for binding of HAV. We are currently investigating whether the mucin-like region of havcr-1 plays any role in HAV receptor function. Second, in this report we showed that MAb 190/4 binds to purified D1-Fc and blocks the binding of HAV, which is consistent with our previous finding that MAb 190/4 protects AGMK GL37 cells against HAV infection by binding to havcr-1 and blocking its interaction with HAV (18). On the basis of these results, we conclude that additional coreceptors are not required for the expression of protective epitope 190/4 and binding of HAV to D1, which strongly suggests that an additional coreceptor(s) is not required for binding of HAV to the cell surface. However, it is possible that other regions of havcr-1 and coreceptors may be necessary to promote a more efficient binding of the virus to the cell, which could be required for cell entry. Therefore, the possibility that coreceptors are required for HAV infection (23) cannot be entirely ruled out at present. Finally, our neutralization studies demonstrate that the HAV–havcr-1 interaction required for infection can be prevented by treatment with D1-Fc, which supports the concept that havcr-1 is a functional HAV receptor. Absolute proof that havcr-1 is a functional receptor for HAV will require the isolation of an elusive receptor-negative but replication-permissive cell line that could be made susceptible to HAV infection upon expression of havcr-1.
Stable intermediates of uncoating have been described for several picornaviruses but not for HAV. In this work we show that D1-Fc binds and neutralizes HAV; however, we have not determined whether D1-Fc can induce uncoating of the genomic RNA and formation of empty capsids or other uncoating intermediates. Because of the poor growth characteristics of HAV, such experiments are not trivial and will require further investigation. The availability of active soluble forms of havcr-1 will allow us to use biochemistry and genetics to further understand the mechanism of cell entry by HAV. The isolation of HAV mutants resistant to neutralization with soluble receptors, as was done for poliovirus (17), and the determination of the structure of HAV complexed with soluble forms of havcr-1 will advance our understanding of the HAV–havcr-1 interaction.
ACKNOWLEDGMENTS
We thank Stephen Feinstone for encouragement and helpful advice and Barry Falgout and Hira Nakhasi for comments on the manuscript. We thank Michael Klutch for sequencing of DNA samples.
This research was supported in part by the appointment of E.S. to the Postgraduate Research Participation Program at the Center for Biologics Evaluation and Research administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the U.S. Food and Drug Administration.
REFERENCES
- 1.Arita M, Horie H, Nomoto A. Interaction of poliovirus with its receptor affords a high level of infectivity to the virion in poliovirus infections mediated by the Fc receptor. J Virol. 1999;73:1066–1074. doi: 10.1128/jvi.73.2.1066-1074.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernhardt G, Harber J, Zibert A, deCrombrugghe M, Wimmer E. The poliovirus receptor: identification of domains and amino acid residues critical for virus binding. Virology. 1994;203:344–356. doi: 10.1006/viro.1994.1493. [DOI] [PubMed] [Google Scholar]
- 3.Chamow S M, Ashkenazi A. Immunoadhesins: principles and applications. Trends Biotechnol. 1996;14:52–60. doi: 10.1016/0167-7799(96)80921-8. [DOI] [PubMed] [Google Scholar]
