

Abstract
The molecular organization of the human neocortex has been historically studied in the context of its histological layers. However, emerging spatial transcriptomic technologies have enabled unbiased identification of transcriptionally-defined spatial domains that move beyond classic cytoarchitecture. Here we used the Visium spatial gene expression platform to generate a data-driven molecular neuroanatomical atlas across the anterior-posterior axis of the human dorsolateral prefrontal cortex (DLPFC). Integration with paired single nucleus RNA-sequencing data revealed distinct cell type compositions and cell-cell interactions across spatial domains. Using PsychENCODE and publicly available data, we map the enrichment of cell types and genes associated with neuropsychiatric disorders to discrete spatial domains.
Summary:
Generation of a molecular neuroanatomical map of the human prefrontal cortex reveals features relevant for psychiatric disease.
The emergence of single cell and spatially-resolved transcriptomics has facilitated the generation of integrated molecular anatomical maps across a variety of tissues in both rodents and humans (1–4). Data-driven unsupervised approaches to identify spatial domains in these large datasets, particularly in the rodent brain, have refined our understanding of the spatial organization of tissues beyond classical cytoarchitectural boundaries (2), and computational models have helped reveal new insights into cellular diversification during development (5). However, efforts to generate spatially-resolved transcriptomics data in the human brain at the scale and size necessary to employ these approaches with sufficient statistical power, have lagged behind.
We previously characterized the spatial topography of gene expression in the human dorsolateral prefrontal cortex (DLPFC), a brain region associated with several neurodevelopmental and psychiatric disorders (6–11), by manually annotating the six histological layers and white matter of the neocortex in a small cohort of 3 neurotypical adult donors (12). Although we identified robust layer-enriched gene expression with this approach, recent computational tools for unsupervised clustering (13–17) and cell to cell communication (18) combined with the ability to expand the generation of molecular neuroanatomical maps across a larger donor pool has enabled data-driven identification of higher resolution spatial domains. Applying these data-driven approaches to larger-scale studies might facilitate the demarcation of fine cortical sublayers, which currently lack molecular annotations. They also could enable identification of non-laminar spatial domains associated with anatomical or topographical features in the human brain, including vasculature. Here we sought to develop and validate a framework for application of unsupervised spatial clustering approaches in human brain tissue across different anterior-posterior positions of the DLPFC and generate an analytical roadmap that can be extended to other brain areas with less well-characterized neuroanatomical architecture in the future.
Recent single nucleus RNA-sequencing (snRNA-seq) analyses are defining transcriptionally distinct DLPFC cell types and revealing cell type-specific changes associated with neurodevelopmental and neuropsychiatric disorders, such as schizophrenia (SCZ), autism spectrum disorder (ASD), major depressive disorder (MDD) and post-traumatic stress disorder (PTSD) (19–23). However, snRNA-seq data lack spatial context, which when retained during molecular profiling, can provide important insights into cell-cell communication and disease pathogenesis. To facilitate this type of analysis, we generated large-scale, unsupervised spatial transcriptomic molecular maps of the human DLPFC from ten neurotypical brain donors, which we integrated with snRNA-seq data across a variety of brain disorders. This spatially-resolved, molecular atlas of gene expression architecture in the human brain is provided as an interactive data resource for the scientific community to help reveal molecular mechanisms associated with psychiatric illness.
Study Overview
Here we applied single cell and spatial transcriptomic approaches to create a large-scale, data-driven spatial map of gene expression at single cell resolution in the adult human DLPFC in order to identify spatial domains, define cell-cell communication (CCC) patterns, and perform spatial registration of cell types across brain disorders (Fig. 1A–B). Using the Visium spatial transcriptomics platform (24), we measured spatial gene expression in fresh frozen postmortem human tissue blocks from 10 neurotypical adult donors (Table S1) in three positions spanning the rostral-caudal axis of the DLPFC (anterior [Ant], middle [Mid] and posterior [Post]) for a total of 30 tissue sections [n=10 per position] (Fig. 1A). In parallel, we performed snRNA-seq (10x Genomics 3’ gene expression) on a subset of the same DLPFC samples (n=1–2 blocks per donor) to generate matched snRNA-seq and spatial transcriptomic data for 19 tissue blocks (Fig. 1A–C). To preserve Layer (L)1, blocks were microdissected across sulci in the plane perpendicular to the pia that extended to the gray-white matter junction. The morphology of each tissue block was assessed with RNAscope multiplex single molecule fluorescent in situ hybridization (smFISH) using regional and laminar marker genes to ensure dissection consistency (Fig. 1D). Sample orientation was confirmed by expression of genes enriched in the gray matter (SNAP25), white matter (WM; [MBP]), and L5 (PCP4; Fig. 1C, Fig S2, Fig S3, Fig S4). For Visium, 4,866 (4.1%) spots with low library size were excluded (Fig S5), resulting in a total of 113,927 spots across 30 tissue blocks and 10 donors. Downstream analyses at the gene-level (Fig S6) and spot-level (Fig S7) were not impacted by tissue artifacts, including wrinkles, shears, and folds (see Methods: Evaluating the impact of histology artifacts on Visium H&E data). For snRNA-seq, 54,394 nuclei across 19 tissue blocks from 10 donors passed quality control and were included in the study (Fig. 1C). Using these integrated datasets, we performed several analyses including unsupervised clustering, spot deconvolution, CCC analyses, and spatial registration of PsychENCODE single cell datasets (Fig. 1B).
Figure 1. Study design to generate paired single nucleus RNA-sequencing (snRNA-seq) and spatially-resolved transcriptomic data across DLPFC.
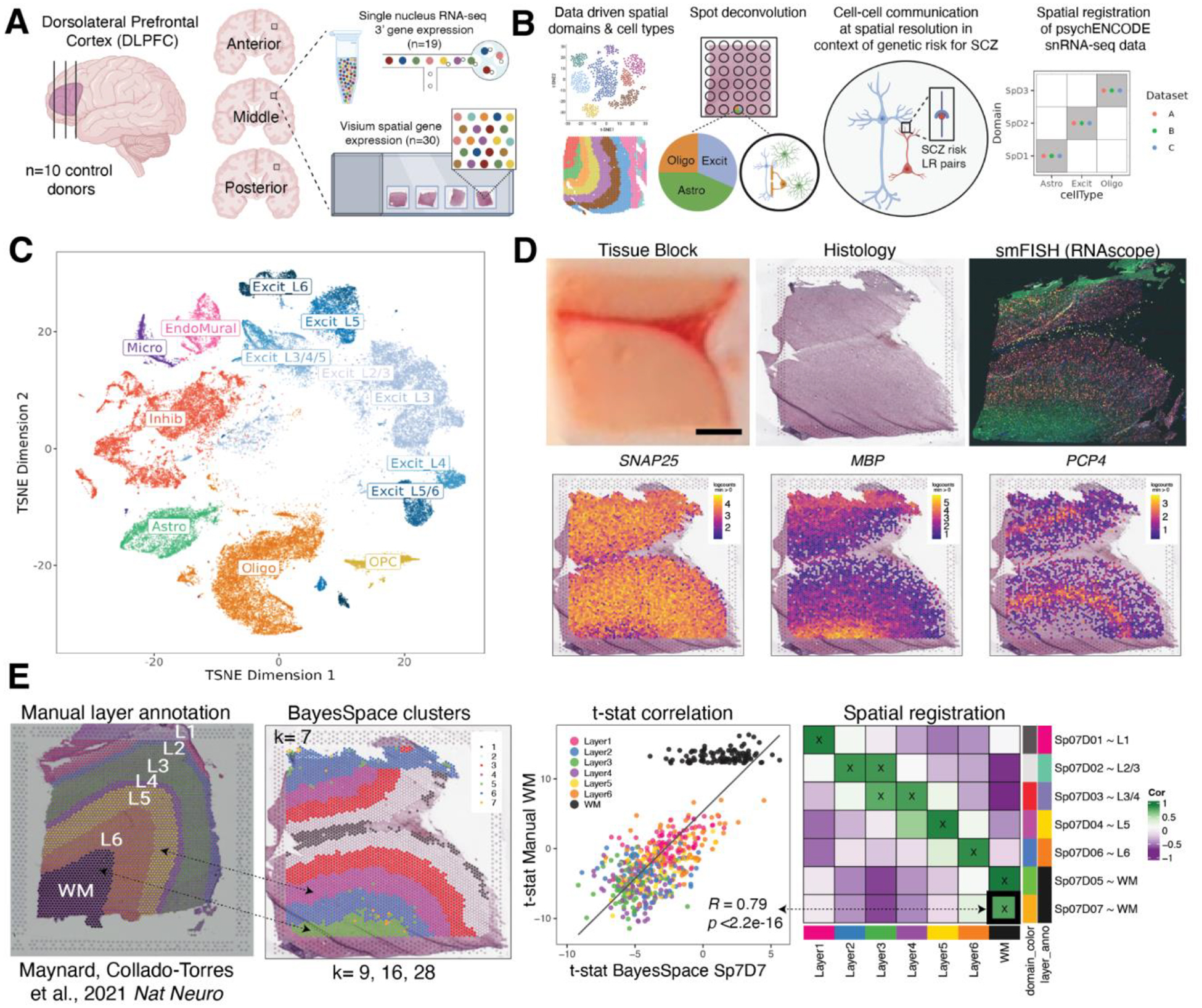
(A) DLPFC tissue blocks were dissected across the rostral-caudal axis from 10 adult neurotypical control postmortem human brains, including anterior (Ant), middle (Mid), and posterior (Post) positions (n=3 blocks per donor, n=30 blocks total). The same tissue blocks were used for snRNA-seq (10x Genomics 3’ gene expression assay, n=1–2 blocks per donor, n=19 samples) and spatial transcriptomics (10x Genomics Visium spatial gene expression assay, n=3 blocks per donor, n=30 samples). (B) Paired snRNA-seq and Visium data were used to identify data-driven spatial domains (SpDs) and cell types, perform spot deconvolution, conduct cell-cell communication analyses, and spatially register companion PsychENCODE snRNA-seq DLPFC data. (C) t-distributed stochastic neighbor embedding (t-SNE) summarizing layer resolution cell types identified by snRNA-seq. (D) Tissue block orientation and morphology was confirmed by hematoxylin and eosin (H&E) staining and single molecule fluorescent in situ hybridization (smFISH) with RNAscope (SLC17A7 marking excitatory neurons in pink, MBP marking white matter (WM) in green, RELN marking layer (L)1 in yellow, and NR4A2 marking L6 in orange). Scale bar is 2mm. Spotplots depicting log transformed normalized expression (logcounts) of SNAP25, MBP, and PCP4 in the Visium data confirm the presence of gray matter, WM, and cortical layers, respectively (see also Fig S2–Fig S4). (E) Schematic of unsupervised SpD identification and registration using BayesSpace SpDs at k=7. Enrichment t-statistics computed on BayesSpace SpDs were correlated with manual histological layer annotations from (12) to map SpDs to known histological layers. The heatmap of correlation values summarizes the relationship between BayesSpace SpDs and classic histological layers. Higher confidence annotations (cor > 0.25, merge ratio = 0.1, see Methods: Spatial registration of Spatial Domains) are marked with an “X”.
