

Abstract
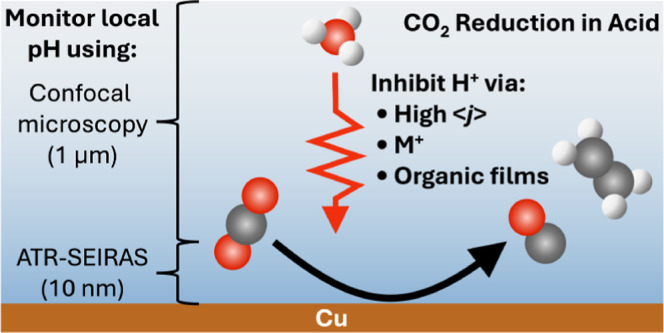
Inspired by recent advances in electrochemical CO2 reduction (CO2R) under acidic conditions, herein we leverage in situ spectroscopy to inform the optimization of CO2R at low pH. Using attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) and fluorescent confocal laser scanning microscopy, we investigate the role that alkali cations (M+) play on electrochemical CO2R. This study hence provides important information related to the local electrode surface pH under bulk acidic conditions for CO2R, both in the presence and absence of an organic film layer, at variable [M+]. We show that in an acidic electrolyte, an appropriate current density can enable CO2R in the absence of metal cations. In situ local pH measurements suggest the local [H+] must be sufficiently depleted to promote H2O reduction as the competing reaction with CO2R. Incrementally incorporating [K+] leads to increases in the local pH that promotes CO2R but only at proton consumption rates sufficient to drive the pH up dramatically. Stark tuning measurements and analysis of surface water structure reveal no change in the electric field with [M+] and a desorption of interfacial water, indicating that improved CO2R performance is driven by suppression of H+ mass transport and modification of the interfacial solvation structure. In situ pH measurements confirm increasing local pH, and therefore decreased local [CO2], with [M+], motivating alternate means of modulating proton transport. We show that an organic film formed via in situ electrodeposition of an organic additive provides a means to achieve selective CO2R (FECO2R ∼ 65%) over hydrogen evolution reaction in the presence of strong acid (pH 1) and low cation concentrations (≤0.1 M) at both low and high current densities.
Introduction
Electrochemical CO2 reduction (CO2R) is an appealing approach to convert a cheap and abundant precursor into value-added fuels and chemicals through the use of renewably sourced electricity.1−3 The majority of CO2R studies are conducted in neutral and basic electrolytes to help favor the selectivity for CO2R over the hydrogen evolution reaction (HER).3,4 However, these electrolytes suffer from low CO2 utilization due to parasitic (bi)carbonate formation and subsequent migration to the anolyte.5,6 Strategies to mitigate this problem include the use of artificial membrane-electrode assemblies with bipolar membranes or solid electrolytes7−10 and performing CO2R in acidic electrolytes.11−13
Despite the fact that the operation of CO2R at low pH is a promising method for minimizing (bi)carbonate formation, HER becomes a more significant challenge to overcome. Many studies have shown that using electrolytes with high alkali cation concentrations ([M+]) can suppress HER and promote CO2R.12−20 This strategy relies on high [M+] to effectively screen the surface potential over a very short distance (the Debye length) in order to suppress H+ transport to the cathode, thereby reducing HER. Recent work from our group and that of other laboratories has demonstrated selective CO2R under acidic conditions without high [M+] or any M+ at all,11,21−24 which raises questions regarding the role(s) cations play in enabling CO2R, especially in acidic media. The primary mechanistic hypotheses that have been proposed include:
-
(i)
Partially dehydrated M+ ions assist in the CO2 adsorption/activation steps, via short-range electrostatic or direct bonding interactions with adsorbed CO2R intermediates.25−27
-
(ii)
M+ ions accumulated at the outer Helmholtz plane enhance the local electric field to stabilize adsorbed polar intermediates to promote CO2R; these accumulated M+ cations also screen the electric field generated from the cathode to suppress transport of H+ and hence competing HER.12,16,21,23,28
-
(iii)
Solvated M+ ions buffer the local pH based on the pKa of the coordinated H2O, which mitigates carbonate formation and maintains appropriate local [CO2] to facilitate CO2R.29
In the present study, we experimentally address the local electrode surface pH under bulk acidic conditions for CO2R, both in the presence and absence of an organic film layer, at a variable [M+]. We investigate systems where the Debye length is extended due to low M+ concentrations, enabling investigation into the effect of proton transport across both the diffusion layer (ca. 100 μm) and the double layer (1–20 nm).30 By employing attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) and fluorescent confocal laser scanning microscopy (CLSM), we show that interfacial [H+] depletion is essential toward enhancing CO2R in acidic media. We demonstrate that this condition can be achieved by high surface proton consumption, the presence of metal cations, or the application of an organic coating (Figure 1). Specifically, time-resolved in situ pH measurements were performed to interrogate the changes in the microenvironment at the electrode–electrolyte interface using ATR-SEIRAS and within the concentration diffusion layer using CLSM in each case.
Figure 1.

Strategies to access and promote CO2R in acidic electrolytes are probed in the study. We find that for CO2R to occur on Cu under bulk acidic conditions, the local [H+] must be depleted. Such a microenvironment may be induced by (a) high current; (b) added [M+] to generate a local electric field; and (c) organic film-coated electrodes. Gray, red, white, and blue spheres represent C, O, H, and K atoms, respectively. Black arrows represent the relative H+ transport to the cathode surface for each scenario.
Our studies indicate that in the absence of M+, a current density high enough to deplete local [H+], promoting H2O reduction over H+ reduction, enables CO2R to compete favorably with the HER. By increasing [M+], we observe improved selectivity for CO2R products. SEIRAS measurements reveal a constant local electric field within the [K+] range studied, pointing to improved selectivity correlating with longer range cation-induced transport effects, including interfacial solvation and H+ diffusion. Additionally, CLSM pH measurements demonstrate the concomitant increase in local pH with the M+ concentration, highlighting the need for an alternative means of local pH control.
Experiments with an organic film-modified electrode, a known proton transport modulator, in the absence of M+, support these findings by demonstrating an order of magnitude increase in FECO2R compared to the uncoated electrode for CO2R. Furthermore, by leveraging short-range electric field effects from small concentrations of metal cations and long-range transport modulation effects from organic films, we identify optimized conditions for performing CO2R in acidic media without high [M+].
Results and Discussion
Quantification of pH Changes at Different Length Scales via ATR-SEIRAS and CLSM
ATR-SEIRAS and CLSM measurements were performed to monitor the reaction interface as a function of the potential and time. The high surface sensitivity of SEIRAS is leveraged to determine the local pH at a length scale relevant to surface catalytic reactions,31 whereas the greater lateral and vertical resolution of CLSM (∼500 nm) is used to differentiate interfacial versus more extended pH phenomena. Herein, the term “interfacial pH” will refer to the pH measured in the electrolyte from the cathode surface to ∼10 nm into the double layer using SEIRAS,31 and the term “local pH” will refer to the pH within 1 μm of the surface measured using CLSM.32 Due to the resolution of CLSM, the pH value reported was averaged across 1 μm and is expected to be less alkaline than that reported by SEIRAS.
