

Abstract
Altered branched chain amino acids (BCAAs), including leucine, isoleucine, and valine, are frequently observed in patients with advanced cancer. We evaluated the efficacy of chimeric antigen receptor (CAR) T cell-mediated cancer cell lysis potential in the immune microenvironment of BCAA supplementation and deletion. BCAA supplementation increased cancer cell killing percentage, while accelerating BCAA catabolism and decreasing BCAA transporter decreased cancer cell lysis efficacy. We thus designed BCKDK engineering CAR T cells for the reprogramming of BCAA metabolism in the tumor microenvironment based on the genotype and phenotype modification. BCKDK overexpression (OE) in CAR-T cells significantly improved cancer cell lysis, while BCKDK knockout (KO) resulted in inferior lysis potential. In an in vivo experiment, BCKDK-OE CAR-T cell treatment significantly prolonged the survival of mice bearing NALM6-GL cancer cells, with the differentiation of central memory cells and an increasing proportion of CAR-T cells in the peripheral circulation. BCKDK-KO CAR-T cell treatment resulted in shorter survival and a decreasing percentage of CAR-T cells in the peripheral circulation. In conclusion, BCKDK-engineered CAR-T cells exert a distinct phenotype for superior anticancer efficiency.
Keywords: chimeric antigen receptor, immunotherapy, branched chain amino acids, cancer metabolism, cancer microenvironment, BCKDK
Graphical abstract
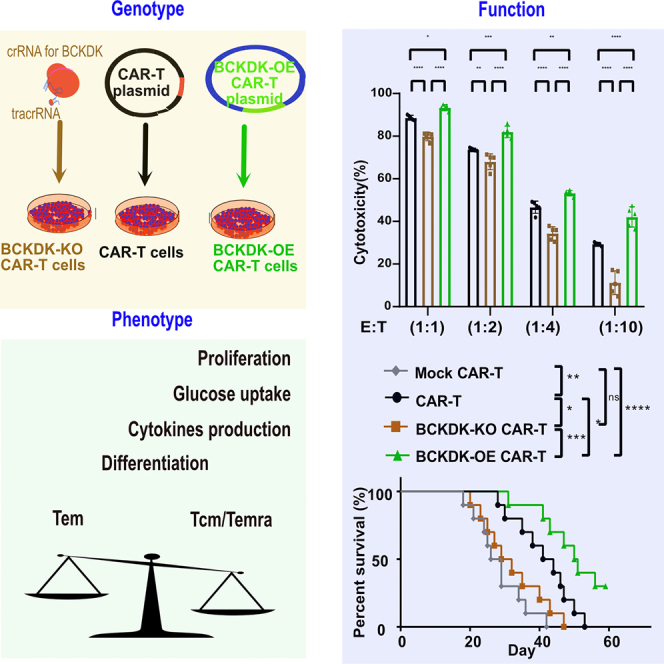
Guo and colleagues investigated the effect of BCAA metabolic reprogramming on the genotype, phenotype, and function of CAR-T cells. BCKDK-engineered CAR-T cells utilize BCAA metabolic reprogramming to enhance their cancer cell killing potential, as evidenced by the differentiation of central memory cells.
Introduction
The superior safety and efficacy of anti-CD19 chimeric antigen receptor (CAR)-T cells for the treatment of relapsed and refractory CD19+ B cell malignancies have resulted in the approval of multiple products by the U.S. Food and Drug Administration.1,2,3 CARs have undergone five generations of structural and functional modifications to improve their anticancer activity.4 However, there are challenges about beneficiary populations and effectiveness with CAR-T therapy. One important aspect is the immunosuppressive tumor microenvironment, which hinders T cell proliferation, suppresses T cell function, and impairs T cell recognition and killing of cancer cells.5,6 Specifically, cancer cells compete for nutrients to support their growth and metastasis.7 Strategies have been explored and developed to boost CAR-T activity, such as modifying co-stimulation, improving conditioning regimens,8 incorporating synthetic elements,9 or manipulating the tumor microenvironment.10
CAR-T cells are genetically engineered T cells that express a unique fusion receptor consisting of an extracellular single-chain variable fragment (scFv) domain, a hinge region, a transmembrane domain, a cytoplasmic sequences of the human costimulatory domain, and a CD3ζ activation domain.4 The extracellular domain contains an scFv that specifically recognizes tumor-associated antigens, facilitating the targeted lysis of cancer cells independent of major histocompatibility complex restriction. Modifications to the costimulatory and activation domains have exhibited distinct phenotypic and functional characteristics. Studies have indicated that the 4-1BB costimulatory domain enhances mitochondrial biogenesis and oxidative metabolism that promote the generation and persistence of memory T cells in vivo,11 compared with the CD28 domain that favors effector T cell generation.12 Additionally, combining CAR-T cells with immunoregulatory factors such as synthetic IL-9R,13 IL-12,14 IL-15,15 D2HG,16 and KYNU,17 have shown enhanced anticancer activity. Therefore, various strategies for modulating CAR function have been proposed to improve the metabolic fitness and intrinsic anticancer activity of engineered CAR- T cells.
Branched chain amino acids (BCAAs) constitute approximately 35% of the essential amino acids present in the bloodstream.18 BCAAs serve not only as crucial nutrients for protein synthesis, but also play a vital role in T cell proliferation, differentiation, and function through the activation of the mammalian target of rapamycin (mTOR) complex 1.19 Insufficient leucine impeded the clonal expansion of CD8+ T cells.20 A recent study demonstrated the important role of BCAA catabolism in preserving stemness in primary human acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL).21 JPH203 treatment targeting the BCAA transporter SLC7A5 was reported to induce anti-leukemia effects in a murine AML model while preserving healthy bone marrow cells.22 Chronically altered plasma BCAA levels are frequently observed in in patients with advanced cancer.23,24,25,26
In the present study, we investigated the effect of BCAA supplementation on the regulation of T cell proliferation, differentiation, and function. Based on the altered BCAA levels in hematological malignancies and the functional characteristics of mTOR in T lymphocytes, we propose integration CAR-T cells with an immune-regulatory functional kinase as a technological strategy to overcome the immunosuppressive tumor microenvironment and address BCAA metabolic dysregulation for the dysfunction of CAR-T cells.
Results
BCAA metabolic disorder decreased the cancer treatment efficacy of CAR-T cells
In this study, we optimized the evaluation strategy to assess the effect of BCAA metabolism on the cancer cell-killing activity of CAR-T cells based on the effect of microenvironment nutrients on CAR-T proliferation and persistence (Figure 1A). The second-generation CD19-targeting scFv (FMC63)-containing CAR plasmid was used for lentivirus transfection and CAR T cell generation. Peripheral blood mononuclear cells (PBMCs) were isolated from healthy donors, and CD8+ T cells were purified from positive selection using the MACS CD8+ T cell isolation kit (Miltenyi Biotec). The killing assay was performed by coculturing CAR-T cells with NALM6-GL cancer cells at different effector-to-target (E:T) ratios. Three BCAAs—leucine, isoleucine, and valine—were added to the media to modulate the BCAA concentration. An inhibitor of BCAA catabolism, BT2, and three inhibitors of BCAA transporters, BCH, V9302, and JPH203, were used to decrease the intracellular BCAA concentration. The concentrations of metabolites and inhibitors are listed in Table S1. The quality and functionality of CAR-T cells were assessed prior to immune microenvironment manipulation. The robust expression of the FLAG tag DYKDDDDK indicated highly purified CAR-T cells (>70%), with consistent reproducibility of killing assay (Figure 1B). The killing assay revealed similar cancer cell lysis ability of control CAR-T cells against NALM-6 GL cells at different E:T ratios from 1:1 to 1:10 after co-culture for 48 h (Figure 1C). BCAA supplementation increased the CAR-T cell killing efficacy against cancer cells, whereas BT2, BCH, KMH-233, and JPH203 consistently decreased CAR-T cell killing efficacy against cancer cells (Figure 1C). CAR-T cells and NALM6-GL cancer cells were analyzed by flow cytometry after 48 h of co-culture. There were similar trends of changes in CAR-T cells and cancer cells at different E:T ratios (Figure 1D). The column plot shows the different effects of BCAA decreased the percentage of resident NALM6-GL cancer cells (Figure 1E) and increased the percentage of CAR T cells (Figure 1F). The four inhibitors BT2, BCH, V9302, and JPH203 decreased CAR-T cell-mediated cancer cell lysis efficiency and increased the percentage of NALM6-GL cancer cells.
Figure 1.

BCAA supplementation improved CAR-T cell-mediated cancer cell lysis effectiveness, while disruption of BCAA metabolism and transport through 4 inhibitors impaired CAR-T cell-mediated antitumor effectiveness
(A) The strategy of evaluating BCAA metabolism on the cancer cell killing potential of CAR T cells. (B) The pseudocolor images of flow cytometry showed CAR percentages that were indicated by the FLAG tag of anti-DYKDDDDK. (C) The grouped scatterplots showed cancer cell lysis percentage from different killing assay at different E:T ratios after co-culture for 48 h with the addition of BCAA, inhibitors of BCAA metabolism, and transport. (D) The pseudocolor images of flow cytometry showed CAR-T cells and cancer cells of NALM6-GL percentages after co-culture for 48 h. (E) The histograms showed the NALM6-GL cancer cells percentages after co-culture for 48 h. (F) The histograms showed the CAR-T cells percentages after co-culture for 48 h. One-way or two-way ANOVA followed by post hoc Tukey’s multiple comparison tests were used for comparing multiple groups. Statistical significance is indicated in all figures by the following annotations: ∗p < 0.05; ∗∗p < 0.01, ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. The original statistical data are included in Tables S4–S6.
