

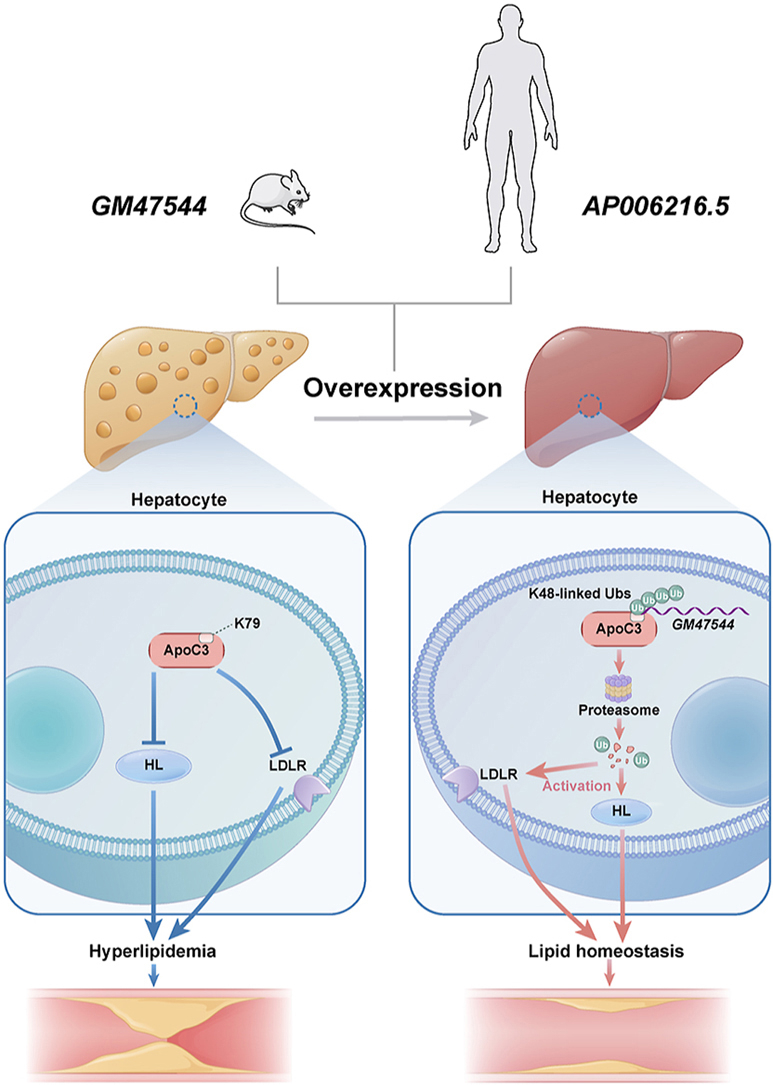
Abstract
Objective
Emerging evidence highlights the pivotal roles of long non-coding RNAs (lncRNAs) in lipid metabolism. Apoprotein C3 (ApoC3) is a well-established therapeutic target for hypertriglyceridemia and exhibits a strong association with cardiovascular disease. However, the exact mechanisms via which the lncRNAs control ApoC3 expression remain unclear.
Methods
We identified a novel long noncoding RNA (lncRNA), GM47544, within the ApoA1/C3/A4/A5 gene cluster. Subsequently, the effect of GM47544 on intracellular triglyceride metabolism was analyzed. The diet-induced mouse models of hyperlipidemia and atherosclerosis were established to explore the effect of GM47544 on dyslipidemia and plaque formation in vivo. The molecular mechanism was explored through RNA sequencing, immunoprecipitation, RNA pull-down assay, and RNA immunoprecipitation.
Results
GM47544 was overexpressed under high-fat stimulation. GM47544 effectively improved hepatic steatosis, reduced blood lipid levels, and alleviated atherosclerosis in vitro and in vivo. Mechanistically, GM47544 directly bound to ApoC3 and facilitated the ubiquitination at lysine 79 in ApoC3, thereby facilitating ApoC3 degradation via the ubiquitin-proteasome pathway. Moreover, we identified AP006216.5 as the human GM47544 transcript, which fulfills a comparable function in human hepatocytes.
Conclusions
The identification of GM47544 as a lncRNA modulator of ApoC3 reveals a novel mechanism of post-translational modification, with significant clinical implications for the treatment of hypertriglyceridemia and atherosclerosis.
Keywords: Atherosclerotic cardiovascular disease, Hypertriglyceridemia, Ubiquitination, APOC3, Long noncoding RNA, GM47544
Graphical abstract
Highlights
-
•
Liver-enriched lncRNA GM47544 plays a critical role in lowering triglyceride levels.
-
•
Overexpression of hepatic GM47544 protects against hyperlipidemia and atherosclerosis.
-
•
GM47544 facilitating ApoC3 degradation by ubiquitin-proteasome pathway.
-
•
GM47544 is highly homologous in human and mice.
1. Introduction
Hypertriglyceridemia (HTG) is frequently observed in clinical settings as the predominant type of dyslipidemia and is linked to a variety of serious disorders, including atherosclerotic cardiovascular disease (ASCVD), nonalcoholic fatty liver disease (NAFLD), acute pancreatitis, as well as lipotoxic cardiomyopathy [[1], [2], [3], [4]]. Dietary triglycerides (exogenous) are transported in the bloodstream as chylomicrons, whereas triglycerides produced by the liver (endogenous) circulate as particles with very low-density lipoprotein (VLDL) [5]. Lipoprotein lipase (LPL) activity serves as the primary mechanism for the hydrolysis of plasma triglycerides, facilitating the effective elimination of triglyceride-rich lipoproteins (TRLs) and remnants [6,7]. TRLs and remnants have been identified as significant contributors to the residual cardiovascular risk in patients receiving optimal LDL-lowering medication [8,9]. Despite the effectiveness of LDL-lowering drugs in decreasing the occurrence of cardiovascular disease, the burden of cardiovascular disease remains high owing to persistent dyslipidemia characterized by hypertriglyceridemia and low HDL levels [10,11].
Recently, APOC3 has become the most promising target for triglyceride-lowering drugs [12]. Significant advancements have been made using antibody, antisense, and RNAi methods [13,14]. Specifically, a monoclonal antibody was found to effectively eliminate lipoprotein-bound human APOC3 in mice expressing this protein, thereby enhancing the breakdown of triglyceride-rich lipoproteins in vivo [15]. AKCEA-APOCIII-LRx (olezarsen) is a liver-targeted antisense oligonucleotide conjugated with N-acetyl galactosamine, designed to specifically suppress the synthesis of APOC3 protein. It has been found to significantly reduce APOC3, triglycerides, and atherogenic lipoproteins in patients [16,17]. Furthermore, ISIS 304801, a novel antisense inhibitor targeting APOC3 synthesis, has demonstrated dose-dependent and sustained reductions of up to 40.0%∼79.6% in triglyceride levels and parallel reduction in plasma APOC3 levels [18]. Given that targeting hepatic APOC3 protein and mRNA has been proven efficacious and safe in decreasing triglyceride (TG) levels, APOC3 is an attractive target for modulating lipid homeostasis [19]. However, the post-translational modification (PTM) patterns of APOC3 protein remain poorly understood.