The SEIRAS-active cathode was fabricated according to previously reported methods.33 Briefly, a polycrystalline gold underlayer was chemically deposited onto a Si ATR prism. The gold film was used to template the Cu electrodeposition,34 which was then used as the cathode. We used the phosphate buffer system as the interfacial pH reporter, as has been previously demonstrated.35,36
The spectra of the phosphate species at different pH values on the Cu film were collected (Figure 2a). To monitor the pH changes during electrolysis, calibration spectra were recorded between pH 5 and 13 by the addition of KOH to the initial KH2PO4 solution and recording a spectrum every ∼0.4 pH units at a constant potential of 0 VRHE (Figure S1). The overlapping bands were deconvoluted to resolve the contributions from the individual bands (Figure S2). The deconvoluted peak contributions are plotted as a function of pH (Figure 2b), where the intensities are derived from va(PO) of H2PO4– and vs(PO) of HPO42– between pH 5 and 9 and va(PO) of both HPO42– and PO43– between pH 9 and 12.35,36 The interfacial pH was calculated by correlating the ratio of phosphate electrolyte peaks in the sample spectra to the calibration spectra.
Figure 2.
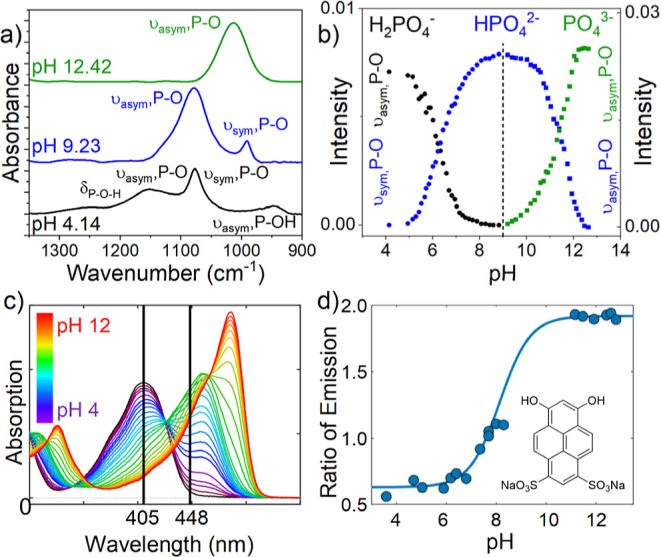
(a) pH-dependent SEIRA spectra of 0.5 M phosphate solutions with corresponding band assignments; (b) phosphate ion speciation used for pH calculations derived from SEIRA spectra absorption intensity; (c) absorption spectrum as a function of pH showing the two excitation wavelengths for fluorescent dye 6,8-dihydroxypyrene-1,3-disulfonic acid disodium salt (DHPDS); and (d) calibration curve for the local pH based on the ratio of emission of the dye and chemical structure of DHPDS.
CLSM measurements were performed using the ratiometric fluorescent dye 6,8-dihydroxypyrene-1,3-disulfonic acid disodium salt (DHPDS) to monitor the local pH near the electrode surface at different ⟨j⟩.32,37,38 The ratio of the DHPDS emission allows for the detection of pH values between 6 and 11.5 (Figure 2c,d). Although DHPDS contains Na+ in the structure, its low concentration in the test solution ([Na+] = 0.05 mM, close to 1 ppm (∼0.03 mM) of M+ in pH 2 H3PO4) is assumed to have a negligible effect on the CO2R results, in accordance with results presented in the following section.
CO2R on Cu Foil without Metal Cations in the Electrolyte
We studied CO2R in the absence of M+ on Cu foil electrodes using controlled-current electrolysis in a CO2-saturated pH 2 H3PO4 electrolyte. Application of low (⟨j⟩ = −1.25 mA/cm2) and moderate (⟨j⟩ = −5.00 mA/cm2) current densities did not yield gas or liquid CO2R products (Table S2). However, at a high current density (⟨j⟩ = −10.0 mA/cm2), gaseous products (CO, CH4) were observed by online gas chromatography, and formic acid was detected in the liquid phase after electrolysis (Table S2), albeit at very low FECO2R = 0.4 ± 0.2%. Evidence for CO2R occurring at high ⟨j⟩ is supported by monitoring the *CO bands of the SEIRA spectra (Figure 3a).
Figure 3.
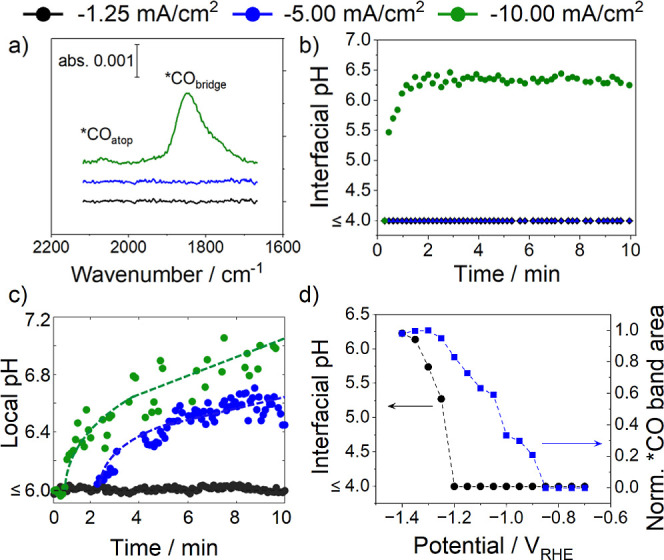
(a) SEIRA spectra of the adsorbed CO region after 5 min of applied ⟨j⟩ in pH 2 H3PO4; (b) interfacial pH as a function of time and ⟨j⟩ in pH 2 H3PO4 extracted from SEIRAS data; (c) local pH as a function of time and ⟨j⟩ in pH 2 H3PO4 extracted from CLSM data; and (d) local pH (black points) and *CO band intensity (blue points) as a function of potential in pH 2 H3PO4 extracted from SEIRAS data. Note pH 4 is the lower limit of detection for the phosphate system, and therefore, pH values < 4 will appear as pH 4, although they could be much lower.
Adsorbed CO (*CO) is the longest-lived CO2R reaction intermediate and is most readily detected using in situ spectroscopy. The detection of *CO on Cu at reducing potentials is indicative of CO2 activation and reduction. The *CO band shape and position are highly sensitive to the reaction microenvironment, making *CO particularly suited to probe the electrocatalytic interface.39
At low (black trace) and moderate (blue trace) currents, no *CO is detected, confirming the absence of CO2 activation and reduction. Applying a high current (green trace) generates two binding modes of *CO, indicating the occurrence of CO2R. *COatop, where the CO carbon is bound to one Cu atom, has been shown to be active for CO2R, while *CObridge, where the CO carbon is bound to two Cu atoms, has been shown to be inactive for CO2R but present in larger quantities on restructured Cu surfaces.39−41 The small intensity of the *COatop peak aligns with the observed trace selectivity for CO2R products, while the large *CObridge band is consistent with high *CO coverage.42
The potential vs time trace for this applied ⟨j⟩ = 10.0 mA/cm2 shows a rapid drop to potentials below −1.40 VRHE (Figure S3). Based on HER studies, once the applied E is more negative than −1.40 VRHE, the local [H+] is depleted so that H2O reduction replaces H+ reduction for HER.43 As H2O reduction is a slower process than H+ reduction and H2O reduction can be inhibited by adsorbed CO, CO2R can occur without added M+. A relevant recent study by Koper and Liu demonstrated that CO2R can compete with H+ reduction with [M+] > 100 mM, yielding an increase in the interfacial pH due to CO2R-generated hydroxide ions consuming H+.44 We find that CO2R is unable to compete with H+ reduction without M+, consistent with CO2R in acidic media requiring M+, or some other means of generating a sufficiently nonacidic interfacial pH.