BT2 is an allosteric inhibitor of BCKDK, which increases BCKDH activity and decreases plasma BCAA levels.27,28,29 BT2 is used directly to decrease plasma BCAA levels.27 BCH is a selective and competitive inhibitor of L amino acid transporter 1 (LAT1) that inhibits BCAA uptake.30 The affinity of BCH for LAT1 suppresses the function of neutral amino acid transporters and creates a BCAA-deficient microenvironment.31 KMH-233 is a selective and reversible inhibitor of LAT1 that has been developed as an antiproliferative drug for cancer treatment.32,33 Treatment with KMH-233 below 25 μM resulted in reduced total protein levels of mTOR and increased apoptosis in LAT1-expressing cancer cells.34 JPH203 is also a selective inhibitor of LAT1 used to inhibit BCAA transport.30 In a phase 1 study in patients with biliary tract cancer, JPH203 decreased the plasma levels of free amino acids, particularly BCAAs.35 Treatment of human T cells with JPH203 resulted in suppressed leucine uptake and decreased cytokine production.36 These four inhibitors of BCAA metabolism and transport collectively demonstrated that decreased intracellular BCAA levels hindered CAR-T cell-mediated anticancer efficacy. BCAA supplementation at referenced concentrations enhanced the efficacy of cancer cell lysis.
Reprogramming of BCAA metabolism regulated CAR-T cell function, proliferation, and glucose uptake
After recognizing cancer antigens, CAR-T cells produce and secrete large amounts of inflammatory cytokines, including interferon (IFN)-γ and tumor necrosis factor (TNF)-α, to induce cancer cell pyroptosis.37 The intracellular staining of IFN-γ and TNF-α represents the function of CAR-T cells in mediating cancer cell pyroptosis.38 After co-culture of CAR-T cells with NALM6-GL cancer cells, the intracellular staining of IFN-γ and TNF-α was not clearly observed according to the flow cytometry results (Figure 2A). The three BCAAs, leucine, isoleucine, and valine, increased the intracellular IFN-γ and TNF-α production, whereas the inhibitors of BCAA metabolism and transport, BT2, BCH, KMH-233, and JPH203, decreased intracellular IFN-γ and TNF-α production (Figure 2B). These results suggest that reprogramming BCAA metabolism regulates CAR-T cell function.
Figure 2.

Reprogramming of BCAA metabolism regulated CAR-T cell function, proliferation, and glucose uptake
(A) The pseudocolor images of flow cytometry showed intracellular staining of IFN-γ and TNF-α after co-culture for 48 h at Effector CAR-T cells: Target cancer cells ratio of 1:1 with the addition of BCAA, inhibitors of BCAA metabolism and transport after co-culture for 2 days. (B) The histograms showed the IFN-γ+ and TNF-α+ CAR-T cell percentages after co-culture for 48 h. (C) The pseudocolor images of flow cytometry showed CFSE+ CAR-T with the addition of BCAA, inhibitors of BCAA metabolism, and transport after culture for 5 days. (D) The histograms showed CAR-T cell proliferation through the analysis of CFSE+ cell percentages. (E) The pseudocolor images of flow cytometry showed 2-NBDG+ CAR-T with the addition of BCAA, inhibitors of BCAA metabolism, and transports after culture for 60 min. (F) The histograms showed CAR-T cell glucose uptake through the analysis of 2-NBDG+ cell percentages. One-way ANOVA followed by post hoc Tukey’s multiple comparison test were used for comparing multiple groups. Statistical significance were indicated in all figures by the following annotations: ∗p < 0.05; ∗∗p < 0.01, ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. The original statistical data are included in Tables S7–S9.
Next, we used the carboxyfluorescein diacetate succinimidyl ester (CFSE) assay to assess the effect of BCAA metabolic reprogramming on CAR-T cell proliferation.39,40 The eight groups showed different expansion capacities (Figure 2C). Compared with the control treatment, supplementation with leucine, isoleucine and valine increased CAR-T cell proliferation (Figure 2D). However, BT2, BCH, V9302, and JPH203 decreased cell proliferation. The metabolic adaptation of CAR-T cells was assessed using a glucose uptake capacity of the 2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-D-glucose (2-NBDG) assay.40 The metabolic environment of the changed BCAA concentrations showed distinct differences in glucose uptake capacity (Figure 2E). Leucine, isoleucine, and valine enhanced glucose uptake functions, while BT2, BCH, KMH-233, and JPH203 inhibited glucose uptake capacity (Figure 2F). This finding indicated that BCAA restriction impairs CAR-T cell proliferation, expansion, and metabolic adaptation.
BCKDK over-expression and knockout CAR-T cell showed distinct function, proliferation, and metabolism
BCAA restriction and supplementation showed different effects on CAR-T cell function, proliferation, and metabolism. Lymphocyte-specific supplementation of BCAAs is ineffective in the clinic due to competition for nutrient metabolites between immune cells, stromal cells, and cancer cells. Additional difficulties include the supplemental amounts of BCAA and the toxicity resulting from bulk supplementation.41 The approach proposed was to couple CAR-T cells with a modified immunoregulatory functional element to drive CAR-T proliferation, persistence, and anticancer activity. CAR T cell quality and fitness was improved based on BCAA pathway analysis (Figure 3A). BCAAs are first converted to branched-chain α-keto acids (BCKAs), such as 3-methyl-2-oxopentanoic acid, 3-methyl-2-oxobutanoic acid, and 4-methyl-2-oxopentanoic acid, by cytosolic branched-chain aminotransferase a (BCAT1) or mitochondrial branched-chain aminotransferase (BCAT2).27 BCKAs are sequentially catabolized by BCKD complex. BCKD complex catabolism is the rate-limiting irreversible step of BCAA catabolism, and the activity of BCKDC is inhibited by the phosphorylation of the E1α subunit by BCKA dehydrogenase kinase (BCKDK). Inhibition of BCKDK results in the phosphorylation and inactivation of BCKDC and a decrease in intracellular BCAA levels. Previous studies have identified BCKDK as a target for the treatment of metabolic diseases and cancer through the regulation of BCAA metabolism.27 After preliminary experimental analyses, we generated BCKDK modified CAR-T cells (Figure 3B). BCKDK overexpression (OE) CAR was produced by coupling BCKDK cDNA (No: NM_005881.4) to the CAR vector via T2A. The plasmids were used for high titer lentivirus preparations and the production of BCKDK-OE CAR-T cells by spinal infection transduction.42 BCKDK knockout (KO) CAR-T cells were generated based on ribonucleoprotein (RNP) delivery of CRISPR-Cas9 editing of BCKDK single guide (sgRNA) into CAR-T cells.43
Figure 3.

BCKDK-modified CAR-T cell function, proliferation, and metabolism
(A) The metabolic pathways of BCAA catabolism and their key enzyme. (B) The strategy of production BCKDK modified CAR-T cells. (C) The pseudocolor images of flow cytometry showed CAR percentages that were indicated by the FLAG tag of anti-DYKDDDDK from BCKDK modified CAR-T cells. (D) The histograms showed BCKDK gene expression at different BCKDK-modified CAR-T cells. (E) The protein expression levels of BCKDK t different BCKDK modified CAR-T cells through Western blot gels (Figure S1). (F) The gel image of endonuclease T7E1 mismatch detection assay showed a significant KO of BCKDK in BCKDK-KO CAR-T cells (Figure S2). (G) The pseudocolor images of flow cytometry and the histograms showed CFSE+ cell percentages from BCKDK modified CAR-T cells after culture for 5 days. (H) The pseudocolor images of flow cytometry and the histograms showed 2-NBDG+ cell percentages from BCKDK modified CAR-T cells after cultured for 60 min. (I) The grouped histograms showed cancer cell lysis percentage from killing assay at different E:T ratios after co-culture for 48 h based on BCKDK modified CAR-T cells. (J) The pseudocolor images of flow cytometry and the histograms showed intracellular staining of IFN-γ and TNF-α after co-culture for 48 h at Effector CAR-T cells: target cancer cells ratio of 1:1 from BCKDK modified CAR-T cells. One-way or two-way ANOVA followed by post hoc Tukey’s multiple comparison tests were used for comparing multiple groups. Statistical significance was indicated in all figures by the following annotations: ∗p < 0.05; ∗∗p < 0.01, ∗∗∗p < 0.001; ∗∗∗p < 0.0001. The original statistical data are included in Tables S10–S15.
The BCKDK-modified CAR-T cells showed high purity after multiple flow sorting, referring to previous studies.16,17 CAR expression was reflected by the FLAG tag of the anti-DYKDDDDK antibody (Figure 3C). The gene expression of BCKDK was significantly different, as BCKDK-OE CAR-T cells showed a 1.9-fold increase in BCKDK levels, whereas BCKDK-KO CAR-T cells showed a 0.4-fold decrease in BCKDK levels (Figure 3D). The protein expression of BCKDK was consistent with its mRNA expression, as BCKDK-OE CAR-T cells showed upregulated BCKDK protein expression, whereas BCKDK-KO CAR-T cells showed downregulated BCKDK protein expression (Figures 3E and S1). The endonuclease T7E1 mismatch detection assay showed a significant decrease in BCKDK expression levels in BCKDK-KO CAR-T cells based on the optimized sgRNA with sequence: 5′-GCCAGCGTAGAGCATCATGG-3′ (Figures 3F and S2). These BCKDK-modified CAR-T cells were then used for phenotype and functional analyses.
