

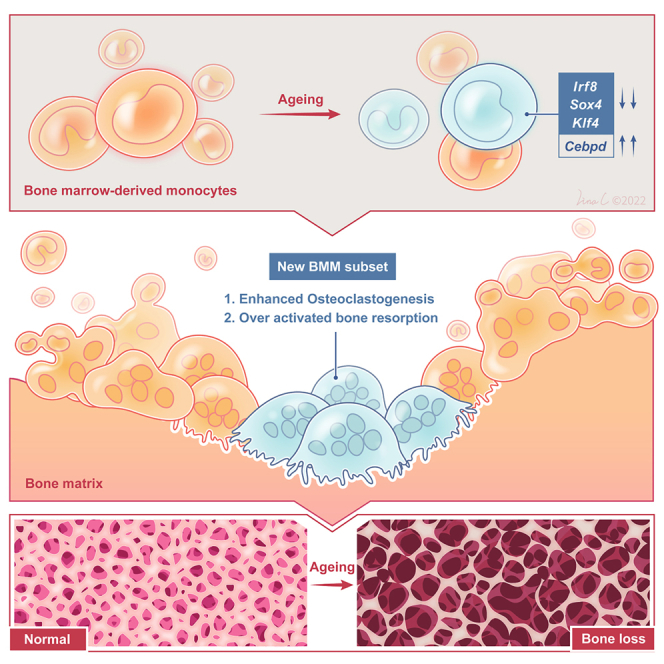
Summary
Age-related osteoporosis manifests as a complex pathology that disrupts bone homeostasis and elevates fracture risk, yet the mechanisms facilitating age-related shifts in bone marrow macrophages/osteoclasts (BMMs/OCs) lineage are not fully understood. To decipher these mechanisms, we conducted an investigation into the determinants controlling BMMs/OCs differentiation. We performed single-cell multi-omics profiling on bone marrow samples from mice of different ages (1, 6, and 20 months) to gain a holistic understanding of cellular changes across time. Our analysis revealed that aging significantly instigates OC differentiation. Importantly, we identified Cebpd as a vital gene for osteoclastogenesis and bone resorption during the aging process. Counterbalancing the effects of Cebpd, we found Irf8, Sox4, and Klf4 to play crucial roles. By thoroughly examining the cellular dynamics underpinning bone aging, our study unveils novel insights into the mechanisms of age-related osteoporosis and presents potential therapeutic targets for future exploration.
Subject areas: Molecular biology, Cell biology, Omics, Transcriptomics
Graphical abstract
Highlights
-
•
Overactivation of osteoclasts is one of the main causes of age-related osteoporosis
-
•
Newly identified subpopulations of osteoclasts are overactivated during aging
-
•
Irf8, Sox4, Klf4, and Cebpd regulate osteoclasts differentiation during aging
Molecular biology; Cell biology; Omics; Transcriptomics
Introduction
Aging has emerged as a significant socioeconomic and scientific concern in recent times.1 With advancing age, various physiological and pathological processes undergo systemic changes that affect all organs and tissues, including the skeletal system. Among the numerous age-related alterations, progressive bone loss stands out as a prominent feature, leading to the development of osteoporosis in the elderly population. This disorder not only hampers daily activities but also elevates the risk of fractures, resulting in increased morbidity, mortality, and diminished quality of life, which escalating healthcare expenditures.2
Age-related osteoporosis is now acknowledged to involve a series of interconnected factors, including heightened bone resorption by osteoclasts (OCs), reduced bone mineral density (BMD), compromised bone microarchitecture, and impaired regenerative capacity. These changes reflect an imbalance in bone homeostasis, with a shift toward enhanced bone resorption orchestrated by activated OCs.3,4,5 Excessive osteoclastogenesis and hyperactivity of OCs constitute primary concerns in the pathogenesis of age-related osteoporosis.6,7,8
In recent years, numerous studies have highlighted the role of macrophage colony stimulating factor (M-CSF) and receptor activator of nuclear factor κB (NF-κB) ligand (RANKL) in initiating osteoclastogenesis in the bone marrow.9 Subsequent to this initiation, a sequence of events occurs involving the proliferation, activation, and fusion of bone marrow monocytes (BMMs), which are finely regulated by various factors.10 The culmination of this process results in the formation of mature OCs with the ability to degrade bone tissue through the action of proteases and acids, including tartrate-resistant acid phosphatase (TRAP), and cathepsin K (CTSK).11,12 During the aging process, it has been observed that BMMs/OCs become overactivated, which may be attributed to low-grade chronic inflammation commonly referred to as “inflammaging”.13,14,15,16 Additionally, components of the cellular senescence-associated secretory phenotype (SASP), such as IL-6 and TNF-α, have proinflammatory properties and can stimulate the maturation and activation of OCs.17,18 Inhibition of SASP production has been shown to increase bone mass, enhance bone strength, improve bone microarchitecture, and counteract bone loss in aged mice.19,20,21 Furthermore, treatment of aged mice with senolytic drugs, such as dasatinib and quercetin, which eliminate senescent cells, has been demonstrated to reduce OC formation and bone loss, enhance mineral reabsorption and thickness, and significantly improve trabecular and cortical bone microarchitecture. Despite these findings, intrinsic changes in BMMs/OCs and their impact on OC differentiation during aging have received limited attention. Moreover, due to the complexity of BMMs fusion and the heterogeneity of BMMs/OCs, the precise mechanisms underlying OC differentiation and activation during aging remain incompletely understood.
Emerging techniques such as single-cell RNA sequencing (scRNA-seq) have provided unprecedented opportunities to investigate cellular dynamics during the aging process, particularly the association between cellular states and OCs, as well as the changes occurring in their progenitors over time. For instance, Mo et al. identified a slow-cycling bone marrow sub-population expressing Notch3, which exhibited close association with vasculatures and key transcriptional networks promoting osteo-chondrogenic differentiation.22 Yahara et al. uncovered an OC precursor population derived from erythromyeloid progenitors (EMPs), contributing to postnatal bone remodeling.23 Ambrosi et al. linked functional decline to reduced transcriptomic diversity in skeletal stem cells of aged mice, thereby contributing to alterations in the bone marrow niche.24 Tsukasaki et al. identified Cbp/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain 2 (Cited2) as a molecular switch triggering terminal OC differentiation.25 Shao et al., through the integration of scRNA-seq and bulk RNA-seq data, identified 11 OC differentiation-related genes (ODRGs) that predict OC survival.26 While unimodal scRNA-seq data provides valuable insights into OC cellular dynamics during aging at the transcriptomic level, it fails to capture cellular dynamics in other modalities such as genetic mutations. Hence, the application of single-cell multi-omics techniques holds promise in comprehensively studying cellular dynamics associated with OCs during the aging process and identifying potential targets for anti-aging interventions.
In this study, we employed a single-cell multi-omics approach to explore the mechanisms underlying the overactivity of OCs and the differentiation and maturation of OCs during aging. Our focus was to profile the changes in BMMs and OCs during the aging process and to investigate the direct impact of aging on OCs and their precursors. We demonstrated that BMMs with dysfunctional telomerase, a hallmark of aging, exhibited a heightened propensity to differentiate into more active OCs, leading to excessive bone resorption and noticeable bone loss in murine models. Moreover, through further single-cell multi-omics analysis, we identified novel subpopulations of BMMs/OCs associated with overactivated osteoclastogenesis during aging. These findings provide novel insights into the maintenance of skeletal tissue homeostasis in aged individuals and enhance our understanding of the physiological behavior of BMMs/OCs. Additionally, the identification of novel regulators of OC differentiation and markers for distinct subpopulations from the single-cell multi-omics data holds promise for the development of therapeutic interventions targeting age-related osteoporosis. By shedding light on the underlying mechanisms driving OC maturation and bone loss during aging, our study contributes to the growing body of knowledge aiming at combating age-related skeletal disorders and improving the quality of life for the aging population.
Results
Terc deficiency induces natural aging-like bone loss phenotypes
OCs play a crucial role in bone resorption, and previous studies have reported that overactivity of OCs is associated with age-related osteoporosis.27 To investigate the bone loss phenotypes during natural aging and assess OC activity, we utilized 20-month-old wild-type (20 m WT) mice as the aging group and 6-month-old (6 m WT) mice as the young control. Micro-CT scanning and TRAP staining of distal femur sections confirmed decreased trabecular bone volume and activated osteoclastogenesis in long bone tissue from naturally aged mice (Figures 1A–1F) with only modest changes in cortical bone in aged mice compared to young mice (Figures 1B and 1C).
Figure 1.

Natural aging and progeria precipitates significant bone loss and activated osteoclastogenesis
See also Figure S1 and Table S1. Parameters of micro-CT scanning of long bones from 6 m WT, 6 m KO and 20 m WT mice, related to Figure 1C and 1E, Table S2. The number of osteoclasts (TRAP+) on unit area of trabecular bone from mice distal femoral section, related to Figure 1F, Table S3. Area of single resorption pit on bone slices (6 m KO vs. WT), related to Figure 1G, Table S4. Expression matrix of 46 DEGs in FULL stage of RANKL stimulation of young and aged groups, related to Figure 1H, Table S5. DEGs between 6 m (WT & KO) and 20 m WT mice, related to Figure 1I, Table S10. Top significant mutation genes between 20 m WT and 6 m KO mice, related to Figure 5C, Table S11. Inferred receptors and key transcription factors that modulate the aged-induced osteoclastogenesis, related to Figure 6D, Table S12. The relative expression of TRAP and CTSK in scramble or knock-down OCs, related to Figure 7A left, Table S13. The relative expression of TRAP and CTSK in scramble or knock-down OCs, related to Figure 7A right, Table S14. Area of single resorption pit on bone slices (scramble and knock-down OCs), related to Figure 7B, Table S15. TRAP positive cells in 4 TFs knock-down in vitro cultured BMMs of 20 m and 6 m mice, related to Figure 7D.
(A) Representative sectional images of micro-CT scanning of distal femur from 6 m, 20 m wild type mice and 6 m Terc−/− mice.
(B) Reconstruction model of femoral cortical bone from young (6 m WT) and aged (20 m WT and 6 m Terc−/−) mice (left).
(C) Cortical bone parameters of micro-CT scanning (right) revealed modest changes in femoral cortical bone between young and aged samples.