The degree of local [H+] depletion was probed by using SEIRAS interfacial pH measurements. The experimentally measured interfacial pH is shown as a function of time for different applied ⟨j⟩ (Figure 3b). Consistent with the applied ⟨j⟩ not sufficiently depleting the local [H+] to allow for CO2R, we did not observe a substantial pH increase for the low (black points) and moderate (blue points) ⟨j⟩ (note: since the detection range of the phosphate system is between pH 4 and 13, pH values < 4 will also appear as pH 4, although they could be lower). However, applying a high current (green points) generated a fast interfacial pH change toward ∼6. CLSM measurements confirm this trend, showing increasing local pH with ⟨j⟩ (Figure 3c). Similar steady-state local and interfacial pH trends for high ⟨j⟩ underscore low OH– and H+ transport barriers within the concentration diffusion layer in pure acidic electrolytes. We caution that the absolute pH values obtained with the two methods cannot be quantitatively correlated due to differences in convection within each spectroelectrochemical cell geometry (the CLSM electrolyte is circulated at a rate of 1 mL/min to mitigate bubble buildup that hinders pH imaging, while the SEIRAS electrolyte is stationary aside from bubbling perpendicular to the cathode at 5 mL/min).
To further probe the occurrence of CO2R without M+, we conducted the same controlled-current electrolysis using Cu gas diffusion electrodes (Cu-GDE) to mitigate CO2 mass transport limitations and obtained FECO2R = 19.8 ± 5.4% in pH 2 H3PO4 at −10.0 mA/cm2 (Figure S5, Table S3). We note that Cu ions dissolved from the Cu catalyst or trace impurities of M+ in H3PO4 may also facilitate CO2R on Cu in the pH 2 H3PO4 electrolyte.27 Impurities of M+ (∼1 ppm) were detectable by inductively coupled plasma mass spectrometry in the H3PO4 electrolyte (Table S1). To assess the effect of these trace metals, we performed electrolysis experiments in the presence of 10 mM 18-crown-6 as a chelate. Similar CO2R performance was observed at −10.0 mA/cm2 in pH 2 H3PO4 with 18-crown-6 (Figure S4), which suggests that the observed CO2R was not due to dissolved Cu or other trace amounts of M+ in the electrolyte.
The depletion of local [H+] at high ⟨j⟩ was cross-validated using constant potential SEIRAS measurements, where an increase in the interfacial pH is not observed until the potential becomes more negative than −1.25 VRHE. We observed constant growth of the *CO peak area with increasing potential (Figure 3d), indicating that generated *CO molecules remain adsorbed on the surface in the absence of M+.24 The high *CO coverage, paired with the low FECO2R, indicates that M+-induced interfacial solvation and/or electric field are essential for promoting *CO consumption.
Enhanced CO2R in Acidic Electrolytes with Varying [M+]
Although CO2R can occur on Cu in the absence of M+, the conditions of high current and overpotential to generate a local pH gradient, as well as poor selectivity, are not ideal for practical implementation. The addition of M+ to the electrolyte alters the electrochemical double layer. M+ can change the interfacial water structure and also induce a stronger local electric field within the Stern layer, which inhibits the transport of H+ to the electrode surface by screening the electric field felt within the diffuse layer.12,21 Inhibited H+ transport is expected to create a local pH gradient, which was quantified using SERIAS.
Constant-current electrolysis experiments were performed in CO2-saturated pH 2 H3PO4 with varying [KCl]. Application of low (⟨j⟩ = −1.25 mA/cm2) current did not yield gas or liquid CO2R products with any of the K+-containing electrolytes (Figure S6, Table S5); relatedly, no change in the local pH from the SEIRA spectra was observed. The low current is insufficient to saturate the double layer with K+ to reduce the level of H+ transport, implying that CO2R cannot occur at acidic local pH values regardless of [K+]. By applying a higher current (⟨j⟩ = −5.00 mA/cm2), we observe CO2R products for all [K+] (Figure 4a) along with similar trends in interfacial and local pH, with pH values increasing to ≥6 for all K+-containing electrolytes (Figures 4c and S7). Again, we note that slightly different steady-state values between the techniques can be attributed to differences in spectroelectrochemical cell geometry for SEIRAS and CLSM. The rate of *CO growth increases with [K+] (Figure 4e), and only 200 mM K+ displays *CO consumption over 10 min. The increasing *CO peak areas over time indicate high *CO coverage. Furthermore, there is not an apparent trend between the *CO peak intensity and the measured interfacial pH.
Figure 4.
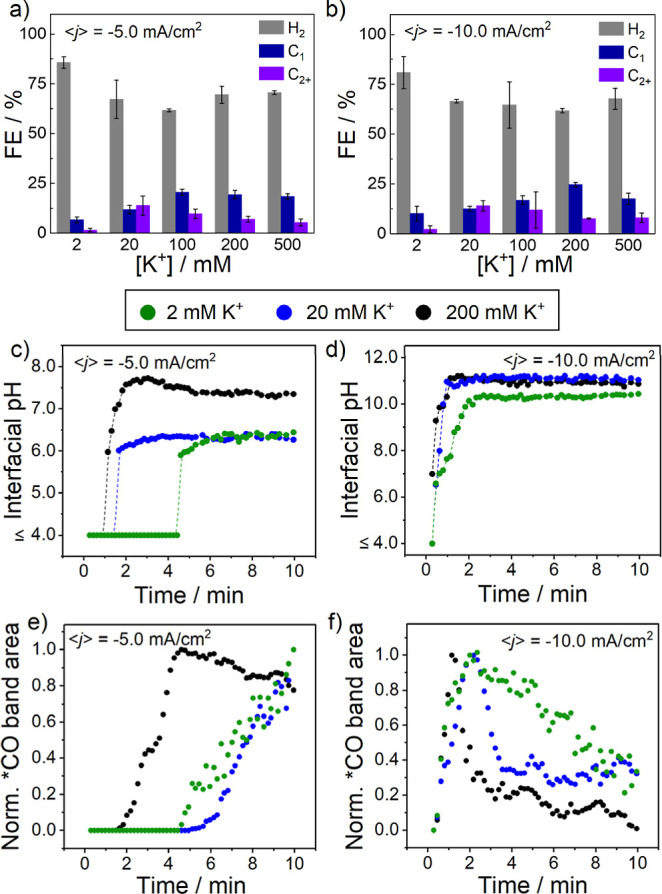
(a) CO2R electrolysis product distribution for different [K+] at ⟨j⟩ = −5.0 mA/cm2 in pH 2 H3PO4; (b) CO2R electrolysis product distribution for different [K+] at ⟨j⟩ = −10.0 mA/cm2; (c) interfacial pH as a function of time for different [K+] at ⟨j⟩ = −5.0 mA/cm2; (d) interfacial pH as a function of time for different [K+] at ⟨j⟩ = −10.0 mA/cm2; (e) time-dependent *CO band intensity for ⟨j⟩ = −5.0 mA/cm2; and (f) time-dependent *CO band intensity for ⟨j⟩ = −10.0 mA/cm2.