Identification of data-driven spatial domains at different resolutions across DLPFC
We selected an unsupervised clustering method for robust identification of laminar spatial domains (SpDs) in Visium data by benchmarking three algorithms, graph-based clustering, SpaGGN and BayesSpace (13, 15, 25, 26), using our previously published DLPFC Visium data (12 sections from 3 donors; (12)). We compared data-driven clustering accuracy against manual layer annotations (Fig S9, Fig S10). Among the algorithms tested, BayesSpace most accurately identified spatial domains (SpDs) consistent with the histological cortical layers. Therefore, we used this clustering algorithm to identify 7 unsupervised SpDs approximating the 6 cortical layers and WM. To relate unsupervised SpDs to the classic histological layers, we pseudo-bulked spots (by gene, sum the expression for all spots in the same SpD in a given sample) within each SpD across individual tissue sections to generate SpD-specific expression profiles and performed differential expression (DE) analysis to identify genes enriched in each SpD. Next, we performed “spatial registration” by correlating the enrichment statistics computed on BayesSpace SpDs with those from manually annotated cortical layers (12) to approximate the most strongly associated histological layer for a given BayesSpace SpD (Fig. 1E). We annotated the association of a specific spatial domain (SpD) at cluster resolution k to a classic histological layer using the term SpkDd~L, where L refers to the histological layer most strongly associated to domain d following cluster registration at resolution k. For example, spatial domain 7 at cluster resolution k=7 (Sp7D7) was mostly strongly associated with white matter (Fig. 1E) and is annotated as Sp7D7~WM. We found that k=7 was not sufficient to fully separate histological layers, especially superficial L2-L4, suggesting the presence of higher resolution data-driven SpDs and highlighting the challenges of manually annotating spatial domains (12).
We next evaluated how increasing cluster resolution (k) influenced the identification and cluster registration of unsupervised SpDs (Fig. 2). As expected, clustering at k=2 reliably separated white and gray matter (Fig S11, Fig S12). Next, we evaluated three clustering resolutions: a broad resolution k=9, a data-driven fine resolution k=16 (Fig S13, Fig S14), and a super-fine resolution k=28. Hereafter, we refer to these SpDs as Sp9D, Sp16D, and Sp28D, respectively. Broad clustering most accurately recapitulated the classic histological layers with clear separation of Sp9Ds enriched in genes expressed in L1–6 and WM (Fig. 2A–B, Fig S15). At fine clustering resolution, SpDs were largely laminar with two or more Sp16Ds registering to a given histological layer, suggesting the presence of molecularly-defined sublayers (Fig. 2A–B, Fig S16). At super-fine resolution, many SpDs lacked a laminar structure, but spots belonging to Sp28Ds frequently mapped back to a single broad or fine resolution spatial domain (Fig S17, Fig S18). To evaluate the unsupervised architecture of cortical layers in the human DLPFC in more depth, we focused on broad and fine SpDs based on the robust laminar features of these domains.
Figure 2. Unsupervised clustering at different resolutions identifying spatial domains (SpDs) and defining molecular anatomy of DLPFC.
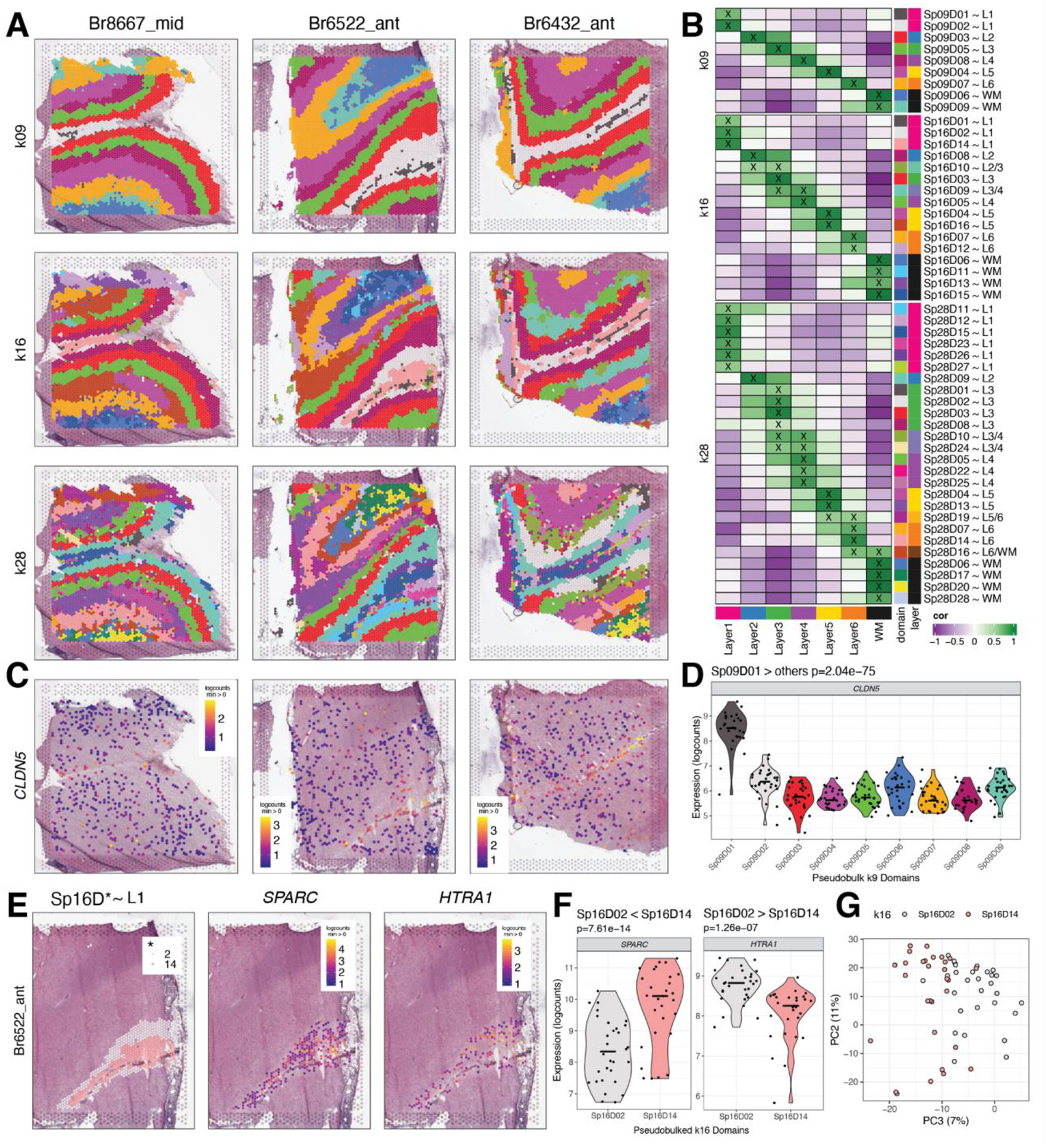
(A) BayesSpace clustering at k=9, 16, and 28 (broad, fine, and super-fine resolution, respectively, which we refer to as SpkDd for domain d from SpDs at k resolution) for three representative DLPFC tissue sections (Br8667_mid, Br6522_ant, Br6432_ant). (B) Heatmap of spatial registration with manually annotated histological layers from (12). BayesSpace identifies laminar SpDs at increasing k with the majority of SpkDs correlating with one or more histological layer(s). SpDs were assigned layer annotations following spatial registration to histological layers. Annotations with high confidence (cor > 0.25, merge ratio = 0.1, see Methods: Spatial registration of Spatial Domains) are marked with an “X”, and this histological layer association is denoted for a given SpkDd by adding “~L,” where L is the most strongly correlated histological layer (or WM). See also Fig S11–Fig S18. (C) Spotplots depicting expression of CLDN5 in vasculature domain 1 at k=9 resolution (Sp9D1). (D) Boxplot confirming enrichment of CLDN5 in Sp9D1 compared to other Sp9Ds across 30 tissue sections. (E) Spotplots of representative section Br6522_ant showing identification of molecularly-defined sublayers for histological L1 at k=16 (Sp16D2 and Sp16D14) and enrichment of HTRA1 and SPARC, respectively. (F) Boxplots quantifying enrichment of SPARC and HTRA1 in Sp16D14 and Sp16D2, respectively, across 30 tissue sections. (G) PCA plot showing separation of Sp16D2 and Sp16D14 supporting identification of molecularly distinct SpDs.
Enrichment of differentially expressed genes in unsupervised spatial domains
To identify the molecular signatures of SpDs within each clustering resolution, we next performed differential expression (DE) analysis with linear mixed-effects modeling using the Sp9D- or Sp16D-specific expression profiles. As previously described, we employed three different statistical models: ANOVA model, enrichment model, and pairwise model (12). As expected, all three models confirmed that unsupervised clustering at broad or fine resolution identified biologically meaningful SpDs with significantly DE of genes across the laminar architecture of the cortex (5,931 FDR<5% unique enriched genes in at least one Sp9D, see Methods: Layer-level data processing and differential expression modeling). Although we did identify 512 unique genes that were differentially enriched across the anterior-posterior axis of the DLPFC, SpD had a much stronger effect on gene expression compared to anatomical position (anterior, middle, posterior, Fig S19).
Analysis of DEGs identified using the enrichment model allowed for characterization of previously unrecognized data-driven SpDs, such as Sp9D1 and Sp16D1 (Fig. 2B). At both k=9 and k=16, domain 1 is adjacent to histological L1 and enriched for genes associated with blood vessels and brain vasculature, such as CLDN5 (p=2.04e-75; Fig. 2C–D). Due to its thinness, this vascular-rich meninges layer was not manually annotated in our previous study (12), demonstrating the utility of unsupervised approaches to robustly identify biologically meaningful SpDs. Data-driven clustering at fine resolution also revealed molecularly-defined sublayers, including two adjacent laminar spatial subdomains (Sp16D14~L1 and Sp16D2~L1; Fig. 2E) enriched in L1 marker genes, including RELN (p=6.98e-17, p=3.19e-19 respectively) and AQP4 (p=9.37e-21, p=9.43e-12 respectively). Pairwise tests across these L1-related subdomains highlighted differential expression of SPARC (enriched in Sp16D14~L1, p=7.61e-14) and HTRA1 (enriched in Sp16D2~L1, p=1.26e-07), and principal component analysis (PCA) further confirmed the unique nature of these molecularly-defined sublayers (Fig. 2E–G). We also identified multiple SpDs associated with histological L4 (Sp16D5-L4 and Sp16D9-L4), L5 (Sp16D4-L5 and Sp16D16-L5), and L6 (Sp16D7-L6 and Sp16D12-L6; Fig. 2B). Taken together, this analysis validated the biological relevance of data-driven SpDs and increased the understanding of the molecular neuroanatomy across the laminar architecture of the adult human DLPFC.
Identification of molecularly and spatially distinct neuronal populations across cortical layers
To add single cell resolution to our molecular maps, we performed snRNA-seq on a subset of the same tissue blocks used for Visium (Fig. 1). Following assessment of quality control metrics (Fig S20, Fig S21), we performed batch correction (Fig S22, Fig S23, Fig S24) and data-driven clustering to generate 29 fine-resolution clusters across 7 broad cell types represented throughout the anterior-posterior DLPFC axis (Fig. 3A, Fig S25). Further, we spatially registered all clusters to the histological layers using manually annotated Visium data from previous work (12) (Fig. 3B). Given that molecularly-defined excitatory neuron populations in the cortex have distinct laminar identities (27), we systematically assigned a histological layer to excitatory neuron clusters (Table S2), resulting in the identification of 13 layer-resolution clusters with distinct marker genes (Fig. 3B–D, Fig S26). At both fine-resolution and layer-resolution, our clusters strongly correlated with those derived from the reference-based mapping tool, Azimuth (https://azimuth.hubmapconsortium.org) (28, 29) (Fig S27).