(D) Reconstruction model of femoral trabecular bone from young (6 m) and aged (20 m) mice (left).
(E) Trabecular bone parameters of micro-CT scanning (right) revealed evident bone loss in trabecular area in aged mice.
(F) TRAP staining of distal femoral sections from young and aged mice. More TRAP+ cells are found in trabecular area from aged mice (left).
(G) Pits array of bone slices by SEM, as each pit’s area is recorded, showed in vitro-cultured OCs from aged mice (6 m Terc−/−) formed more and bigger mature cells (right).
(H) Heatmap of bulk RNA-seq analysis of BMMs transcriptome. 46 differential expression genes are identified to be activated or suppressed between aged and young groups in WT and Terc−/− mice with RANKL stimulation (FULL: 8-day RANKL stimulation or total mature).
(I) Pathway enrichment analysis through comparing 6 m and 20 m WT (top) or 6 m KO group (bottom). Top 25 pathways are listed. In each figure, the direction of effect (down/up-regulation) relates to the effect seen in the 20 m WT or 6 m KO group. Data are shown as mean ± s.e.m. n = 12 (7 WT and 5 KO) for 6 m; n = 4 (WT) for 20 m. For (C) and (E), each dot represents a single individual. p values were determined by two-tailed Student’s t test. Labels of Y axis in (C) and (E) (parameters of bone structure obtained from micro-CT scanning) have been defined further: cortical bone: Po (%): total porosity; Es.Pm (mm): endosteal perimeter; Ps.Pm (mm): periosteal perimeter; Ct.Ar (mm2): cortical bone area; Tt.Ar (mm2): total cortical bone area. Trabecular bone: BV/TV (%): bone volume fraction; Tb.N (1/mm): trabecular number; Tb.Th (mm): trabecular thickness; Tb.Sp (mm): trabecular separation.
Genome instability is known to contribute to aging, and telomeres, crucial for maintaining genome stability, can be elongated by telomerase. Previous studies28,29 have utilized telomerase-deficient mice (Terc−/−) to investigate the mechanisms underlying bone aging. Therefore, in this study, we bred 6-month-old Terc−/− (6 m KO) male mice to explore the presence of bone loss phenotypes and the mechanisms of OC formation and activation under conditions of telomerase dysfunction. Micro-CT analysis of long bones revealed evident bone loss in Terc−/− mice compared to their wild-type counterparts. Similar to the morphological analysis of long bones in naturally aged mice, bone sections from Terc−/− mice exhibited severe trabecular bone loss with cortical bone remaining unchanged between KO and WT mice (Figures 1A–1E; Table S1). Histological analysis of distal femur using TRAP staining showed more TRAP positive cells presenting in aged mice and further confirmed overactivated osteoclastogenesis in KO mice (Figure 1F; Table S2). Subsequently, we harvested bone marrow tissue from mice and cultured BMMs/OCs in vitro on bone slices to observe differences in OC maturation and function between WT and KO mice. Analysis of the bone slice pits revealed that OCs cultured from KO mice formed larger single pit areas, indicating increased osteoclastogenesis and bone resorption function in BMMs and OCs from progeria mice (Figure 1G; Table S3). Thus, Terc deficiency stimulates the formation of OCs, contributing to the trabecular bone loss phenotypes.
To delve into the implications of Terc deficiency in aging, we analyzed gene expression levels in BMMs across five different cohorts: 1-month-old (1 m) WT, 1 m knockout (KO), 6 m WT, 6 m KO, and 20 m WT mice. We were unable to obtain 20 m KO mice due to their low survival rate associated with Terc deficiency. In this assessment, we recognized 46 differentially expressed genes (DEGs) showcasing consistent regulation. A total of 12 DEGs were upregulated and 34 downregulated in the 6 m KO and 20 m WT groups, compared to the 6 m WT group serving as control (Figure 1H; Table S4). Of these 46 DEGs, 24 were significantly differentially regulated (either up or downregulated) in the 6 m KO group relative to the 20 m WT group. In another interesting trend, 36 genes in the 1 m KO group showed significantly differential regulation when compared to the 1 m WT group. These genes followed the same pattern of regulation as observed in the 6 m KO and 20 m WT groups.
To understand the functional implications of these Terc deficiency-associated DEGs, we conducted functional enrichment analysis through ToppGene Suite.30 This involved comparisons between the 6 m WT and 20 m WT mice (Figure 1I top; Figure S1 top; Table S5) as well as the 6 m KO mice (Figure 1I bottom; Figure S1 bottom; Table S5). Our analysis of the top 25 enriched pathways revealed a significant downregulation in pathways tied to collagen synthesis and senescence in the 20 m WT mice. These pathways included collagen biosynthesis and modifying enzymes, assembly of collagen fibrils and other multimeric structures, and genes encoding collagen proteins. Additionally, there was a prominent decrease in the lysosome and OC differentiation pathways in the 6 m KO mice, implying an aged phenotype. Intriguingly, we noted high expression of cell cycle-related pathways in both the 20 m WT and 6 m KO groups.
Our extensive transcriptome analysis of bone marrow derived from young and aged mice, along with phenotype confirmation, reveals the adverse impact of Terc deficiency during aging, which in turn, results in an increased formation and activity of OCs.
Single-cell RNA-seq identifies different cell populations in WT and Terc−/− bone marrow samples during aging
In this study, we aimed to investigate the changes in BMMs and OCs during the aging process through bioinformatics analysis and then validate our observations by experiments (Figure 2A). To explore the changes in cellular classification, we performed scRNA-seq on a total of 25,904 cells from young, healthy old, and mutant old mice after quality control. Our analysis of bone marrow cells from 1 m to 20 m WT and KO mice identified 35 distinct cell populations (clusters) (Figures 2B and S2A; Table S6), which were further annotated based on known markers in the CellMarker database,31 characterizing them into different cell types including hematopoietic stem cells, CMPs (common myeloid progenitors), GMPs (granulocyte-monocyte progenitors), natural killer cells, CLPs (common lymphoid progenitors), B cells, erythroid cells, Ly6g+Mmp9- monocytes, and Mmp9+ BMMs (Figure 2C).
Figure 2.

Different osteoclast progenitor cell populations were identified based on the single-cell transcriptome analysis of bone marrow cells from aging wild-type and Terc−/− mice
See also Figure S2 and Table S6.
(A) The bone marrow cells from 1 m, 6 m, 20 m WT, and 1 m, 6 m Terc−/− mice were profiled with single-cell RNA-seq and DNA-seq. n = 8 (4 WT and 4 KO) for 1 m; n = 8 (4 WT and 4 KO) for 6 m; n = 4 (WT) for 20 m.
(B) is the UMAP plot that presents the cell clusters obtained from Leiden clustering.
(C) All the obtained clusters were annotated with the cell type information by comparing the top differential genes (signature genes) associated with each cluster with known cell type markers.
(D) Shows the cells from different libraries.
(E) Shows the cell compositions of 6 m WT, 20 m WT, and 6 m KO.
(F) List the UMAP plots for selected markers genes that we used for the cell type annotation.
Our results showed cell population of Mmp9+ BMMs increased with aging (Figure 2D; Figure S2B) specifically more Mmp9+ BMMs were identified in 6 m KO than those in 20 m WT. Furthermore, the proportion of Mmp9+ BMMs in 6 m KO (63.07%) was larger than that in 20 m WT (58.36%) (Figure 2E). The proportion of Ly6g+Mmp9- monocytes was increased in 6 m KO and 20 m WT compared to 6 m WT, while the proportion of CMP and GMP was decreased. These findings revealed a trend toward increased BMMs differentiation in aged mice, specifically in 6 m KO and 20 m WT mice, compared to their young counterparts. Moreover, we also observed 21 Terc-associated genes significantly up/downregulated in 6 m KO and 20 m WT groups (Figure S2C). These observations were consistent with our previous findings from bulk RNA sequencing that showed OC differentiation from BMMs was affected during both natural and induced aging (Terc−/−). The markers that we used to annotate cell types are shown. Here, H2-k132 was employed to mark all the cells involved in OC differentiation, H2-q7 was the marker of hematopoietic stem cells; Cd79a33 marked CLP; Elane,34 Ms4a4, and Mpo34 marked cells in transition period of CMP and GMP; Cpox33 marked erythroid cells. Specifically, Ly6g and Mmp9 were employed to mark the BMM progenitors for OCs32,35 (Figure 2F).
Our findings highlight distinct cell populations and changes in OC cell differentiation that occur during the aging process and also reveal the cellular dynamics of murine bone marrow samples, which lay foundation of explorating the molecular mechanisms underlying aging in this tissue.
Cellular trajectory inference identified the dynamic cell population for osteoclast differentiation in mouse bone aging
To assess the differentiation dynamics within bone marrow cell clusters during the aging process, we employed RNA-velocity analysis (scVelo36) to construct an overall cellular trajectory. Clear transcriptional dynamics within the cell clusters was depicted (Figure 3A), with cluster 16 showing unique movement toward mature OCs. The progression from cluster 16 to cluster 17, cluster 2, and finally cluster 12 indicates the key steps involved in OC differentiation. Furthermore, we observed a lineage progression from GMP to Ly6g+Mmp9- monocytes, followed by further development into Mmp9+ BMMs, revealing the origin of OCs. Additionally, the lineage reconstruction technique, partition-based graph abstraction (PAGA37), was employed to capture the characteristics of multi-lineage cell differentiation and connectivity within subcluster populations, based on the differences in cell expression (Figure 3B). The likelihood of the transitions between cluster 16, 17, 2, and 12 is relatively strong (edge confidence > 0.2), and a sequential trajectory from cluster 16 to cluster 12 is observed.
Figure 3.

The cellular trajectory reconstructed from mouse bone marrow aging single-cell RNA-seq data
(A) The RNA-velocity plot that we got from the single-cell RNA-seq data, which infers the cellular trajectory based on RNA velocity.
(B) A cellular trajectory (graph) inferred from the single-cell RNA-seq data by PAGA. This trajectory inference is based on the expression difference between cells, which is complementary to the RNA velocity based trajectory.
(C) Presents a cellular trajectory predicted by SCDIFF (version 2.0), which integrates the RNA velocity-based trajectory with the expression based trajectory (by PAGA). In the combined trajectory, we identified a key cellular transition stage for osteoclast cell differentiation (Cluster 16, 17, 2, and 12), which is marked in green in the plot.