For the highest applied current density (⟨j⟩ = −10.00 mA/cm2), the FECO2R and C1/C2+ ratio are relatively unchanged from the moderate current case (Figure 4b). We find that increased [M+] leads to raised interfacial and local pH values (Figure 4d), underscoring that elevating the alkali metal concentration is an undesirable means of controlling proton transport as it depletes the local [CO2]. We observed that the rate of *CO growth, and in this case also *CO consumption, increased with [K+] (Figure 4f), supporting the hypothesis that the local depletion of H+, and subsequently CO2, is the primary factor in the rate of CO2R in acidic electrolytes.
Vibrational Stark effect measurements of *COatop were performed to assess whether the increased FECO2R and *CO buildup from 2 to 200 mM [K+] was due to differences in the electric field. The Stark tuning slope reflects the relative strength of the electric field in the electrochemical double layer, the change of which induces a shift in vibrational band positions.45 We performed these measurements in the limit of low *CO coverage (post electrolysis at −10.0 mA/cm2) to determine the intrinsic Stark tuning slope.46
Using *CO generated from CO2 allows for an estimation of the average electric field at CO2R active sites.47 The *COatop bands and the calculated Stark tuning slope (dν/dV) do not change for different [K+] values (Figure 5a–c). The spectrometer resolution used in this work is 4 cm–1; therefore, differences in the values observed are not significant. This result is consistent with a previous report demonstrating an [M+]-independent electric field strength for electrolyte cation concentrations from 0.1 to 1 M.48 The value of ∼40 cm–1/V is on the order of what has previously been reported for *CO in K+-containing electrolytes on Cu.49,50 Therefore, the interfacial electric field stabilization of *CO is likely the same for 2–200 mM [K+] at high ⟨j⟩ and is not an important factor toward improved selectivity as [K+] increases. Instead, the role of M+ in boosting acidic CO2R is derived from field-induced transport effects, namely, the local pH gradient generated by increasingly diminished H+ transport with greater [K+].
Figure 5.
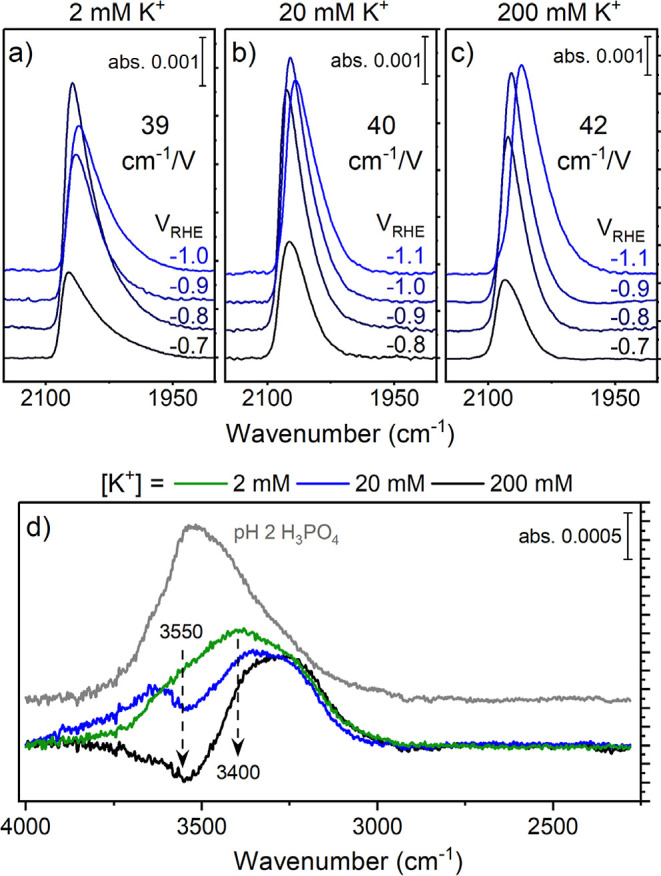
SEIRA spectra of *COatop region as a function of potential and the calculated Stark tuning slopes for (a) 2 mM K+; (b) 20 mM K+; and (c) 200 mM K+. (d) SEIRA spectra for the OH stretching region of the Cu/electrolyte interface measured at OCV for pH 2 H3PO4 (gray trace) with a background of pure water, and various [K+] with a background of pH 2 H3PO4.
We emphasize that for both the moderate and high current cases, we do not observe a trend between the local pH and *CO formation/consumption. Therefore, the cation-induced interfacial solvation structure must also be considered to understand differences between [K+]. Previous studies have shown that the OH stretching mode of H2O can be deconvoluted into three peaks corresponding to strongly H-bonded water at ∼3250 cm–1, asymmetric H-bonded water at 3400 cm–1, and isolated water at 3600 cm–1.51 Addition of K+ to the M+ free pH 2 electrolyte changes the interfacial water structure (Figures 5d and S15). At the open-circuit potential, the features corresponding to asymmetric H-bonded water and isolated water decrease as [K+] increases, while those for strongly H-bonded interfacial water are slightly enhanced. Disruption of the interfacial water structure could be responsible for the relative increase in interfacial pH and different reactivities with [K+]. The presence of strongly H-bonded water with increased [K+] helps explain why we observe ∼60% FEHER even at high ⟨j⟩.
More detailed analyses of the water structure are outside of the scope of this work. However, we can infer that interfacial water displacement can help promote CO2R in acid. This mechanistic picture motivated us to explore the promotion of CO2R under acidic bulk conditions via a strategy whereby both nonacidic microenvironment pH and a desirable interfacial water structure is maintained via an alternative means to M+.
Promotion of CO2R under Acidic Conditions on Organic Film-Modified Cu with Low [M+]
We have recently shown that a film derived from the electrodeposition of N-tolylpyridinium on Cu electrodes mitigates H+ mass transport and thus suppresses HER under strongly acidic conditions, such as 1.0 M H3PO4/KH2PO4.11 This finding inspired us to study film-modified Cu electrodes (Cu/film) using the same N-tolylpyridinium additive (which deposits as a neutral organic species)52 toward CO2R under acidic conditions in the absence of M+, reasoning that the Cu/film microenvironment might sufficiently suppress H+ transport to the interface to observe CO2R.
A series of electrolysis experiments were conducted in CO2-saturated pH 2 H3PO4 with Cu/film electrodes (Table S6). We find CO2R is promoted on Cu/film electrodes even at ⟨j⟩ as low as −0.50 mA/cm2, with a FECO2R that is an order of magnitude higher than is achieved at −10.0 mA/cm2 on a bare Cu electrode (Figure 6a, Table S5). We have elsewhere shown that CO2 is the C-source of the detected products based on 13CO2R experiments using this type of Cu/film electrode.11 In the present study, we also confirmed that an electrolysis at −1.25 mA/cm2 under argon showed no detectable CO2R products (Table S6).
Figure 6.