Figure 3. Spatial registration of fine resolution snRNA-seq clusters defining laminar cell types.
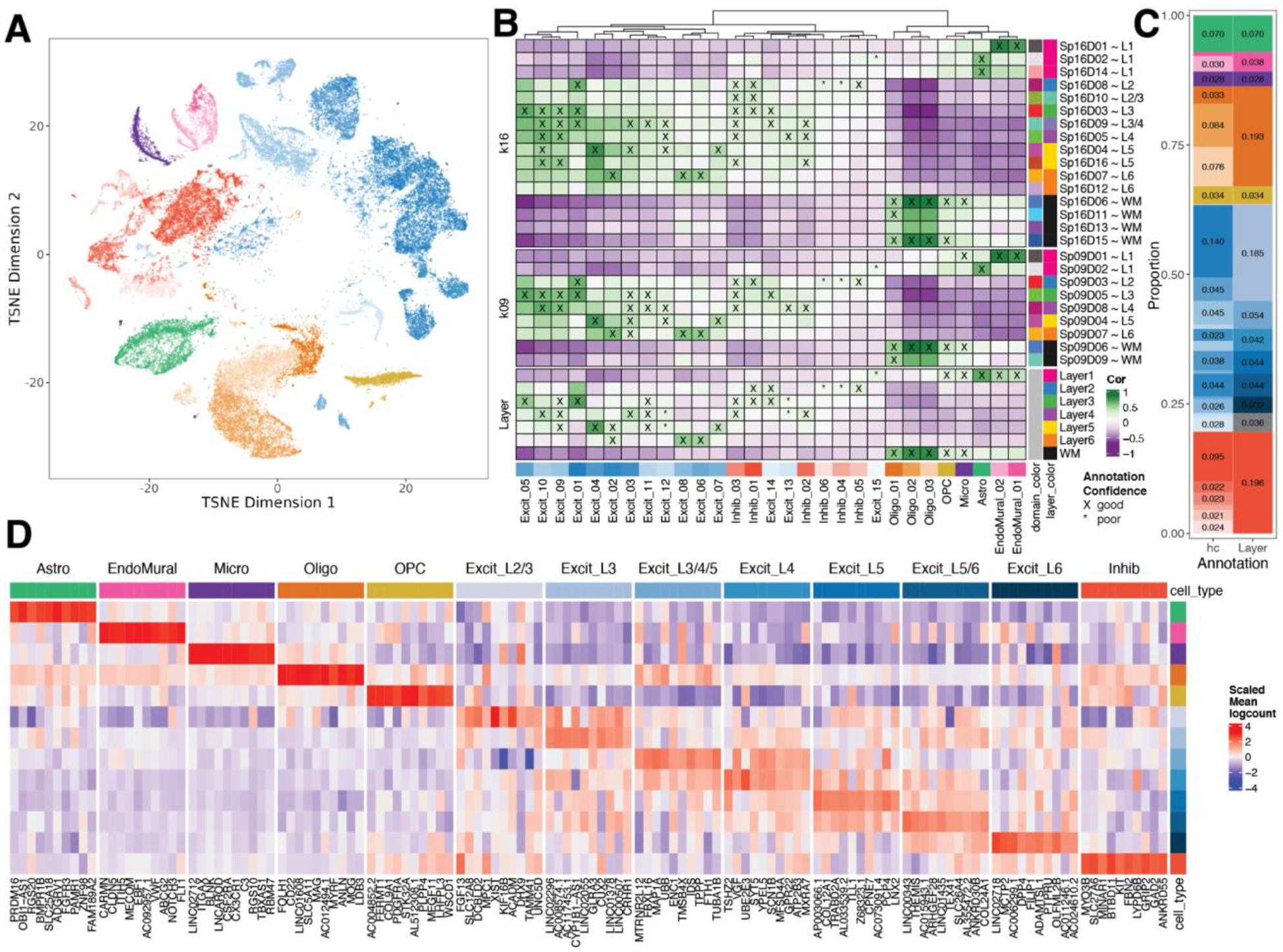
(A) t-distributed stochastic neighbor embedding (t-SNE) plot of 56,447 nuclei across 29 cell type-annotated fine resolution hierarchical clusters (hc; related to Fig S25A). (B) Spatial registration heatmap showing correlation between snRNA-seq hierarchical clusters (hc) and manually annotated histological layers from (12) as well as unsupervised BayesSpace clusters at k=9 and 16 (Sp9Ds and Sp16Ds). Hierarchical clusters for excitatory neurons (Excit) were assigned layer-level annotations following spatial registration to histological layers (cor > 0.25, merge ratio = 0.25, see Methods: snRNA-seq spatial registration). For Sp9Ds and Sp16Ds, annotations with good confidence (cor > 0.25, merge ratio = 0.1) are marked with “X” and poor confidence are marked with “*”. (C) Summary barplot of cell type composition for hc and layer level resolutions (related to Fig S25B & Fig S26) (D) Heatmap of the scaled mean pseudo-bulked logcounts for the top 10 marker genes identified for each cell type at layer-level resolution.
To gain further insight into the relationship between our snRNA-seq clusters and Visium unsupervised SpDs, we performed spatial registration of fine resolution snRNA-seq clusters with Visium SpDs at k=9 and k=16 (Fig. 3B). We refined the spatial positioning of our 29 fine resolution snRNA-seq clusters and validated the laminar associations of broad and fine unsupervised SpDs. For example, we showed that snRNA-seq excitatory neuron clusters Excit_06 and Excit_08, which spatially registered to histological L6, were also highly correlated with Visium Sp9D7, a domain enriched for L6-associated genes (Fig. 2B). These snRNA-seq clusters were also highly correlated with a single spatial domain (Sp16D7), thereby further refining their anatomical position to upper L6. Inhibitory GABAergic populations were also assigned to specific spatial locations. For example, Inhib_05 uniquely registered to histological L2, which was confirmed with strong correlations to Sp9D3 and Sp16D8 showing enrichment for L2-associated genes (Fig. 2B). We also showed registration of snRNA-seq endothelial cell populations to vascular spatial domains (Sp9D1 and Sp16D1) enriched in HBA1 and CLDN5. Whereas endothelial cells showed the strongest correlation with vascular-associated SpDs and glial cells showed the strongest correlation with L1 and WM-associated SpDs (Fig. 2B), these cell types are indeed distributed across the cortical layers necessitating higher resolution spatial mapping approaches, such as spot deconvolution.
Defining cell type composition of unsupervised spatial domains using spot deconvolution
Given that individual Visium spots in the human DLPFC contain an average of 3 cells per spot (12), we used our paired snRNA-seq data to perform cellular deconvolution of Visium spots to better understand the cell type composition of unsupervised SpDs. First, we benchmarked 3 spot-level deconvolution algorithms, SPOTlight, Tangram, and Cell2location (14, 30, 31), using a gold standard reference dataset acquired with the Visium Spatial Proteogenomics (Visium-SPG) assay. Visium-SPG replaces H&E histology with immunofluorescence staining, enabling us to label and quantify 4 broad cell types across the DLPFC, including NeuN (neurons), OLIG2 (oligodendrocytes), GFAP (astrocytes), and TMEM119 (microglia) (Fig. 4A, Fig S28). After verifying marker genes for each snRNA-seq cluster (Fig S29) and confirming the utility of these genes for spot deconvolution (Fig. 4B, Fig S30, Fig S31, Fig S32), we applied SPOTlight, Tangram, and Cell2location to our Visium-SPG data and calculated the predicted cell type counts per spot at broad and fine resolution (Fig. 4B, Fig S33).
Figure 4. Integration of snRNA-seq and Visium data to benchmark spot deconvolution algorithms and define cellular composition across spatial domains.
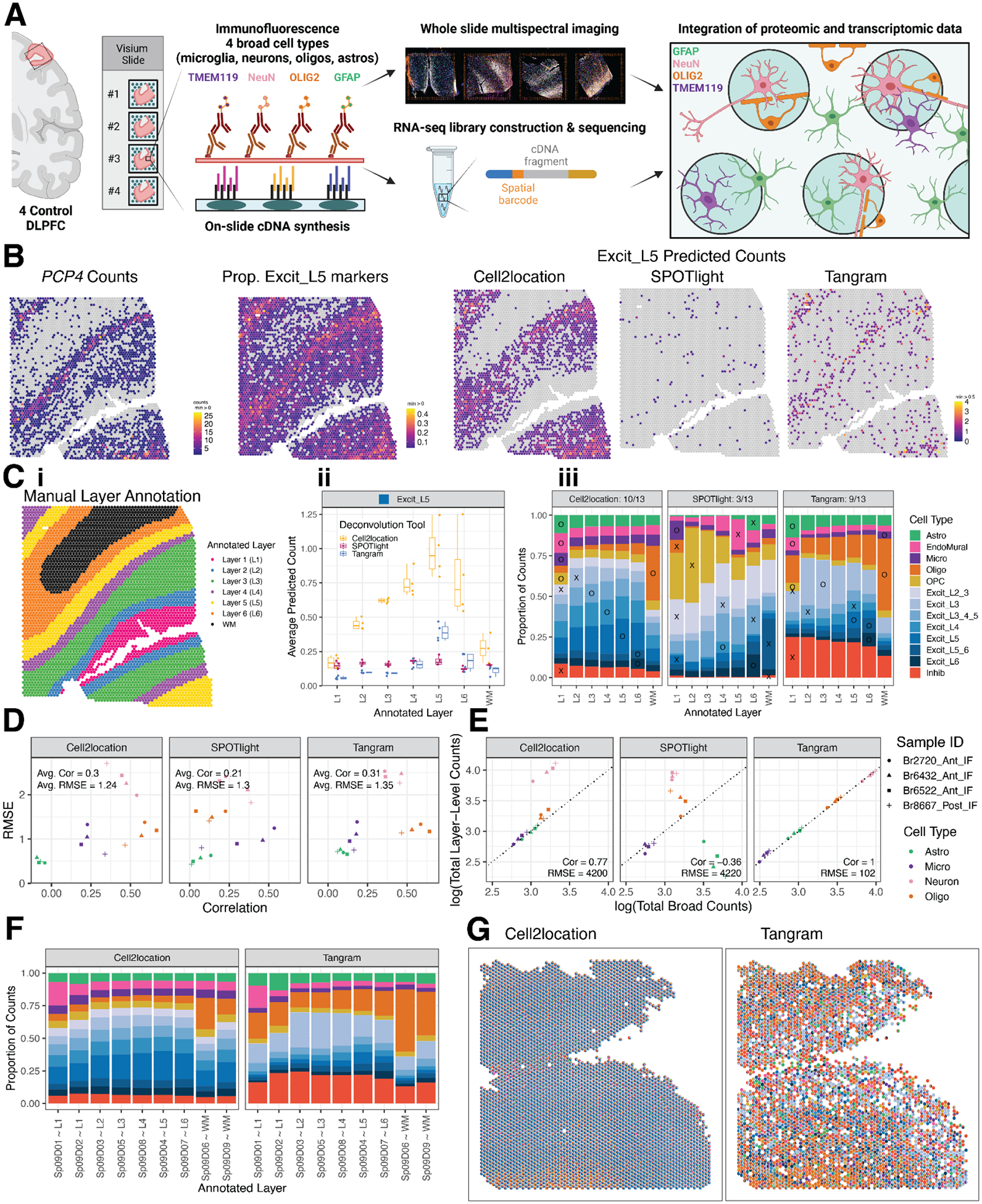
(A) Schematic of the Visium-SPG protocol. (B) For Br6522_Ant_IF, counts for L5 marker gene PCP4 are compared to the proportion of Excit_L5 marker genes with nonzero expression as well as the counts of Excit_L5 cells as predicted by the 3 evaluated deconvolution algorithms. (C) Example of manually annotated layer assignments for Br6522_Ant_IF (i), which are used to benchmark predicted cell type composition across layers. Using Excit_L5 as an example, predicted Excit_L5 counts for each method are averaged across all spots within each annotated layer for each tissue section (ii). These data are summarized across layers and tissue sections for the 13 cell types using a bar plot (iii). An “X” or “O” is placed on the layer with maximal proportion; an “O” is placed for a “correct” match for the given cell type, and an “X” is placed otherwise. For example, Tangram correctly predicts the maximal proportion of Excit_L5 cells in L5 annotated spots, leading to the placement of an “O” for Excit_L5. The “O”s are tallied for each method to generate a summary score in each facet’s title (for example, 9 of 13 cell types were correctly predicted to the expected layer using Tangram). (D) Predicted counts for a given method, section, and layer-level cell type are collapsed and compared against the corresponding CART predictions by computing the Pearson correlation and RMSE, forming a single point in the scatterplot (Supplemental Methods: Evaluating performance of spot-deconvolution methods). Each of these values is then averaged to generate a single correlation and RMSE value for each method, indicated in the top left inside each plot facet. (E) Section-wide counts for each cell type are compared between broad and layer-level resolutions, collapsed onto the cell-type resolution used by the CART, where values theoretically should precisely match. (F) The predicted proportion of cells in each Sp9D, deconvoluted by Cell2location and Tangram, are averaged across all Visium samples (n=30). (G) Cell composition of each Visium spot for Br8667_mid, deconvoluted by Cell2location and Tangram, revealing differences in cell composition prediction.