Long noncoding RNA (lncRNA) transcript length exceeds 200 nt, most of which have no protein-coding potential [20]. LncRNAs facilitate DNA-protein interactions, sequester microRNAs and act as decoys for proteins. This allows them to modify mRNA and proteins to control the processes of methylation, phosphorylation, and ubiquitination, which affect a protein's stability [[21], [22], [23]]. Many functional lncRNAs driven by genome-wide association study (GWAS) loci participate in the regulation of epigenetic signatures and gene expression and, thus, susceptibility to human diseases [24,25]. GWAS has identified the APOA1/C3/A4/A5 cluster in the 11q23.3 region as one of the most powerful gene loci for regulating plasma triglyceride levels [26,27]. Genetic variants within the APOA1/C3/A4/A5 cluster exert a significant influence on plasma triglyceride levels and contribute to the risk and severity of coronary artery disease (CAD) [28]. However, the function and working mode of most lncRNAs within the APOA1/C3/A4/A5 cluster remain to be elucidated.
Here, we identified a novel lncRNA named GM47544, which was highly expressed in the livers of high-fat-fed mice. Subsequently, we investigated the potential involvement of GM47544 in TG metabolism and its regulatory role in modulating the expression levels of the ApoA1/C3/A4/A5 cluster.
2. Methods & protocols
2.1. Gain- and loss-of-function studies
To generate GM47544-overexpressed AML-12 cells, GM47544 cDNAs were obtained from Augct Biotech and inserted into the pLKO.1-TRC Cloning Vector (Addgene) and then transfected into HEK293T cells along with packaging vectors psPAX2 and pMD2.G (Addgene). We treated AML-12 cells with the collected culture media after 48 h. Subsequently, puromycin selection (5 μg/mL, Thermo Fisher Scientific) was employed to isolate the desired cell population. After confirming the overexpression efficiency by RT-qPCR, the cells were used for subsequent experiments. To knockdown GM47544 in AML-12 cells, an Antisense oligonucleotide against GM47544 gene (ASO-GM47544, RiboBio) was transfected using Lipofectamine 3000 (Thermo Fisher Scientific), and the lncRNA ASO Negative Control was used. The sequence of ASO-GM47544 was 5′-CTTCCAACAAAAGCTCTCAT-3′.
2.2. Cell culture
HEK293T, AML-12, and THLE-3 cells were obtained from the Shanghai Life Academy of Sciences Cell Library. Primary mouse hepatocytes were isolated in accordance with previously described methods [29,30]. The cells were cultured at 37 °C in a 5% CO2 atmosphere, using DMEM (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific) plus antibiotics.
2.3. Mice and diets
Male C57BL/6 mice, aged 8 weeks, were obtained from Beijing Vital River Laboratory and randomly divided into groups. Mice were fed a chow diet (CD, Research Diets, D10001) or a high-fat diet (HFD, D12492, research diets, Jiangsu Xietong Pharmaceutical Bio-engineering) for 16 weeks and then euthanized to examine the expression levels of lncRNAs within the ApoA1/C3/A4/A5 gene cluster in the liver.
Male C57BL/6 mice aged 8 weeks were purchased from the same laboratory and randomly grouped. Mice were fed an HFD for 3 weeks before receiving treatment with AAV8-GM47544 (AAV-GM47544) or AAV8-GFP (AAV-Ctrl) recombinant adeno-associated virus (AAVs). Mice were kept on an HFD for another 16 weeks before being euthanized to analyze lipids in their serum and liver.
Apoe−/− mice were purchased from the Beijing Vital River Laboratory at the age of 8 weeks and randomly grouped. Mice were treated with either AAV8-GM47544 (AAV-GM47544) or AAV8-GFP (AAV-Ctrl) recombinant AAVs and fed a high-cholesterol diet (HCD, MD12015, Research Diets, Biopike) for 16 weeks. The mice were euthanized, with lipids in their serum, liver, and atherosclerotic lesions.
AAVs can efficiently transduce host cells, allowing stable and sustained expression of exogenous genes over extended durations, with remarkable safety profiles and minimal immunogenicity. All the AAVs were obtained from Weizhen Biosciences (China). The mice were housed in SPF rooms with a 12 h light/12 h dark cycle. They were given unrestricted access to food and water. All experimental procedures adhered to the guidelines set by the National Institutes of Health (NIH) and received approval from the Animal Care and Use Committee of Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China, IACUC Number: 3281).
2.4. Quantification of atherosclerotic lesions in the aorta
The control and GM47544-overexpressed Apoe−/− mice were euthanized following 16 weeks of high cholesterol diet (HCD) feeding. The entire aorta was isolated and incubated in 4% paraformaldehyde for 24 h. The aorta was opened longitudinally from its origin at the aortic root to its division at the iliac bifurcation after carefully removing the surrounding adipose tissue using a dissection microscope. The aorta was then treated with 60% isopropanol for 10 min and stained with Oil Red O solution for 15 min. To eliminate background staining, destaining was performed using 60% isopropanol for an additional 5 min. The complete length of the aortic tree was then secured onto a black rubber plate and images were captured accordingly. All heart samples were rapidly frozen with liquid nitrogen and maintained at −80 °C for further study. Frozen sections obtained from the region around the aortic root were prepared and subsequently subjected to Oil Red O staining. ImageJ software was used to measure the area of Oil Red O-stained atherosclerotic lesions throughout the length of the aorta and aortic root.
2.5. Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.0, and data were expressed as the mean ± standard deviation. A two-tailed Student's t-test was used for comparison between the two groups. Two-way analysis of variance (ANOVA) followed by Tukey's post hoc test and two-way ANOVA with Sidak's multiple comparison test were used to compare multiple groups. Differences were considered statistically significant at P < 0.05.
3. Results
3.1. Identification of GM47544 as a hepatic regulator of TG metabolism
Given that the ApoA1/C3/A4/A5 gene cluster serves as critical component in regulatinglipoprotein [28], we hypothesis that lncRNAs derived from this gene cluster may participate in lipoprotein metabolism by modulating those genes expression. To explore the potential functionality of lncRNAs within the ApoA1/C3/A4/A5 cluster, we queried the Ensembl Genome Browser (www.ensembl.org/) database and obtained the gene information of all 12 annotated lncRNAs (Figure 1A,B). Liver tissues obtained from mice exposed to a high-fat diet (HFD) were subjected to Quantitative Real-time PCR (RT-qPCR) analysis, and tissues from chow-diet (CD)-fed mice were used for comparison. Among these (12 lncRNAs), the expression level of GM47544, a previously uncharacterized lncRNA, has increased significantly, which attracted our attention (Figure 1A). GM47544 is derived from the natural antisense transcript of ApoA5 located on chromosome 9 of the mouse genome (Figure 1B). Moreover, higher GM47544 expression was observed in the livers of db/db mice and in palmitic acid (PA)-treated AML-12 liver cells (Figure 1C,D). The findings indicated that GM47544 potentially serves as a regulator of lipid metabolism.
Figure 1.

Identification of GM47544 as a hepatic regulator of lipid metabolism.
(A) Representative heatmap showing the response of 12 differentially expressed lncRNAs in mice liver to HFD-feeding. Four highly expressed mRNAs (fold change > 2; FDR-adjusted P < 0.05) located in the ApoA1/C3/A4/A5 gene cluster in the livers of HFD mice. The heatmap was drawn based on normalized expression levels (n = 5).
(B) Schematic representation of the mouse ApoA1/C3/A4/A5 gene cluster locus.
(C) Relative RNA expression of GM47544 in the livers of wide type (WT) or db/db male mice (n = 8).