(D) Shows the gene expression of top predicted transcription factors across different cell clusters of the osteoclast cellular trajectory.
(E) Shows the top signature genes associated with each of those clusters.
To further elucidate the OC cell differentiation process, we utilized SCDIFF2,38 a single-cell trajectory inference method that we previously developed, which combines RNA velocity results with expression-based trajectory inferred from PAGA. This analysis identified a crucial developmental stage from cluster 16 to cluster 12 in OC cell differentiation (Figure 3C). The reconstructed trajectories also revealed transcription factors (TFs) that modulate the cellular trajectories (Figure 3D). Furthermore, we dissected the marker gene expression patterns associated with the transition stages of the subclusters. The marker genes related to different cell clusters (Figure 3E) showed high expression in the early stage and contrasting patterns in the terminal stage of OC differentiation. Collectively, these results revealed a cellular trajectory of OC differentiation during aging, which derived from CMP, GMP to Ly6g+Mmp9- monocytes, and further to Mmp9+ BMMs.
Gene regulatory networks identified key regulators that modulate osteoclast differentiation
To gain further insights into how gene regulatory networks in the bone marrow are altered during aging, we utilized the SCDIFF2 method to examine the OC and its progenitors and identified TFs involved.
In addition to the trajectory inference analysis discussed earlier, SCDIFF2 can identify critical regulators that govern cellular trajectories. We identified a list of TFs that modulate OC cell differentiation within clusters 16, 17, 2, and 12 (Figure 4A). Examples of these regulators include Cenpa, Irf8, Gabpa, and Cebpe, all of which exhibited cluster-specific enrichments along the OC differentiation trajectory. Notably, Sox4, Klf4, Irf8, and Cebpd were predicted to be key regulators of OC differentiation. While some of these TFs have been previously studied and shown potential links to OC differentiation during aging, direct evidence supporting their roles in the activation of osteoclastogenesis during aging is currently lacking. For instance, Klf4 expression disruption in aged macrophages is associated with the disruption of circadian innate immune homeostasis.39 OC differentiation is stabilized by DNA methylation at Irf8 mediated by DNA methyltransferase 3a (Dnmt3a), which suppresses Irf8 gene expression.40 It has also been reported that HnRNP A2 acts as a coactivator with nuclear TFs cRel and Cebpd for Ctsk promoter activation under hypoxic conditions.41 Furthermore, using the interactive dynamic event minor (iDREM42) approach that we developed, we identified key regulators. All four selected TFs were captured by the iDREM-based gene regulatory network inference, further confirming their roles in modulating OC differentiation during the aging process.
Figure 4.

Gene regulatory network inference from the single-cell RNA-seq data identifies 4 key regulators that modulate the critical cellular transitions to OCs
See also Figure S3.
(A) The cellular state transitions (tree) from the progenitors to the BMMs and the transcription factors underlying each of the edges, identified by SCDIFF2.
(B) The gene regulatory network reconstructed by the iDREM tool shows that Cebpd, Sox4, and Irf8 are regulating the gene expression dynamics between cluster 16 to cluster 12, the critical state transitions from the progenitors to BMMs.
(C) The log-normalized expression of the 4 predicted key regulators (Cebpd, Irf8, Sox4, and Klf4) are monotonically changing along with the trajectory.
(D) The expression changes of top predicted transcription factors along with the trajectory.
(E)Top differential genes associated with the critical cellular state transitions for OCs.
By integrating mRNA expression data and time-series single-cell RNA transcriptomic data, we visualized the predicted paths (a, b, c, and d) based on different combinations of genes, with the top related genes also listed (Figure 4B left and middle). Path a and b exhibited increased genes’ expression, while path c and d were on the contrary. It was also identified the significant up or downregulation of top related genes’ expression within trajectory from cluster 16 to cluster 12 (Figure 4B middle). Specifically, Cebpd acted as a controller of paths a and d, while Sox4 regulated path a and Irf8 was involved in path c (Figure 4B right). These three TFs displayed distinct trajectory regulation patterns, and we further quantified their absolute expression levels in the four clusters. We observed an increasing trend of Cebpd expression during OC differentiation, while the remaining TFs exhibited the opposite pattern (Figure 4C). Moreover, these four TFs also exhibited important regulatory effects in RNA trajectory inference. It is showed the four key TFs and other top predicted TFs’ average expression level and cell fraction in each cluster along trajectory (Figure 4D). We observed significant upregulation of Cebpd and downregulation of Sox4, Irf8, and Klf4. To elucidate the TFs controlling cellular trajectories, we identified TFs that were enriched in different clusters, noting their high expression in cluster 16 and decreased expression in clusters 17, 2, and 12 (Figure 4E). Additionally, we determined the cell composition of the four clusters and found that aged cells (20 m WT and 6 m KO) predominantly populated this OC-committed trajectory (Figure S3A). Cluster 17, 2, and 12 exhibited significant enrichment of aged cells, while cluster 16 was significantly enriched in 6 m WT cells (Figure S3B). Throughout the trajectory, the expression levels of Terc-related genes generally decreased (Figure S3C). These results elucidate the gene regulatory networks that control cellular trajectories and highlight four important TFs (Sox4, Irf8, Cebpd, and Klf4) that modulate the age-induced OC differentiation.
Single-cell DNA-seq analysis show the mutation feature of osteoclastogenesis process during mouse bone marrow aging
To investigate whether the genetic mutation is associated with OCs, we implement whole genome sequencing (WGS) and identified single-nucleotide variants in five age groups (1 m WT, 1 m KO, 6 m WT, 6 m KO, 20 m WT). As expected, more candidates somatic SNVs were detected in KO group (average 8802 per cell) than WT groups (average 1374 per cell). The number of SNVs of 6 m KO group was relatively larger than that of 6 m WT group (p = 0.097). Similarly, the number of SNVs of 1 m KO group was significantly larger than that of 1 m WT group (p = 0.0014). The number of SNVs between 20 m WT group and 6 m KO was significantly different (p = 0.031) (Figure 5A; Table S7). This finding suggested that Terc deficiency induced more genetic mutations. We also analyzed the point mutation spectra in these 5 age groups. We found high contribution of C>T transitions in each group and the contribution was increased with age (Figure 5B; Table S7). Previous study has reported that these mutations are known to be accumulated during aging, which are induced by DNA methylation.43 However, in our result, the contribution of C>T in 6 m KO group was significantly lower than that in 6 m WT. T>C contribution was the opposite, suggesting that Terc depletion reduced C>T transitions but induced T>C transitions. The SNPs identified between 6 m WT and 6 m KO are similar to the ones between 20 m WT and 6 m KO, which are distinct from the SNPs detected between 1 m WT and 1 m KO (Figure 5C; Table S8. Top significant mutation genes between 1 m WT and 1 m KO mice, related to Figure 5C, Table S9. Top significant mutation genes between 6 m WT and 6 m KO mice, related to Figure 5C, Table S10. Top significant mutation genes between 20 m WT and 6 m KO mice, related to Figure 5C). In addition, the genes associated with those SNPs were significantly enriched with OC differentiation and IL6-mediated pathways (p < 0.05), which indicates that the osteoclastogenesis process is significantly affected by the aging relevant mutations (Figure 5D; Figure S3D). Together, these results revealed that the genetic mutations are strongly associated with OC differentiation during aging.
Figure 5.

Genetic mutation inference from single-cell DNA-seq data supports the change of osteoclastogenesis during aging
See also Figure S3 and Table S7. Number and types of SNVs in bulk and single-cell groups, related to Figure 5A and 5B, Table S8. Top significant mutation genes between 1 m WT and 1 m KO mice, related to Figure 5C, Table S9. Top significant mutation genes between 6 m WT and 6 m KO mice, related to Figure 5C, Table S10. Top significant mutation genes between 20 m WT and 6 m KO mice, related to Figure 5C.
(A) Number of somatic SNVs per cell in five age groups.
(B) Contribution of the indicated six mutation types to the point mutation spectrum for the five age groups.
(C) A heatmap of mutation score (see STAR Methods for details) associated with the SNPs detected in the proximity of genes. The SNPs identified between 6 m WT vs. 6 m KO are similar to the ones between 20 m WT and 6 m KO, which are distinct from the SNPs detected between 1 m WT and 1 m KO.
(D) Pathway enrichment analysis suggests that the genes associated with those SNPs are significantly enriched with osteoclast differentiation, which indicates that the osteoclastogenesis process is significantly affected by the aging relevant mutations. Data are shown as mean ± s.d. n = 6 (3 WT and 3 KO) for 1 m; n = 8 (4 WT and 4 KO) for 6 m; n = 4 (WT) for 20 m. For (A), each dot represents a single individual. p values were determined by two-tailed Student’s t test.
Cell-cell interaction inference and the reconstructed signaling network support the identified regulators for osteoclastogenesis during aging
To gain insights into the intercellular communication within the bone marrow, we employed the CellChat approach, which infers and analyzes cell-cell interactions involving ligands, receptors, and cofactors.44 By computing the number of inferred interactions between each cell type and the rest of the cell populations, we identified strong interactions between CMP-GMP and GMP-Mmp9+ BMMs (Figure 6A). Notably, CMP and Mmp9+ BMMs exhibited more interactions compared to that of CMP with other groups, emphasizing the OC cellular trajectory from the progenitor (GMP) to the highly activated BMMs during aging. Furthermore, we quantified the likelihood of different signal inputs and outputs, and we found the most significant interaction in the GMP-Mmp9+ BMMs and Ly6g+ Mmp9- monocyte-Mmp9+ BMMs was Anxa1-Fpr2 pair(p < 0.05, Commun.Prob = max) (Figure 6B). The second significant one was C3-(Itgam+Itgb2). We assessed the expression of the receptors in Figure 6B, revealing 6 of them including Fpr2, Itgam, and Itgb2 were high expressed in Mmp9+ BMMs, which is consistent with the strong interactions predicted in Figure 6B (Figure 6C). Previous studies45,46 showed that Anxa1 was an agonist of Fpr2, and Anxa1-Fpr2 together inhibited OC differentiation. However, other researchers reported that C3 stimulated osteoblast differentiation.47,48 Itgam was positively regulated during bone resorption,49 and Itgb2 was identified as upregulated DEG related to osteoarthritis.50 In our study, the expression of them were increased along with OC differentiation trajectory, which means that the stimulation effect should be greater than the inhibition effect. As a result, osteoclastogenesis was promoted.