To quantify algorithm performance, we took 2 complementary approaches: 1) evaluating the localization of laminar cell types to their expected cortical layer (Fig. 4C, Fig S34, Fig S35) and 2) comparing predicted cell type counts to those obtained from immunofluorescent images using a classification and regression tree (CART) strategy to categorize nuclei into the 4 immunolabeled cell types (Fig. 4D, Fig S36, Fig S37, Fig S38, Fig S39). Using the first approach, we found that Tangram and Cell2location performed best across all cell types, but SPOTlight failed to accurately map excitatory neuron subtypes to the correct layer (Fig. 4B). Using the second approach, we found that the predicted counts from Tangram and Cell2location also had the highest correlation to CART-calculated counts (Fig. 4D) and Tangram showed the most consistent performance at both broad and layer level resolution across all cell types and samples (Fig. 4E). Finally, we applied Tangram and Cell2location to our H&E Visium dataset to predict the cellular composition of SpDs across the anterior-posterior axis of the DLPFC (Fig. 4F–G, Fig S40, Fig S41) and found that Tangram and Cell2location showed differences in the predicted cell counts per spot (Fig. 4F) whereas, for both tools, the predicted cellular composition of SpDs was consistent across samples at both broad (k=9) and fine (k=16) resolution regardless of DLPFC position (anterior, middle, posterior).
Spatial mapping of ligand-receptor (LR) interactions associated with schizophrenia (SCZ)
To add clinical relevance to this integrated DLPFC dataset, we next sought to identify interacting cell types and spatially map ligand receptor (LR) interactions associated with neuropsychiatric disorders. We focused on genetic risk for schizophrenia (SCZ) because receptors occur more frequently in SCZ risk genes than would be expected of a brain-expressed gene list of this size (p<0.0001, Fig S42A). First, we identified interacting cell types using cell-cell communication (CCC) analysis (18), which uses a data-driven approach to predict crosstalk between sender and receiver cells based on known LR interactions (Fig. 5A, Table S3). In parallel, using the OpenTargets and Omnipath databases (32, 33), we identified 834 LR pairs (Table S4) associated with genetic risk for SCZ. Of these, 90 interactions overlapped with those identified in CCC analysis where at least one of the interactors was associated with SCZ risk (Table S5).
Figure 5. Schizophrenia (SCZ)-associated ligand-receptor (LR) interactions identified by integrative analysis of snRNA-seq and Visium data.
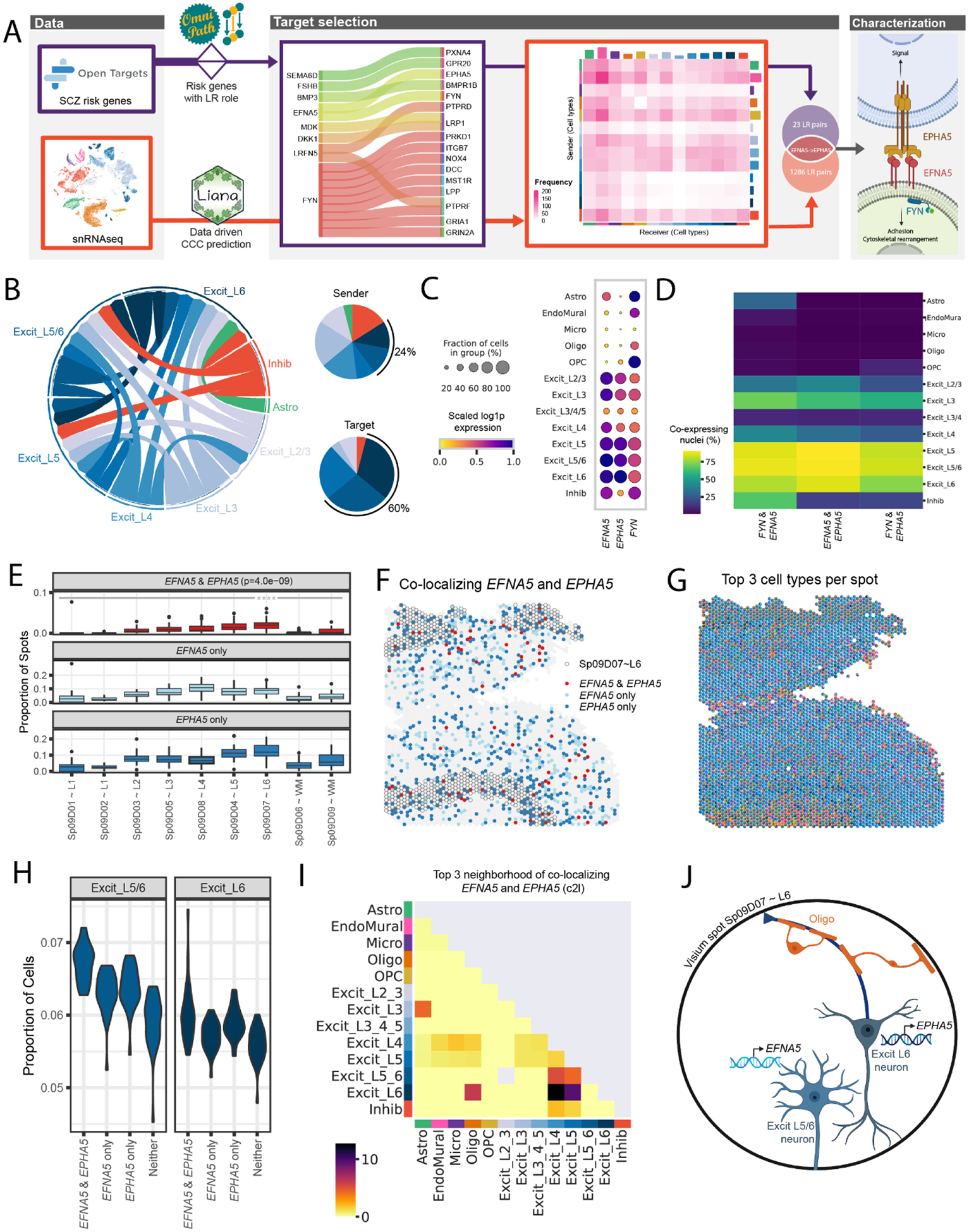
(A) The LR interaction between membrane-bound ligand ephrin A5 (EFNA5) and ephrin type-A receptor 5 (EPHA5) is a consensus target identified in both data-driven (Table S3) and clinical risk-driven LR (Table S4) analyses. Notably, this interaction also requires an intracellular interaction between EFNA5 and protein tyrosine kinase (FYN), which was also identified among clinical risk targets. (B) Cell-cell communication analysis predicts the sender/receiver cross-talk pattern of EFNA5-EPHA5 between layer-level cell types visualized in a circular plot. Excit_L5/6 and Excit_L6 neurons account for 24% of the cross-talk as senders and 60% as targets compared to other cell types shown in the pie charts. (C-D) Downstream analysis of snRNA-seq data characterizes FYN-EFNA5-EPHA5 signaling pathway, showing these genes are highly enriched (C) and co-expressed (D) in excitatory neuron populations. (E) Across all 30 tissue sections, EFNA5 and EPHA5 are co-expressed in a statistically higher proportion of spots in Sp9D7 (median (interquartile range) = 0.0196 (0.0137), p = 4.0e-09) compared to other Sp9Ds (Sp9D1 = 0 (0), Sp9D2 = 0 (0), Sp9D3 = 0.0052 (0.0078), Sp9D4 = 0.0140 (0.0142), Sp9D5 = 0.0091 (0.0102), Sp9D6 = 0 (0.0016), Sp9D8 = 0.0077 (0.0123), Sp9D8 = 0.0016 (0.0104)). (F) Spotplot of EFNA5 and EPHA5 co-expression in Br8667_mid. (G) Spotplot with spot-level pie charts for Br8667_mid showing the top 3 dominant cell types in each Visium spot predicted by Cell2location (c2l). (H) Visium spots co-expressing EFNA5 and EPHA5 have higher proportions of predicted Excit_L5/6 neurons (p=1.8e-12) and Excit_L6 (p=3.9e-4) compared to non-coexpressing spots, consistent with snRNA-seq specificity analyses (Fig S42). Few other cell types show this relationship (Fig S43). Complementary analyses of EFNA5 and FYN co-expression are shown in Fig S42. (I) Spatial network analysis of all 30 tissue sections, using top 3 dominant c2l cell types in each spot (exemplified in G with Br8667_mid), confirms EFNA5 and EPHA5 co-expression occurs frequently in spots containing Excit_L6 neurons. Complementary analyses using top 6 dominant c2l cell types as well as Tangram predictions are reported in Fig S42. (J) Schematic of a Visium spot depicting EFNA5-EPHA5 interactions between Excit_L5/6 neurons and Excit_L6. The high colocalization score in the spatial network analysis in (I) suggests oligodendrocytes also likely co-exist with Excit_L6 neurons.
We chose to prioritize 18 inter- and intra-cellular interactions where both counterparts showed disease association, including 9 interactions involving the protein tyrosine kinase, FYN (Fig. 5A, Fig S42). A consensus LR pair was identified between these complementary data-driven and clinical risk-driven approaches: the membrane-bound ligand ephrin A5 (EFNA5) and its receptor ephrin type-A receptor 5 (EPHA5). As part of this signaling cascade, we also evaluated the intracellular interaction between EFNA5 and FYN, which was one of the 18 SCZ-associated LR pairs (Fig. 5A).