(D) Relative RNA expression of GM47544 in AML-12 cells with the treatment of PA (250 μM) for 24 h.
(E) Relative RNA expression levels of GM47544 in multiple mouse tissues (n = 6).
(F) RNA FISH analysis of GM47544 in AML-12 cells revealed that the majority of GM47544 is located in the cytoplasm. Red: GM47544; blue: DAPI staining. Scale bar, 0.5 μm.
(G) Detection of GM47544 in the cytoplasm and nucleus of AML-12 cells by qPCR. As a control, GAPDH was used for the cytoplasm and U6 was used for the nucleolus.
(H) Relative expression levels of GM47544 in AML-12 cell lines stably overexpressed GM47544 (OE-GM47544) and the negative control (OE-Ctrl).
(I) Gene set enriched expression (GSEA) plots for fatty acid metabolic pathways. GSEA was performed based on average GM47544 expression (OE-GM47544 versus OE-Ctrl).
(J) Oil Red O staining and hematoxylin staining of lipid droplets in control and GM47544-overexpressed AML-12 cells treated with PA (250 μM, 24 h). Scale bar, 100 μm.
(K) Intracellular TG and TC levels of control and GM47544-overexpressed AML-12 cells with or without PA treatment (250 μM, 24 h).
(L) Left: Dil-LDL uptake was quantified under fluorescence microscopy in control and GM47544-overexpressed AML-12 cells treated with Dil-LDL (15 μg/mL) for 24 h. Red: Dil-LDL; blue: DAPI staining. Scale bar, 100 μm. Right: quantification of the relative fluorescence intensity of internalized DiI-LDL in control and GM47544-overexpressed AML-12 cells. (M) Cellular HL activity in control and GM47544-overexpressed AML-12 cells was detected.
Data are presented as mean ± SD in C, D, H, J, K, L, and M, with statistical significance determined with unpaired two-tailed Student's t-test. Data are presented as mean ± SD in E and G.
We subsequently analyzed the protein-coding potential of GM47544 using ORF Finder, which indicated the presence of two open reading frames (ORFs) (Supplementary Fig. 1A). However, both were confirmed to have no protein-coding ability, suggesting that GM47544 is a noncoding transcript (Supplementary Figs. 1B–D). To determine the distribution of GM47544 in tissues, we performed RT-qPCR, which showed that GM47544 was widely distributed in multiple organs and tissues and extremely abundant in the liver (Figure 1E). Using fluorescence in situ hybridization (FISH) assays, we found that GM47544 was mainly located in the cytoplasm (Figure 1F), which was further confirmed by RT-qPCR analysis of nuclear and cytosolic extracts (Figure 1G).
To determine GM47544's function, we first generated AML-12 cell lines stably overexpressing GM47544 (OE-GM47544) and negative control (OE-Ctrl) using lentiviral vectors (Figure 1H). RNA-seq analysis revealed that GM47544 is implicated in adipogenesis, cholesterol homeostasis, and fatty acid metabolism (Figure 1I and Supplementary Fig. 1E). Subsequently, oil red staining and intracellular lipid content detection were employed to compare OE-GM47544 and OE-Ctrl cells. The results indicated that overexpression of GM47544 significantly downregulates intracellular TG content and reduces the number of lipid droplets within the cells (Figure 1J,K). Furthermore, we also observed that in cells overexpressing GM47544, the ability of low-density lipoprotein receptor (LDLR) to uptake LDL is enhanced, while the activity of intracellular hepatic lipase (HL) is also markedly increased, which demonstrated the lipid-lowering function of GM47544 in facilitating lipid uptake and clearance (Figure 1L,M). By contrast, GM47544 knockdown by ASO in AML-12 cells did not alter the levels of intracellular triglyceride or cholesterol (Supplementary Figs. 1F–H). These findings suggest that the overexpression of GM47544 confers resistance to fatty acid-induced steatosis, highlighting its potential significance in hepatic triglyceride metabolism.
3.2. GM47544 overexpression profoundly prevented diet-induced hypertriglyceridemia and hepatic steatosis
To better understand the contribution of GM47544 to lipid metabolism in vivo, we generated adenovirus (Ad-GM47544) and adeno-associated virus 8 (AAV-GM47544) carrying GM47544, which were intravenously injected into C57BL/6 mice to induce GM47544 overexpression in the liver. Initially, we conducted a lipid tolerance test in mice treated with Ad-GM47544 by administering olive oil orally and monitoring plasma TG levels for a duration of 4 h. Ad-GM47544 mice exhibited a notable reduction in the fluctuation of plasma TG levels (Figure 2A and Supplementary Fig. 2A).
Figure 2.

GM47544 overexpression profoundly prevented diet-induced hypertriglyceridemia and hepatic steatosis.
(A) 8-week WT male mice were injected with adenovirus harbored GFP or GM47544 (Ad-Ctrl or Ad-GM47544) via the tail vein. After 5 days of chow diet feeding, the mice orally received olive oil. Blood samples were collected each hour for 4 h post-oil administration. Plasma TG levels at indicated times in control and GM47544-overexpressed mice (n = 6).
(B) 11-week WT male mice were injected with AAV-Ctrl (Control) or AAV-GM47544 (GM47544 overexpression) via the tail vein after HFD feeding for 3 weeks. Blood samples were collected every 2 weeks. After continuous HFD feeding for 19 weeks, these mice were sacrificed. Dynamic changes in plasma TG and TC levels of control and GM47544-overexpressed mice during the first 11 weeks of HFD feeding (n = 7).
(C) Plasma TG and TC levels of control and GM47544-overexpressed mice after 19 weeks HFD (n = 7).
(D) Fast protein liquid chromatography (FPLC) analysis was performed on pooled plasma from control and GM47544-overexpressing mice, and triglyceride and cholesterol were determined in each of the eluted fractions.
(E) Liver weight and liver-to-body weight ratio of control and GM47544-overexpressed mice.
(F) Hepatic TG and TC levels of control and GM47544-overexpressed mice after 19 weeks HFD (n = 7).
(G) Macroscopic appearance of liver, H&E, and Oil Red O staining of liver sections from control and GM47544-overexpressed mice. Scale bar, 2 cm (upper), 100 μm (lower).
Data are presented as mean ± SD in C, E, and F, with statistical significance determined with unpaired two-tailed Student's t-test. Data are presented as mean ± SD in A and B, with statistical significance determined using a two-way analysis of variance (ANOVA).
Next, we sought to determine whether GM47544 regulated plasma lipid levels and liver lipid deposition in long-term HFD-fed mice. Compared with the control group, GM47544 overexpressed mice showed lower plasma TG and total cholesterol (TC) contents from the 2 nd week of AAV-GM47544 injection in the HFD-fed group, and these effects lasted until 16 weeks of HFD feeding (Figure 2B,C). The fast protein liquid chromatography (FPLC) analysis of plasma revealed a significant reduction in circulating cholesterol and triglycerides specifically within the very-low-density lipoprotein fraction of GM47544 overexpressed mice (Figure 2D). Interestingly, the GM47544 overexpression group showed significant improvements in fatty liver parameters, such as body weight and liver weight (Figure 2E and Supplementary Fig. 2B). Lower levels of triglycerides were detected in the livers of GM47544 overexpression group, whereas the levels of TC in the liver were similar in both groups (Figure 2F). Moreover, improvement in hepatic steatosis was confirmed by histological analysis of liver size and Oil Red O staining (Figure 2G). Hence, GM47544 is a potential target for the advancement of innovative therapeutic strategies for the management of hyperlipidemia.