Figure 6.

Cell-cell interaction analysis infers potential critical ligand-receptors associated with aged-induced osteoclastogenesis
See also Table S11.
(A) The cell-cell interactions between different cell populations inferred by cellchat.
(B) The top ligand-receptors predicted between different cell populations.
(C) Gene expression of different receptors across different cell populations.
(D) Signaling network that connects the inferred receptors and key transcription factors that modulate the aged-induced osteoclastogenesis. Red, green, and blue square indicate ligand-receptors, signature genes, and TFs, respectively.
(E) Shows the top 20 enriched pathways of genes in (D).
To further explore the interactions between ligands, receptors, and TFs, we constructed a signaling network (Figure 6D; Table S11). Signature gene Akt1 was inferred as the hub gene that located in center of the network. Akt1 is often known as protein kinase B, whose activation relies on the PI3K pathway. Previous study has reported PI3K/AKT pathway was involved in metabolism, aging51 and osteoblast differentiation.52 The four TFs located in the inner circle (Sox4) and the outer circle (Irf8, Cebpd, and Klf4), exhibited the direct and indirect relationship with Akt1. It had been reported Sox4, Irf8, Cebpd, and Klf4 played a role in tumorigenesis and associated with dysregulation of Akt1.53,54,55,56 The four TFs might be involved in the process of aging. We also observed that the ligand-receptor interactions are strongly associated with Akt1 and the four core TFs, as evidenced by multiple gene-TF relationships. Our findings suggest the crucial roles of the four identified TFs in the aging process. We then performed pathway enrichment of genes in the network and found they were significantly enriched with aging related pathways such as PI3K-Akt-mTOR pathway,51 TNF-α- NF-κB pathway,57 and IL6-JAK-STAT3 pathway mentioned previously (p < 0.05) (Figure 6E).
The results of our analysis revealed strong interactions between CMP-GMP and GMP-Mmp9+ BMMs underscore the OC cellular trajectory during aging. Moreover, the identification of specific ligand-receptor interactions, such as Anxa1-Fpr2, and the expression profiles of receptors and TFs identified signaling network that governs the aging process. These findings further emphasize the importance of the four TFs in the aging-associated cellular dynamics within the bone marrow.
Experimental validation of the effect of key translational factors on regulating osteoclastogenesis of in vitro BMMs
To validate the effects of the candidate TFs identified in our single-cell sequencing analysis, we utilized RNA interference (RNAi) to knock down the four TFs in vitro-cultured BMMs. We observed that the expression of Cebpd increased in aged BMMs, while the expression of Irf8, Sox4, and Klf4 decreased during aging. Therefore, we knocked down Cebpd in BMMs derived from 20 m WT male mice, while Irf8, Sox4, and Klf4 were knocked down in BMMs derived from 6 m WT male mice using a shRNA-lentivirus system with the zsGreen reporter (Figure S4).
To assess the effects of TF knockdown on OC differentiation, we measured the expression of TRAP and CTSK, which are canonical markers of mature OCs, using RT-qPCR before and after 3 and 8 days of RANKL stimulation. These time points represent sequential stages of OC differentiation. Our results showed that the expression of TRAP and CTSK decreased in TF knockdown BMMs from 20 m mice and increased in TF knockdown BMMs from 6 m mice after 8 days of RANKL stimulation (Figure 7A; Tables S12 and S13). Additionally, pits array analysis revealed that aged BMMs formed smaller pits after Cebpd knockdown, while young BMMs with TF knockdown formed larger pits (Figure 7B; Table S14). Furthermore, TRAP staining of in vitro-cultured OCs confirmed that blockage of Cebpd in aged BMMs prevented the generation of mature OCs, whereas knockdown of Irf8, Sox4, and Klf4 in young BMMs led to an increased number of mature OCs. (Figures 7C–7E; Table S15). Collectively, our findings confirm that Cebpd promotes osteoclastogenesis and bone resorption during aging, while Irf8, Sox4, and Klf4 inhibit these processes. The knockdown experiments provide functional validation for the roles of these TFs in regulating OC differentiation and bone resorption dynamics during aging.
Figure 7.

Validation of the effects of key translational factors on regulating osteoclastogenesis of in vitro BMMs
See also Figure S4 and Table S12. The relative expression of TRAP and CTSK in scramble or knock-down OCs, related to Figure 7A left, Table S13. The relative expression of TRAP and CTSK in scramble or knock-down OCs, related to Figure 7A right, Table S14. Area of single resorption pit on bone slices (scramble and knock-down OCs), related to Figure 7B, Table S15. TRAP positive cells in 4 TFs knock-down in vitro cultured BMMs of 20 m and 6 m mice, related to Figure 7D.
(A) The expression of TRAP and CTSK, the canonical makers of mature OC, were measured by RT-qPCR before and after 3 days and 8 days the stimulation of RANKL, which are sequential time points along the differentiation of OC. The expression of TRAP and CTSK increased in TFs knockdown BMMs of 6 m and decreased in TFs knockdown BMMs of 20 m mice after 8 days stimulation of RANKL. The two upper plots showed the expression of CTSK and the lower plots showed the expression of TRAP; the two plots on the left demonstrated results from 20 m mice while the other two plots on the right showed the results from 6 m mice.
(B) Pits array analysis by SEM scanning showed smaller pits formed by aged (20 m) TF knockdown BMMs and bigger pits formed by young (6 m) TF knockdown BMMs.
(C–E) Images and representative inserts of TRAP staining of in vitro-cultured OC. TFs knockdown BMMs generated more TRAP positive cells in young (6 m) BMMs after Irf8, Sox4, and Klf4 were knocked down. On the contrary, aged (20 m) BMMs couldn’t form mature OCs after Cebpd was blocked. Data are shown as mean ± s.e.m. In (A), n = 116 (26 Scr, 36 Cebpd, 18 Sox4, 18 Irf8, 18 Klf4) for TRAP group; n = 116 (26 Scr, 36 Cebpd, 18 Sox4, 18 Irf8, 18 Klf4) for CTSK group, each dot represents a single individual. In (B), n = 10 (1 Scr, 2 Cebpd) for 20 m group; n = 7 (1 Scr, 2 Sox4, 2 Irf8, and 2 Klf4) for 6 m group. In (D), n = 9 (3 Scr, 6Cebpd) for 20 m. In (E), n = 28 (4 Scr, 8Sox4, 8 Irf8, 8 Klf4) for 6 m, each dot represents a single individual. p values were determined by two-tailed Student’s t test. “Scr” stands for scramble.
Discussion
In this study, we comprehensively profile the cellular states of bone marrow cells with single-cell multi-omics techniques, which enables the deep examination of different cell populations of bone marrow cells along with the aging process. Specifically, we clustered and annotated all the cells based on the single-cell RNA-seq measurements across different age groups (WT 6 months, WT 20 months, Terc−/− 6 months). We also demonstrated that the dysfunction of the telomerase can induce aged-related osteoporosis. As showed in the joint single-cell RNA-seq analysis, the OC differentiation in Terc−/− mice is significantly enhanced compared to its WT counterparts. As a result, 6-month Terc−/− mice show more evident bone aging phenotypes than 20 m wild-type mice.
Telomere and telomerase have gained increasing concern because telomere attrition and telomerase dysfunctions accelerate the speed of aging.58 One of the first and best characterized mechanisms of aging is genome instability caused by telomere complex dysfunction.59 Animal models with dysfunctional telomerase have been extensively used to explore the mechanism of aging and age-related diseases. The phenotypes of osteoporosis was observed and confirmed in an accelerated model of aging in mice where WS helicase and telomerase were genetically removed.60 It was later reported that single mutation in the telomerase gene (Terc) and double mutants of WS helicase and telomerase (Wern−/−Terc−/−) showed accelerated age-related osteoporosis.29 Mutations of telomerase complex genes are also associated with the phenotype of osteoporosis.61 These studies suggested the dysfunctions of the telomerase can induce aged-related osteoporosis. It has been revealed that telomerase has telomere length independent functions. The telomerase protein subunit is reported to regulate the expression of several genes including NF-κB, Myc, SF1, VEGF, and DNA methyltransferases, and most relevant to the regulation is inflammation.62 However, the link between telomerase dysfunction and osteoporosis and the mechanisms of regulation of BMMs/OCs differentiation and maturation during aging is not fully elucidated.
Through enrichment pathway analysis of different cell populations across different age groups, we identified dynamic cellular changes of BMMs/OCs during the aging process. Further examination of these dynamic cell populations revealed that the OCs differentiation substantially shifted during aging. Single-cell trajectory as well as the underlying gene regulatory and signaling network inference analyses identified critical TFs to dictate OCs differentiation. The identified TFs are differentially expressed as demonstrated in the reconstructed gene regulatory network. From single-cell DNA-seq data, we identified significant mutation DEGs between different aging groups. These genes (found in aged but not in adult mice) were enriched with the functions in OC differentiation, which supports our discovery that OC differentiation is substantially altered during the aging process. Finally, we validated our model predictions with in vitro knockdown experiments. A list of four TFs (Klf4, Cebpd, Irf8, and Sox4) out of the model top predictions was selected for experimental validation. Knocking down the predicted four TFs, respectively, has disrupted the differentiation of OC cells, which demonstrates the potential of these identified TFs in manipulating the cell fates of OCs. To our best knowledge, our study is the very first one that verifies the roles of the four TFs in regulating osteoclastogenesis during the aging process. The identification and validation of those four novel regulators clearly demonstrate the power of the single-cell multi-omics and also suggests potential interventions targets of age-related osteoporosis.
Limitations of the study
This study observed that the OC differentiation is significantly enhanced during aging in Terc−/− mice. However, the link between telomerase dysfunction and age-related osteoporosis is not fully elucidated. The possible explanation about this problem is that aging alters the microenvironment of bone marrow, which causes the expression changes of these four TFs. While the other possibility is that telomerase has telomere length independent functions, which have direct interactions with the four key TFs in BMMs/OCs. These mechanisms need further investigations. Moreover, the single-cell DNA-seq measurements performed in this study remains relatively low-resolution. We plan to increase the scale of single-cell DNA-seq and another single-cell omic measurement (e.g., single-cell ATAC-seq) to enable a more accurate and comprehensive single-cell profiling of the bone marrow during the aging process. Finally, the effects of these four TFs on BMMs/OCs during aging should be experimentally verified in vivo. We plan to develop corresponding animal models to conduct further studies for elucidating the mechanism.