- 4.Cohen J I, Rosenblum B, Feinstone S M, Ticehurst J, Purcell R H. Attenuation and cell culture adaptation of hepatitis A virus (HAV): a genetic analysis with HAV cDNA. J Virol. 1989;63:5364–5370. doi: 10.1128/jvi.63.12.5364-5370.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen J I, Rosenblum B, Ticehurst J R, Daemer R J, Feinstone S M, Purcell R H. Complete nucleotide sequence of an attenuated hepatitis A virus: comparison with wild-type virus. Proc Natl Acad Sci USA. 1987;84:2497–2501. doi: 10.1073/pnas.84.8.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen J I, Ticehurst J R, Feinstone S M, Rosenblum B, Purcell R H. Hepatitis A virus cDNA and its RNA transcripts are infectious in cell culture. J Virol. 1987;61:3035–3039. doi: 10.1128/jvi.61.10.3035-3039.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crevat D, Crance J M, Chevrinais A M, Passagot J, Biziagos E, Somme G, Deloince R. Monoclonal antibodies against an immunodominant and neutralizing epitope on hepatitis A virus antigen. Arch Virol. 1990;113:95–98. doi: 10.1007/BF01318357. [DOI] [PubMed] [Google Scholar]
- 8.Daemer R J, Feinstone S M, Gust I D, Purcell R H. Propagation of human hepatitis A virus in African green monkey kidney cell culture: primary isolation and serial passage. Infect Immun. 1981;32:388–393. doi: 10.1128/iai.32.1.388-393.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dotzauer A, Feinstone S M, Kaplan G. Susceptibility of nonprimate cell lines to hepatitis A virus infection. J Virol. 1994;68:6064–6068. doi: 10.1128/jvi.68.9.6064-6068.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emerson S U, Huang Y K, McRill C, Lewis M, Purcell R H. Mutations in both the 2B and 2C genes of hepatitis A virus are involved in adaptation to growth in cell culture. J Virol. 1992;66:650–654. doi: 10.1128/jvi.66.2.650-654.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emerson S U, Huang Y K, McRill C, Lewis M, Shapiro M, London W T, Purcell R H. Molecular basis of virulence and growth of hepatitis A virus in cell culture. Vaccine. 1992;10:S36–S39. doi: 10.1016/0264-410x(92)90539-v. [DOI] [PubMed] [Google Scholar]
- 12.Emerson S U, Huang Y K, Purcell R H. 2B and 2C mutations are essential but mutations throughout the genome of HAV contribute to adaptation to cell culture. Virology. 1993;194:475–480. doi: 10.1006/viro.1993.1286. [DOI] [PubMed] [Google Scholar]
- 13.Evans D J, Almond J W. Cell receptors for picornaviruses as determinants of cell tropism and pathogenesis. Trends Microbiol. 1998;6:198–202. doi: 10.1016/s0966-842x(98)01263-3. [DOI] [PubMed] [Google Scholar]
- 14.Feigelstock D, Thompson P, Mattoo P, Kaplan G G. Polymorphisms of the hepatitis A virus cellular receptor 1 in African green monkey kidney cells result in antigenic variants that do not react with protective monoclonal antibody 190/4. J Virol. 1998;72:6218–6222. doi: 10.1128/jvi.72.7.6218-6222.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Funkhouser A W, Purcell R H, D'Hondt E, Emerson S U. Attenuated hepatitis A virus: genetic determinants of adaptation to growth in MRC-5 cells. J Virol. 1994;68:148–157. doi: 10.1128/jvi.68.1.148-157.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hollinger F B, Ticehurst J R. Hepatitis A virus. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. 3rd ed. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 735–782. [Google Scholar]
- 17.Kaplan G, Peters D, Racaniello V R. Poliovirus mutants resistant to neutralization with soluble cell receptors. Science. 1990;250:1596–1599. doi: 10.1126/science.2177226. [DOI] [PubMed] [Google Scholar]
- 18.Kaplan G, Totsuka A, Thompson P, Akatsuka T, Moritsugu Y, Feinstone S M. Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J. 1996;15:4282–4296. [PMC free article] [PubMed] [Google Scholar]
- 19.Koike S, Ise I, Nomoto A. Functional domains of the poliovirus receptor. Proc Natl Acad Sci USA. 1991;88:4104–4108. doi: 10.1073/pnas.88.10.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koike S, Ise I, Sato Y, Mitsui K, Horie H, Umeyama H, Nomoto A. Early events of poliovirus infection. In: Nomoto A, editor. Cellular receptors for virus infection. Vol. 3. New York, N.Y: Saunders Scientific Publications/Academic Press; 1992. pp. 109–115. [Google Scholar]
- 21.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 22.Lemon S M, Binn L N. Incomplete neutralization of hepatitis A virus in vitro due to lipid-associated virions. J Gen Virol. 1985;66:2501–2505. doi: 10.1099/0022-1317-66-11-2501. [DOI] [PubMed] [Google Scholar]