3.3. GM47544 attenuated the development of atherogenesis
To investigate whether the overexpression of GM47544 can attenuate the progression of atherosclerosis, Apoe−/− mice were treated with AAV8-GM47544 (AAV-GM47544) or AAV8-Control (AAV-Ctrl) after high-cholesterol diet-feeding (HCD) for 16 weeks, followed by the assessment of plasma lipids and atherosclerotic lesions in the aorta. Compared to AAV-Ctrl Apoe−/− mice, AAV-GM47544 Apoe−/− mice displayed lower serum cholesterol, free plasma cholesterol, and triglyceride levels (Figure 3A,B). FPLC analysis demonstrated that GM47544 exhibited the potential to effectively decrease the plasma levels of total cholesterol and triglycerides, particularly in very low-density lipoprotein fractions (Figure 3C). Consistent with the observed decrease in circulating cholesterol levels, AAV-GM47544 Apoe−/− mice demonstrated a 50% decrease in atherosclerotic plaques throughout the entire aorta and a 40% decrease in lesion areas within the aortic root (Figure 3D,E), indicating their enhanced resistance to atherosclerosis progression. In addition, GM47544 relieved hepatic steatosis as evidenced by the reduced number of lipid droplets (LDs) and the decreased hepatic TG and TC content (Figure 3F,G). Taken together, these results indicate that GM47544 overexpression improves liver steatosis and atherosclerotic plaque formation by lowering plasma lipid levels.
Figure 3.

GM47544 attenuated atherogenesis in Apoe−/−mice.
(A) 8-week Apoe−/− mice were injected with AAV8-Ctrl (Control) or AAV8-GM47544 (GM47544 overexpression) via the tail vein. After 16 weeks of HCD feeding, the mice were sacrificed. Plasma TC and free plasma cholesterol levels of control and GM47544-overexpressed Apoe−/− mice (n = 7).
(B) Plasma TG levels of control and GM47544-overexpressed Apoe−/− mice (n = 7).
(C) Fast protein liquid chromatography (FPLC) analysis was performed on pooled plasma from control and GM47544-overexpressed Apoe−/− mice, and triglyceride and cholesterol were determined in each of the eluted fractions.
(D) Representative images of en face aortas and quantification of plaque area in control and GM47544-overexpressed Apoe−/− mice.
(E) Oil-Red O staining and quantification of plaque in aortic root sections of control and GM47544-overexpressed Apoe−/− mice (n = 7). Scale bar = 200 μm.
(F) H&E and Oil-Red O staining of the liver in control and GM47544-overexpressed Apoe−/− mice. Scale bar = 100 μm.
(G) Hepatic TC and TG levels of control and GM47544-overexpressed Apoe−/− mice after 16 weeks of HCD feeding (n = 7).
Data are presented as mean ± SD in A, B, D, E, F, and G, with statistical significance determined with unpaired two-tailed Student's t-test.
3.4. GM47544 enhances plasma TG clearance by suppressing ApoC3 expression
Because lncRNAs can regulate the expression of adjacent genes, we hypothesized that GM47544 may affect the expression of the ApoA1/C3/A4/A5 cluster and consequently regulate lipid metabolism. Intriguingly, we found that overexpression of GM47544 markedly abolished ApoC3 protein levels in AML-12 cells, whereas the mRNA levels of the ApoA1/C3/A4/A5 cluster were barely changed in the GM47544 overexpressed cells, confirming that GM47544 regulates ApoC3 expression at the translational level (Figure 4A,B). The regulating effect of GM47544 on APOC3 is concentration-dependent, with high expression levels of GM47544 effectively downregulating APOC3 protein levels (Supplementary Figs. 3A and B). Furthermore, a significant reduction in ApoC3 protein levels was also detected in GM47544 overexpressed primary hepatocytes, as well as in the livers of AAV-GM47544 C57BL/6 and AAV-GM47544 Apoe−/− mice fed with a high-fat diet, without changes at the transcriptional level (Figure 4C–H). By contrast, GM47544 knockdown by ASO in AML-12 cells did not exert any significant impact on the ApoA1/C3/A4/A5 cluster (Supplementary Figs. 3C–E). The above results indicated that GM47544 significantly downregulates the protein expression level of ApoC3 within hepatocytes, both in vivo and in vitro.
Figure 4.

GM47544 activated HL and LDLR by suppressing ApoC3 to enhance plasma TG clearance.
(A) Relative mRNA expression of GM47544 and ApoA1/C3/A4/A5 gene cluster measured by RT-qPCR.
(B) The ApoA1/C3/A4/A5 protein level was measured by western blot in control and GM47544-overexpressed AML-12 cells.
(C) Relative mRNA expression of GM47544 and ApoA1/C3/A4/A5 gene cluster measured by RT-qPCR.
(D) The ApoA1/C3/A4/A5 protein level was measured by western blot in the primary mouse hepatocytes transfected with empty vector (OE-Ctrl) and GM47544 overexpression plasmids (OE-GM47544).
(E) Relative mRNA expression of GM47544 and ApoA1/C3/A4/A5 gene cluster measured by RT-qPCR.
(F) The ApoA1/C3/A4/A5 protein level was measured by western blot in the livers of control and GM47544-overexpressed mice.
(G) Relative mRNA expression of GM47544 and ApoA1/C3/A4/A5 gene cluster measured by RT-qPCR.
(H) The ApoA1/C3/A4/A5 protein level was measured by western blot in the livers of control and GM47544-overexpressed Apoe−/− mice.
(I) Plasma ApoC3 protein levels in control and GM47544-overexpressed mice.
(J) Plasma LPL activity stimulation in control and GM47544-overexpressed mice.
(K) Plasma ApoC3 protein levels in control and GM47544-overexpressed Apoe−/− mice.
(L) Plasma LPL activity stimulation in control and GM47544-overexpressed Apoe−/− mice.
Data are presented as mean ± SD in A, B, C, D, E, F, G, H, I, J, K, and L, with statistical significance determined using an unpaired two-tailed Student's t-test.
The liver is the primary organ responsible for ApoC3 synthesis, and it also serves as the primary source of circulating ApoC3 [13]. We examined and found that the plasma ApoC3 protein levels were markedly reduced in AAV-GM47544 C57BL/6 and AAV-GM47544 Apoe−/− mice compared to controls (Figure 4I,K). Meanwhile, we observed that exogenous LPL activity was strongly elevated in the plasma of AAV-GM47544 C57BL/6 and AAV-GM47544 Apoe−/− mice (Figure 4J,L). This activation of LPL is associated with a pronounced enhancement in systemic triglyceride (TG) clearance, leading to reduced circulating TG levels. Collectively, these findings provide robust support for the role of GM47544 in reducing ApoC3 protein levels in the liver and plasma, thereby activating LPL, and subsequently enhancing plasma TG clearance.