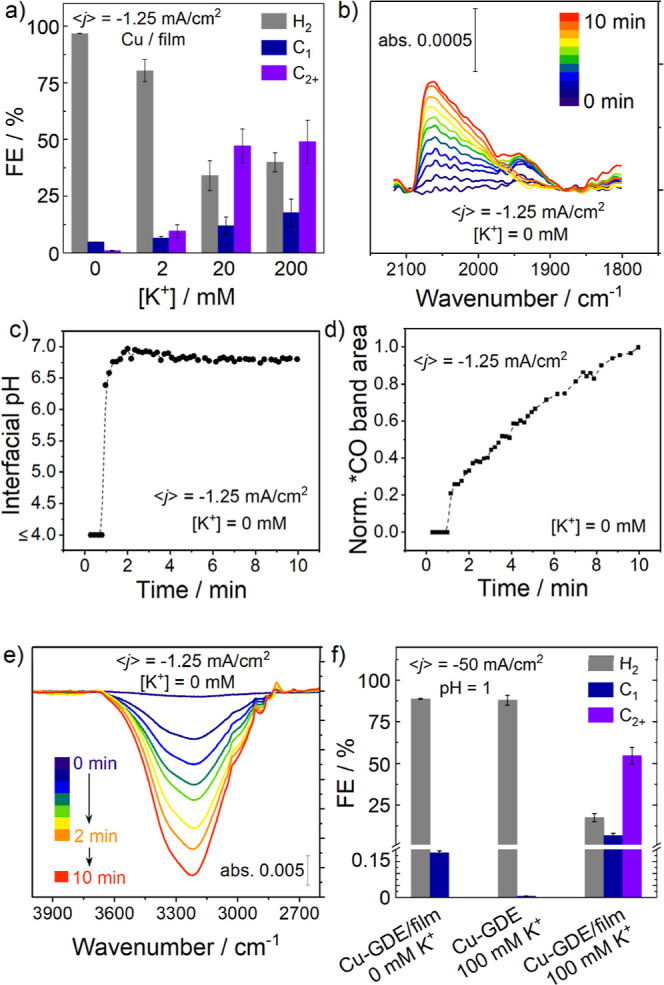
(a) Product selectivity at −1.25 mA/cm2 on Cu/film electrodes in pH 2 H3PO4 at different [K+]; (b) time-dependent *COatop formation on Cu/film electrodes at −1.25 mA/cm2 in pH 2 H3PO4 at 0 mM K+; (c) interfacial pH as a function of time for Cu/film electrodes at −1.25 mA/cm2 in pH 2 H3PO4 at 0 mM K+; (d) time-dependent *CO band area increase on Cu/film at −1.25 mA/cm2 in pH 2 H3PO4 at 0 mM K+; (e) time-dependent OH stretching region of SEIRA spectra during film deposition on Cu at −1.25 mA/cm2, demonstrating film-induced interfacial water desorption; and (f) product selectivity at −50 mA/cm2 on Cu-GDE and Cu-GDE/film electrodes in pH 1 H3PO4 at 0 and 100 mM K+.
In SEIRAS experiments, we observe *COatop formation without any M+ at low current (Figure 6b), which further supports that the reduction of CO2 does not require M+ and instead relies on interfacial [H+] depletion. The enhancement of CO2R selectivity is attributed to the ability of the film to block H+ transport to the electrode, generating an interfacial pH gradient, even at low ⟨j⟩.11,53 This idea is supported by SEIRAS interfacial pH measurements (Figure 6c), showing a substantial increase in the pH compared to the bulk at low ⟨j⟩.
Using low ⟨j⟩ did not result in a pH change on bare Cu using the same acidic electrolyte (Figure 3a). The *CO band intensity increases over 10 min, indicating high *CO coverage (Figure 6d). Additionally, a small shoulder is observed at ∼1950 cm–1, which has previously been assigned to two *COatop molecules interacting with two surface hydroxide molecules.54,55 The difference between the *CO band shapes and types of *CO for the Cu/film interface compared to the bare Cu interface (Figures 3a and 6b), while the interfacial pH is nearly identical (Figures 3b and 6c), demonstrates that there is a unique microenvironment induced by the organic film.
Although the FECO2R is improved from the bare Cu low current case, the *CO band growth without notable consumption demonstrates that the film alone cannot effectively stabilize further reduced CO2R intermediates to promote CO2R over HER. We also cannot rule out the possibility that the water in the M+ solvation shell is required as a donor to promote *CO reduction.
The OH stretching region of the SEIRA spectrum upon deposition of the film in pH 2 H3PO4 shows strong displacement of interfacial water as the film is deposited (Figure 6e). In particular, we see that the film induces desorption of strongly H-bonded and asymmetric H-bonded water at 3250 and 3400 cm–1. The film does not displace or increase isolated interfacial water at 3600 cm–1, which could explain why HER still dominates over CO2R with 0 mM K+ in pH 2 H3PO4.
Adding a small amount of K+ (20 mM) into pH 2 H3PO4 dramatically boosts the FECO2R from ∼6 to ∼60% on Cu/film electrodes at −1.25 mA/cm2, while no CO2R is observed on bare Cu electrodes at the same [K+] (Figure 6a, Table S7). We note that these are the only conditions discussed thus far in this study that led to the CO2R becoming dominant versus the HER. We can rationalize this result to cooperative effects between the film and M+ as follows. The film induces long-range H+ transport suppression and modulates the interfacial water structure, while the M+ can stabilize and/or donate water to reaction intermediates and further suppress H+ transport. This cooperation promotes CO2R over HER under acidic conditions. In addition, we note that promoted selectivity toward C2+ products (∼50% FEC2+) on Cu/film electrodes under acidic electrolyte conditions at low [M+] (Figure 6a, Table S8) supports the hypothesis that organic films can affect the concentration of surface-bound *CO intermediates to favor the C–C coupling pathway, which is a subject of ongoing investigation.11,52,56
To evaluate the Cu/film system at even lower pH, the CO2R in pH 1 H3PO4 was tested using a Cu gas diffusion electrode (Cu-GDE) (Table S11). In such a strongly acidic electrolyte, no CO2R products are observed at ⟨j⟩ = −50 mA/cm2, or even up to −200 mA/cm2, on a Cu-GDE in absence of the film (Figure S11). Adding 100 mM K+ leads to trace CO2R at −50 mA/cm2 (Figure 6f), consistent with results reported under similar conditions.13 For comparison, a Cu-GDE/film electrode displayed a very low FECO2R in M+ free pH 1 H3PO4 (Figure 6f). However, once 100 mM K+ was added, remarkable suppression of HER (FEHER = 17.5 ± 2.4%) and enhanced selectivity toward C2+ products (FEC2+ = 54.5 ± 5.1%) were observed at −50 mA/cm2 (Figure 6f). This result is striking and is comparable to FEC2+ values obtained by using much higher [M+] (>1 M) in combination with very high operating ⟨j⟩ (>500 mA/cm2) in previous studies.13 This comparison highlights the distinct properties of film-modified electrodes for CO2R under acidic conditions as a means of modifying interfacial water and H+ transport to mitigate acidity near the electrode. Synergistic effects arise from the presence of a modest [M+] concentration to generate an enhanced electric field, compared to the M+-free electrolyte, to further reduce interfacial H+ transport.