The CFSE assay proliferation experiment results showed that BCKDK-modified CAR-T cells had different expansion capacities (Figure 3G). BCKDK-OE CAR-T cells showed increased proliferation capacity, whereas BCKDK-KO CAR-T cells showed decreased proliferation capacity. The 2-NBDG assay-based glucose uptake experiment also showed that BCKDK-modified CAR-T cells had different metabolic adaptive capacities (Figure 3H). BCKDK-OE CAR-T cells showed increased glucose uptake capacity, whereas BCKDK-KO CAR-T cells showed decreased glucose uptake capacity.
The killing assay results of BCKDK-modified CAR-T cells co-cultured with NALM6-GL cancer cells for 24 h at different E:T ratios showed different cytotoxicities. The gradient decrease in the percentage of cancer cells killed by CAR-T cells gradually decreased, indicating a suitable functional verification system (Figure 3I). BCKDK-OE CAR-T cells showed increasing cytotoxicity against cancer cells, whereas BCKDK-KO CAR-T cells showed decreasing cancer cell lysis capacity compared with that of control CAR-T cells. The functional assay results of intracellular IFN-γ and TNF-α production were consistent with the killing assay results (Figure 3J). BCKDK-OE CAR-T cells showed increased intracellular IFN-γ and TNF-α production, whereas BCKDK-KO CAR-T cells showed decreased intracellular IFN-γ and TNF-α production. These results indicate that reprogramming BCAA metabolism by BCKDK modification has the potential to regulate CAR-T cell function.
BCKDK-OE CAR-T cells increase cancer cell apoptosis and inhibit CAR-T cell apoptosis
CAR-T cells and NALM6-GL cancer cells were analyzed after co-culture for 48 h at different E:T ratios. BCKDK-KO CAR-T cells were less preserved, and BCKDK-OE CAR-T cells were obviously enriched according to the flow cytometry results (Figure 4A). There was an increasing percentage of NALM6-GL cancer cells as the E:T ratio increased from 1:1 to 1:10. Compared with the CAR-T group, the BCKDK-KO CAR-T group showed a relatively high preservation of NALM6-GL cancer cells, while the percentage of CAR-T cells was low. The BCKDK-OE CAR-T group showed a relatively low percentage of NALM6-GL cancer cells and a high percentage of CAR-T cells (Figure 4B). The apoptosis of NALM6-GL cancer cells and CAR-T cells at an E:T ratio of 1:1 showed consistent results (Figure 4C). The BCKDK-KO CAR-T group showed less apoptosis of NALM6-GL cancer cells and more apoptosis of CAR-T cells than the control group. The BCKDK-OE CAR-T group showed more apoptosis of NALM6-GL cancer cells and less apoptosis of CAR-T cells (Figure 4D).
Figure 4.

BCKDK-OE CAR-T cells increase cancer cell apoptosis and inhibit CAR-T cell apoptosis
(A) The pseudocolor images of flow cytometry showed BCKDK-modified CAR-T cells and NALM6-GL cancer cells percentages after co-culture for 48 h. (B) The histograms showed the CAR-T cell and the NALM6-GL cancer cell percentages after co-culture for 48 h from BCKDK-modified CAR-T cells. (C) The pseudocolor images of flow cytometry showed apoptosis cell percentages of BCKDK-modified CAR-T cells and NALM6-GL cancer cells. (D) The histograms showed apoptosis cell percentages of the NALM6-GL cancer cells and the BCKDK-modified CAR-T cells. (E) The pseudocolor images of flow cytometry showed markers PD-1 and LAG-3 expression of BCKDK modified CAR-T cells after co-culture with NALM6-GL cancer cells at an E:T ratio of 1:1 for 48 h with the addition of 10 ng/mL IFN-γ. (F) The histograms showed expression of PD-1 and LAG-3 in BCKDK modified CAR-T cells after co-culture with NALM6-GL cancer cells at an E:T ratio of 1:1 for 48 h. One-way ANOVA or two-way ANOVA followed by post hoc Tukey’s multiple comparison tests were used for comparing multiple groups. Statistical significance is indicated in all figures by the following annotations: ∗p < 0.05; ∗∗p < 0.01, ∗∗∗p < 0.001; ∗∗∗p < 0.0001. The original statistical data are included Tables S16–S18.
To reveal the mechanism by which BCKDK-modified CAR-T cells exert different apoptotic and cancer cell lysis effects, we co-cultured CAR-T cells with NALM6-GL cancer cells at an E:T ratio of 1:1 for 48 h with the addition of 10 ng/mL IFN-γ. The differentiation markers programmed cell death (PD-1) and LAF-3 were evaluated (Figure 4E). The BCKDK-KO CAR-T group showed high expression of PD-1 and LAG-3 compared with the control group. The BCKDK-KO CAR-T group showed lower expression of PD-1 and LAG-3 (Figure 4F). These results indicated that BCKDK modified CAR-T cells may have distinct functional characteristics.
BCKDK-modified CAR-T cells showed distinct metabolic features and differentiation phenotypes
To reveal the distinct metabolic features of BCKDK-modified CAR-T cells, we used metabolomics to visualize intracellular metabolite reprogramming. The heatmap of metabolites and samples showed that the abundance of 19 metabolites was significantly changed in BCKDK-modified CAR-T cells (Figure 5A). BCAAs such as leucine, isoleucine, and valine were enriched in BCKDK-OE CAR-T cells and depleted in BCKDK-KO CAR-T cells. The biplot shows the first two principal components, with samples discriminated by metabolite abundance (Figure 5B). Pathway analysis showed that the top altered metabolic processes included aminoacyl-tRNA biosynthesis, arginine biosynthesis, alanine, aspartate, and glutamate metabolism; valine, leucine, and isoleucine biosynthesis; arginine and proline metabolism; glycine and glutathione metabolism; glyoxylate and dicarboxylate metabolism; beta-alanine metabolism; histidine metabolism; pantothenate and co-enzyme A biosynthesis; cysteine and methionine metabolism; and the citric acid (TCA) cycle (Figure 5C). Based on the function of BCAAs and the regulatory mechanism of mTOR,44,45 we compared the phosphorylation levels of mTOR, 4E-BP1, and P70S6K using Western blotting (Figures 5D and S3). Interestingly, BCKDK-KO CAR-T cells showed low phosphorylation levels of mTOR, 4E-BP1, and P70S6K, whereas BCKDK-OE CAR-T cells showed high phosphorylation levels of mTOR, 4E-BP1, and P70S6K. These results suggest that BCAA supplementation promotes the protein synthesis and proliferation functions of CAR-T cells.
Figure 5.

BCKDK modified CAR-T cells showed distinct metabolic features and differentiation phenotypes
(A) The heatmap of metabolomics showed metabolic distinction of BCKDK-modified CAR-T cells based on 19 metabolites. (B) The biplot showed that two principal components discriminated BCKDK-modified CAR-T cells and their relevant metabolites. (C) The pathway plot showed the main metabolic pathway resulted from BCKDK modification. (D) The protein expression levels of mTOR, p70S6K, 4EBP1, and their phosphorylation expression. The histograms showed expression phosphorylation levels of mTOR, p70S6K, and 4EBP1 at BCKDK-modified CAR-T cells (Figure S3). (E) The pseudocolor images of flow cytometry showed memory differentiation markers CD45RA and CD62L from BCKDK modified CAR-T cells after co-culture with NALM6-GL cancer cells at an E:T ratio of 1:1 for 2 days and 14 days. (F) The histograms showed percentages of Tscm (CD45RA+ CD62L+), Tcm (CD45RA− CD62L+), Tcm (CD45RA− CD62L−), and Temra (CD45RA+ CD62L−) from BCKDK-modified CAR-T cells after co-culture with NALM6-GL cancer cells at an E:T ratio of 1:1 for 2 days and 14 days. One-way or two-way ANOVA or two-way ANOVA followed by post hoc Tukey’s multiple comparison tests were used for comparing multiple groups. Statistical significance were indicated in all figures by the following annotations: ∗p < 0.05; ∗∗p < 0.01, ∗∗∗p < 0.001; ∗∗∗p < 0.0001. The original statistical data are included in Tables S19 and S20.
Extensive studies have shown that modulation of mTOR signaling triggers the generation of stem cell-like memory T cells, and mTOR has emerged as a critical integrator of cues from the immune microenvironment.45,46 Antigen-activated T cells rapidly switch to aerobic glycolysis and glutaminolysis metabolic programs to sustain cell proliferation, expansion, and survival. After antigen clearance, most effector T cells are degraded by apoptosis, while partial CAR-T cells remain to form a subpopulation of memory T cells.47 T cells adjust their metabolomic processing of amino acids, lipids, and nucleotides according to genotypic and phenotypic immunonutritional needs. Therefore, we co-cultured CAR-T cells with NALM6-GL cancer cells at an E:T ratio of 1:1 for 7 days. At this stage, the cancer cells were almost completely lysed, and the resident CAR-T cells were differentiated. We enriched CAR-T cells for phenotypic analysis (Figure 5E). CAR-T cells showed a 29% effector memory phenotype (Tem, CD45RA− CD62L−), 27% central memory phenotype (Tcm, CD45RA− CD62L+), and 24% terminally differentiated Tem (Temra, CD45RA+ CD62L−) on day 2. After 14 days of co-culture, the cancer cells were lysed, and the resident CAR-T cells showed a distinct memory phenotype, with an increase in Tem (from 29% to 71%) and a decrease in Temra (from 24% to 2.1%). On both day 2 and day 14, BCKDK-KO CAR-T cells showed lower percentages of Tcm cells and higher percentages of Temra cells than the control CAR-T cells (Figure 5F). BCKDK-OE CAR-T cells showed higher percentages of Tcm cells and lower percentages of Temra cells than the control CAR-T cells. These results suggest that BCKDK promotes the generation of memory T cells by regulating of mTOR and reprogramming of BCAA metabolism. BCKDK-OE CAR-T cells possess superior functional characteristics, such as proliferation, memory differentiation, and function, for adoptive CAR-T therapy.