3.5. GM47544 promotes ubiquitin-dependent degradation of hepatic ApoC3
The ubiquitin–proteasome system is the principal proteolytic machinery responsible for regulating protein degradation in eukaryotic cells, which is essential for maintaining intracellular protein homeostasis and function [31]. Thus, we further explored the possibility that GM47544 regulates ApoC3 protein through a ubiquitination modification pathway. Firstly, we demonstrated that in AML-12 cells overexpressing GM47544, the overall ubiquitin levels were markedly higher than those in the control group (Figure 5A). Additionally, we identified that ApoC3 has the potential to bind ubiquitin (Ub) molecules and can be degraded further via the ubiquitin-proteasome pathway (Supplementary Figs. 4A–B). Subsequently, we confirmed the interaction between Ub and ApoC3 by co-immunoprecipitation with or without GM47544 treatment. As anticipated, overexpression of GM47544 facilitated the ubiquitination of ApoC3 (Figure 5B). Subsequently, the effect of GM47544 on ApoC3 stability was evaluated using the protein synthesis inhibitor CHX. After treatment with CHX, a significant decrease in ApoC3 protein synthesis was observed across all groups, indicating that overexpression of GM47544 did not impact ApoC3 protein synthesis (Figure 5C). In addition, the proteasome inhibitor MG132 reversed the GM47544 overexpression-induced ApoC3 protein decrease in a concentration-dependent manner, unlike the lysosome inhibitor NH4Cl, suggesting that GM47544 regulated ApoC3 stability in a proteasome-mediated manner (Figure 5D–E). These results suggest that GM47544 facilitates the ubiquitination of ApoC3 after translation and is then degraded through the proteasome pathway.
Figure 5.

GM47544 promotes ubiquitin-dependent degradation of hepatic ApoC3.
(A) Overall ubiquitin levels in the control and GM47544-overexpressed AML-12 cells.
(B) IP assays in AML-12 cells transfected with vectors expressing FLAG-ApoC3 were carried out using anti-FLAG antibodies, followed by western blotting analysis with antibodies against ubiquitin and ApoC3.
(C) ApoC3 protein levels in the control and GM47544-overexpressed AML-12 cells after treatment with cycloheximide (CHX, 20 μM).
(D) The control and GM47544-overexpressed AML-12 cells were cultured in the medium containing MG132 with the indicated dose for 6 h. Protein levels of ApoC3 were detected by western blot.
(E) The control and GM47544-overexpressed AML-12 cells were cultured in the medium containing NH₄Cl with the indicated dose for 6 h. Protein levels of ApoC3 were detected by western blot.
(F–G) The AML-12 cells, both control and GM47544-overexpressed, were transfected with the specified plasmids for 24 h. Afterward, ubiquitination assays were conducted using the indicated antibodies. WT, wild type; KR, K is mutated to R; KO, K only.
(H) AML-12 cells were transfected with the specified plasmids for 24 h before conducting ubiquitination assays using the designated antibodies.
(I) The AML-12 cells, both control and GM47544-overexpressed, were transfected with the specified plasmids for 24 h. Afterward, ubiquitination assays were conducted using the indicated antibodies.
(J) ApoC3 protein levels in the control and GM47544-overexpressed AML-12 cells transfected with the indicated plasmids for 24 h. IP, immunoprecipitation; IB, immunoblotting; IgG, immunoglobulin G.
Data are presented as mean ± SD in C, D, E, F, G, I and J, with statistical significance determined with unpaired two-tailed Student's t-test.
Next, we explored the types of polyubiquitin chains linked to ApoC3 by GM47544. By co-transfecting ApoC3 with ubiquitin mutants in which only one lysine residue was mutated to arginine (KR), we observed that GM47544 failed to conjugate the polyubiquitin chains and ApoC3 exclusively when K48 was mutated to arginine (Figure 5F). By co-transfecting ApoC3 with ubiquitin mutants containing only a single lysine residue (KO), we found that GM47544 facilitates the interaction between K48-linked polyubiquitin moieties and ApoC3 (Figure 5G). These results suggest that GM47544 markedly promotes the K48-linked ubiquitination of ApoC3.
We further endeavored to identify the specific residues of ApoC3 that were modified by ubiquitination. To achieve this, we individually substituted all four lysine residues, which were predicted as potential sites for ApoC3 ubiquitination, with arginine and then assessed their modifications through ubiquitination assays. As shown in Figure 5H, there was a significant reduction in the ubiquitination of the ApoC3K79R mutant compared to the other four mutants. Further investigations revealed that GM47544 facilitated the ubiquitination of wild-type ApoC3 but failed to enhance the basal level of ubiquitination of the ApoC3K79R mutant (Figure 5I). Consistently, GM47544 downregulated the level of wild-type ApoC3, but not ApoC3K79R (Figure 5J). Collectively, ApoC3K79R is the critical ubiquitination site for ApoC3.
3.6. GM47544 stem-loop 2 directly interacted with ApoC3 and promoted the degradation of ApoC3
To further probe the functional domain of GM47544, we constructed three truncated GM47544 vectors and performed RNA pull-down experiments based on the distribution principle of stem-loop (SL) structure (Figure 6A). Only the full-length SL2 (142-278 nt) of GM47544 bound closely to ApoC3 (Figure 6B). RNA immunoprecipitation (RIP) also supported an interaction between GM47544 SL2 and ApoC3 (Figure 6C). We constructed plasmids overexpressing GM47544 SL2 and transfected them into AML-12 cells. Similar to the full-length GM47544, GM47544 SL2 had no impact on the mRNA expression of the ApoC3-cluster, but it could significantly degrade the ApoC3 protein (Figure 6D–F). In line with this, the ubiquitylation level of the ApoC3 protein was increased by SL2 (Figure 6G,H). By contrast, GM47544 SL2 did not regulate the protein content or ubiquitin levels of ApoC3 when K79 was mutated to arginine (K79R) (Figure 6I). The collective findings suggest that SL2 is the key region of GM47544 that mediates the ubiquitination of APOC3 K79 and its subsequent degradation.
Figure 6.

GM47544 SL2 directly interacted with ApoC3 and promoted the degradation of ApoC3.
(A) The predicted secondary structure of GM47544 RNA.
(B) Invitro-synthesized full-length (FL), stem-loop structure1 (SL1), stem-loop structure2 (SL2) and stem-loop structure3 (SL3) fragments of GM47544 were incubated with AML-12 cell protein lysates. Subsequently, RNA pull-down and Western blotting assays were performed.
(C) RIP assays were performed using an anti-Flag antibody in AML-12 cells stably transfected with Flag-ApoC3. RT-qPCR analysis was employed to measure the enrichment of GM47544 and its fragments.
(D–E) ApoC3 protein levels were measured by western blot in AML-12 cells transfected with pc3.1 (OE-Ctrl) or pc3.1-GM47544 SL2 (OE-SL2).
(F) Relative expression of GM47544 and ApoA1/C3/A4/A5 were measured by RT-qPCR in control and GM47544 SL2-overexpressed AML-12 cells.
(G) IP assays were performed on AML-12 cells transfected with vectors expressing FLAG-ApoC3 and pc3.1-GM47544 SL2 using anti-FLAG antibodies, followed by western blotting analysis with antibodies against ubiquitin.
(H) IP assays were performed on AML-12 cells transfected with vectors expressing FLAG-ApoC3, pc3.1-GM47544, or pc3.1-GM47544 SL2 using anti-FLAG antibodies, followed by western blotting analysis with antibodies against ubiquitin.
(I) With or without GM47544 full length or SL2, ApoC3 proteins were measured by western blot in AML-12 cells transfected with ApoC3 WT or ApoC3 K79R.
Data are presented as mean ± SD in C, E, F, and I, with statistical significance determined with unpaired two-tailed Student's t-test.