Conclusions
In conclusion, our study of CO2R in pH 2 H3PO4 with varying [M+], combined with in situ SEIRAS and CLSM measurements, reveal a mechanistic correlation between an interfacial pH gradient and the onset of CO2R. We find that while M+ is not required for CO2R to occur, accumulation of M+ at the electrode surface uniquely produces an interfacial barrier toward proton transport. Even the presence of an organic film, a known H+ transport modulator, is insufficient to significantly bias selectivity for CO2R over HER. Only in the combination of the two strategies is high selectivity for CO2R achieved. Through SEIRAS and CLSM experiments, we highlight that increasing metal salt concentration is a deleterious strategy toward local pH management due to the depletion of local CO2 from (bi)carbonate formation. We show that organic films expand the current density window where CO2R can occur, and that selective CO2R over HER can be obtained by combining with low [M+]. Together, this study establishes a promising strategy to promote selective CO2R under acidic conditions using film-decorated electrodes and low concentrations of metal salts.
Acknowledgments
The Resnick Sustainability Institute at Caltech is acknowledged for its support of enabling infrastructure and facilities. We thank Dr. Yung-Chieh Lai for ICP-MS measurement assistance. We thank Dr. Nicholas B. Watkins and Prof. Wilson A. Smith for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c09512.
Complete experimental details, CO2R product selectivity, time-dependent electrode potential traces, and additional SEIRA spectra (PDF)
Author Contributions
∥ M.H.H. and W.N. contributed equally to this study. All authors contributed to the formulation of this project and have given approval to the final version of the manuscript.
This material is based on work performed by the Liquid Sunlight Alliance, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Fuels from Sunlight Hub under award no. DE-SC0021266.
The authors declare no competing financial interest.
Supplementary Material
References
- Jordaan S. M.; Wang C. Electrocatalytic Conversion of Carbon Dioxide for the Paris Goals. Nat. Catal. 2021, 4, 915–920. 10.1038/s41929-021-00704-z. [DOI] [Google Scholar]
- De Luna P.; Hahn C.; Higgins D.; Jaffer S. A.; Jaramillo T. F.; Sargent E. H. What Would It Take for Renewably Powered Electrosynthesis to Displace Petrochemical Processes?. Science 2019, 364, 3506. 10.1126/science.aav3506. [DOI] [PubMed] [Google Scholar]
- Nitopi S.; Bertheussen E.; Scott S. B.; Liu X.; Engstfeld A. K.; Horch S.; Seger B.; Stephens I. E. L.; Chan K.; Hahn C.; Nørskov J. K.; Jaramillo T. F.; Chorkendorff I. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. 10.1021/acs.chemrev.8b00705. [DOI] [PubMed] [Google Scholar]
- Birdja Y. Y.; Pérez-Gallent E.; Figueiredo M. C.; Göttle A. J.; Calle-Vallejo F.; Koper M. T. M. Advances and Challenges in Understanding the Electrocatalytic Conversion of Carbon Dioxide to Fuels. Nat. Energy 2019, 4, 732–745. 10.1038/s41560-019-0450-y. [DOI] [Google Scholar]
- Rabinowitz J. A.; Kanan M. W. The Future of Low-Temperature Carbon Dioxide Electrolysis Depends on Solving One Basic Problem. Nat. Commun. 2020, 11, 5231. 10.1038/s41467-020-19135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alerte T.; Edwards J. P.; Gabardo C. M.; O’Brien C. P.; Gaona A.; Wicks J.; Obradović A.; Sarkar A.; Jaffer S. A.; MacLean H. L.; Sinton D.; Sargent E. H. Downstream of the CO2 Electrolyzer: Assessing the Energy Intensity of Product Separation. ACS Energy Lett. 2021, 6, 4405–4412. 10.1021/acsenergylett.1c02263. [DOI] [Google Scholar]
- Perazio A.; Creissen C. E.; Rivera de la Cruz J. G.; Schreiber M. W.; Fontecave M. Acidic Electroreduction of CO2 to Multi-Carbon Products with CO2 Recovery and Recycling from Carbonate. ACS Energy Lett. 2023, 8, 2979–2985. 10.1021/acsenergylett.3c00901. [DOI] [Google Scholar]
- Xu Y.; Miao R. K.; Edwards J. P.; Liu S.; O’Brien C. P.; Gabardo C. M.; Fan M.; Huang J. E.; Robb A.; Sargent E. H.; Sinton D. A Microchanneled Solid Electrolyte for Carbon-Efficient CO2 Electrolysis. Joule 2022, 6, 1333–1343. 10.1016/j.joule.2022.04.023. [DOI] [Google Scholar]
- Kim J. Y. T.; Zhu P.; Chen F.-Y.; Wu Z.-Y.; Cullen D. A.; Wang H. Recovering Carbon Losses in CO2 Electrolysis Using a Solid Electrolyte Reactor. Nat. Catal. 2022, 5, 288–299. 10.1038/s41929-022-00763-w. [DOI] [Google Scholar]
- Xie K.; Miao R. K.; Ozden A.; Liu S.; Chen Z.; Dinh C.-T.; Huang J. E.; Xu Q.; Gabardo C. M.; Lee G.; Edwards J. P.; O’Brien C. P.; Boettcher S. W.; Sinton D.; Sargent E. H. Bipolar Membrane Electrolyzers Enable High Single-Pass CO2 Electroreduction to Multicarbon Products. Nat. Commun. 2022, 13, 3609. 10.1038/s41467-022-31295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie W.; Heim G. P.; Watkins N. B.; Agapie T.; Peters J. C. Organic Additive-Derived Films on Cu Electrodes Promote Electrochemical CO2 Reduction to C2+ Products Under Strongly Acidic Conditions. Angew. Chem., Int. Ed. 2023, 62, 202216102. 10.1002/anie.202216102. [DOI] [PubMed] [Google Scholar]
- Gu J.; Liu S.; Ni W.; Ren W.; Haussener S.; Hu X. Modulating Electric Field Distribution by Alkali Cations for CO2 Electroreduction in Strongly Acidic Medium. Nat. Catal. 2022, 5, 268–276. 10.1038/s41929-022-00761-y. [DOI] [Google Scholar]
- Huang J. E.; Li F.; Ozden A.; Sedighian Rasouli A.; García de Arquer F. P.; Liu S.; Zhang S.; Luo M.; Wang X.; Lum Y.; Xu Y.; Bertens K.; Miao R. K.; Dinh C.-T.; Sinton D.; Sargent E. H. CO2 Electrolysis to Multicarbon Products in Strong Acid. Science 2021, 372, 1074–1078. 10.1126/science.abg6582. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Hao L.; Ozden A.; Liu S.; Miao R. K.; Ou P.; Alkayyali T.; Zhang S.; Ning J.; Liang Y.; Xu Y.; Fan M.; Chen Y.; Huang J. E.; Xie K.; Zhang J.; O’Brien C. P.; Li F.; Sargent E. H.; Sinton D. Conversion of CO2 to Multicarbon Products in Strong Acid by Controlling the Catalyst Microenvironment. Nat. Synth. 2023, 2, 403–412. 10.1038/s44160-022-00234-x. [DOI] [Google Scholar]