The Ephrin/Eph signaling system is critical for neuronal wiring during brain development and neural plasticity and synaptic homeostasis in adulthood (34). To better understand the role of this SCZ-associated signaling pathway in the adult DLPFC, we next characterized the cell types mediating this interaction using our snRNA-seq data. We identified enrichment of EFNA5, EPHA5, and FYN in excitatory neuron populations (Fig. 5C), which are the dominant sender and receiver cells for this LR interaction (Fig. 5B). Particularly, Excit_L5/6 neurons most specifically expressed the ligand EFNA5 (Specificity Measure, SPM = 0.6847) and its intracellular partner FYN (SPM = 0.5345), whereas Excit_L6 neurons most specifically expressed the receptor EPHA5 (SPM = 0.6508, Fig S42B). Furthermore, Excit_L5 and L6 neurons showed the highest co-expression of FYN and EFNA5, co-expressing in 87.25% of this population (Fig. 5D).
Since EFNA5-EPHA5 is a contact-dependent interaction (34), we used the Visium data to spatially map sites of likely EFNA5 and EPHA5 crosstalk. Across data-driven SpDs, the highest proportion of spots co-expressing EFNA5 and EPHA5 localized to Sp9D7~L6 (median (interquartile range) = 0.0196 (0.0137), p=4.0e-09, Fig. 5E–F) compared to other Sp9Ds (Sp9D1 = 0 (0), Sp9D2 = 0 (0), Sp9D3 = 0.0052 (0.0078), Sp9D4 = 0.014 (0.0142), Sp9D5 = 0.0091 (0.0102), Sp9D6 = 0 (0.0016), Sp9D8 = 0.0077 (0.0123), Sp9D8 = 0.0016 (0.0104)). Consistent with snRNA-seq specificity analysis (Fig S42B–C), spots co-expressing EFNA5 and EPHA5 showed a higher predicted proportion of Excit_L5/6 neurons and Excit_L6 neurons compared to spots lacking co-expression (Fig. 5H, Fig S43). Although Excit_L5 is the dominant cell2location predicted cell type among all spots (Fig S41, Fig S43B), it is even higher among these co-expression spots (median predicted proportion = 0.211) than among other spots (Fig S43A–B). Spatial network analyses further supported that co-localization of EFNA5 and EPHA5 occurs frequently in spots containing Excit_L6 neurons - with strongest co-localization relationships between Excit_L6/Excit_L5 neurons, Excit_L6/Excit_L4 neurons and Excit_L6/oligodendrocytes (Fig. 5I, Fig S42D). Spatial mapping of EFNA5 and FYN interactions also showed significant co-expression of these genes in Sp9D7~L6 (p=0.0046, Fig S42F–G) with frequent co-localization between Excit_L5/L6 and Excit_4 neurons (Fig S42H–I). In summary, we demonstrate the utility of this integrated single cell and spatial transcriptomic data for identifying and mapping disease-associated interactions in spatially localized cell types across the human DLPFC.
Spatial registration of cell populations across neuropsychiatric disorders
To leverage the large amount of snRNA-seq data collected across the PsychENCODE consortium (PEC) (35, 36), we spatially registered eight DLPFC snRNA-seq datasets generated in the context of several brain disorders (Autism Spectrum Disorder [ASD], SCZ, bipolar disorder, and Williams Syndrome) to both the histological layers and unsupervised SpDs annotated in Visium data (Fig. 6A, Fig S44). Across the consortium, in neurotypical controls, we found that excitatory neuron subtypes with a laminar annotation spatially register to the relevant histological layers and converge on the same unsupervised SpDs. As expected, most inhibitory populations registered to multiple histological layers and unsupervised SpDs, with the exception of Pvalb and VLMC subtypes, which mapped to Sp9D8~L4 and Sp9D1~L1, respectively. Finally, glial populations also showed expected spatial registration with astrocytes strongly mapping to L1-associated SpDs, oligodendrocytes and OPCs strongly mapping to the WM, and endothelial, pericyte (PC), and smooth muscle cells (SMC) mapping to the newly characterized vascular domain Sp9D1.
Figure 6. Spatial enrichment of cell types and genes associated with neurodevelopmental and neuropsychiatric disorders.

(A) Dot plot summarizing spatial registration results for eight PsychENCODE (PEC) snRNA-seq datasets from human DLPFC. snRNA-seq data was uniformly processed through the same pipeline and annotated with common nomenclature based on work from Allen Brain Institute (35, 69). Registration was performed for control donors only (see Fig S44 for full dataset) across manually annotated histological layers from (12) as well as unsupervised BayesSpace clusters at k=9 and k=16 (Sp9Ds and Sp16Ds, respectively). Each dot shows the histological layer(s) or SpD(s) where a dataset’s cell type was annotated during spatial registration. Solid dots show good confidence in the spatial annotation, translucent dots show poor confidence in the annotation. IT, intratelencephalon-projecting; ET, extratelencephalon-projecting; CT, corticothalamic-projecting; NP, near-projecting; VLMC, vascular lepotomeningeal cell; OPC, oligodendrocyte precursor cell; PC, pericyte; SMC, smooth muscle cell. (B) Spatial registration of cell type populations from control samples from (20) against unsupervised BayesSpace clusters at k=9 (Sp9Ds). Higher confidence annotations (cor > 0.25, merge ratio = 0.1, Supplemental Methods: Spatial registration of PsychENCODE and other external snRNA-seq datasets) are marked with an “X”. (C) Enrichment analysis using Fisher’s exact test for Sp9D- enriched statistics versus differentially expressed genes (DEGs, FDR < 0.05) in Autism spectrum disorder (ASD) for each cell type population. The values are the odds ratios (ORs) for the enrichment in significant (FDR < 0.001) blocks of the heatmap, and the color scale indicates −log10(p-value) for the enrichment test. The top bar plot shows the number of DEGs for each cell type. (D) Enrichment analysis using Fisher’s exact test for Sp9D- enriched statistics versus differentially expressed genes (DEGs, FDR < 0.05) in Post Traumatic Stress Disorder (PTSD) and/or Major Depressive Disorder (MDD) in bulk RNA-seq of DLPFC and medial prefrontal cortex (mPFC) (23). Top bar plot shows the number of DEGs for each DE test. Left bar plot shows the number of significantly enriched genes for each Sp9D in both enrichment analyses.
Next, we spatially registered snRNA-seq data from 30,147 nuclei derived from the human prefrontal cortex (Brodmann areas unspecified) from control donors in a study of ASD (Fig. 6B) (20). As expected, glial populations and laminar excitatory cell types mapped to the relevant SpDs (L2/3 neurons mapped to Sp9D3~L2 and Sp9D5~L3). We were also able to provide laminar assignments to some cell populations, such as mapping NRGN neuronal subtypes to Sp9D5~L3 and Sp9D8~L4. For each cell type, we next used clinical gene set enrichment analyses to assess the SpD enrichment of cell type-specific DEGs between individuals with ASD compared to neurotypical controls (Fig. 6C). Across many cell types, we observed multiple Sp9Ds enriched for ASD DEGs. Sp9D3~L2 showed significant enrichment of genes differentially expressed in L2/3 nuclei between individuals with ASD and neurotypical controls (p=2.60e-11), highlighting that these L2/3 DEGs are core Sp9D3~L2 marker genes. We also identified spatial enrichments for DEGs expressed in inhibitory neuron and neurogranin populations, including Sp9D4~L5 for SV2C inhibitory neurons and Sp9D7~L6 for Neu_NRGN_I neurons. Finally, to demonstrate how this large-scale dataset can be used to provide spatial information about genes associated with neuropsychiatric disease, we performed gene set enrichment analysis of bulk RNA-seq DEGs identified in a companion PEC study of PTSD and MDD (PEC study 6) (23). For both DLPFC and ventral medial prefrontal cortex (mPFC), we demonstrated that vasculature domain Sp9D1~L1 and L1-associated domain Sp9D2~L1 are enriched in DEGs associated with both PTSD and MDD. This is consistent with previous studies implicating neuroimmune signaling in PTSD (37), and current PEC single cell analyses that implicate glial and vascular- cells in both MDD and PTSD (PEC study 6) (23). Together, these spatial registration and clinical gene set enrichment analyses add anatomical context to cell type identities and provide valuable biological insights into molecular changes associated with brain disorders, including ASD, MDD, and PTSD.
Discussion
Here we generated a large-scale, transcriptome-wide, data-driven molecular map across the anterior-posterior axis of adult human DLPFC from ten neurotypical control donors. This highly integrated single cell and spatial gene expression reference dataset enabled identification of unsupervised SpDs, which were characterized in terms of both cellular composition and domain-enriched genes, at different resolutions across the DLPFC. We provide a landmark molecular neuroanatomical atlas that complements our understanding of classic cortical cytoarchitecture through identification and characterization of discrete molecularly-defined layers and sublayers. In particular, we annotated a vasculature-rich meninges layer and several molecularly distinct subdomains in histological L1, 4, 5 and 6. An advantage of Visium over snRNA-seq approaches is the ability to capture transcripts in the cell cytoplasm and neuropil, which we speculate may influence identification of higher resolution spatial domains, particularly for demarcating laminar transitions and at the gray/white matter junction (38–40).
In this study, we provide a roadmap for the implementation and biological validation of unsupervised spatial clustering approaches in human brain tissue. Whereas manual annotation of spatial domains is feasible for a limited number of samples in brain regions where neuroanatomical boundaries are well-characterized (12, 22), the application of data-driven clustering methods is critical for future studies that aim to analyze spatial gene expression changes across diagnostic cohorts groups to identify changes in spatially-resolved cell types. Furthermore, unsupervised approaches will be essential for spatial profiling in brain regions that lack clear molecular or histological boundaries since they allow for identification of unknown or unexpected SpDs as well as SpDs that may be technically difficult to manually annotate (for example the meninges). Although here we evaluated several of the spatial clustering algorithms, including SpaGCN and BayesSpace (13, 15), there are more tools coming online for both spatially variable gene detection, including nnSVG (41), and also spatial domain identification, including GraphST and PRECAST (16, 17). Large-scale, integrated datasets, including the present study, continue to offer developers of computational tools opportunities to develop methods scalable to atlas-level data, while also extracting meaningful biological information.
Visium offers transcriptome-wide information at spatial resolution; however, a limitation of the platform is that spots often contain multiple cells and cell types. We believe that this might be overcome using spot-level deconvolution tools, as discussed here. We rigorously benchmarked the utility of these tools against Visium-SPG data in the DLPFC, where we manually assigned spots to the histological layers while also immunolabeling 4 broad cell populations in the same tissue sections. From these data we predicted the proportion of cell types in each spot, allowing us to achieve cellular resolution for our spatio-molecular map. We note that although spatial registration correlation approaches can be informative for broadly mapping cell types to SpDs, spot deconvolution approaches provide higher resolution mapping across the cortical layers. This is especially evident for glial cell populations, which show the strongest spatial registration correlations to L1 and WM, but are indeed localized to all other SpDs when performing spot deconvolution. This is also true for endothelial cells, which are strongly associated with vasculature-enriched Sp9D1~L1, but are similarly distributed across the cortex when utilizing spot deconvolution approaches. As spot deconvolution tools continue to improve with the emergence of more large-scale single cell and spatial transcriptomic datasets, we expect that algorithms will be able to map finer resolution and rarer cell types.