Resource availability
Lead contact
Additional information and requests for resources and reagents should be directed to the lead contact, Hui Wang (huiwang@shsmu.edu.cn).
Materials availability
This study did not generate any unique reagents.
Data and code availability
-
•
Data: Raw single-cell DNA-seq and bulk DNA-seq data have been deposited in NCBI database. Raw single-cell RNA-seq and bulk RNA-seq data are available in GEO database. Accession numbers are listed in the key resources table.
-
•
Code: All other data of bioinformatics analysis are available at http://dinglab.rimuhc.ca:8080/projects/aging_shsmu/.
-
•
Additional information: Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
We thank all the enrolled participants who contributed specimens and data to this study. This study was supported by grants from the National Natural Science Foundation of China (82030099, 32270202, 82002339, and 81820108020), the MOST Key R&D 805 Program of China (2022YFC2304703 and 2020YFA0907200), National Key R&D Program of China (2022YFD2101500), the Science and Technology Commission of Shanghai Municipality (22DZ2303000), Shanghai Frontiers Science Center of Degeneration and Regeneration in Skeletal System (BJ1-9000-22-4002), Shanghai Public Health System Construction Three-Year Action Plan (GWVI-11.1-43), Medicine and Engineering Interdisciplinary Research Fund of Shanghai Jiao Tong University (YG2019QNB39 and YG2020YQ19), Innovative Research Team of High-level Local Universities in Shanghai, Shanghai Municipal Hospital Orthopedic Specialist Alliance, Shanghai Municipal Health Commission Key Priority Discipline Project, Shanghai Spinal Disease and Trauma Orthopedics Research Center (2022ZZ01014), CIHR PJT-180505, FRQS 295298 and 295299, NSERC RGPIN2022-04399. We thank NovelBio Co., Ltd for technical support of single-cell RNA sequencing.
Author contributions
H.W., C.-Q.Z., J.-J.G., N.-N.L. and J.D. conceived and designed the study. H.L. and J.-C.T. performed the experiments. J.D., H.L., W.-X.X., and Y.-M.Z. performed the data analysis. M.L., X.-Q.Z., J.H., B.-P.Z., and J.-A.Z. provided animals, facilities, and technical guidance. J.D., H.L., W.-X.X., J.-C.T., and Y.-M.H. drafted the manuscript. J.-J.G., N.-N.L., J.D., H.L., and W.-X.X. revised the manuscript. All authors discussed the experiments and results and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Mouse bone marrow | Collected from long bones (femurs and tibias) of C57BL/6J and B6.Cg-Terctm1Rdp/J strains. | N/A |
| Critical commercial assays | ||
| Chromium Single Cell 3′ V3.1 Reagent Kits | 10X Genomics | 1000121 |
| Leukocyte Acid Phosphatase (TRAP) Kit | Sigma-Aldrich | 387A |
| NucleoSpin RNA II | MN(Macherey-Nagel) | 740955.50 |
| REPLI-g UltraFast Mini Kit | Qiagen | 150035 |
| QIAamp DNA mini kit | Qiagen | 51306 |
| Deposited data | ||
| Raw single-cell DNA-seq and bulk DNA-seq data | This paper | PRJNA906340 |
| Raw single-cell RNA-seq and bulk RNA-seq data | This paper | GSE190535; GSE169608; GSE224119 |
| Mouse reference genome mm10 | UCSC Genome Browser | http://genome.ucsc.edu/cgi-bin/hgGateway?db=mm10 |
| Experimental models: organisms/strains | ||
| C57BL/6J, mus musculus | The Jackson Laboratory | IMSR_JAX:000664 |
| B6.Cg-Terctm1Rdp/J,mus musculus | The Jackson Laboratory | IMSR_JAX:004132 |
| Oligonucleotides | ||
| shRNA targeting sequence: Irf8 #1: GGACATTTCTGAGCCATATAA | This paper | N/A |
| shRNA targeting sequence: Irf8 #2: GCCTATGACACACACCATTCA | This paper | N/A |
| shRNA targeting sequence: Cebpd #1: GCTGGCCTCCGGCAGTTCTTC | This paper | N/A |
| shRNA targeting sequence: Cebpd #2: GCTCCACGACTCCTGCCATGT | This paper | N/A |
| shRNA targeting sequence: Klf4 #1: GACCTAGACTTTATCCTTTC | This paper | N/A |
| shRNA targeting sequence: Klf4 #2: GGTCATCAGTGTTAGCAAAGG | This paper | N/A |
| shRNA targeting sequence: Sox4 #1: GCGACAAGATTCCGTTCATCC | This paper | N/A |
| shRNA targeting sequence: Sox4 #2: GCTCAAGGACAGCGACAAGAT | This paper | N/A |
| Recombinant DNA | ||
| pGMLV-zsGreen-vshRNA-TFs | Genomeditech | N/A |
| Software and algorithms | ||
| DESeq2 (v1.38.3) | Love et al.63 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| fastp (v0.21.0) | Chen et al.64 | https://github.com/OpenGene/fastp |
| CellRanger (v5.0.0) | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-pression/software/pipelines/latest/what-is-cell-ranger |
| Scanpy (v1.8.2) | Wolf et al.65 | https://github.com/scverse/scanpy |
| velocyto (v.17.17) | La Manno et al.66 | http://velocyto.org/ |
| SCDIFF (v2) | Ding et al.38 | https://github.com/phoenixding/scdiff2 |
| CellChat (v1.5.0) | Jin et al.44 | https://github.com/sqjin/CellChat |
| HISAT2 (v2.2.0) | Kim et al.67 | http://daehwankimlab.github.io/hisat2/ |
| SCcaller (v2.0.0) | Dong et al.68 | https://github.com/biosinodx/SCcaller |
| bwa (v0.7.17) | Li et al.69 | https://github.com/lh3/bwa |
| GATK (v4.1.6) | McKenna et al.70 | https://gatk.broadinstitute.org/hc/en-us |
| ToppGene Suite | Chen et al.30 | https://toppgene.cchmc.org/ |
| GraphPad Prism 9 | Graphpad Software | https://www.graphpad.com/ |
Experimental model and study participant details
Animals
All animal breeding and sample harvest procedures were approved by ethics committee of Shanghai Sixth People’s Hospital Affiliated to Shanghai Jiao Tong University School of medicine (approval number: 2020-0355). The C57BL/6J Terc knockout (mTRKO) animals71 (JAX stock #004132) were a kind gift from Prof. Ming Lei. All mice were kept in C57BL/6J background and maintained in standard, specific pathogen-free facility at a controlled temperature (22°C–24°C), with a 12 h dark/light cycle (07:00 to 19:00 light on), with standard food and water provided ad libitum and environmental enrichments. The mice are healthy and used for in-house mating to generate enough mice for experiments. Only male mice were used for experiments and the littermate controls were used. The genotypes of mice were determined by PCR. The primers of genotyping for Terc−/− mice are: Terc F- ATTTGTCACGTCCTGCACGACG; WT F-GCACTCCTTACAAGGGACGA; common R-CTTCAATTTCCTTGGCTTCG. Wild type mice were raised up to 6 months and 20 months. Terc−/− mice was generated by crossing the Terc+/−. Terc−/− mice and wild type littermates were raised up to 6 months.
Method details
Cell isolation and culture
After being completely anesthetized, mice were euthanatized by cervical vertebral dislocation. Mice bone marrow tissue was harvested, and bone marrow derived monocytes were obtained to generate mature OC in vitro. BMMs were cultured in Minimum Essential Medium α (MEM α, Gibco, Cat. 12571071) with 10% fetal bovine serum (FBS). Osteoclastogenesis assays were carried out. The BMMs were cultured in complete α-MEM supplemented with 25 ng/ml M-CSF in 24-well plates and 96-well plates with 4x104 cells per well or 1x104 cells per well respectively. In both instances, cells were stimulated with 100 ng/ml RANKL through culture medium changing at different time points (1, 3, 5 days and so on). After the big multinucleated cell presenting, the cells were either extracted for RNA lysates (24-well plates) or fixed with 4% PFA (96-well plates) for further analysis.
Single cell isolation for scRNA-seq
Single-cell RNA-seq experiment was performed by experimental personnel in the laboratory of NovelBio Bio-Pharm Technology Co.,Ltd. The tissues were surgically removed and kept in MACS Tissue Storage Solution (Miltenyi Biotec) until processing. The tissue samples were processed as described below. Briefly, Bones were harvested, samples were first washed with phosphate-buffered saline (PBS), bone marrow was flushed, samples were sieved through a 40μm cell strainer, and centrifuged at 300g for 5 min. After the supernatant was removed, the pelleted cells were suspended in red blood cell lysis buffer (Miltenyi Biotec) to lyse red blood cells. After washing with PBS containing 0.04% BSA, the cell pellets were re-suspended in PBS containing 0.04% BSA and re-filtered through a 35 μm cell strainer. Dissociated single cells were then stained with AO/PI for viability assessment using Countstar Fluorescence Cell Analyzer.
scRNA-seq sample and library preparation
The scRNA-Seq libraries were generated using the 10X Genomics Chromium Controller Instrument and Chromium Single Cell 3′ V3.1 Reagent Kits (10X Genomics, Pleasanton, CA, 1000121). Briefly, cells were concentrated to 1000 cells/uL and approximately 8,000 cells were loaded into each channel to generate single-cell Gel Bead-In-Emulsions (GEMs), which results into expected mRNA barcoding of 6,000 single-cells for each sample. After the RT step, GEMs were broken and barcoded-cDNA was purified and amplified. The amplified barcoded cDNA was fragmented, A-tailed, ligated with adaptors and index PCR amplified. The final libraries were quantified using the Qubit High Sensitivity DNA assay (Thermo Fisher Scientific) and the size distribution of the libraries were determined using a High Sensitivity DNA chip on a Bioanalyzer 2200 (Agilent). All libraries were sequenced by illumina sequencer (Illumina, San Diego, CA) on a 150 bp paired-end run.