- Jiang Z.; Zhang Z.; Li H.; Tang Y.; Yuan Y.; Zao J.; Zheng H.; Liang Y. Molecular Catalyst with Near 100% Selectivity for CO2 Reduction in Acidic Electrolytes. Adv. Energy Mater. 2023, 13, 2203603. 10.1002/aenm.202203603. [DOI] [Google Scholar]
- Ren W.; Xu A.; Chan K.; Hu X. A Cation Concentration Gradient Approach to Tune the Selectivity and Activity of CO2 Electroreduction. Angew. Chem., Int. Ed. 2022, 61, 202214173. 10.1002/anie.202214173. [DOI] [PubMed] [Google Scholar]
- Qiao Y.; Lai W.; Huang K.; Yu T.; Wang Q.; Gao L.; Yang Z.; Ma Z.; Sun T.; Liu M.; Lian C.; Huang H. Engineering the Local Microenvironment over Bi Nanosheets for Highly Selective Electrocatalytic Conversion of CO2 to HCOOH in Strong Acid. ACS Catal. 2022, 12, 2357–2364. 10.1021/acscatal.1c05135. [DOI] [Google Scholar]
- Ma Z.; Yang Z.; Lai W.; Wang Q.; Qiao Y.; Tao H.; Lian C.; Liu M.; Ma C.; Pan A.; Huang H. CO2 Electroreduction to Multicarbon Products in Strongly Acidic Electrolyte via Synergistically Modulating the Local Microenvironment. Nat. Commun. 2022, 13, 7596. 10.1038/s41467-022-35415-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro M. C. O.; Philips M. F.; Schouten K. J. P.; Koper M. T. M. Efficiency and Selectivity of CO2 Reduction to CO on Gold Gas Diffusion Electrodes in Acidic Media. Nat. Commun. 2021, 12, 4943. 10.1038/s41467-021-24936-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondue C. J.; Graf M.; Goyal A.; Koper M. T. M. Suppression of Hydrogen Evolution in Acidic Electrolytes by Electrochemical CO2 Reduction. J. Am. Chem. Soc. 2021, 143, 279–285. 10.1021/jacs.0c10397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H.-G.; Li F.-Z.; Du Y.-F.; Yang L.-F.; Wang H.; Bai Y.-Y.; Lin M.; Gu J. Quantitative Understanding of Cation Effects on the Electrochemical Reduction of CO2 and H+ in Acidic Solution. ACS Catal. 2023, 13, 916–926. 10.1021/acscatal.2c04875. [DOI] [Google Scholar]
- Weng S.; Toh W. L.; Surendranath Y. Weakly Coordinating Organic Cations Are Intrinsically Capable of Supporting CO2 Reduction Catalysis. J. Am. Chem. Soc. 2023, 145, 16787–16795. 10.1021/jacs.3c04769. [DOI] [PubMed] [Google Scholar]
- Qin H.-G.; Du Y.-F.; Bai Y.-Y.; Li F.-Z.; Yue X.; Wang H.; Peng J.-Z.; Gu J. Surface-Immobilized Cross-Linked Cationic Polyelectrolyte Enables CO2 Reduction with Metal Cation-Free Acidic Electrolyte. Nat. Commun. 2023, 14, 5640. 10.1038/s41467-023-41396-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrashekar S.; van Montfort H. P. I.; Bohra D.; Filonenko G.; Geerlings H.; Burdyny T.; Smith W. A. Investigating the Role of Potassium Cations during Electrochemical CO2 Reduction. Nanoscale 2022, 14, 14185–14190. 10.1039/D2NR03438G. [DOI] [PubMed] [Google Scholar]
- Shin S.-J.; Choi H.; Ringe S.; Won D. H.; Oh H.-S.; Kim D. H.; Lee T.; Nam D.-H.; Kim H.; Choi C. H. A Unifying Mechanism for Cation Effect Modulating C1 and C2 Productions from CO2 Electroreduction. Nat. Commun. 2022, 13, 5482. 10.1038/s41467-022-33199-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro M. C. O.; Dattila F.; López N.; Koper M. T. M. The Role of Cation Acidity on the Competition between Hydrogen Evolution and CO2 Reduction on Gold Electrodes. J. Am. Chem. Soc. 2022, 144, 1589–1602. 10.1021/jacs.1c10171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro M. C. O.; Dattila F.; Hagedoorn B.; García-Muelas R.; López N.; Koper M. T. M. Absence of CO2 Electroreduction on Copper, Gold and Silver Electrodes without Metal Cations in Solution. Nat. Catal. 2021, 4, 654–662. 10.1038/s41929-021-00655-5. [DOI] [Google Scholar]
- Resasco J.; Chen L. D.; Clark E.; Tsai C.; Hahn C.; Jaramillo T. F.; Chan K.; Bell A. T. Promoter Effects of Alkali Metal Cations on the Electrochemical Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2017, 139, 11277–11287. 10.1021/jacs.7b06765. [DOI] [PubMed] [Google Scholar]
- Singh M. R.; Kwon Y.; Lum Y.; Ager J. W.; Bell A. T. Hydrolysis of Electrolyte Cations Enhances the Electrochemical Reduction of CO2 over Ag and Cu. J. Am. Chem. Soc. 2016, 138, 13006–13012. 10.1021/jacs.6b07612. [DOI] [PubMed] [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons, 2000. [Google Scholar]
- Osawa M. Dynamic Processes in Electrochemical Reactions Studied by Surface-Enhanced Infrared Absorption Spectroscopy (SEIRAS). Bull. Chem. Soc. Jpn. 1997, 70 (12), 2861–2880. 10.1246/bcsj.70.2861. [DOI] [Google Scholar]
- Welch A. J.; Fenwick A. Q.; Böhme A.; Chen H.-Y.; Sullivan I.; Li X.; DuChene J. S.; Xiang C.; Atwater H. A. Operando Local pH Measurement within Gas Diffusion Electrodes Performing Electrochemical Carbon Dioxide Reduction. J. Phys. Chem. C 2021, 125, 20896–20904. 10.1021/acs.jpcc.1c06265. [DOI] [Google Scholar]
- Miyake H.; Ye S.; Osawa M. Electroless Deposition of Gold Thin Films on Silicon for Surface-Enhanced Infrared Spectroelectrochemistry. Electrochem. Commun. 2002, 4 (12), 973–977. 10.1016/S1388-2481(02)00510-6. [DOI] [Google Scholar]
- Chang X.; Malkani A.; Yang X.; Xu B. Mechanistic Insights into Electroreductive C-C Coupling between CO and Acetaldehyde into Multicarbon Products. J. Am. Chem. Soc. 2020, 142 (6), 2975–2983. 10.1021/jacs.9b11817. [DOI] [PubMed] [Google Scholar]