BCKDK-OE CAR-T cells possess superior cancer cell lysis activity, and BCKDK-KO CAR-T cell treatment results in shorter survival
To validate the cancer cell killing ability, we designed an in vivo experiment using a mouse model bearing NALM6-GL cancer cells (Figure 6A). NALM6-GL cancer cells were inoculated 3 days prior to CAR-T adaptive transfer treatment. Then, 2 million CAR-T cells as well as mock CAR-T cells and BCKDK-modified CAR-T cells were injected into the tail vein on days 0, 3, and 6. Disease progression was followed, and overall survival was calculated (Figure 6B). The median survival of the mock CAR-T group was 27.5 days, and the median survival of the CAR-T cell group was 42.5 days. The BCKDK-KO CAR-T group showed a shortened median survival of 30.5 days, while the BCKDK-OE CAR-T group showed a prolonged median survival of 50.5 days. The significantly improved survival of the BCKDK-OE CAR-T cells and the worsened survival of the BCKDK-KO CAR-T cells indicated that the BCKDK-modified CAR-T cells have different cancer cell killing abilities. Flow cytometry analysis revealed resident circulating cancer cells and immune CAR-T cells on day 14 (Figure 6C). In the mock CAR-T cell group, there were a number of NALM6-GL cancer cells (Figure 6D). CAR-T cells and BCKDK-modified CAR-T cells showed different percentages of resident NALM6-GL cancer cells. BCKDK-KO CAR-T cells showed relatively more NALM6-GL cells, and BCKDK-OE CAR-T cells showed relatively fewer NALM6-GL cells. In contrast, BCKDK-KO CAR-T cells showed relatively fewer CAR-T cells, and BCKDK-OE CAR-T cells showed relatively more CAR-T cells. Serum levels of the cytokines IFN-γ and TNF-α were assessed by ELISA on day 28 (Figure 6E). The BCKDK-KO CAR-T cells produced and released significantly decreased levels of IFN-γ and TNF-α, whereas the BCKDK-OE CAR-T cells produced and released increased levels of IFN-γ and TNF-α. The differentiation phenotype of CAR-T cells was examined at days 14 and 28 (Figure 6F). On days 14 and 28, CAR-T cells mainly showed an Tem and a partial Tcm. Compared with the CAR-T group, the BCKDK-KO CAR-T group showed an increase in the percentage of effector memory cells and an increase in the percentage of terminally differentiated effector memory cells (Figure 6G). There was a significant decrease in the percentage of central memory cells in the BCKDK-KO CAR-T group at both days 14 and 28. The BCKDK-OE CAR-T group showed opposite differentiation phenotypes, as the BCKDK-OE CAR-T group showed fewer effector memory cells and more terminally differentiated effector memory cells than the CAR-T group at days 14 and 28. The increase in terminally differentiated effector memory cells indicated the killing process of CAR-T cells. The BCKDK-OE CAR-T cells showed an increasing Tcm at both days 14 and 28. These results suggested that BCKDK-modified CAR-T cells not only exert superior cancer cell lysis activity, but also possess a distinct memory T cell phenotype.
Figure 6.

BCKDK-OE CAR-T cells exert superior cancer cell lysis ability, while BCKDK-KO CAR-T cell treatment resulted in shorter survival
(A) The in vivo strategy of BCKDK modified CAR-T cells for the treatment cancer mouse bearing NALM6-GL cells and the disease progression following. (B) The survival plot of BCKDK modified CAR-T cells for the treatment cancer mouse bearing NALM6-GL cells. (C) The pseudocolor images of flow cytometry showed CAR-T cells percentages and NALM6-GL cancer cells percentages from BCKDK modified CAR-T cell-treated mice bearing NALM6-GL cancer cells at day 14. (D) The histograms showed NALM6-GL cancer cells percentage and CAR-T cell percentages from BCKDK modified CAR-T cell-treated mice bearing NALM6-GL cancer cells at day14. (E) The histograms showed sera IFN-γ and TNF-α levels from BCKDK-modified CAR-T cell-treated mice bearing NALM6-GL cancer cells at day 14. (F) The pseudocolor images of flow cytometry showed CD45RA and CD62L expression in CAR-T cells from BCKDK modified CAR-T cell-treated mice bearing NALM6-GL cancer cells at days 14 and 28. (G) The histograms showed percentages of Tscm (CD45RA+ CD62L+), Tcm (CD45RA− CD62L+), Tcm (CD45RA− CD62L−), and Temra (CD45RA+ CD62L−) from BCKDK modified CAR-T cell-treated mice bearing NALM6-GL cancer cells at days 14 and 28. Survival analysis was conducted using the Kaplan-Meier method, and significance was determined using the log rank test. One-way or two-way ANOVA or two-way ANOVA followed by post hoc Tukey’s multiple comparison tests were used for comparing multiple groups. Statistical significance were indicated in all figures by the following annotations: ∗p < 0.05; ∗∗p < 0.01, ∗∗∗p < 0.001; ∗∗∗p < 0.0001. The original statistical data are included in Tables S21–S23.
Discussion
In the present study, we evaluated the effects of BCAA supplementation and inhibition of BCAA metabolism on the phenotype and function of CAR-T cells. BCAA supplementation enhanced cancer cell killing efficacy, while accelerating BCAA catabolism through BCKDK inhibition and decreasing BCAA transport through LAT1 inhibitors diminished the efficacy of cancer cell lysis. BCAA supplementation stimulated the proliferation and glucose uptake of CAR-T cells. We then engineered BCKDK-modified CAR-T cells and assessed their genotype, phenotype, and function. BCKDK-OE CAR-T cells demonstrated heightened cancer cell lysis capability. Conversely, BCKDK-KO CAR-T cells displayed reduced cancer cell lysis activity. The in vivo experiments revealed that BCKDK-OE CAR-T cells significantly prolonged the survival of mice harboring NALM6-GL cancer cells, whereas treatment with BCKDK-KO CAR-T cells led to shorter survival. The enhanced cancer cell lysis capacity of BCKDK-OE CAR-T cells was linked to central memory T cell differentiation and the proportion of resident CAR-T cells in the peripheral blood circulation.
BCAA metabolic dysfunction is prevalent in multiple advanced hematological malignancies.23,24,25,26 Previous studies have shown that BCAT1, a catalytic enzyme for BCAA, is abnormally activated and functionally crucial for CML in humans and in mouse models of CML.24 Mechanically, epigenetic modifications involving EZH2 and oncogenic NRAS collaboratively reconfigure BCAA metabolism and induce the leukemic transformation of myeloproliferative neoplasms.48 The activation of BCAT1 enhances BCAA metabolism, while mTOR signaling drives leukemic transformation and leukemogenesis.49 Human AML and ALL cells rely on BCAT1-mediated BCAA metabolism for sustaining energy production.21 Leukemia-initiating cells depend on activated BCAA metabolism to maintain their leukemic stemness, irrespective of their lineage origin. BCKDK is a key negative regulator of BCAA catabolism that inhibits the dehydrogenase activity of BCKDC by dephosphorylating the E1 component of the complex.28 BCKDC is the rate-limiting enzyme in the catabolism of BCAA, which plays an important role in many serious human diseases.27 Experimental findings based on a compensatory mechanism revealed that the deletion of Bckdk results in impaired TCA cycle function and embryonic lethality.50 Therefore, we evaluated the effect of BCAA catabolism on the metabolism, phenotype, and function of CAR-T cells. By modulating BCAA catabolism, we engineered BCKDK-modified CAR-T cells and observed that BCKDK-OE CAR-T cells exhibited enhanced survival, superior cancer cell lysis activity, and reprogrammed BCAA metabolism.
BCAAs play a crucial role in regulating T cell fate through the activation of the mTOR pathway. mTOR is a component of a highly conserved complex with a protein synthesis pathway that connects cell growth and metabolism to external stimuli in all eukaryotes.44 T cells rely on mTOR signaling to perceive and integrate immune signals (such as antigenic signals, costimulatory molecules, and cytokines), environmental stimuli (such as growth factors), and nutrients.51 Dysfunction of mTOR function impairs the development and maintenance of T cells.48 In the present study, we identified a distinctive metabolic profile in BCKDK-modified CAR-T cells. BCKDK-OE CAR-T cells exhibited elevated levels of intracellular BCAAs, including leucine, isoleucine, and valine. These heightened levels were evident in the cellular metabolism, proliferation, differentiation, and functionality of the CAR-T cells. Moreover, BCKDK-OE CAR-T cells displayed increased mTOR phosphorylation and a shift in phenotypic differentiation toward memory T cells. Conversely, BCKDK-KO CAR-T cells demonstrated reduced mTOR phosphorylation and lower levels of intracellular BCAA. This decrease manifested in delayed proliferation, compromised cancer cell cytotoxicity, and impaired phenotypic differentiation that featured as increasingly short-lived Temra and effector cells in BCKDK-KO CAR-T cells.