Single cell and bulk DNA library preparation and whole genome sequencing
Femurs and tibias were dissected and cut at both ends with sharp sterile scissors. Bone marrow cells were flushed out using pre-warmed DMEM (Meilunbio, MA0591) until the flow through turned white, filtered with a cell strainer (Falcon, 70 μm, 352350) to remove debris and bone fragment and washed with PBS 2 times. Red cells were removed using red cells lysis buffer (Beyotime, C3702). Bone marrow cells were resuspended in pre-warmed DMEM so that the cells were ready for single cell isolation and bulk DNA extraction.
Single cells were prepared using the CellRaft array (Cell Microsystems) based on the methods described in Dong et al.68 In brief, Poly-D-lysine (Sigma, P6407) were added into the CellRaft array and incubate at 37°C overnight. A total of 1.6 mL pre-warmed DMEM medium were added and the cell solution (4,000 cells in 600 μL) was added evenly onto the CellRaft array. The array was centrifuged for 30 s at 200 g and observed under microscope (Nikon, ECLIPSE-Ci-L) to confirm all cells were detached. Remove the supernatant carefully and add 4 mL HBSS (Thermo, 14155063) (containing 2.5% FBS) to the array gently.
Single rafts containing one cell (as observed by microscopy with a 10× objective) were transferred with the magnetic wand supplied with the CellRaft system into 0.2-mL PCR tubes containing 3 μL PBS. By observing the PCR tubes under a magnifying glass and the CellRaft array under a microscope before and after the transfer, only one cell per tube was validated. Tubes containing single rafts were frozen immediately on crushed dry ice and kept at −80°C until use.
Single-Cell Multiple Displacement Amplification (SCMDA) was performed using Exo-Resistant Random Primer (Thermo Scientific, SO181) and REPLI-g UltraFast Mini Kit (Qiagen, 150035) according to the methods described in Dong et al.68 Amplified DNA was purified with AMPureXP-beads (Beckman Coulter, A63880), and the concentration was measured with a Qubit High Sensitivity dsDNA Kit (Invitrogen Life Science, Q32854). As a positive control for the amplification, 1 ng of mice genomic DNA in 3 μL PBS was also amplified, and 3 μL of PBS without any template was used as a negative control.
DNA from bulk cell populations were prepared with QIAamp DNA mini kit (Qiagen, 51306) and libraries were prepared with NEB Next Ultra DNA Library Prep Kit for Illumina (NEB). All the bulk and single-cell libraries were sequenced on Illumina NovaSeq 6000 with 2 × 150-bp paired-end reads.
Bulk RNA extraction and sequencing
Male C57BL/6J mice were euthanized under anesthesia. Bone marrow was harvested from long bones by rinsing with culture medium. Nucleospin RNA II kits was used for total RNA extraction. Subsequent denaturation by magnetic oligo (dT) beads was performed before the RNA sample purification. After the first-strand cDNA being obtained by reverse transcription of purified mRNA, and a second cDNA being synthesized, fragmented DNA samples were blunt ended and adenylated at the 3′ ends. Adaptors were ligated to construct a library. DNA samples quantification was performed by Qubit (Invitrogen), which were then sequenced by an Illumina Nova-seq 6000 instrument from Genergy Bio (Shanghai) after cBot cluster generation.
Knock-down validations
To silence the expression of four TFs, Klf4, Sox4, Irf8 and Cebpd BMMs, we constructed four shRNA lentiviral vectors based on the shRNAi vector pGMLV (pGMLV-zsGreen-vshRNA, Genomeditech, Shanghai, China). The four shRNA-targeting sequences for these TFs were as follows:
Irf8-shRNA1:GGACATTTCTGAGCCATATAA;
Irf8-shRNA2:GCCTATGACACACACCATTCA;
Cebpd-shRNA1:GCTGGCCTCCGGCAGTTCTTC;
Cebpd-shRNA2:GCTCCACGACTCCTGCCATGT;
Klf4-shRNA1:GACCTAGACTTTATCCTTTC;
Klf4-shRNA2:GGTCATCAGTGTTAGCAAAGG;
Sox4-shRNA1:GCGACAAGATTCCGTTCATCC;
Sox4-shRNA2:GCTCAAGGACAGCGACAAGAT.
The recombinant pGMLV-zsGreen-vshRNA-TFs were identified by PCR and DNA sequencing. Lentivirus packaging was conducted in 293T cells, followed by transfection with the shRNA-TFs into A375 cells. A scramble lentiviral vector was used as a negative control. The optimum shRNA fragment against TFs was determined by real-time PCR. The interference of this selected shRNA on TFs was verified by fluorescent array.
Quantification and statistical analysis
Single-cell RNA data preprocessing
scRNA-seq data analysis was performed by NovelBio Bio-Pharm Technology Co.,Ltd. with NovelBrain Cloud Analysis Platform. We applied fastp v0.21.064 with default parameter filtering the adaptor sequence and removed the low-quality reads to achieve the clean data. Then the feature-barcode matrices were obtained by aligning reads to the mouse genome (mm10) using CellRanger v5.0.0. We applied the down sample analysis among samples sequenced according to the mapped barcoded reads per cell of each sample and finally achieved the aggregated matrix. Cells contained over 200 expressed genes and mitochondria UMI rate below 20% passed the cell quality filtering and mitochondria genes were removed in the expression table. The filtered cell-by-gene matrices will be further analyzed using Scanpy v1.8.2.65
Single-cell RNA-seq data processing and clustering
With the raw cell-by-gene expression matrix, we further filtered the genes with a small dispersion (i.e., 0.05) of gene expression or with very small expression (e.g., 0.01). The filtered cell-by-gene gene expression matrices will be further normalized and log1p transformed using the Scanpy tool. The final normalized gene expression matrices were then dimensionality reduced with PCA72 and then were clustered using the Leiden approach73 in the Scanpy tool. Here, a default resolution parameter of 1.0 and a neighborhood graph (k = 15) were used for the Leiden clustering of all the cells in the sample. Next, the top differential genes (signatures) associated with each cluster were identified by comparing them with the cells in all other clusters. All the obtained clusters were annotated with the cell type by comparing the top signature genes of each cluster with the known cell type marker genes from databases such as CellMarker31 and PanglaoDB.74 To visualize all the cells and clusters in the data, here Uniform Manifold Approximation and Project (UMAP) methods have been employed to map the cells into the 2D space.
Reconstruction of cellular dynamics trajectory and underlying gene regulatory network
We employed multiple strategies to robustly infer the cellular trajectory that represents the cellular state transitions between different cell populations. Namely, the final cellular trajectory is built by combining the cellular trajectories inferred from RNA velocity (e.g., velocyto) and from between-cell cellular similarity (e.g., PAGA).
The RNA velocity is the time derivative of the gene expression state, which can be estimated from the unspliced and spliced mRNAs profiled in the single-cell RNA-seq. RNA velocity is a high-dimensional vector that predicts the future state of individual cells on a timescale of hours. Therefore, RNA velocity presents unique opportunities (from the perspective of future change of individual cells) to reconstruct the cellular dynamics (in the form of cellular trajectories). In this study, we used the velocyto v.17.1766 method to infer the RNA velocity for all the cells in the sample.
Partition-based graph abstraction
Another common trick to infer cellular dynamics is to compare the expression between different cells and clusters. Partition-based graph abstraction (PAGA)37 presents an interpretable graph-like map of the single-cell data manifold, inferred from estimating the connectivity of different partitions of the manifold. The node in the graph denotes different cell populations (cell clusters) while the edges between nodes represent the strength of the connection between clusters. Such a graph representation represents the cellular transitions between different cell populations (i.e., cell clusters). Here we employed the PAGA function in the Scanpy toolset for the inference of the PAGA graph, which can take the inferred RNA velocity vectors as inputs. Such a joint strategy will deliver a trajectory graph that integrates both the RNA velocity and between-cell expression comparison and thus could more accurately depict the cellular state transitions in the single-cell data.
Reconstruction of Gene regulatory networks that dictate the gene expression dynamics in the sample
To identify potential strategies to manipulate the cell fates in the studied biological process, here we also reconstruct the gene regulatory network that modulates the cellular trajectories for all cell populations in the data. To achieve such an objective, here we extend and apply the SCDIFF38 that we previously developed to this mouse bone aging single-cell data. The extended SCDIFF (named SCDIFF2) can directly take the clustering and trajectory inference results from the Scanpy tool (in the format of H5AD) as the inputs to infer the gene regulatory network that modulates the transition between cell populations (edges in the inferred PAGA graph). For each edge in the graph, we will infer the differential genes using the Wilcoxson rank-sums test.75 The transcription factors for each edge in the trajectory graph will be given by the SCDIFF2 method. The transcription factors and their target genes (differential genes) associated with the graph edges will constitute the gene regulatory networks that dictate the cellular dynamics, which should also be the candidate targets for cell fate manipulations.
Inference of the cell-cell interactions between different cellular populations
The cross-talking between different cell populations is also critical for regulating cellular dynamics as reported in previous studies and thus cell-cell interaction inference is an indispensable piece of the cellular dynamics puzzle in mouse bone aging, particularly if we are interested in finding potentially effective cell fate manipulation strategies. In this work, we utilized CellChat v1.5.044 to infer cell-cell interactions in the data. The cell clusters, together with the cell type annotation, from the Scanpy tool, were fed into the CellChat method to infer the interaction between different cell populations. Significant Ligand-Receptor pairs will also be provided between each cluster pair.
Bulk RNA-seq data analysis
The bulk RNA-seq reads were trimmed to remove the adapter and segments of bad quality. The trimmed reads were then mapped to the mouse reference genome mm10 using HISAT2 v2.2.0.67 HTSeq-count were then used to quantify the raw gene expression count matrices for all samples. The raw gene expression count matrices were then normalized using the DESeq2 v1.38.3,63 which also identify the differential genes between samples of different conditions (e.g., WT vs. Terc−/−). Here we used an FDR cutoff (q < 0.05) and log2 fold change (|log2fc| > 1) to call significant differential genes from DESeq2 output. Furthermore, the up-regulated (q < 0.05 & log2fc > 1) and down-regulated genes (q < 0.05 & log2fc < −1) were analyzed separately in the downstream analyses.