- Yang K.; Kas R.; Smith W. A. In Situ Infrared Spectroscopy Reveals Persistent Alkalinity near Electrode Surfaces during CO2 Electroreduction. J. Am. Chem. Soc. 2019, 141 (40), 15891–15900. 10.1021/jacs.9b07000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corson E. R.; Guo J.; Tarpeh W. A. ATR-SEIRAS Method to Measure Interfacial pH during Electrocatalytic Nitrate Reduction on Cu. J. Electrochem. Soc. 2024, 171 (4), 046503. 10.1149/1945-7111/ad3a22. [DOI] [Google Scholar]
- Hakonen A.; Hulth S. A High-Performance Fluorosensor for pH Measurements between 6 and 9. Talanta 2010, 80, 1964–1969. 10.1016/j.talanta.2009.10.055. [DOI] [PubMed] [Google Scholar]
- Böhme A.; Bui J. C.; Fenwick A. Q.; Bhide R.; Feltenberger C. N.; Welch A. J.; King A. J.; Bell A. T.; Weber A. Z.; Ardo S.; Atwater H. A. Direct Observation of the Local Microenvironment in Inhomogeneous CO2 Reduction Gas Diffusion Electrodes via Versatile pOH Imaging. Energy Environ. Sci. 2023, 16, 1783–1795. 10.1039/D2EE02607D. [DOI] [Google Scholar]
- Gunathunge C. M.; Li J.; Li X.; Waegele M. M. Surface-Adsorbed CO as an Infrared Probe of Electrocatalytic Interfaces. ACS Catal. 2020, 10 (20), 11700–11711. 10.1021/acscatal.0c03316. [DOI] [Google Scholar]
- Chou T.-C.; Chang C.-C.; Yu H.-L.; Yu W.-Y.; Dong C.-L.; Velasco-Vélez J. J.; Chuang C.-H.; Chen L.-C.; Lee J.-F.; Chen J.-M.; Wu H.-L. Controlling the Oxidation State of the Cu Electrode and Reaction Intermediates for Electrochemical CO2 Reduction to Ethylene. J. Am. Chem. Soc. 2020, 142 (6), 2857–2867. 10.1021/jacs.9b11126. [DOI] [PubMed] [Google Scholar]
- Gunathunge C. M.; Li X.; Li J.; Hicks R. P.; Ovalle V. J.; Waegele M. M. Spectroscopic Observation of Reversible Surface Reconstruction of Copper Electrodes under CO2 Reduction. J. Phys. Chem. C 2017, 121 (22), 12337–12344. 10.1021/acs.jpcc.7b03910. [DOI] [Google Scholar]
- Salimon J.; Hernández-Romero R. M.; Kalaji M. The Dynamics of the Conversion of Linear to Bridge Bonded CO on Cu. J. Electroanal. Chem. 2002, 538–539, 99–108. 10.1016/S0022-0728(02)01052-5. [DOI] [Google Scholar]
- Ooka H.; Figueiredo M. C.; Koper M. T. M. Competition between Hydrogen Evolution and Carbon Dioxide Reduction on Copper Electrodes in Mildly Acidic Media. Langmuir 2017, 33, 9307–9313. 10.1021/acs.langmuir.7b00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Koper M. T. M. Tuning the Interfacial Reaction Environment for CO2 Electroreduction to CO in Mildly Acidic Media. J. Am. Chem. Soc. 2024, 146 (8), 5242–5251. 10.1021/jacs.3c11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge A.; Videla P. E.; Lee G. L.; Rudshteyn B.; Song J.; Kubiak C. P.; Batista V. S.; Lian T. Interfacial Structure and Electric Field Probed by in Situ Electrochemical Vibrational Stark Effect Spectroscopy and Computational Modeling. J. Phys. Chem. C 2017, 121 (34), 18674–18682. 10.1021/acs.jpcc.7b05563. [DOI] [Google Scholar]
- Chang X.; Xiong H.; Xu Y.; Zhao Y.; Lu Q.; Xu B. Determining Intrinsic Stark Tuning Rates of Adsorbed CO on Copper Surfaces. Catal. Sci. Technol. 2021, 11 (20), 6825–6831. 10.1039/D1CY01090E. [DOI] [Google Scholar]
- Rebstock J. A.; Zhu Q.; Baker L. R. Comparing Interfacial Cation Hydration at Catalytic Active Sites and Spectator Sites on Gold Electrodes: Understanding Structure Sensitive CO2 Reduction Kinetics. Chem. Sci. 2022, 13 (25), 7634–7643. 10.1039/D2SC01878K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Wu D.; Malkani A. S.; Chang X.; Cheng M.-J.; Xu B.; Lu Q. Hydroxide Is Not a Promoter of C2+ Product Formation in the Electrochemical Reduction of CO on Copper. Angew. Chem., Int. Ed. 2020, 59 (11), 4464–4469. 10.1002/anie.201912412. [DOI] [PubMed] [Google Scholar]
- Malkani A. S.; Li J.; Oliveira N. J.; He M.; Chang X.; Xu B.; Lu Q. Understanding the Electric and Nonelectric Field Components of the Cation Effect on the Electrochemical CO Reduction Reaction. Sci. Adv. 2020, 6 (45), eabd2569 10.1126/sciadv.abd2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunathunge C. M.; Ovalle V. J.; Waegele M. M. Probing Promoting Effects of Alkali Cations on the Reduction of CO at the Aqueous Electrolyte/Copper Interface. Phys. Chem. Chem. Phys. 2017, 19 (44), 30166–30172. 10.1039/C7CP06087D. [DOI] [PubMed] [Google Scholar]
- Ataka K.; Yotsuyanagi T.; Osawa M. Potential-Dependent Reorientation of Water Molecules at an Electrode/Electrolyte Interface Studied by Surface-Enhanced Infrared Absorption Spectroscopy. J. Phys. Chem. 1996, 100 (25), 10664–10672. 10.1021/jp953636z. [DOI] [Google Scholar]
- Han Z.; Kortlever R.; Chen H.-Y.; Peters J. C.; Agapie T. CO2 Reduction Selective for C ≥ 2 Products on Polycrystalline Copper with N-Substituted Pyridinium Additives. ACS Cent. Sci. 2017, 3, 853–859. 10.1021/acscentsci.7b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovalle V. J.; Waegele M. M. Understanding the Impact of N-Arylpyridinium Ions on the Selectivity of CO2 Reduction at the Cu/Electrolyte Interface. J. Phys. Chem. C 2019, 123, 24453–24460. 10.1021/acs.jpcc.9b08666. [DOI] [Google Scholar]
- Cao Y.; Chen Z.; Li P.; Ozden A.; Ou P.; Ni W.; Abed J.; Shirzadi E.; Zhang J.; Sinton D.; Ge J.; Sargent E. H. Surface Hydroxide Promotes CO2 Electrolysis to Ethylene in Acidic Conditions. Nat. Commun. 2023, 14 (1), 2387. 10.1038/s41467-023-37898-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima G.; Inomata T.; Yamaguchi H.; Ito M.; Masuda H. Role of a Hydroxide Layer on Cu Electrodes in Electrochemical CO2 Reduction. ACS Catal. 2019, 9 (7), 6305–6319. 10.1021/acscatal.9b00896. [DOI] [Google Scholar]
- Watkins N. B.; Schiffer Z. J.; Lai Y.; Musgrave C. B. III; Atwater H. A.; Goddard W. A. III; Agapie T.; Peters J. C.; Gregoire J. M. Hydrodynamics Change Tafel Slopes in Electrochemical CO2 Reduction on Copper. ACS Energy Lett. 2023, 8, 2185–2192. 10.1021/acsenergylett.3c00442. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.