While other imaging-based, spatially-resolved transcriptomics platforms, such as Xenium and MERFISH (42, 43), directly measure transcripts in individual cells, only a limited number of genes can currently be probed. In contrast, the discovery-based approach afforded by Visium, as well as its scalability to a large number of samples, allowed for robust identification of previously unrecognized spatial marker genes across many donors. These Visium-identified genes can be followed up at single cell resolution in smaller cohorts using probe-based approaches, such as MERFISH, which was recently applied to the middle and superior temporal gyri in the human brain (44). As spatial transcriptomics technologies continue to evolve, it will be important to functionally validate these spatial domains and consider the biological significance of these molecularly-defined compartments. Indeed, previous spatial transcriptomic studies in the mouse brain identified unique astrocyte layers that diverge from classic excitatory neuron layers (45), suggesting that the cortex likely has a more complex laminar architecture than previously appreciated. Future studies should evaluate the conservation of laminar spatial domains between rodent and human brain and explore their functional and clinical relevance.
In terms of clinical relevance, alterations in neural activity patterns within the DLPFC are noted in several neurodevelopmental and neuropsychiatric disorders (46–49), and it is hypothesized that changes in molecular signaling cascades may contribute to these alterations in activity. To gain insight into molecular dysfunction in DLPFC in the context of disease, we used our integrated molecular atlas to spatially map cell type-specific LR interactions that are associated with genetic risk for SCZ. For example, we highlight the interaction between EFNA5 and EPHA5 in excitatory L5/6 and L6 neuron subtypes in deep cortical layers, which is consistent with results from the most recently released SCZ genome-wide association study (GWAS) that identified enrichment of SCZ risk genes in glutamatergic neurons (50). Not only is EFNA5 the locus of a GWAS-identified common SCZ risk variant, but it is also differentially expressed between individuals with SCZ and neurotypical controls in specific cell types (21). Spatially mapping disease-relevant LR pairs, which are often highly specific and druggable targets, can provide valuable insights into pathophysiology and can help prioritize spatially restricted targets for therapeutic development. In combination with our interactive web resources, this highly integrated single cell and spatial transcriptomic data from neurotypical control DLPFC can be used to accelerate research across a variety of brain disorders by allowing researchers to search for relevant genes of interest, spatially register clinical gene sets, and explore disease-associated cell types for complementary assays, such as in vitro disease models.
Finally, spatial registration of eight DLPFC snRNA-seq datasets collected across the PsychENCODE consortium in the context of different neuropsychiatric disorders (35) revealed a convergence of excitatory, inhibitory, and non-neuronal cell types in relevant spatial domains. We observed increased confidence of inhibitory neuron mapping in our current expanded study compared to (12), likely due to the larger donor/sample number and data-driven clustering approach, which allowed for identification of finer resolution SpDs. Furthermore using ASD as an example (20), we demonstrated the utility of our data-driven molecular atlas for localizing cell-type specific DEGs to specific SpDs. For example, ASD DEGs in VIP inhibitory neurons were enriched in L3, L5, and L6-associated spatial domains, while those in SV2C inhibitory neurons were enriched only in the L5-associated domain. Together, this analysis provides anatomical context for cell type-specific gene expression changes and molecular mechanisms associated with neurodevelopmental disorders and psychiatric illness.
In summary, we provide a large-scale, highly integrated single cell and spatial transcriptomics resource for understanding the molecular neuroanatomy of the human DLPFC. We share web-based tools for the scientific community to interact with these datasets for further interrogation of molecular pathways associated with brain disorders.
Materials and methods summary
Detailed materials and methods can be found in the supplementary materials. Three tissue blocks from the anterior, middle, and posterior positions along the rostral-caudal axis of human DLPFC were microdissected from the post-mortem brains of 10 donors for a total of 30 tissue blocks. The 10x Genomics Visium Spatial Gene Expression protocol was performed on all blocks (n = 30). The 10x Genomics Visium SPG and 3′ Single Cell Gene Expression protocols were performed on a subset of blocks (n = 4 and 19, respectively). Visium and Visium-SPG data was processed with 10x Genomics SpaceRanger (51). Dimensionality reduction was performed by using scater (52), and batch correction was performed with Harmony (53). Unsupervised clustering was performed on the Visium data with BayesSpace (13) defining data-driven SpDs. An optimal number of SpDs was determined with fasthplus (54). DEGs between SpDs were found with ANOVA, enrichment, and pairwise models with tools from spatialLIBD (55), powered by limma (56). snRNA-seq data were processed with 10x Genomics CellRanger (57). Reduced dimensions were calculated with GLM-PCA (58), and batch correction was performed with Harmony (53). Graph-based and hierarchical clustering described in (59) identified cell type populations, which were annotated by using established marker genes (59, 60). Spatial registration was applied to snRNA-seq and SpDs with tools from spatialLIBD (55). Images from Visium and Visium-SPG were processed with VistoSeg (61). Nuclei were segmented with Cellpose (62). A decision-tree cell type classifier was built with scikit-learn (63) on the basis of a training set classified by an expert. Cell segmentation and classification from Visium-SPG was used to benchmark spot deconvolution algorithms Tangram, Cell2Location, and SPOTlight (14, 30, 31). Spot deconvolution on the 30 Visium samples was performed with both Tangram and Cell2Location. CCC was performed with LIANA (18). The SCZ risk gene list was obtained from OpenTargets (32). Spatial registration was performed on the uniformly processed PsychENCODE snRNA-seq datasets (35). Enrichment statistics were calculated with spatialLIBD for the DEGs in an ASD snRNA-seq dataset (20) and bulk RNAseq PTSD and MDD dataset (23).
Supplementary Material
Acknowledgements
Data were generated as part of the PsychENCODE Consortium, supported by: U01DA048279, U01MH103339, U01MH103340, U01MH103346, U01MH103365, U01MH103392, U01MH116438, U01MH116441, U01MH116442, U01MH116488, U01MH116489, U01MH116492, U01MH122590, U01MH122591, U01MH122592, U01MH122849, U01MH122678, U01MH122681, U01MH116487, U01MH122509, R01MH094714, R01MH105472, R01MH105898, R01MH109677, R01MH109715, R01MH110905, R01MH110920, R01MH110921, R01MH110926, R01MH110927, R01MH110928, R01MH111721, R01MH117291, R01MH117292, R01MH117293, R21MH102791, R21MH103877, R21MH105853, R21MH105881, R21MH109956, R56MH114899, R56MH114901, R56MH114911, R01MH125516, R01MH126459, R01MH129301, R01MH126393, R01MH121521, R01MH116529, R01MH129817, R01MH117406, and P50MH106934 awarded to: Alexej Abyzov, Nadav Ahituv, Schahram Akbarian, Kristen Brennand, Andrew Chess, Gregory Cooper, Gregory Crawford, Stella Dracheva, Peggy Farnham, Michael Gandal, Mark Gerstein, Daniel Geschwind, Fernando Goes, Joachim F. Hallmayer, Vahram Haroutunian, Thomas M. Hyde, Andrew Jaffe, Peng Jin, Manolis Kellis, Joel Kleinman, James A. Knowles, Arnold Kriegstein, Chunyu Liu, Christopher E. Mason, Keri Martinowich, Eran Mukamel, Richard Myers, Charles Nemeroff, Mette Peters, Dalila Pinto, Katherine Pollard, Kerry Ressler, Panos Roussos, Stephan Sanders, Nenad Sestan, Pamela Sklar, Michael P. Snyder, Matthew State, Jason Stein, Patrick Sullivan, Alexander E. Urban, Flora Vaccarino, Stephen Warren, Daniel Weinberger, Sherman Weissman, Zhiping Weng, Kevin White, A. Jeremy Willsey, Hyejung Won, and Peter Zandi. Additional data were provided to the PsychENCODE Consortium, supported by 2015 and 2018 NARSAD Young Investigator grants from Brain & Behavior Research Foundation awarded to: Nikolaos Daskalakis.
We would like to thank Linda Orzolek and the Johns Hopkins Single Cell and Transcriptomics Core facility for executing all sequencing. We also acknowledge Elizabeth Engle and the Johns Hopkins Tumor Microenvironment Lab core facility for assistance with the Vectra Polaris slide scanner. We would like to thank the Joint High Performance Computing Exchange (JHPCE) for providing computing resources for these analyses. While an Investigator at LIBD, Andrew E. Jaffe helped secure funding for this work, and contributed to the conceptualization of the spatial registration framework. We thank William S. Ulrich for his assistance in deploying the Samui interactive websites. We thank R. A. Miller for assistance with uploading data to the NIMH Data Archive. We thank Daniel R. Weinberger and members of the PsychENCODE consortium for feedback on the manuscript. We thank Amy Deep-Soboslay and her diagnostic team for curation of brain samples. We thank the neuropathology team, especially James Tooke, for assistance with tissue dissection. We thank the physicians and staff at the brain donation sites and the generosity of donor families for supporting our research efforts. Finally, we thank the families of Connie and Stephen Lieber and Milton and Tamar Maltz for their generous support of this work. Schematic illustrations were generated using Biorender. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institute on Aging, the National Institutes of Health, the Department of Health and Human Services, or the United States government. A preprint of this work is available at https://doi.org/10.1101/2023.02.15.528722. For the purpose of open access, the authors have applied a CC BY public copyright license to all Author Accepted Manuscripts arising from this submission.
Funding
Funding for these studies was provided by the Lieber Institute for Brain Development (LIBD) and National Institute of Mental Health (NIH) grants U01MH122849 and R01MH123183. AB was supported by NIH National Institute of General Medical Sciences (NIGMS) grant R35GM139580. MGP and MR were funded by Aligning Science Across Parkinson’s [Grant numbers: ASAP-000478 and ASAP-000509] through the Michael J. Fox Foundation for Parkinson’s Research (MJFF).
Footnotes
Competing Interests
AB was a consultant for Third Rock Ventures and a shareholder in Alphabet, Inc. Joel E. Kleinman is a consultant on a Data Monitoring Committee for an antipsychotic drug trial for Merck & Co., Inc.
Data and materials availability
The source data described in this manuscript are available from the National Institute of Mental Health (NIMH) Data Archive (https://nda.nih.gov/) (64). The source data are also publicly available from the Globus endpoint ‘jhpce#spatialDLPFC’ and ‘jhpce#DLPFC_snRNAseq’ that are also listed at http://research.libd.org/globus. The raw data provided through Globus include all the FASTQ files and raw image files. Processed data are publicly available from the Bioconductor package spatialLIBD version 1.11.7 (55) through the fetch_data() function. All source code is publicly available through GitHub and permanently archived through Zenodo at https://github.com/LieberInstitute/spatialDLPFC (65) and https://github.com/LieberInstitute/DLPFC_snRNAseq (66).
We provide several interactive web applications to explore this highly integrated DLPFC dataset as listed at http://research.libd.org/spatialDLPFC/#interactive-websites. First, we developed two spatialLIBD apps (55) that allow users to analyze Visium gene expression, cell segmentation, spot deconvolution, and clinical gene set enrichment results at k=9 and k=16. We added a third spatialLIBD app for the position differential expression results and a fourth for the Visium-SPG data. Second, both our Visium H&E (n=30 tissue sections) and Visium-SPG (n=4 tissue sections) data is available through a newly created performant web-based interactive visualization tool, called Samui Browser (67), which allows rapid loading, visualization, and custom annotation of high resolution images and corresponding gene expression. Finally, snRNA-seq data at both fine and laminar resolution as well as pseudo-bulked Visium data (k=9, 16, and 28) are available through iSEE apps (68) that allows users to visualize expression levels for genes of interest through violin and heatmap plots.