In the present study, we examined the effect of BCAA on the cytotoxic activity of CAR-T cells against cancer cells. The addition of BCAAs enhanced the efficacy of CAR-T cells in killing cancer cells. However, the clinical advantages of high BCAA supplementation for advanced cancer patients are subject to debate. We also tested four inhibitors of BCAA metabolism and transport for their cytotoxic effects on cancer cells. BT2 functions as an allosteric inhibitor of BCKDK, boosting BCKDH activity while decreasing plasma BCAA levels.27,28,29 BCH serves as a selective and competitive inhibitor of LAT1, hindering BCAA uptake and establishing a BCAA-deficient microenvironment.30 KMH-233, a selective and reversible LAT1 inhibitor, has been developed as an antiproliferative agent for cancer therapy.32,33 JPH203, another selective LAT1 inhibitor, is utilized to block BCAA transport and decrease plasma BCAA levels.35 These four inhibitors collectively exhibited their ability to decrease intracellular BCAA levels. It was interesting that accelerating BCAA catabolism through BCKDK inhibition and decreasing BCAA transport through LAT1 inhibitors showed a common impairment in the CAR-T cell-mediated killing efficacy against NALM6 cancer cells. These results indicated the importance of BCAA metabolism on CAR-T cell phenotype and function. The engineered BCKDK-modified CAR-T cells have distinct genotype, phenotype, and function for their superior anti-cancer potential in the immunosuppressive metabolic microenvironment.
The typically immunosuppressive tumor microenvironment comprises immune cells, stromal cells, the extracellular matrix, and other secreted molecules, such as growth factors, cytokines, chemokines, and extracellular vesicles.7 The competition for metabolic resources by cancer cells hampers the metabolic adaptation of infiltrating lymphocytes. However, supplementing with high levels of BCAAs can lead to neurotoxicity and metabolic disorders. Therefore, we engineered BCKDK for reprogramming BCAA metabolism to enhance the metabolic adaptation, phenotype, and function of CAR-T cells. Integrated or multifaceted therapies may address the challenges of CAR-T cell therapy of the beneficiary population and effectiveness with CAR-T therapy. Our study confirmed that modifying CAR-T cells with BCKDK enhances metabolic adaptation of CD8+ T lymphocytes within the immunosuppressive tumor microenvironment. The findings illustrated the phenotype and function of BCKDK-modified CAR-T cells and their potential application in cancer patients with BCAA metabolic dysfunction. The clinical validation of the therapeutic capacity of BCKDK-OE CAR-T cells is still pending. Altered BCAA concentration in cancer patients and BCAA metabolic reprogramming are common in solid tumors. The research and application of BCAA metabolism in solid tumor immunotherapy will be of wide interest to oncologists, hematologists, biochemists, nutritionists, researchers, and scientists.
In conclusion, BCAAs are essential amino acids that influence the phenotype and function of T cells. BCKDK-modified CAR-T cells reprogrammed BCAA metabolism and altered the proliferation, differentiation, and cancer cell killing effects. Treatment with BCKDK-OE CAR-T cells significantly improved the survival of mice bearing NALM6-GL cancer cells. The superior cancer cell lysis ability of BCKDK-OE CAR-T cells was associated with central memory cell differentiation and the percentage of resident CAR-T cells in the peripheral circulation.
Methods
Patient samples for primary human T lymphocyte isolation and expansion
This study was approved by the Human Research Ethics Committee of Shanghai Sixth People’s Hospital affiliated with Shanghai Jiao Tong University School of Medicine (No.:2021-YS-0215). PBMCs were isolated from heparinized blood samples of healthy donors by Ficoll-Hypaque density gradient (Sigma). CD8+ T lymphocytes were positively selected using the MACS CD8+ T cell isolation kit (Miltenyi Biotec). To generate sufficient CD8+ T lymphocytes, T lymphocytes were stimulated with anti-CD3 and anti-CD28 antibodies (Thermo Fisher Scientific) or anti-CD3/-CD28 Dynabeads (Gibco). The supernatant was replaced with RPMI-1640 (Gibco) containing 10% fetal bovine serum (FBS) (Gibco), 2 mmol/L L-glutamine (Gibco), 100 U/mL penicillin and streptomycin (Gibco), supplemented with 100 U/mL hIL2 and 0.1% β-mercaptoethanol (Thermo Fisher Scientific).
Cell lines
Mycoplasma testing was performed according to the manufacturer’s instructions to confirm mycoplasma-free status. Low-passage HEK-293T cells were cultured in DMEM (Gibco) supplemented with 10% FBS (Gibco), 5 mM L-glutamine (Gibco), 10 mM HEPES, 2 mM glutamine, and 1% penicillin-streptomycin. NALM6 lines were cultured in complete RPMI-1640 medium (Gibco) supplemented with 10% FBS, 2 mM L-glutamine (Gibco), 10 mM HEPES, 2 mM glutamine, and 1% penicillin-streptomycin. All cells were cultured at 37°C in a 5% carbon dioxide atmosphere. NALM6 cells were infected with a lentivirus carrying Luciferase and GFP by spinoculation. After incubation for 2 days, GFP+ cells (NALM6-GL) were sorted, and stable NALM6-GL cell lines were generated.
Production of CAR-T cells and BCKDK-OE CAR-T cells
We used the second-generation recombinant lentiviral plasmid backbone for the production of CAR-T cells and BCKDK-OE CAR-T cells. The CAR construct was designed with the anti-CD19 scFv (FMC-63), the human IgD hinge, the transmembrane sequence of human CD8α, the cytoplasmic sequences of the human CD137 costimulatory moiety, and the CD3ζ activation domain. The FLAG tag sequence was inserted before the CD8a leader peptide fragment and was used to detect the CAR. The BCKDK-OE CAR construct was designed with by engineered Human BCKDK (NM_005881.4) with CAR construct using a self-cleavable 2A linear sequence. Both the plasmid backbone was designed under the regulation of the EF-1α promoter. Lentiviruses were produced from HEK293T cells as previously described with packaging plasmids (psPAX2 and pMD2.G). Supernatants lentiviral particles were collected 48 h after transfection, isolated by centrifugation at 500×g for 5 min and then passed through a 0.45-μM filter. Lentiviral particles were concentrated using AmiconUltra 100-kDa ultrafiltration centrifugation tubes (Millipore). All viruses were resuspended in 500 μL of serum-free RPMI and stored at −80°C until use. Viral titer was determined by titration in HEK-293T cells.
To integrate the CAR and BCKDK-OE CAR-modified DNA cassette into the genomic DNA of CD8+ T cells, spinfection was used to enhance the transduction efficiency of the T cells. Briefly, 3 days before spinfection, T cells were activated for 16 h with a 1:1 ratio of human anti-CD3/anti-CD28 beads (CD3/CD28 Dynabeads; Thermo Fisher), which were later removed using a magnetic separation rack. The purity of the positive cell fraction was assessed by flow cytometric staining using a fixable viability stain (Live/Dead Near-Infrared, Life Sciences) and anti-CD8 and anti-Flag tag antibody. T cells were incubated for 2 days. CD8 T cells were then prepared at a density of 2 × 106 cells/mL in medium and seeded in 0.8 mL in each well of a 24-well plate. For each 0.8 mL of cell suspension, 0.2 mL of concentrated lentivirus and 1.5 μL of polybrene stock (8 mg/mL) were added, and the solution was mixed well by pipetting up and down several times. The plate was then spun at 900×g for 60 min at 37°C and cultured for 16 h. The next day, all cells were counted and resuspended in RPMI 1640 medium supplemented with 5% FBS and recombinant human IL-2 (30 U/mL). Every 2 or 3 days, fresh medium was added, and T cells were seeded into wells at a density ranging from 0.5–2 × 106 cells/mL.
Production of BCKDK-KO CAR-T cells
BCKDK-KO CAR-T cells were generated based on RNP delivery of CRISPR-Cas9 of BCKDK sgRNA into CAR-T cells. The gRNA sequences for BCKDK KO were designed CRISPick (https://portals.broadinstitute.org/gppx/crispick/public). The optimal gRNA sequences for BCKDK was identified as 5′-GCCAGCGTAGAGCATCATGG-3′ based on a preliminary analysis of the top 5 sgRNAs. tracrRNA and crRNA were obtained from Genscript Biotech. Each sgRNA was formed by mixing tracrRNA and the corresponding crRNA at a 1:1 ratio through an annealing process. Stocked Cas9 protein (Genscript Biotech) was mixed with sgRNA at a molar ratio of 1:1. The RNP complex was formed after incubation at room temperature for 15 min. T cells were prepared at a density of 5 × 106 cells per 50-μL tip reaction in electroporation Buffer R (Neon Transfection System Kits). BCKDK KO was performed via RNP-based delivery through electroporation at 1,600 V, 10 ms, and 3 pulses. Immediately, the cells were transferred into RPMI 1640 medium supplemented with 5% FBS and recombinant human IL-2 (30 U/mL) for continued incubation.