Single-cell DNA-seq data analysis
The raw single-cell DNA-seq reads were trimmed with fastp v0.21.0.64 Then we used bwa v0.7.1769 to align the reads to the mouse mm10 reference genome. We performed somatic SNV calling through GATK v4.1.670 and SCcaller v2.0.0.68 The single-cell mutation identification is notoriously noisy and suffers from enormous false positives. To address this issue, here we employed statistical measurements to remove potential false positives by leveraging known mutations reported in public databases. Specially, we score each candidate mutation based on the following aspects. First, the mutation will be assigned a higher score if it is already reported in a public mutation database such as snp142 common.76 Second, A mutation will be given a higher weight if it was shared by multiple samples in our study (e.g., 1 m WT and 6 m WT). Third, a mutation will be given a higher score if the mutation quality score (QUAL) is higher. Here, if mutation i is found in public mutation database, otherwise P(i) will be assigned as the default value (e.g., 0.5). Similarly, if a mutation i is shared by multiple samples, it will be given a score of 1, otherwise a default 0.5 will be assigned. Q(i) will be normalized score of the mutation quality (in range [0,1]). All these mutations will be used to estimate a null background, which will be used to calculate a significance p-value for all each mutation. Similarly, we can also calculate a mutation significance p-value for each gene by summarizing all the mutations within the region (and normalized by the gene length). A null background score distribution will need to be learned for the gene mutation score and the genes with scores that are significantly larger than the background will be considered as significant mutation genes.
Gene set enrichment analysis
All the gene sets (e.g., differential genes), whichever data in this study it comes from, were analyzed with the functional enrichment analysis. Here, we used the ToppGene Suite30 for the GO term and pathway enrichment analyses. All the functional terms (GO terms or pathway) with FDR < 0.05 will be considered as significant.
Published: August 18, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110734.
Contributor Information
Jun Ding, Email: jun.ding@mcgill.ca.
Ning-Ning Liu, Email: liuningning@shsmu.edu.cn.
Jun-Jie Gao, Email: colingjj@163.com.
Chang-Qing Zhang, Email: zhangcq@sjtu.edu.cn.
Hui Wang, Email: huiwang@shsmu.edu.cn.
Supplemental information
References
- 1.Partridge L., Deelen J., Slagboom P.E. Facing up to the global challenges of ageing. Nature. 2018;561:45–56. doi: 10.1038/s41586-018-0457-8. [DOI] [PubMed] [Google Scholar]
- 2.Rachner T.D., Khosla S., Hofbauer L.C. Osteoporosis: now and the future. Lancet. 2011;377:1276–1287. doi: 10.1016/S0140-6736(10)62349-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perkins S.L., Gibbons R., Kling S., Kahn A.J. Age-related bone loss in mice is associated with an increased osteoclast progenitor pool. Bone. 1994;15:65–72. doi: 10.1016/8756-3282(94)90893-1. [DOI] [PubMed] [Google Scholar]
- 4.Cao J.J., Wronski T.J., Iwaniec U., Phleger L., Kurimoto P., Boudignon B., Halloran B.P. Aging increases stromal/osteoblastic cell-induced osteoclastogenesis and alters the osteoclast precursor pool in the mouse. J. Bone Miner. Res. 2005;20:1659–1668. doi: 10.1359/JBMR.050503. [DOI] [PubMed] [Google Scholar]
- 5.Sui B., Hu C., Liao L., Chen Y., Zhang X., Fu X., Zheng C., Li M., Wu L., Zhao X., Jin Y. Mesenchymal progenitors in osteopenias of diverse pathologies: differential characteristics in the common shift from osteoblastogenesis to adipogenesis. Sci. Rep. 2016;6 doi: 10.1038/srep30186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yasuda H., Shima N., Nakagawa N., Mochizuki S.I., Yano K., Fujise N., Sato Y., Goto M., Yamaguchi K., Kuriyama M., et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139:1329–1337. doi: 10.1210/endo.139.3.5837. [DOI] [PubMed] [Google Scholar]
- 7.Kong Y.Y., Yoshida H., Sarosi I., Tan H.L., Timms E., Capparelli C., Morony S., Oliveira-dos-Santos A.J., Van G., Itie A., et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 8.Xiong J., Onal M., Jilka R.L., Weinstein R.S., Manolagas S.C., O'Brien C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011;17:1235–1241. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soltanoff C.S., Yang S., Chen W., Li Y.P. Signaling networks that control the lineage commitment and differentiation of bone cells. Crit. Rev. Eukaryot. Gene Expr. 2009;19:1–46. doi: 10.1615/critreveukargeneexpr.v19.i1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asagiri M., Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40:251–264. doi: 10.1016/j.bone.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 11.Tsukasaki M., Hamada K., Okamoto K., Nagashima K., Terashima A., Komatsu N., Win S.J., Okamura T., Nitta T., Yasuda H., et al. LOX Fails to Substitute for RANKL in Osteoclastogenesis. J. Bone Miner. Res. 2017;32:434–439. doi: 10.1002/jbmr.2990. [DOI] [PubMed] [Google Scholar]
- 12.Li Y.P., Alexander M., Wucherpfennig A.L., Yelick P., Chen W., Stashenko P. Cloning and complete coding sequence of a novel human cathepsin expressed in giant cells of osteoclastomas. J. Bone Miner. Res. 1995;10:1197–1202. doi: 10.1002/jbmr.5650100809. [DOI] [PubMed] [Google Scholar]
- 13.Franceschi C., Bonafè M., Valensin S., Olivieri F., De Luca M., Ottaviani E., De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 14.Jung Y.K., Kang Y.M., Han S. Osteoclasts in the Inflammatory Arthritis: Implications for Pathologic Osteolysis. Immune Netw. 2019;19 doi: 10.4110/in.2019.19.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coppé J.P., Patil C.K., Rodier F., Sun Y., Munoz D.P., Goldstein J., Nelson P.S., Desprez P.Y., Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farr J.N., Fraser D.G., Wang H., Jaehn K., Ogrodnik M.B., Weivoda M.M., Drake M.T., Tchkonia T., LeBrasseur N.K., Kirkland J.L., et al. Identification of Senescent Cells in the Bone Microenvironment. J. Bone Miner. Res. 2016;31:1920–1929. doi: 10.1002/jbmr.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Brien W., Fissel B.M., Maeda Y., Yan J., Ge X., Gravallese E.M., Aliprantis A.O., Charles J.F. RANK-Independent Osteoclast Formation and Bone Erosion in Inflammatory Arthritis. Arthritis Rheumatol. 2016;68:2889–2900. doi: 10.1002/art.39837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yokota K., Sato K., Miyazaki T., Kitaura H., Kayama H., Miyoshi F., Araki Y., Akiyama Y., Takeda K., Mimura T. Combination of tumor necrosis factor alpha and interleukin-6 induces mouse osteoclast-like cells with bone resorption activity both in vitro and in vivo. Arthritis Rheumatol. 2014;66:121–129. doi: 10.1002/art.38218. [DOI] [PubMed] [Google Scholar]
- 19.Xu M., Palmer A.K., Ding H., Weivoda M.M., Pirtskhalava T., White T.A., Sepe A., Johnson K.O., Stout M.B., Giorgadze N., et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife. 2015;4 doi: 10.7554/eLife.12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu M., Tchkonia T., Ding H., Ogrodnik M., Lubbers E.R., Pirtskhalava T., White T.A., Johnson K.O., Stout M.B., Mezera V., et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA. 2015;112:E6301–E6310. doi: 10.1073/pnas.1515386112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farr J.N., Xu M., Weivoda M.M., Monroe D.G., Fraser D.G., Onken J.L., Negley B.A., Sfeir J.G., Ogrodnik M.B., Hachfeld C.M., et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017;23:1072–1079. doi: 10.1038/nm.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mo C., Guo J., Qin J., Zhang X., Sun Y., Wei H., Cao D., Zhang Y., Zhao C., Xiong Y., et al. Single-cell transcriptomics of LepR-positive skeletal cells reveals heterogeneous stress-dependent stem and progenitor pools. EMBO J. 2022;41 doi: 10.15252/embj.2021108415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yahara Y., Barrientos T., Tang Y.J., Puviindran V., Nadesan P., Zhang H., Gibson J.R., Gregory S.G., Diao Y., Xiang Y., et al. Erythromyeloid progenitors give rise to a population of osteoclasts that contribute to bone homeostasis and repair. Nat. Cell Biol. 2020;22:49–59. doi: 10.1038/s41556-019-0437-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ambrosi T.H., Marecic O., McArdle A., Sinha R., Gulati G.S., Tong X., Wang Y., Steininger H.M., Hoover M.Y., Koepke L.S., et al. Aged skeletal stem cells generate an inflammatory degenerative niche. Nature. 2021;597:256–262. doi: 10.1038/s41586-021-03795-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsukasaki M., Huynh N.C.N., Okamoto K., Muro R., Terashima A., Kurikawa Y., Komatsu N., Pluemsakunthai W., Nitta T., Abe T., et al. Stepwise cell fate decision pathways during osteoclastogenesis at single-cell resolution. Nat. Metab. 2020;2:1382–1390. doi: 10.1038/s42255-020-00318-y. [DOI] [PubMed] [Google Scholar]
- 26.Shao H., Ge M., Zhang J., Zhao T., Zhang S. Osteoclasts differential-related prognostic biomarker for osteosarcoma based on single cell, bulk cell and gene expression datasets. BMC Cancer. 2022;22:288. doi: 10.1186/s12885-022-09380-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdallah B.M., Haack-Sørensen M., Fink T., Kassem M. Inhibition of osteoblast differentiation but not adipocyte differentiation of mesenchymal stem cells by sera obtained from aged females. Bone. 2006;39:181–188. doi: 10.1016/j.bone.2005.12.082. [DOI] [PubMed] [Google Scholar]
- 28.Saeed H., Abdallah B.M., Ditzel N., Catala-Lehnen P., Qiu W., Amling M., Kassem M. Telomerase-deficient mice exhibit bone loss owing to defects in osteoblasts and increased osteoclastogenesis by inflammatory microenvironment. J. Bone Miner. Res. 2011;26:1494–1505. doi: 10.1002/jbmr.349. [DOI] [PubMed] [Google Scholar]