References
- 1.Madissoon E, Oliver AJ, Kleshchevnikov V, Wilbrey-Clark A, Polanski K, Richoz N, Ribeiro Orsi A, Mamanova L, Bolt L, Elmentaite R, Pett JP, Huang N, Xu C, He P, Dabrowska M, Pritchard S, Tuck L, Prigmore E, Perera S, Knights A, Meyer KB, A spatially resolved atlas of the human lung characterizes a gland-associated immune niche. Nat. Genet 55, 66–77 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ortiz C, Navarro JF, Jurek A, Märtin A, Lundeberg J, Meletis K, Molecular atlas of the adult mouse brain. Sci. Adv 6, eabb3446 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asp M, Giacomello S, Larsson L, Wu C, Fürth D, Qian X, Wärdell E, Custodio J, Reimegård J, Salmén F, Österholm C, Ståhl PL, Sundström E, Åkesson E, Bergmann O, Bienko M, Månsson-Broberg A, Nilsson M, Sylvén C, Lundeberg J, A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell. 179, 1647–1660.e19 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Hildebrandt F, Andersson A, Saarenpää S, Larsson L, Van Hul N, Kanatani S, Masek J, Ellis E, Barragan A, Mollbrink A, Andersson ER, Lundeberg J, Ankarklev J, Spatial Transcriptomics to define transcriptional patterns of zonation and structural components in the mouse liver. Nat. Commun 12, 7046 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Bella DJ, Habibi E, Stickels RR, Scalia G, Brown J, Yadollahpour P, Yang SM, Abbate C, Biancalani T, Macosko EZ, Chen F, Regev A, Arlotta P, Molecular logic of cellular diversification in the mouse cerebral cortex. Nature. 595, 554–559 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Volk DW, Lewis DA, Prefrontal cortical circuits in schizophrenia. Curr. Top. Behav. Neurosci 4, 485–508 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Weinberger DR, Schizophrenia, the prefrontal cortex, and a mechanism of genetic susceptibility. Eur. Psychiatry 17 Suppl 4, 355s–362s (2002). [DOI] [PubMed] [Google Scholar]
- 8.Koenigs M, Grafman J, The functional neuroanatomy of depression: distinct roles for ventromedial and dorsolateral prefrontal cortex. Behav. Brain Res 201, 239–243 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gamo NJ, Arnsten AFT, Molecular modulation of prefrontal cortex: rational development of treatments for psychiatric disorders. Behav. Neurosci 125, 282–296 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cerullo MA, Adler CM, Delbello MP, Strakowski SM, The functional neuroanatomy of bipolar disorder. Int. Rev. Psychiatry 21, 314–322 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Willsey HR, Willsey AJ, Wang B, State MW, Genomics, convergent neuroscience and progress in understanding autism spectrum disorder. Nat. Rev. Neurosci 23, 323–341 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maynard KR, Collado-Torres L, Weber LM, Uytingco C, Barry BK, Williams SR, Catallini JL, Tran MN, Besich Z, Tippani M, Chew J, Yin Y, Kleinman JE, Hyde TM, Rao N, Hicks SC, Martinowich K, Jaffe AE, Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci 24, 425–436 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao E, Stone MR, Ren X, Guenthoer J, Smythe KS, Pulliam T, Williams SR, Uytingco CR, Taylor SEB, Nghiem P, Bielas JH, Gottardo R, Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol 39, 1375–1384 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biancalani T, Scalia G, Buffoni L, Avasthi R, Lu Z, Sanger A, Tokcan N, Vanderburg CR, Segerstolpe Å, Zhang M, Avraham-Davidi I, Vickovic S, Nitzan M, Ma S, Subramanian A, Lipinski M, Buenrostro J, Brown NB, Fanelli D, Zhuang X, Regev A, Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 18, 1352–1362 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu J, Li X, Coleman K, Schroeder A, Ma N, Irwin DJ, Lee EB, Shinohara RT, Li M, SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 18, 1342–1351 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Liu W, Liao X, Luo Z, Yang Y, Lau MC, Jiao Y, Shi X, Zhai W, Ji H, Yeong J, Liu J, Probabilistic embedding, clustering, and alignment for integrating spatial transcriptomics data with PRECAST. Nat. Commun 14, 296 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long Y, Ang KS, Li M, Chong KLK, Sethi R, Zhong C, Xu H, Ong Z, Sachaphibulkij K, Chen A, Zeng L, Fu H, Wu M, Lim LHK, Liu L, Chen J, Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with GraphST. Nat. Commun 14, 1155 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dimitrov D, Türei D, Garrido-Rodriguez M, Burmedi PL, Nagai JS, Boys C, Ramirez Flores RO, Kim H, Szalai B, Costa IG, Valdeolivas A, Dugourd A, Saez-Rodriguez J, Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat. Commun 13, 3224 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagy C, Maitra M, Tanti A, Suderman M, Théroux J-F, Davoli MA, Perlman K, Yerko V, Wang YC, Tripathy SJ, Pavlidis P, Mechawar N, Ragoussis J, Turecki G, Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat. Neurosci 23, 771–781 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, Bhaduri A, Goyal N, Rowitch DH, Kriegstein AR, Single-cell genomics identifies cell type-specific molecular changes in autism. Science. 364, 685–689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruzicka WB, Mohammadi S, Fullard JF, Davila-Velderrain J, Subburaju S, Tso DR, Hourihan M, Jiang S, Lee H-C, Bendl J, PsychENCODE Consortium, Voloudakis G, Haroutunian V, Hoffman GE, Roussos P, Kellis M, Single-cell multi-cohort dissection of the schizophrenia transcriptome. medRxiv (2022), doi: 10.1101/2022.08.31.22279406. [DOI] [PubMed] [Google Scholar]
- 22.Batiuk MY, Tyler T, Dragicevic K, Mei S, Rydbirk R, Petukhov V, Deviatiiarov R, Sedmak D, Frank E, Feher V, Habek N, Hu Q, Igolkina A, Roszik L, Pfisterer U, Garcia-Gonzalez D, Petanjek Z, Adorjan I, Kharchenko PV, Khodosevich K, Upper cortical layer-driven network impairment in schizophrenia. Sci. Adv 8, eabn8367 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daskalakis NP, Iatrou A, Chatzinakos C, Jajoo A, Snijders C, DiPietro CP, Lugenbühl JF, Soliva-Estruch M, Wylie D, Arasappan D, Bharadwaj RA, Chen C-Y, Collado-Torres L, Wuchty S, Dammer EB, Deep-Soboslay A, Duong DM, Eagles NJ, French K, Huuki-Myers LA, Ressler KJ, Systems Biology Dissection of PTSD and MDD across brain regions, cell-types and blood. Submitted to Science (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.10x Genomics, Spatial Gene Expression, (available at https://www.10xgenomics.com/products/spatial-gene-expression).
- 25.Csardi G, Nepusz T, The igraph software package for complex network research. InterJournal. Complex Systems, 1695 (2006). [Google Scholar]
- 26.Lun ATL, McCarthy DJ, Marioni JC, A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. [version 2; peer review: 3 approved, 2 approved with reservations]. F1000Res. 5, 2122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, Yao Z, Eggermont J, Höllt T, Levi BP, Shehata SI, Aevermann B, Beller A, Bertagnolli D, Brouner K, Casper T, Lein ES, Conserved cell types with divergent features in human versus mouse cortex. Nature. 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, Satija R, Integrated analysis of multimodal single-cell data. Cell. 184, 3573–3587. (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bakken TE, Jorstad NL, Hu Q, Lake BB, Tian W, Kalmbach BE, Crow M, Hodge RD, Krienen FM, Sorensen SA, Eggermont J, Yao Z, Aevermann BD, Aldridge AI, Bartlett A, Bertagnolli D, Casper T, Castanon RG, Crichton K, Daigle TL, Lein ES, Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature. 598, 111–119 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elosua-Bayes M, Nieto P, Mereu E, Gut I, Heyn H, SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 49, e50 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleshchevnikov V, Shmatko A, Dann E, Aivazidis A, King HW, Li T, Elmentaite R, Lomakin A, Kedlian V, Gayoso A, Jain MS, Park JS, Ramona L, Tuck E, Arutyunyan A, Vento-Tormo R, Gerstung M, James L, Stegle O, Bayraktar OA, Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol 40, 661–671 (2022). [DOI] [PubMed] [Google Scholar]
- 32.Ochoa D, Hercules A, Carmona M, Suveges D, Baker J, Malangone C, Lopez I, Miranda A, Cruz-Castillo C, Fumis L, Bernal-Llinares M, Tsukanov K, Cornu H, Tsirigos K, Razuvayevskaya O, Buniello A, Schwartzentruber J, Karim M, Ariano B, Martinez Osorio RE, McDonagh EM, The next-generation Open Targets Platform: reimagined, redesigned, rebuilt. Nucleic Acids Res. 51, D1353–D1359 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Türei D, Korcsmáros T, Saez-Rodriguez J, OmniPath: guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 13, 966–967 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Kania A, Klein R, Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol 17, 240–256 (2016). [DOI] [PubMed] [Google Scholar]
- 35.P. C. (PEC), PyschENCODE Consortium (PEC) Capstone II Cross-study Harmonized Data. Synapse (2023), doi: 10.7303/syn51111084.1. [DOI] [Google Scholar]
- 36.PsychENCODE Consortium, Clark D, Dursun C, Emani P, Gupta C, Jensen M, Lee CY, Liu J, Lou S, Meng R, Warrell J, Xu S, Chen Y, Chu Z, Galeev T, Hwang A, Ni P, Zhou X, Bakken T, Bicks L, Gerstein M, Single-cell genomics & regulatory networks for 388 human brains. Submitted to Science (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaffe AE, Tao R, Page SC, Maynard KR, Pattie EA, Nguyen CV, Deep-Soboslay A, Bharadwaj R, Young KA, Friedman MJ, Williamson DE, Traumatic Stress Brain Research Group, Shin JH, Hyde TM, Martinowich K, Kleinman JE, Decoding Shared Versus Divergent Transcriptomic Signatures Across Cortico-Amygdala Circuitry in PTSD and Depressive Disorders. Am. J. Psychiatry 179, 673–686 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fertuzinhos S, Li M, Kawasawa YI, Ivic V, Franjic D, Singh D, Crair M, Sestan N, Laminar and temporal expression dynamics of coding and noncoding RNAs in the mouse neocortex. Cell Rep. 6, 938–950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paul A, Crow M, Raudales R, He M, Gillis J, Huang ZJ, Transcriptional architecture of synaptic communication delineates GABAergic neuron identity. Cell. 171, 522–539.e20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perez JD, Dieck ST, Alvarez-Castelao B, Tushev G, Chan IC, Schuman EM, Subcellular sequencing of single neurons reveals the dendritic transcriptome of GABAergic interneurons. eLife. 10 (2021), doi: 10.7554/eLife.63092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weber LM, Saha A, Datta A, Hansen KD, Hicks SC, nnSVG for the scalable identification of spatially variable genes using nearest-neighbor Gaussian processes. Nat. Commun 14, 4059 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janesick A, Shelansky R, Gottscho A, Wagner F, Rouault M, Beliakoff G, Faria de Oliveira M, Kohlway A, Abousoud J, Morrison C, Drennon TY, Mohabbat S, Williams S, 10x Development Teams, Taylor S, High resolution mapping of the breast cancer tumor microenvironment using integrated single cell, spatial and in situ analysis of FFPE tissue. BioRxiv (2022), doi: 10.1101/2022.10.06.510405. [DOI] [Google Scholar]
- 43.Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X, Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fang R, Xia C, Close JL, Zhang M, He J, Huang Z, Halpern AR, Long B, Miller JA, Lein ES, Zhuang X, Conservation and divergence of cortical cell organization in human and mouse revealed by MERFISH. Science. 377, 56–62 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bayraktar OA, Bartels T, Holmqvist S, Kleshchevnikov V, Martirosyan A, Polioudakis D, Ben Haim L, Young AMH, Batiuk MY, Prakash K, Brown A, Roberts K, Paredes MF, Kawaguchi R, Stockley JH, Sabeur K, Chang SM, Huang E, Hutchinson P, Ullian EM, Rowitch DH, Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat. Neurosci 23, 500–509 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collado-Torres L, Burke EE, Peterson A, Shin J, Straub RE, Rajpurohit A, Semick SA, Ulrich WS, BrainSeq Consortium, Price AJ, Valencia C, Tao R, Deep-Soboslay A, Hyde TM, Kleinman JE, Weinberger DR, Jaffe AE, Regional Heterogeneity in Gene Expression, Regulation, and Coherence in the Frontal Cortex and Hippocampus across Development and Schizophrenia. Neuron. 103, 203–216.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dienel SJ, Schoonover KE, Lewis DA, Cognitive dysfunction and prefrontal cortical circuit alterations in schizophrenia: developmental trajectories. Biol. Psychiatry 92, 450–459 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smucny J, Dienel SJ, Lewis DA, Carter CS, Mechanisms underlying dorsolateral prefrontal cortex contributions to cognitive dysfunction in schizophrenia. Neuropsychopharmacology. 47, 292–308 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller JA, Scult MA, Conley ED, Chen Q, Weinberger DR, Hariri AR, Effects of schizophrenia polygenic risk scores on brain activity and performance during working memory subprocesses in healthy young adults. Schizophr. Bull 44, 844–853 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trubetskoy V, Pardiñas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, Bryois J, Chen C-Y, Dennison CA, Hall LS, Lam M, Watanabe K, Frei O, Ge T, Harwood JC, Koopmans F, Magnusson S, Richards AL, Sidorenko J, Wu Y, et al. , Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. 604, 502–508 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.10x Genomics, spaceranger count, 10x Genomics, (available at https://support.10xgenomics.com/spatial-gene-expression/software/pipelines/latest/using/count).