3.7. AP006216.5, human ortholog of GM47544, promoted the degradation of APOC3 and thus exerted lipid-lowering effect
By inspecting the syntenic regions of the human genome, we identified three potentially homologous long noncoding RNAs (Figure 7A). Similar to GM47544, the expression levels of these lncRNAs increased under PA stimulation in the human liver cells THLE-3 (Figure 7B). To further explore the ability of these lncRNAs to modulate APOC3 expression, we overexpressed them in THLE-3 cells using plasmids respectively. Notably, AP006216.5 specifically reduced APOC3 protein levels rather than the mRNA levels of the APOA1/C3/A4/A5 cluster (Figure 7C,D and Supplementary Figs. 5A–D) and decreased the deposition of lipids in hepatic cells, especially the intracellular TG level (Figure E,F). AP006216.5 also functioned in intracellular lipid regulation by upregulating LDLR and HL activity (Figure G,H).
Figure 7.

AP006216.5, a human ortholog of GM47544, promoted the degradation of ApoC3 and thus exerted a lipid-lowering effect.
(A) Schematic representation of the human APOA1/C3/A4/A5 locus.
(B) Relative expression levels of ENST00000439104.1, AP006216.5, and ENST000000457746.1 under PA stimulations in THLE-3 cells were detected by RT-qPCR analysis.
(C) Relative expression of APOA1/C3/A4/A5 in control and AP006216.5-overexpressed THLE-3 cells was measured by RT-qPCR.
(D) APOA1/C3/A4/A5 protein levels in control and AP006216.5-overexpressed THLE-3 cells were measured by western blot.
(E) Oil Red O staining of control and AP006216.5-overexpressed THLE-3 cells.
(F) Intracellular TG and TC (F) levels of control and AP006216.5-overexpressed THLE-3 cells with PA treatment (250 μM, 24 h).
(G) Dil-LDL uptake was quantified under fluorescence microscopy in control and AP006216.5-overexpressed THLE-3 cells treated with Dil-LDL (15 μg/mL) for 24 h. Quantification of the relative fluorescence intensity of internalized DiI-LDL in control and C cells. Red: Dil-LDL; blue: DAPI staining. Scale bar, 100 μm.
(H) Cellular HL activity in control and AP006216.5-overexpressed THLE-3 cells was detected.
(I) THLE-3 was transfected with vectors expressing the FLAG-APOC3, with or without vectors expressing the AP006216.5. IP assays were performed using anti-FLAG antibodies, followed by western blotting analysis with antibodies against ubiquitin.
Data are presented as mean ± SD in B, C, E, F, G, and H, with statistical significance determined with unpaired two-tailed Student's t-test.
We next tested whether AP006216.5 could improve the ubiquitin-mediated degradation of APOC3 in human hepatic cells. Co-IP revealed that overexpressing AP006216.5 significantly increased the ubiquitination of APOC3 (Figure 7I). Collectively, these observations indicated that the function of GM47544 in TG regulation is conserved, at least in humans and mice. AP006216.5, a human ortholog of GM47544, could potentially be used to develop novel therapeutic strategies for the management of human hyperlipidemia.
4. Discussion
In the present study, we identified GM47544, a previously uncharacterized functional lncRNA derived from the ApoA1/C3/A4/A5 cluster, is a crucial regulator of ApoC3. We found that GM47544 abruptly decreased plasma triglyceride levels, thereby alleviating NAFLD and atherosclerosis in mice with acute and chronic lipid overload. Consistent with this finding, overexpression of GM47544 significantly attenuated lipid accumulation in vitro. Furthermore, we found that SL2 of GM47544 interacted directly with ApoC3 and facilitated the ubiquitination of K79 of ApoC3, which successively led to the degradation of the ApoC3 protein through the proteasome pathway. The inhibition of ApoC3, in turn, activates LPL/HL and LDLR, profoundly enhancing plasma TG clearance to attenuate hyperglyceridemia, NAFLD, and atherosclerosis. Interestingly, we identified AP006216.5, the human ortholog of GM47544, which promotes the degradation of ApoC3 and thus exerts a lipid-lowering effect in humans. Collectively, these findings suggest that GM47544 plays a pivotal role in triglyceride regulation, promoting the decomposition of triglycerides through the ApoC3-LPL/HL pathway and uptake of triglycerides and cholesterol in the liver through the ApoC3-LDLR pathway.
The APOA1/C3/A4/A5 cluster is one of the most promising targets for the regulation of hyperglyceridemia. Several lncRNAs have been identified as regulators of the APOA1/C3/A4/A5 gene cluster. APOA1-AS, located in the antisense strand of APOA1, epigenetically controls the expression of this apolipoprotein gene cluster by recruiting histone-modifying enzymes [32]. Similarly, APOA4-AS, which interacts with mRNA-stabilizing protein HuR to enhance the stability of APOA4 mRNA, led to reduced levels of plasma TG and TC in ob/ob mice [33]. In our study, within the ApoA1/C3/A4/A5 cluster, the newly identified lncRNA GM47544 interacted directly with ApoC3. GM47544 facilitates ApoC3 degradation via the ubiquitin-proteasome pathway, ultimately lowering plasma and cellular TG accumulation. These results improve our understanding concerning studies of the biological functions of the APOA1/C3/A4/A5 cluster.
TRLs, particularly triglyceride-rich lipoproteins, are considered causal components of atherogenesis. They have been proposed as ideal targets for diet- or drug-based interventions to prevent hyperglyceridemia and ASCVD [34]. Novel targets for TRL-lowering therapy have emerged, including APOC3, angiopoietin-like proteins 3 and 4 (ANGPTL3/4), APOA5, and ATP citrate lyase [35,36]. Interestingly, based on the most important triglyceride regulatory gene loci of the APOA1/C3/A4/A5 cluster, APOC3 has been described as a classic triglyceride regulator that modulates hepatic TRL clearance through lipase-dependent and lipase-independent mechanisms [16,17,37]. Individuals with loss-of-function mutations in APOC3 exhibit a noteworthy 39% reduction in triglyceride levels, coupled with a substantial 40% decrease in the risk of ASCVD compared to noncarriers [38]. Moreover, epidemiological investigations have consistently demonstrated that APOC3 levels possess predictive value for ASCVD risk and cardiovascular mortality [17]. Targeting APOC3 is one of the most unique and potent approaches, resulting in a substantial reduction in TRLs [13,[16], [17], [18]]. In the current study, targeting ApoC3 by overexpression of GM47544 strikingly decreased plasma triglyceride levels by up to 40% in HFD mice at 8 weeks, which conferred protection against hepatic lipid accumulation, hypertriglyceridemia, and atherosclerosis in HFD mice. In terms of mechanisms, we provided evidence that GM47544 overexpression lost the inhibition of HL activity due to ApoC3 degradation, suggesting an association between liver GM47544 and plasma triglyceride levels in a lipase-dependent manner. In addition, GM47544 overexpression facilitated the assimilation of Dil-LDL in AML-12 cells, which represents a lipase-independent way of enhancing the uptake of TG in the liver, leading to the clearance of plasma TG. In summary, the effectiveness of GM47544 in reducing plasma triglyceride levels underscores the importance of targeting ApoC3 and downstream lipase-dependent or lipase-independent pathways. However, we also observed that GM47544 could not influence lipid metabolism at low concentrations or knock-down states. This may be attributed to the interaction between GM47544 and ApoC3. Similar phenomena have been reported in previous studies. For example, overexpression of the lncRNA HOTAIR can upregulate EZH2 expression, whereas intervention with shHOTAIR does not alter EZH2 protein levels. This discrepancy may be due to the dysregulation of the interaction between HOTAIR and EZH2 [39]. Therefore, we speculate that under basal conditions or when knocked down, GM47544 may scarcely bind to ApoC3. Conversely, GM47544 can effectively bind to ApoC3 and trigger ApoC3 degradation process only when its expression level is significantly higher than the basal level.