T7E1 surveyor assay
BCKDK-KO CAR-T cells were lysed with QuickExtract Buffer according to the manufacturer’s recommendations. One microliter of genomic DNA was used for PCR with a 50-μL reaction volume that contained DreamTaq Green 2X PCR Master Mix (Thermo Fisher), primer pairs and nuclease-free water. The PCR conditions were 1 cycle of 3 min at 95°C; 36 cycles of 30 s at 95°C, 30 s at 60°C, and 45 s at 72°C; and, finally, a 4-min incubation at 72°C. The PCR products were 550 and 800 bp. Then, 200 ng PCR products obtained from wild-type and KO samples were included in a 19-μL heteroduplex reaction by heating at 95°C for 5 min in a thermocycler following an annealing process. Heteroduplex digestion was performed by adding 1 μL T7 endonuclease I (T7E1; NEB) and incubating at 37°C for 45 min. The digested heteroduplexes (20 μL) were run on a 2% agarose gel stained with ethidium bromide, and the 100-bp DNA ladder was run with the sample for reference.
Chemical treatment
Leucine, isoleucine, and valine were purchased from Sigma Aldrich. BT2 (HY-114855), BCH (HY-108540), and KMH-233 (HY-120139) were from MedChemExpress. JPH203 (S8667) was purchased from Selleck. The concentration of these chemical treatments were showed in the supplementary material of Table S1.
Flow cytometry
Cultured cells and mouse blood samples were collected, processed, and resuspended in fluorescence-activated cell sorting (FACS) buffer (1%–2% BSA in PBS). The specified cell surface markers were incubated at 4°C for 30 min, followed by two washes with FACS buffer. To ensure consistent live cell counts across all conditions, events were collected from an equivalent fixed volume for each sample. The gating strategies encompassed the exclusion of subcellular debris, singlet discrimination, and live:dead staining. All antibodies were applied in accordance with the manufacturer’s guidelines. Cell analysis was conducted using a CytoFLEX Flow Cytometer (Beckman Coulter), and data were processed using FlowJo software (FlowJo).
For the identification of CAR T cell subsets, cells were dual-stained with anti-CD8-PE (BioLegend) at 1:200 and anti-DYKDDDDK (FLAG tag)-APC (BioLegend) at 1:200. NALM6-GL cancer cells were detected using a fluorescence channel. To assess CAR-T cell activation, anti-PD-1-BV421 (Biolegend) at 1:200 and anti-LAG-3-Percp/Cy5.5 (Biolegend) at 1:200 were employed in conjunction with CD8 and the FLAG tag. Memory differentiation of CAR T cells was evaluated using anti-CD45RA-Percy/Cy5.5 (Biolegend) at 1:150 and anti-CD62L-BV421 (Biolegend) at 1:200 along with CD8 and the FLAG tag. Each antibody underwent validation on cells known to be both negative and positive for the target antigen.
Intracellular cytokine staining
Intracellular cytokines production of IFN-γ and TNF-α were assessed following the co-culture of effector CAR-T cells with target NALM6-GL cancer cells. On the day of the experiment, cells were collected and combined in 200 μL media containing the protein transport inhibitors brefeldin (10 μM) and monensin (2 μM). After centrifugation to remove the culture supernatant, the cells were resuspended in a live/dead near-infrared viability stain. Subsequently, the cells were incubated for 5 min at room temperature and then resuspended in 150 μL of fixation/permeabilization buffer (eBiosciences) for a 30-min incubation at room temperature. Staining was performed using anti-CD8-PE (BioLegend) at 1:200 and anti-DYKDDDDDK (FLAG tag)-APC (BioLegend) at 1:200 to identify CAR-T cells. Anti-IFN-γ-PE/Cy7 (BioLegend) at 1:150 and TNF-α-FITC at 1:100 (BioLegend) were utilized to characterize the functionality of CAR-T cells after co-culture. Different populations of intracellular IFN-γ and TNF-α cytokines were analyzed using FlowJo software (FlowJo).
Proliferation assays
Proliferation assays were assessed by incubating CAR-T cells with a 5-μM CFSE staining solution (Thermo Fisher Scientific) at 37°C for 8 min. After staining, cells underwent three washes in MACS buffer and were then allowed to rest in complete media for 10 min. Subsequently, the labeled cells were co-cultured in 96-well flat-bottomed plates with NALM6-GL target cells for 5 days. Following the co-culture period, cells were harvested in 200 μL MACS buffer for the proliferation assay using a CytoFLEX flow cytometer (Beckman Coulter). Histograms displaying distinct proliferating cell populations were generated post subcellular debris exclusion, singlet gating, and live:dead staining in CAR-T cells.
2-NBDG glucose uptake assay
The 2-NBDG glucose uptake assay was assessed by incubating CAR-T cells with the 2-NBDG reagent (Thermo Fisher) at a final concentration of 200 μg/mL for 60 min at 37°C. Subsequently, the cells were collected in 200 μL MACS buffer for the glucose uptake assay using a CytoFLEX flow cytometer (Beckman Coulter). Histograms displaying distinct populations of 2-NBDG-FITC+ proliferating cells were generated after excluding subcellular debris, applying singlet gating, and conducting live:dead staining in CAR-T cells.
Apoptosis assay
The apoptosis assay was accessed by evaluating Annexin V/propidium iodide staining following the co-culture of effector CAR-T cells with target NALM6-GL cancer cells. For the experiment, cells were centrifuged and suspended in 1× Annexin V Binding Buffer supplemented with 5 μL Annexin V-APC working solution and 10 μL of propidium iodide staining solution. Subsequently, the plate was incubated at 37°C for 15 min, followed by the addition of 200 μL of 1× binding buffer to each well. Distinct populations of apoptotic cells were identified through the exclusion of subcellular debris, singlet gating, and live:dead staining in both CAR-T cells and NALM6-GL cancer cells.
Luciferase-based cytotoxicity assays
The cytotoxicity assay to assess the killing efficiency of CAR-T cells against NALM6-GL target cells was performed using the Synergy Multifunctional Microplate Assay following the manufacturer’s instructions (BioTek). Prior to the experiment, 100 μL 2 × 105 NALM6-GL cells (target cells) were fixed and seeded in each well of flat-bottom 96-well plate. The target cells were allowed to settle at 37°C and 5% CO2 for no more than 24 h. Subsequently, modified CAR-T cells (effector cells) were added to each well at the specified ratio and incubated for 24 or 48 h. Target cell viability was assessed by adding 10 μL 150 μg/mL D-Luciferin (LUCK-1G, Goldbio) per well. The fluorescence signal was measured within 10 min, with background luminescence being minimal (<1% of the signal from wells containing only target cells). Cytotoxicity for each sample was calculated using the formula: 100 × (1 − [sample-minimum]/[maximum − minimum]). The minimum value was determined by adding Tween to the cancer cell culture, while the maximum value was determined by applying no treatment. The parallel killing assay experiments were maintained through the compaction of the control CAR-T cells targeted NALM6-GL cells. The remaining viable cells were collected and stained for CAR and CD8 markers to differentiate various subsets, followed by quantitative analysis using flow cytometry.
ELISA
Blood was then collected from the mice in heparinized EP tubes, and plasma was prepared by centrifugation for 10 min at room temperature. The released cytokines IFN-γ (DIF50C) and TNF-α (DTA00D) levels were measured using ELISA kits (R&D Systems) according to the kit instructions. The absorbance was read at 450 nm with background suppression at 650 nm.
RT-qPCR
Total RNA was isolated using TRIzol Reagent (Invitrogen) according to the manufacturer’s protocol. The synthesis of complementary DNA was completed by reverse transcription reaction using SuperScript IV Reverse Transcriptase (Invitrogen). After normalizing the concentrations of complementary DNA with nuclease-free water. For quantification of gene expression, quantitative PCR was performed using SYBR Green Master Mix on the Applied Biosystems StepOne Plus Real-Time PCR system (Applied Biosystems). The internal positive control used the housekeeping gene beta-actin. Gene copy numbers were calculated using the 2-ΔCT method. ΔCT = CT (a target gene) − CT (house keeping gene).
Western blotting analysis
Total protein extraction from CAR cells was performed with ice-cold 1× RIPA buffer containing protease inhibitors (Beyotime Biotech). The protein was adjusted to the same concentration by adding 1× RIPA buffer after detection of the protein concentration. Equal quantities of protein from each sample were separated on a 10% sodium dodecyl sulfate-polyacrylamide gel and transferred to a polyvinylidene fluoride membrane. The membranes were blocked at room temperature for 1 h using 5% BSA in TBST buffer. The primary antibodies were diluted with 5% BSA in TBST buffer at a ratio of 1:800. The membranes were then incubated with the indicated primary antibody at 4°C overnight, washed three times with TBST, incubated with the indicated horseradish peroxidase-conjugated secondary antibody, and washed three times with TBST. Chemiluminescent substrate was mixed and added to the membranes for the detection of protein expression. The blots were imaged using an ECL detection solution (Biotanon).
Metabolomics analysis
Two million CAR-T cells were extracted in 1,000 μL of prechilled 80% MS-grade methanol. After centrifugation at 14,000×g for 15 min, the supernatants were transferred to polypropylene tubes and evaporated under vacuum. The residues were reconstituted with 50 μL methanol-water (90%:10%, v/v), and 10 μL was injected into an Agilent 6490 Triple Quadrupole liquid chromatography mass spectrometry System. Liquid chromatography was optimized with a Kinetex 2.6-μm PS C18, LC Column 150 × 2.1 mm (Phenomenex). Each sample was tested using three replicates. Multiple reaction monitoring was used for the qualitative and quantitative analysis of purified standards. The metabolomics data are provided in Table S2 and the variable importance in projection from partial least squares discriminant analysis is provided in Table S3. The spectral features were extracted using Agilent Mass Hunter Qualitative Analysis Software (version B 6.0.633.0).