- 52.McCarthy DJ, Campbell KR, Lun ATL, Wills QF, Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 33, 1179–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P-R, Raychaudhuri S, Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dyjack N, Baker DN, Braverman V, Langmead B, Hicks SC, A scalable and unbiased discordance metric with H. Biostatistics. 25, 188–202 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pardo B, Spangler A, Weber LM, Page SC, Hicks SC, Jaffe AE, Martinowich K, Maynard KR, Collado-Torres L, spatialLIBD: an R/Bioconductor package to visualize spatially-resolved transcriptomics data. BMC Genomics. 23, 434 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.10x Genomics, cellranger count, 10x Genomics, (available at https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/count).
- 58.Townes FW, Hicks SC, Aryee MJ, Irizarry RA, Feature selection and dimension reduction for single-cell RNA-Seq based on a multinomial model. Genome Biol. 20, 295 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tran MN, Maynard KR, Spangler A, Huuki LA, Montgomery KD, Sadashivaiah V, Tippani M, Barry BK, Hancock DB, Hicks SC, Kleinman JE, Hyde TM, Collado-Torres L, Jaffe AE, Martinowich K, Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain. Neuron. 109, 3088–3103.e5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai L-H, Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 570, 332–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tippani M, Divecha HR, Catallini JL, Kwon SH, Weber LM, Spangler A, Jaffe AE, Hyde TM, Kleinman JE, Hicks SC, Martinowich K, Collado-Torres L, Page SC, Maynard KR, VistoSeg : processing utilities for high resolution images for spatially resolved transcriptomics data. Biol. Imaging, 1–21 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stringer C, Wang T, Michaelos M, Pachitariu M, Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106 (2021). [DOI] [PubMed] [Google Scholar]
- 63.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E, Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research. 12, 2825–2830 (2011). [Google Scholar]
- 64.Martinowich K, Maynard K, Collado-Torres L, Synapse DataSet Only T: Zintel, Integrated single cell and unsupervised spatial transcriptomic analysis defines molecular anatomy of the human dorsolateral prefrontal cortex. Synapse (2023), doi: 10.7303/syn51032055.1. [DOI] [Google Scholar]
- 65.Nick-Eagles, Spangler A, Collado-Torres L, Huuki-Myers L, Tippani M, Divecha H, Seyedian A, Guo B, Kmaynard12, LieberInstitute/spatialDLPFC: v0_prerelease. Zenodo (2023), doi: 10.5281/zenodo.7616121. [DOI] [Google Scholar]
- 66.Huuki-Myers L, Collado-Torres L, Guo B, Nick-Eagles MN Tran, LieberInstitute/DLPFC_snRNAseq: v0_prerelease. Zenodo (2023), doi: 10.5281/zenodo.7616050. [DOI] [Google Scholar]
- 67.Sriworarat C, Nguyen A, Eagles NJ, Collado-Torres L, Martinowich K, Maynard KR, Hicks SC, Performant web-based interactive visualization tool for spatially-resolved transcriptomics experiments. Biol. Imaging, 1–10 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rue-Albrecht K, Marini F, Soneson C, Lun ATL, iSEE: Interactive SummarizedExperiment Explorer. [version 1; peer review: 3 approved]. F1000Res. 7, 741 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ma S, Skarica M, Li Q, Xu C, Risgaard RD, Tebbenkamp ATN, Mato-Blanco X, Kovner R, Krsnik Ž, de Martin X, Luria V, Martí-Pérez X, Liang D, Karger A, Schmidt DK, Gomez-Sanchez Z, Qi C, Gobeske KT, Pochareddy S, Debnath A, Sestan N, Molecular and cellular evolution of the primate dorsolateral prefrontal cortex. Science. 377, eabo7257 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lipska BK, Deep-Soboslay A, Weickert CS, Hyde TM, Martin CE, Herman MM, Kleinman JE, Critical factors in gene expression in postmortem human brain: Focus on studies in schizophrenia. Biol. Psychiatry 60, 650–658 (2006). [DOI] [PubMed] [Google Scholar]
- 71.Maynard KR, Tippani M, Takahashi Y, Phan BN, Hyde TM, Jaffe AE, Martinowich K, dotdotdot: an automated approach to quantify multiplex single molecule fluorescent in situ hybridization (smFISH) images in complex tissues. Nucleic Acids Res. 48, e66 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Righelli D, Weber LM, Crowell HL, Pardo B, Collado-Torres L, Ghazanfar S, Lun ATL, Hicks SC, Risso D, SpatialExperiment: infrastructure for spatially-resolved transcriptomics data in R using Bioconductor. Bioinformatics. 38, 3128–3131 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pachitariu M, Stringer C, Cellpose 2.0: how to train your own model. Nat. Methods 19, 1634–1641 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van der Walt S, Schönberger JL, Nunez-Iglesias J, Boulogne F, Warner JD, Yager N, Gouillart E, Yu T, scikit-image contributors, scikit-image: image processing in Python. PeerJ. 2, e453 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hafemeister C, Satija R, Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Choudhary S, Satija R, Comparison and evaluation of statistical error models for scRNA-seq. Genome Biol. 23, 27 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Robinson MD, Oshlack A, A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lun ATL, Riesenfeld S, Andrews T, Dao TP, Gomes T, participants in the 1st Human Cell Atlas Jamboree, J. C. Marioni, EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 20, 63 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Griffiths JA, Richard AC, Bach K, Lun ATL, Marioni JC, Detection and removal of barcode swapping in single-cell RNA-seq data. Nat. Commun 9, 2667 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Germain P-L, Lun A, Macnair W, Robinson MD, Doublet identification in single-cell sequencing data using scDblFinder. F1000Res. 10, 979 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miller JA, Gouwens NW, Tasic B, Collman F, van Velthoven CT, Bakken TE, Hawrylycz MJ, Zeng H, Lein ES, Bernard A, Common cell type nomenclature for the mammalian brain. eLife. 9 (2020), doi: 10.7554/eLife.59928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yan L, Sun X, Benchmarking and integration of methods for deconvoluting spatial transcriptomic data. Bioinformatics. 39 (2023), doi: 10.1093/bioinformatics/btac805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li B, Zhang W, Guo C, Xu H, Li L, Fang M, Hu Y, Zhang X, Yao X, Tang M, Liu K, Zhao X, Lin J, Cheng L, Chen F, Xue T, Qu K, Benchmarking spatial and single-cell transcriptomics integration methods for transcript distribution prediction and cell type deconvolution. Nat. Methods 19, 662–670 (2022). [DOI] [PubMed] [Google Scholar]
- 85.Breiman L, Friedman JH, Olshen RA, Stone CJ, Classification and regression trees (Wadsworth & Brooks/Cole Advanced Books & Software, Monterey, CA, 1984), The Wadsworth statistics/probability series. [Google Scholar]
- 86.Gu Z, Eils R, Schlesner M, Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 32, 2847–2849 (2016). [DOI] [PubMed] [Google Scholar]
- 87.Gu Z, Gu L, Eils R, Schlesner M, Brors B, circlize Implements and enhances circular visualization in R. Bioinformatics. 30, 2811–2812 (2014). [DOI] [PubMed] [Google Scholar]
- 88.Guo B, Huuki-Myers LA, Grant-Peters M, Collado-Torres L, Hicks SC, escheR: unified multi-dimensional visualizations with Gestalt principles. Bioinformatics Advances. 3, vbad179 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source data described in this manuscript are available from the National Institute of Mental Health (NIMH) Data Archive (https://nda.nih.gov/) (64). The source data are also publicly available from the Globus endpoint ‘jhpce#spatialDLPFC’ and ‘jhpce#DLPFC_snRNAseq’ that are also listed at http://research.libd.org/globus. The raw data provided through Globus include all the FASTQ files and raw image files. Processed data are publicly available from the Bioconductor package spatialLIBD version 1.11.7 (55) through the fetch_data() function. All source code is publicly available through GitHub and permanently archived through Zenodo at https://github.com/LieberInstitute/spatialDLPFC (65) and https://github.com/LieberInstitute/DLPFC_snRNAseq (66).
We provide several interactive web applications to explore this highly integrated DLPFC dataset as listed at http://research.libd.org/spatialDLPFC/#interactive-websites. First, we developed two spatialLIBD apps (55) that allow users to analyze Visium gene expression, cell segmentation, spot deconvolution, and clinical gene set enrichment results at k=9 and k=16. We added a third spatialLIBD app for the position differential expression results and a fourth for the Visium-SPG data. Second, both our Visium H&E (n=30 tissue sections) and Visium-SPG (n=4 tissue sections) data is available through a newly created performant web-based interactive visualization tool, called Samui Browser (67), which allows rapid loading, visualization, and custom annotation of high resolution images and corresponding gene expression. Finally, snRNA-seq data at both fine and laminar resolution as well as pseudo-bulked Visium data (k=9, 16, and 28) are available through iSEE apps (68) that allows users to visualize expression levels for genes of interest through violin and heatmap plots.