Targeted protein degradation (TPD) is an emerging and rapidly expanding strategy in drug discovery that utilizes small molecules to facilitate the rapid breakdown of disease-causing proteins [40]. TPD exploits the endogenous proteolytic pathways of cells to selectively eliminate proteins of interest (POIs), including disease-causing proteins, from cellular and tissue environments [41]. These technologies hold promise as novel therapies for unmet medical needs. They are much smaller and may have a higher membrane permeability and better cellular uptake [42]. In terms of lncRNA GM47544, we identified that SL2 of GM47544 could directly bind to K79 of ApoC3, which promotes the ubiquitination of ApoC3, thus inducing anti-hyperglyceridemia and atherogenesis [43]. This represents a brand-new direction of research rather than the traditional strategies for lowering plasma triglycerides by targeting ApoC3 mRNA.
In summary, we demonstrated that lncRNA GM47544 can directly bind to ApoC3 and promote its degradation of ApoC3 through the ubiquitin-proteasome pathway, which strikingly lowered hyperlipidemia, attenuated NAFLD, and prevented atherosclerosis. These findings have set the stage for new therapeutic approaches aimed at lipoproteins and promise to advance CVD management substantially.
CRediT authorship contribution statement
Qianqian Xiao: Writing – review & editing, Writing – original draft, Visualization, Resources, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Luyun Wang: Writing – review & editing, Writing – original draft, Resources, Project administration, Conceptualization. Jing Wang: Methodology, Investigation, Data curation. Man Wang: Methodology, Investigation, Data curation. Dao Wen Wang: Writing – review & editing, Supervision, Resources, Project administration. Hu Ding: Writing – review & editing, Supervision, Project administration, Data curation, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (grant numbers: 2021YFC2500600 and 2021YFC2500604) and the National Natural Science Foundation of China (grant numbers: 82170348, 81974047, and 82170283).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2024.102011.
Contributor Information
Dao Wen Wang, Email: dwwang@tjh.tjmu.edu.cn.
Hu Ding, Email: huding@tjh.tjmu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Supplementary information containing the supplementary methods and protocols, and supplementary figures for this study is available online. The primer sequences used in this study can be found in Supplementary Table 1. All the antibodies employed in this study were listed in Supplementary Table 2.
Data availability
Data will be made available on request.
References
- 1.Ginsberg H.N., Packard C.J., Chapman M.J., Boren J., Aguilar-Salinas C.A., Averna M., et al. Triglyceride-rich lipoproteins and their remnants: metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur Heart J. 2021;42(47):4791–4806. doi: 10.1093/eurheartj/ehab551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nordestgaard B.G. Triglyceride-rich lipoproteins and atherosclerotic cardiovascular disease: new insights from epidemiology, genetics, and biology. Circ Res. 2016;118(4):547–563. doi: 10.1161/CIRCRESAHA.115.306249. [DOI] [PubMed] [Google Scholar]
- 3.Laufs U., Parhofer K.G., Ginsberg H.N., Hegele R.A. Clinical review on triglycerides. Eur Heart J. 2020;41(1) doi: 10.1093/eurheartj/ehz785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahebkar A., Chew G.T., Watts G.F. Recent advances in pharmacotherapy for hypertriglyceridemia. Prog Lipid Res. 2014;56:47–66. doi: 10.1016/j.plipres.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Sandesara P.B., Virani S.S., Fazio S., Shapiro M.D. The forgotten lipids: triglycerides, remnant cholesterol, and atherosclerotic cardiovascular disease risk. Endocr Rev. 2019;40(2):537–557. doi: 10.1210/er.2018-00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olivecrona G. Role of lipoprotein lipase in lipid metabolism. Curr Opin Lipidol. 2016;27(3):233–241. doi: 10.1097/MOL.0000000000000297. [DOI] [PubMed] [Google Scholar]
- 7.Malick W.A., Do R., Rosenson R.S. Severe hypertriglyceridemia: existing and emerging therapies. Pharmacol Therapeut. 2023;251 doi: 10.1016/j.pharmthera.2023.108544. [DOI] [PubMed] [Google Scholar]
- 8.Sampson U.K., Fazio S., Linton M.F. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: the evidence, etiology, and therapeutic challenges. Curr Atherosclerosis Rep. 2012;14(1):1–10. doi: 10.1007/s11883-011-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quispe R., Martin S.S., Michos E.D., Lamba I., Blumenthal R.S., Saeed A., et al. Remnant cholesterol predicts cardiovascular disease beyond LDL and ApoB: a primary prevention study. Eur Heart J. 2021;42(42):4324–4332. doi: 10.1093/eurheartj/ehab432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quispe R., Sweeney T., Varma B., Agarwala A., Michos E.D. Recent updates in hypertriglyceridemia management for cardiovascular disease prevention. Curr Atherosclerosis Rep. 2022;24(10):767–778. doi: 10.1007/s11883-022-01052-4. [DOI] [PubMed] [Google Scholar]
- 11.Ganda O.P. Triglyceride-rich lipoproteins, remnant-cholesterol, and atherosclerotic cardiovascular disease. Curr Opin Lipidol. 2023;34(3):105–113. doi: 10.1097/MOL.0000000000000875. [DOI] [PubMed] [Google Scholar]
- 12.Gouni-Berthold I., Schwarz J., Berthold H.K. Updates in drug treatment of severe hypertriglyceridemia. Curr Atherosclerosis Rep. 2023;25(10):701–709. doi: 10.1007/s11883-023-01140-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D'Erasmo L., Di Costanzo A., Gallo A., Bruckert E., Arca M. ApoCIII: a multifaceted protein in cardiometabolic disease. Metabolism. 2020;113 doi: 10.1016/j.metabol.2020.154395. [DOI] [PubMed] [Google Scholar]
- 14.Tall A.R., Thomas D.G., Gonzalez-Cabodevilla A.G., Goldberg I.J. Addressing dyslipidemic risk beyond LDL-cholesterol. J Clin Invest. 2022;132(1) doi: 10.1172/JCI148559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khetarpal S.A., Zeng X., Millar J.S., Vitali C., Somasundara A.V.H., Zanoni P., et al. A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat Med. 2017;23(9):1086–1094. doi: 10.1038/nm.4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexander V.J., Xia S., Hurh E., Hughes S.G., O'Dea L., Geary R.S., et al. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur Heart J. 2019;40(33):2785–2796. doi: 10.1093/eurheartj/ehz209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tardif J.C., Karwatowska-Prokopczuk E., St Amour E., Ballantyne C.M., Shapiro M.D., Moriarty P.M., et al. Apolipoprotein C-III reduction in subjects with moderate hypertriglyceridaemia and at high cardiovascular risk. Eur Heart J. 2022;43(14):1401–1412. doi: 10.1093/eurheartj/ehab820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaudet D., Alexander V.J., Baker B.F., Brisson D., Tremblay K., Singleton W., et al. Antisense inhibition of apolipoprotein C-iii in patients with hypertriglyceridemia. N Engl J Med. 2015;373(5):438–447. doi: 10.1056/NEJMoa1400283. [DOI] [PubMed] [Google Scholar]