In vivo anticancer xenograft models
The animal experiments in this study were approved by the Animal Care Committee of Shanghai Sixth People’s Hospital Affiliated Shanghai Jiao Tong University School of Medicine (Protocol No.: 2021-0697). All of the animal experiments were conducted in the laboratory animal center and were approved by the Institutional Animal Care and Use Committee. Female M-NSG mice (NOD-PrkdcscidIl2rgem1/Smoc) were purchased from Shanghai Modelorg Biotechnology Co., LTD at 8 week s of age. After 1 week of acclimatization, the animals were maintained in specific pathogen-free cages under a standard 12:12 light:dark cycle, at a temperature of 20°C–26°C and humidity between 30% and 70%. To develop the xenograft models, 0.5 × 106 NALM6-GL cells in 100 μL PBS were intravenously inoculated into M-NSG mice via the tail vein on day −3. The mice were then randomized into four groups for eight cages. The percentage of CAR-positive cells, which varied between the CAR constructs, was equalized by counting the number of CAR-T cells prior to injection. Then, 1 × 106 control CAR-T cells, mock CAR-T cells, BCKDK-OE CAR-T cells, and BCKDK-KO CAR-T cells in 100 μL RPMI 1640 medium were injected via the tail vein on days 0, 3, and 6 after injection of NALM6-GL cells. Survival and disease status were recorded according to the veterinarian’s instructions. On days 14 and 28 after CAR T cell injection, peripheral blood was collected from five mice in each group for flow cytometry and ELISA. Body weight was measured daily.
Statistics
The statistical analysis of the experiment was detailed in the figure panel, figure legend, and the supplementary material. The results were expressed as the mean ± SD for biological replicates, with the sample size at least independently triplication that indicated in the figure legend for each experiment. Normality of variable distribution was assessed using the Kolmogorov-Smirnov test, and homogeneity of variances was checked with Levene’s test. Two-tailed unpaired Student’s t test was employed for comparing two groups, while one-way ANOVA or two-way ANOVA followed by post hoc Tukey’s multiple comparison test were used for comparing multiple groups. Survival analysis was conducted using the Kaplan-Meier method, and significance was determined using the log rank test. Statistical analyses were carried out using GraphPad Prism 8.0. Statistical significance were indicated in all figures by the following annotations: ∗p < 0.05; ∗∗p < 0.01, ∗∗∗p < 0.001; and ∗∗∗p < 0.0001. The original statistical data are included in Tables S4–S23.
Data and code availability
Data will be made available on request.
Acknowledgments
We appreciate Instrumental Analysis Center Shanghai Jiao Tong University for the help with flow cytometry and metabolomics analysis. This work was supported by a grant from National Natural Science Foundation of China (No.: 82272925, 81872494 and 82274151), Excellent Young Scientists Fund of the Natural Science Foundation of China (No.: 82322020), and Shanghai Pujiang Talent Project (No.: 21PJ1411900).
Author contributions
Q.Y. and C.G. designed the experiments. Y.H. supervised the project. J.H. and T.Y. provided experimental materials. Q.Y., X.Z., L.H., R.G., B.X., Y.T., S.Z., and J.H. performed the experiments. P.H., X.Z., C.L., and L.S. analyzed the data. Q.Y., J.H., C.L., L.Z., and L.S. did the data curation and visualization. X.Z., Q.Y., L.S., and C.G. wrote the manuscript.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2024.05.017.
Contributor Information
Lei Shi, Email: rm001276@whu.edu.cn.
Cheng Guo, Email: guopharm@126.com.
Supplemental information
References
- 1.Mullard A. FDA approves fourth CAR-T cell therapy. Nat. Rev. Drug Discov. 2021;20:166. doi: 10.1038/d41573-021-00031-9. [DOI] [PubMed] [Google Scholar]
- 2.Lin X., Lee S., Sharma P., George B., Scott J. Summary of US Food and Drug Administration Chimeric Antigen Receptor (CAR) T-Cell Biologics License Application Approvals From a Statistical Perspective. J. Clin. Oncol. 2022;40:3501–3509. doi: 10.1200/JCO.21.02558. [DOI] [PubMed] [Google Scholar]
- 3.Granit V., Benatar M., Kurtoglu M., Miljković M.D., Chahin N., Sahagian G., Feinberg M.H., Slansky A., Vu T., Jewell C.M., et al. Safety and clinical activity of autologous RNA chimeric antigen receptor T-cell therapy in myasthenia gravis (MG-001): a prospective, multicentre, open-label, non-randomised phase 1b/2a study. Lancet Neurol. 2023;22:578–590. doi: 10.1016/S1474-4422(23)00194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keshavarz A., Salehi A., Khosravi S., Shariati Y., Nasrabadi N., Kahrizi M.S., Maghsoodi S., Mardi A., Azizi R., Jamali S., Fotovat F. Recent findings on chimeric antigen receptor (CAR)-engineered immune cell therapy in solid tumors and hematological malignancies. Stem Cell Res. Ther. 2022;13:482. doi: 10.1186/s13287-022-03163-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Z., Zhou Z., Dang Q., Xu H., Lv J., Li H., Han X. Immunosuppression in tumor immune microenvironment and its optimization from CAR-T cell therapy. Theranostics. 2022;12:6273–6290. doi: 10.7150/thno.76854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng J.J., Wang L., Li Z., Ku C.L., Ho P.C. Metabolic challenges and interventions in CAR T cell therapy. Sci. Immunol. 2023;8 doi: 10.1126/sciimmunol.abq3016. [DOI] [PubMed] [Google Scholar]
- 7.Arner E.N., Rathmell J.C. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell. 2023;41:421–433. doi: 10.1016/j.ccell.2023.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng Z., Wei R., Ma Q., Shi L., He F., Shi Z., Jin T., Xie R., Wei B., Chen J., et al. In Vivo Expansion and Antitumor Activity of Coinfused CD28- and 4-1BB-Engineered CAR-T Cells in Patients with B Cell Leukemia. Mol. Ther. 2018;26:976–985. doi: 10.1016/j.ymthe.2018.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lanitis E., Coukos G., Irving M. All systems go: converging synthetic biology and combinatorial treatment for CAR-T cell therapy. Curr. Opin. Biotechnol. 2020;65:75–87. doi: 10.1016/j.copbio.2020.01.009. [DOI] [PubMed] [Google Scholar]
- 10.Freitas K.A., Belk J.A., Sotillo E., Quinn P.J., Ramello M.C., Malipatlolla M., Daniel B., Sandor K., Klysz D., Bjelajac J., et al. Enhanced T cell effector activity by targeting the Mediator kinase module. Science. 2022;378 doi: 10.1126/science.abn5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawalekar O.U., O' Connor R.S., Fraietta J.A., Guo L., McGettigan S.E., Posey A.D., Jr., Patel P.R., Guedan S., Scholler J., Keith B., et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44:712. doi: 10.1016/j.immuni.2016.02.023. [DOI] [PubMed] [Google Scholar]
- 12.Zhao X., Yang J., Zhang X., Lu X.-A., Xiong M., Zhang J., Zhou X., Qi F., He T., Ding Y., et al. Efficacy and safety of CD28-or 4-1BB-based CD19 CAR-T cells in B cell acute lymphoblastic leukemia. Mol. Ther. Oncolytics. 2020;18:272–281. doi: 10.1016/j.omto.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalbasi A., Siurala M., Su L.L., Tariveranmoshabad M., Picton L.K., Ravikumar P., Li P., Lin J.X., Escuin-Ordinas H., Da T., et al. Potentiating adoptive cell therapy using synthetic IL-9 receptors. Nature. 2022;607:360–365. doi: 10.1038/s41586-022-04801-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agliardi G., Liuzzi A.R., Hotblack A., De Feo D., Núñez N., Stowe C.L., Friebel E., Nannini F., Rindlisbacher L., Roberts T.A., et al. Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat. Commun. 2021;12:444. doi: 10.1038/s41467-020-20599-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alizadeh D., Wong R.A., Yang X., Wang D., Pecoraro J.R., Kuo C.-F., Aguilar B., Qi Y., Ann D.K., Starr R., et al. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol. Res. 2019;7:759–772. doi: 10.1158/2326-6066.CIR-18-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Q., Hao J., Chi M., Wang Y., Li J., Huang J., Zhang J., Zhang M., Lu J., Zhou S., et al. D2HGDH-mediated D2HG catabolism enhances the anti-tumor activities of CAR-T cells in an immunosuppressive microenvironment. Mol. Ther. 2022;30:1188–1200. doi: 10.1016/j.ymthe.2022.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Q., Hao J., Chi M., Wang Y., Xin B., Huang J., Lu J., Li J., Sun X., Li C., et al. Superior antitumor immunotherapy efficacy of kynureninase modified CAR-T cells through targeting kynurenine metabolism. Oncoimmunology. 2022;11 doi: 10.1080/2162402X.2022.2055703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang L., Chu Z., Liu M., Zou Q., Li J., Liu Q., Wang Y., Wang T., Xiang J., Wang B. Amino acid metabolism in immune cells: essential regulators of the effector functions, and promising opportunities to enhance cancer immunotherapy. J. Hematol. Oncol. 2023;16:59. doi: 10.1186/s13045-023-01453-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ananieva E.A., Powell J.D., Hutson S.M. Leucine Metabolism in T Cell Activation: mTOR Signaling and Beyond. Adv. Nutr. 2016;7:798S–805S. doi: 10.3945/an.115.011221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sinclair L.V., Rolf J., Emslie E., Shi Y.B., Taylor P.M., Cantrell D.A. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 2013;14:500–508. doi: 10.1038/ni.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kikushige Y., Miyamoto T., Kochi Y., Semba Y., Ohishi M., Irifune H., Hatakeyama K., Kunisaki Y., Sugio T., Sakoda T., et al. Human acute leukemia uses branched-chain amino acid catabolism to maintain stemness through regulating PRC2 function. Blood Adv. 2023;7:3592–3603. doi: 10.1182/bloodadvances.2022008242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y.W., Velasco-Hernandez T., Mess J., Lalioti M.E., Romero-Mulero M.C., Obier N., Karantzelis N., Rettkowski J., Schönberger K., Karabacz N., et al. GPRC5C Drives Branched-Chain Amino Acid Metabolism in Leukemogenesis. Blood Adv. 2023;7:7525–7538. doi: 10.1182/bloodadvances.2023010460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Q.J., Zhao J.R., Hao J., Li B., Huo Y., Han Y.L., Wan L.L., Li J., Huang J., Lu J., et al. Serum and urine metabolomics study reveals a distinct diagnostic model for cancer cachexia. J. Cachexia Sarcopenia Muscle. 2018;9:71–85. doi: 10.1002/jcsm.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hattori A., Tsunoda M., Konuma T., Kobayashi M., Nagy T., Glushka J., Tayyari F., McSkimming D., Kannan N., Tojo A., et al. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature. 2017;545:500–504. doi: 10.1038/nature22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Follett A., Figueroa L., Boyer M., Ananieva E. The Oncogene MYC Regulates the Branched Chain Amino Acid Metabolism and mTOR Signaling in Diffuse Large B Cell Lymphoma. FASEB J. 2022;36 [Google Scholar]
- 26.Yu Z., Qiu B., Zhou H., Li L., Niu T. Characterization and application of a lactate and branched chain amino acid metabolism related gene signature in a prognosis risk model for multiple myeloma. Cancer Cell Int. 2023;23:169. doi: 10.1186/s12935-023-03007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.East M.P., Laitinen T., Asquith C.R.M. BCKDK: an emerging kinase target for metabolic diseases and cancer. Nat. Rev. Drug Discov. 2021;20:498. doi: 10.1038/d41573-021-00107-6. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y., Xiao J., Jiang W., Zuo D., Wang X., Jin Y., Qiao L., An H., Yang L., Dumoulin D.W., et al. BCKDK alters the metabolism of non-small cell lung cancer. Transl. Lung Cancer Res. 2021;10:4459–4476. doi: 10.21037/tlcr-21-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tian Q., Yuan P., Quan C., Li M., Xiao J., Zhang L., Lu H., Ma T., Zou L., Wang F., et al. Phosphorylation of BCKDK of BCAA catabolism at Y246 by Src promotes metastasis of colorectal cancer. Oncogene. 2020;39:3980–3996. doi: 10.1038/s41388-020-1262-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan R., Zhao X., Lei J., Zhou Q. Structure of the human LAT1-4F2hc heteromeric amino acid transporter complex. Nature. 2019;568:127–130. doi: 10.1038/s41586-019-1011-z. [DOI] [PubMed] [Google Scholar]
- 31.Fitzgerald E., Roberts J., Tennant D.A., Boardman J.P., Drake A.J. Metabolic adaptations to hypoxia in the neonatal mouse forebrain can occur independently of the transporters SLC7A5 and SLC3A2. Sci. Rep. 2021;11:9092. doi: 10.1038/s41598-021-88757-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huttunen K.M., Gynther M., Huttunen J., Puris E., Spicer J.A., Denny W.A. A Selective and Slowly Reversible Inhibitor of l-Type Amino Acid Transporter 1 (LAT1) Potentiates Antiproliferative Drug Efficacy in Cancer Cells. J. Med. Chem. 2016;59:5740–5751. doi: 10.1021/acs.jmedchem.6b00190. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y., Qin L., Chen W., Chen Q., Sun J., Wang G. Novel strategies to improve tumour therapy by targeting the proteins MCT1, MCT4 and LAT1. Eur. J. Med. Chem. 2021;226 doi: 10.1016/j.ejmech.2021.113806. [DOI] [PubMed] [Google Scholar]
- 34.Markowicz-Piasecka M., Huttunen J., Montaser A., Huttunen K.M. Hemocompatible LAT1-inhibitor can induce apoptosis in cancer cells without affecting brain amino acid homeostasis. Apoptosis. 2020;25:426–440. doi: 10.1007/s10495-020-01603-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okano N., Hana K., Naruge D., Kawai K., Kobayashi T., Nagashima F., Endou H., Furuse J. Biomarker Analyses in Patients With Advanced Solid Tumors Treated With the LAT1 Inhibitor JPH203. In Vivo. 2020;34:2595–2606. doi: 10.21873/invivo.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayashi K., Jutabha P., Endou H., Sagara H., Anzai N. LAT1 is a critical transporter of essential amino acids for immune reactions in activated human T cells. J. Immunol. 2013;191:4080–4085. doi: 10.4049/jimmunol.1300923. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y., Fang Y., Chen X., Wang Z., Liang X., Zhang T., Liu M., Zhou N., Lv J., Tang K., et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 2020;5 doi: 10.1126/sciimmunol.aax7969. [DOI] [PubMed] [Google Scholar]
- 38.Zhang N., Liu X., Qin J., Sun Y., Xiong H., Lin B., Liu K., Tan B., Zhang C., Huang C., et al. LIGHT/TNFSF14 promotes CAR-T cell trafficking and cytotoxicity through reversing immunosuppressive tumor microenvironment. Mol. Ther. 2023;31:2575–2590. doi: 10.1016/j.ymthe.2023.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Golumba-Nagy V., Kuehle J., Hombach A.A., Abken H. CD28-zeta CAR T Cells Resist TGF-beta Repression through IL-2 Signaling, Which Can Be Mimicked by an Engineered IL-7 Autocrine Loop. Mol. Ther. 2018;26:2218–2230. doi: 10.1016/j.ymthe.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ochi T., Maruta M., Tanimoto K., Kondo F., Yamamoto T., Kurata M., Fujiwara H., Masumoto J., Takenaka K., Yasukawa M. A single-chain antibody generation system yielding CAR-T cells with superior antitumor function. Commun. Biol. 2021;4:273. doi: 10.1038/s42003-021-01791-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halaby M.J., McGaha T.L. Amino Acid Transport and Metabolism in Myeloid Function. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.695238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Renauer P., Park J.J., Bai M., Acosta A., Lee W.-H., Lin G.H., Zhang Y., Dai X., Wang G., Errami Y. Immunogenetic metabolomics reveals key enzymes that modulate CAR T-cell metabolism and function. Cancer Immunol. Res. 2023;11:1068–1084. doi: 10.1158/2326-6066.CIR-22-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye L., Park J.J., Peng L., Yang Q., Chow R.D., Dong M.B., Lam S.Z., Guo J., Tang E., Zhang Y., et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. 2022;34:595–614.e14. doi: 10.1016/j.cmet.2022.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ericksen R.E., Lim S.L., McDonnell E., Shuen W.H., Vadiveloo M., White P.J., Ding Z., Kwok R., Lee P., Radda G.K., et al. Loss of BCAA Catabolism during Carcinogenesis Enhances mTORC1 Activity and Promotes Tumor Development and Progression. Cell Metab. 2019;29:1151–1165.e6. doi: 10.1016/j.cmet.2018.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou Z., Tao T., Li H., Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 2020;10:31. doi: 10.1186/s13578-020-00396-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scholz G., Jandus C., Zhang L., Grandclément C., Lopez-Mejia I.C., Soneson C., Delorenzi M., Fajas L., Held W., Dormond O., Romero P. Modulation of mTOR Signalling Triggers the Formation of Stem Cell-like Memory T Cells. EBioMedicine. 2016;4:50–61. doi: 10.1016/j.ebiom.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meyran D., Zhu J.J., Butler J., Tantalo D., MacDonald S., Nguyen T.N., Wang M., Thio N., D’Souza C., Qin V.M., et al. TSTEM-like CAR-T cells exhibit improved persistence and tumor control compared with conventional CAR-T cells in preclinical models. Sci. Transl. Med. 2023;15 doi: 10.1126/scitranslmed.abk1900. [DOI] [PubMed] [Google Scholar]
- 48.Gu Z., Liu Y., Cai F., Patrick M., Zmajkovic J., Cao H., Zhang Y., Tasdogan A., Chen M., Qi L., et al. Loss of EZH2 Reprograms BCAA Metabolism to Drive Leukemic Transformation. Cancer Discov. 2019;9:1228–1247. doi: 10.1158/2159-8290.CD-19-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sivanand S., Vander Heiden M.G. Emerging Roles for Branched-Chain Amino Acid Metabolism in Cancer. Cancer Cell. 2020;37:147–156. doi: 10.1016/j.ccell.2019.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heinemann-Yerushalmi L., Bentovim L., Felsenthal N., Vinestock R.C., Michaeli N., Krief S., Silberman A., Cohen M., Ben-Dor S., Brenner O., et al. BCKDK regulates the TCA cycle through PDC in the absence of PDK family during embryonic development. Dev. Cell. 2021;56:1182–1194.e6. doi: 10.1016/j.devcel.2021.03.007. [DOI] [PubMed] [Google Scholar]
- 51.Maiti A., Daver N.G. Lowering mTORC1 Drives CAR T-Cells Home in Acute Myeloid Leukemia. Clin. Cancer Res. 2021;27:5739–5741. doi: 10.1158/1078-0432.CCR-21-2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.