- 29.Brennan T.A., Egan K.P., Lindborg C.M., Chen Q., Sweetwyne M.T., Hankenson K.D., Xie S.X., Johnson F.B., Pignolo R.J. Mouse models of telomere dysfunction phenocopy skeletal changes found in human age-related osteoporosis. Dis. Model. Mech. 2014;7:583–592. doi: 10.1242/dmm.014928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen J., Bardes E.E., Aronow B.J., Jegga A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–W311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X., Lan Y., Xu J., Quan F., Zhao E., Deng C., Luo T., Xu L., Liao G., Yan M., et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019;47:D721–D728. doi: 10.1093/nar/gky900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giladi A., Paul F., Herzog Y., Lubling Y., Weiner A., Yofe I., Jaitin D., Cabezas-Wallscheid N., Dress R., Ginhoux F., et al. Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat. Cell Biol. 2018;20:836–846. doi: 10.1038/s41556-018-0121-4. [DOI] [PubMed] [Google Scholar]
- 33.Tusi B.K., Wolock S.L., Weinreb C., Hwang Y., Hidalgo D., Zilionis R., Waisman A., Huh J.R., Klein A.M., Socolovsky M. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature. 2018;555:54–60. doi: 10.1038/nature25741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen T., Delano M.J., Chen K., Sperry J.L., Namas R.A., Lamparello A.J., Deng M., Conroy J., Moldawer L.L., Efron P.A., et al. A road map from single-cell transcriptome to patient classification for the immune response to trauma. JCI Insight. 2021;6 doi: 10.1172/jci.insight.145108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jahchan N.S., Mujal A.M., Pollack J.L., Binnewies M., Sriram V., Reyno L., Krummel M.F. Tuning the Tumor Myeloid Microenvironment to Fight Cancer. Front. Immunol. 2019;10:1611. doi: 10.3389/fimmu.2019.01611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bergen V., Lange M., Peidli S., Wolf F.A., Theis F.J. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol. 2020;38:1408–1414. doi: 10.1038/s41587-020-0591-3. [DOI] [PubMed] [Google Scholar]
- 37.Wolf F.A., Hamey F.K., Plass M., Solana J., Dahlin J.S., Göttgens B., Rajewsky N., Simon L., Theis F.J. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019;20:59. doi: 10.1186/s13059-019-1663-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding J., Aronow B.J., Kaminski N., Kitzmiller J., Whitsett J.A., Bar-Joseph Z. Reconstructing differentiation networks and their regulation from time series single-cell expression data. Genome Res. 2018;28:383–395. doi: 10.1101/gr.225979.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blacher E., Tsai C., Litichevskiy L., Shipony Z., Iweka C.A., Schneider K.M., Chuluun B., Heller H.C., Menon V., Thaiss C.A., Andreasson K.I. Aging disrupts circadian gene regulation and function in macrophages. Nat. Immunol. 2022;23:229–236. doi: 10.1038/s41590-021-01083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ivashkiv L.B. Metabolic-epigenetic coupling in osteoclast differentiation. Nat. Med. 2015;21:212–213. doi: 10.1038/nm.3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guha M., Srinivasan S., Koenigstein A., Zaidi M., Avadhani N.G. Enhanced osteoclastogenesis by mitochondrial retrograde signaling through transcriptional activation of the cathepsin K gene. Ann. N. Y. Acad. Sci. 2016;1364:52–61. doi: 10.1111/nyas.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding J., Hagood J.S., Ambalavanan N., Kaminski N., Bar-Joseph Z. iDREM: Interactive visualization of dynamic regulatory networks. PLoS Comput. Biol. 2018;14 doi: 10.1371/journal.pcbi.1006019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ermolaeva M., Neri F., Ori A., Rudolph K.L. Cellular and epigenetic drivers of stem cell ageing. Nat. Rev. Mol. Cell Biol. 2018;19:594–610. doi: 10.1038/s41580-018-0020-3. [DOI] [PubMed] [Google Scholar]
- 44.Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.H., Myung P., Plikus M.V., Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021;12:1088. doi: 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kao W., Gu R., Jia Y., Wei X., Fan H., Harris J., Zhang Z., Quinn J., Morand E.F., Yang Y.H. A formyl peptide receptor agonist suppresses inflammation and bone damage in arthritis. Br. J. Pharmacol. 2014;171:4087–4096. doi: 10.1111/bph.12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alhasan H., Terkawi M.A., Matsumae G., Ebata T., Tian Y., Shimizu T., Nishida Y., Yokota S., Garcia-Martin F., Abd Elwakil M., et al. Inhibitory role of Annexin A1 in pathological bone resorption and therapeutic implications in periprosthetic osteolysis. Nat. Commun. 2022;13:3919. doi: 10.1038/s41467-022-31646-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsuoka K., Park K.A., Ito M., Ikeda K., Takeshita S. Osteoclast-derived complement component 3a stimulates osteoblast differentiation. J. Bone Miner. Res. 2014;29:1522–1530. doi: 10.1002/jbmr.2187. [DOI] [PubMed] [Google Scholar]
- 48.Tu Z., Bu H., Dennis J.E., Lin F. Efficient osteoclast differentiation requires local complement activation. Blood. 2010;116:4456–4463. doi: 10.1182/blood-2010-01-263590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herbert B.A., Steinkamp H.M., Gaestel M., Kirkwood K.L. Mitogen-Activated Protein Kinase 2 Signaling Shapes Macrophage Plasticity in Aggregatibacter actinomycetemcomitans-Induced Bone Loss. Infect. Immun. 2017;85 doi: 10.1128/IAI.00552-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fan Y., Chen J., Yang Y., Lin J., Wu Z. Genome-wide expression and methylation profiling reveal candidate genes in osteoarthritis. Clin. Exp. Rheumatol. 2017;35:983–990. [PubMed] [Google Scholar]
- 51.Ji Z., Liu G.H., Qu J. Mitochondrial sirtuins, metabolism, and aging. J. Genet. Genomics. 2022;49:287–298. doi: 10.1016/j.jgg.2021.11.005. [DOI] [PubMed] [Google Scholar]
- 52.Mukherjee A., Rotwein P. Akt promotes BMP2-mediated osteoblast differentiation and bone development. J. Cell Sci. 2009;122:716–726. doi: 10.1242/jcs.042770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang J., Jiang H., Shao J., Mao R., Liu J., Ma Y., Fang X., Zhao N., Zheng S., Lin B. SOX4 inhibits GBM cell growth and induces G0/G1 cell cycle arrest through Akt-p53 axis. BMC Neurol. 2014;14:207. doi: 10.1186/s12883-014-0207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang L., Zhou Q., Qiu Q., Hou L., Wu M., Li J., Li X., Lu B., Cheng X., Liu P., et al. CircPLEKHM3 acts as a tumor suppressor through regulation of the miR-9/BRCA1/DNAJB6/KLF4/AKT1 axis in ovarian cancer. Mol. Cancer. 2019;18:144. doi: 10.1186/s12943-019-1080-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bernard V., Villa C., Auguste A., Lamothe S., Guillou A., Martin A., Caburet S., Young J., Veitia R.A., Binart N. Natural and molecular history of prolactinoma: insights from a Prlr(-/-) mouse model. Oncotarget. 2018;9:6144–6155. doi: 10.18632/oncotarget.23713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shlapatska L.M., Kovalevska L.M., Gordiienko I.M., Sidorenko S.P. Intrinsic defect in B-lymphoblastoid cell lines from patients with X-linked lymphoproliferative disease type 1. II. receptor-mediated Akt/PKB and ERK1/2 activation and transcription factors expression profile. Exp. Oncol. 2014;36:162–169. [PubMed] [Google Scholar]
- 57.Li J., Yi X., Yao Z., Chakkalakal J.V., Xing L., Boyce B.F. TNF Receptor-Associated Factor 6 Mediates TNFalpha-Induced Skeletal Muscle Atrophy in Mice During Aging. J. Bone Miner. Res. 2020;35:1535–1548. doi: 10.1002/jbmr.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simm A., Campisi J. Stress and aging. Exp. Gerontol. 2014;59:1–2. doi: 10.1016/j.exger.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 59.Campisi J. From cells to organisms: can we learn about aging from cells in culture? Exp. Gerontol. 2001;36:607–618. doi: 10.1016/s0531-5565(00)00230-8. [DOI] [PubMed] [Google Scholar]
- 60.Pignolo R.J., Suda R.K., McMillan E.A., Shen J., Lee S.H., Choi Y., Wright A.C., Johnson F.B. Defects in telomere maintenance molecules impair osteoblast differentiation and promote osteoporosis. Aging Cell. 2008;7:23–31. doi: 10.1111/j.1474-9726.2007.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vulliamy T., Beswick R., Kirwan M., Marrone A., Digweed M., Walne A., Dokal I. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc. Natl. Acad. Sci. USA. 2008;105:8073–8078. doi: 10.1073/pnas.0800042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ségal-Bendirdjian E., Geli V. Non-canonical Roles of Telomerase: Unraveling the Imbroglio. Front. Cell Dev. Biol. 2019;7:332. doi: 10.3389/fcell.2019.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A., et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dong X., Zhang L., Milholland B., Lee M., Maslov A.Y., Wang T., Vijg J. Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat. Methods. 2017;14:491–493. doi: 10.1038/nmeth.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blasco M.A., Lee H.W., Hande M.P., Samper E., Lansdorp P.M., DePinho R.A., Greider C.W. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- 72.Pearson K. LIII. On lines and planes of closest fit to systems of points in space. The London, Edinburgh Dublin Philosophical Magazine J. Sci. 1901;2:559–572. doi: 10.1080/14786440109462720. [DOI] [Google Scholar]
- 73.Traag V.A., Waltman L., van Eck N.J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 2019;9:5233. doi: 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Franzén O., Gan L.M., Bjorkegren J.L.M. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database. 2019;2019:baz046. doi: 10.1093/database/baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wilcoxon F. Individual Comparisons by Ranking Methods. Biometrics Bulletin. 1945;1:80–83. doi: 10.2307/3001968. [DOI] [Google Scholar]
- 76.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: Raw single-cell DNA-seq and bulk DNA-seq data have been deposited in NCBI database. Raw single-cell RNA-seq and bulk RNA-seq data are available in GEO database. Accession numbers are listed in the key resources table.
-
•
Code: All other data of bioinformatics analysis are available at http://dinglab.rimuhc.ca:8080/projects/aging_shsmu/.
-
•
Additional information: Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.