- 23.Locarnini S. The attachment receptor for hepatitis A virus. Trends Microbiol. 1997;5:45–47. doi: 10.1016/S0966-842X(96)30043-7. [DOI] [PubMed] [Google Scholar]
- 24.MacGregor A, Kornitschuk M, Hurrell J G, Lehmann N I, Coulepis A G, Locarnini S A, Gust I D. Monoclonal antibodies against hepatitis A virus. J Clin Microbiol. 1983;18:1237–1243. doi: 10.1128/jcm.18.5.1237-1243.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin S, Casasnovas J M, Staunton D E, Springer T A. Efficient neutralization and disruption of rhinovirus by chimeric ICAM-1/immunoglobulin molecules. J Virol. 1993;67:3561–3568. doi: 10.1128/jvi.67.6.3561-3568.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitchell P J, Carothers A M, Han J H, Harding J D, Kas E, Venolia L, Chasin L A. Multiple transcription start sites, DNase I-hypersensitive sites, and an opposite-strand exon in the 5′ region of the CHO dhfr gene. Mol Cell Biol. 1986;6:425–440. doi: 10.1128/mcb.6.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neutra M R, Forstner J F. Gastrointestinal mucus: synthesis, secretion, and function. In: Johnson L R, editor. Physiology of the gastrointestinal tract. New York, N.Y: Raven Press; 1987. pp. 975–1009. [Google Scholar]
- 28.Ping L H, Lemon S M. Antigenic structure of human hepatitis A virus defined by analysis of escape mutants selected against murine monoclonal antibodies. J Virol. 1992;66:2208–2216. doi: 10.1128/jvi.66.4.2208-2216.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Powell R M, Ward T, Evans D J, Almond J W. Interaction between echovirus 7 and its receptor, decay-accelerating factor (CD55): evidence for a secondary cellular factor in A-particle formation. J Virol. 1997;71:9306–9312. doi: 10.1128/jvi.71.12.9306-9312.1997. . (Erratum, 72:890, 1998.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Provost P J, Hilleman M R. Propagation of human hepatitis A virus in cell culture in vitro. Proc Soc Exp Biol Med. 1979;160:213–221. doi: 10.3181/00379727-160-40422. [DOI] [PubMed] [Google Scholar]
- 31.Reed L J, Muench H. A simple method of estimating fifty per cent end points. Am J Hyg. 1938;27:493–497. [Google Scholar]
- 32.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 33.Selinka H C, Zibert A, Wimmer E. Poliovirus can enter and infect mammalian cells by way of an intercellular adhesion molecule 1 pathway. Proc Natl Acad Sci USA. 1991;88:3598–3602. doi: 10.1073/pnas.88.9.3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stapleton J T, Lemon S M. Neutralization escape mutants define a dominant immunogenic neutralization site on hepatitis A virus. J Virol. 1987;61:491–498. doi: 10.1128/jvi.61.2.491-498.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson P, Lu J, Kaplan G G. The Cys-rich region of hepatitis A virus cellular receptor 1 is required for binding of hepatitis A virus and protective monoclonal antibody 190/4. J Virol. 1998;72:3751–3761. doi: 10.1128/jvi.72.5.3751-3761.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Totsuka A, Moritsugu Y. Hepatitis A vaccine development in Japan. In: Nishioka K, Suzuki H, Mishiro S, Oda T, editors. Viral hepatitis and liver disease. Tokyo, Japan: Springer-Verlag; 1994. pp. 509–513. [Google Scholar]
- 37.Ward T, Powell R M, Pipkin P A, Evans D J, Minor P D, Almond J W. Role for beta2-microglobulin in echovirus infection of rhabdomyosarcoma cells. J Virol. 1998;72:5360–5365. doi: 10.1128/jvi.72.7.5360-5365.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xing L, Tjarnlund K, Lindqvist B, Kaplan G G, Feigelstock D, Cheng R H, Casasnovas J M. Distinct cellular receptor interactions in poliovirus and rhinoviruses. EMBO J. 2000;19:1207–1216. doi: 10.1093/emboj/19.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Kaplan G G. Characterization of replication-competent hepatitis A virus constructs containing insertions at the N terminus of the polyprotein. J Virol. 1998;72:349–357. doi: 10.1128/jvi.72.1.349-357.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]