- 19.Kim K., Ginsberg H.N., Choi S.H. New, novel lipid-lowering agents for reducing cardiovascular risk: beyond statins. Diabetes & Metabolism Journal. 2022;46(4):517–532. doi: 10.4093/dmj.2022.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bridges M.C., Daulagala A.C., Kourtidis A. LNCcation: lncRNA localization and function. J Cell Biol. 2021;220(2) doi: 10.1083/jcb.202009045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaath H., Vishnubalaji R., Elango R., Kardousha A., Islam Z., Qureshi R., et al. Long non-coding RNA and RNA-binding protein interactions in cancer: experimental and machine learning approaches. Semin Cancer Biol. 2022;86(Pt 3):325–345. doi: 10.1016/j.semcancer.2022.05.013. [DOI] [PubMed] [Google Scholar]
- 22.Herman A.B., Tsitsipatis D., Gorospe M. Integrated lncRNA function upon genomic and epigenomic regulation. Mol Cell. 2022;82(12):2252–2266. doi: 10.1016/j.molcel.2022.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao Z.-T., Yang Y.-M., Sun M.-M., He Y., Liao L., Chen K.-S., et al. New insights into the interplay between long non-coding RNAs and RNA-binding proteins in cancer. Cancer Commun. 2022;42(2):117–140. doi: 10.1002/cac2.12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian X., Zhao J., Yeung P.Y., Zhang Q.C., Kwok C.K. Revealing lncRNA structures and interactions by sequencing-based approaches. Trends Biochem Sci. 2019;44(1):33–52. doi: 10.1016/j.tibs.2018.09.012. [DOI] [PubMed] [Google Scholar]
- 25.Cao M., Luo H., Li D., Wang S., Xuan L., Sun L. Research advances on circulating long noncoding RNAs as biomarkers of cardiovascular diseases. Int J Cardiol. 2022;353(1):109–117. doi: 10.1016/j.ijcard.2022.01.070. [DOI] [PubMed] [Google Scholar]
- 26.Bai W., Kou C., Zhang L., You Y., Yu W., Hua W., et al. Functional polymorphisms of the APOA1/C3/A4/A5-ZPR1-BUD13 gene cluster are associated with dyslipidemia in a sex-specific pattern. PeerJ. 2019;6(1) doi: 10.7717/peerj.6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lai C.-Q., Parnell L.D., Ordovas J.M. The APOA1/C3/A4/A5 gene cluster, lipid metabolism and cardiovascular disease risk. Curr Opin Lipidol. 2005;16(2):153–166. doi: 10.1097/01.mol.0000162320.54795.68. [DOI] [PubMed] [Google Scholar]
- 28.Cui G., Li Z., Li R., Huang J., Wang H., Zhang L., et al. A functional variant in APOA5/A4/C3/A1 gene cluster contributes to elevated triglycerides and severity of CAD by interfering with microRNA 3201 binding efficiency. J Am Coll Cardiol. 2014;64(3):267–277. doi: 10.1016/j.jacc.2014.03.050. [DOI] [PubMed] [Google Scholar]
- 29.Jiang Y., Peng J., Song J., He J., Jiang M., Wang J., et al. Loss of Hilnc prevents diet-induced hepatic steatosis through binding of IGF2BP2. Nat Metab. 2021;3(11):1569–1584. doi: 10.1038/s42255-021-00488-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y., Castellani L.W., Sinal C.J., Gonzalez F.J., Edwards P.A. Peroxisome proliferator-activated receptor-gamma coactivator 1alpha (PGC-1alpha) regulates triglyceride metabolism by activation of the nuclear receptor FXR. Genes Dev. 2004;18(2):157–169. doi: 10.1101/gad.1138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bard J.A.M., Goodall E.A., Greene E.R., Jonsson E., Dong K.C., Martin A. Structure and function of the 26S proteasome. Annu Rev Biochem. 2018;87:697–724. doi: 10.1146/annurev-biochem-062917-011931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halley P., Kadakkuzha B.M., Faghihi M.A., Magistri M., Zeier Z., Khorkova O., et al. Regulation of the apolipoprotein gene cluster by a long noncoding RNA. Cell Rep. 2014;6(1):222–230. doi: 10.1016/j.celrep.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qin W., Li X., Xie L., Li S., Liu J., Jia L., et al. A long non-coding RNA, APOA4-AS, regulates APOA4 expression depending on HuR in mice. Nucleic Acids Res. 2016;44(13):6423–6433. doi: 10.1093/nar/gkw341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boren J., Taskinen M.R., Bjornson E., Packard C.J. Metabolism of triglyceride-rich lipoproteins in health and dyslipidaemia. Nat Rev Cardiol. 2022;19(9):577–592. doi: 10.1038/s41569-022-00676-y. [DOI] [PubMed] [Google Scholar]
- 35.Tokgözoğlu L., Libby P. The dawn of a new era of targeted lipid-lowering therapies. Eur Heart J. 2022;43(34):3198–3208. doi: 10.1093/eurheartj/ehab841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenson R.S., Gaudet D., Ballantyne C.M., Baum S.J., Bergeron J., Kershaw E.E., et al. Evinacumab in severe hypertriglyceridemia with or without lipoprotein lipase pathway mutations: a phase 2 randomized trial. Nat Med. 2023;29(3):729–737. doi: 10.1038/s41591-023-02222-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gaudet D., Brisson D., Tremblay K., Alexander V.J., Singleton W., Hughes S.G., et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med. 2014;371(23):2200–2206. doi: 10.1056/NEJMoa1400284. [DOI] [PubMed] [Google Scholar]
- 38.Crosby J., Peloso G.M., Auer P.L., Crosslin D.R., Stitziel N.O., Lange L.A., et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371(1):22–31. doi: 10.1056/NEJMoa1307095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuo F.-C., Neville M.J., Sabaratnam R., Wesolowska-Andersen A., Phillips D., Wittemans L.B.L., et al. HOTAIR interacts with PRC2 complex regulating the regional preadipocyte transcriptome and human fat distribution. Cell Rep. 2022;40(4) doi: 10.1016/j.celrep.2022.111136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao L., Zhao J., Zhong K., Tong A., Jia D. Targeted protein degradation: mechanisms, strategies and application. Signal Transduct Targeted Ther. 2022;7(1):113. doi: 10.1038/s41392-022-00966-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sathe G., Sapkota G.P. Proteomic approaches advancing targeted protein degradation. Trends Pharmacol Sci. 2023;44(11):786–801. doi: 10.1016/j.tips.2023.08.007. [DOI] [PubMed] [Google Scholar]
- 42.Sasso J.M., Tenchov R., Wang D., Johnson L.S., Wang X., Zhou Q.A. Molecular glues: the adhesive connecting targeted protein degradation to the clinic. Biochemistry. 2022;62(3):601–623. doi: 10.1021/acs.biochem.2c00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Domostegui A., Nieto-Barrado L., Perez-Lopez C., Mayor-Ruiz C. Chasing molecular glue degraders: screening approaches. Chem Soc Rev. 2022;51(13):5498–5517. doi: 10.1039/d2cs00197g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information containing the supplementary methods and protocols, and supplementary figures for this study is available online. The primer sequences used in this study can be found in Supplementary Table 1. All the antibodies employed in this study were listed in Supplementary Table 2.
Data Availability Statement
Data will be made available on request.