

Abstract
Triple-negative breast cancer (TNBC) is associated with poor prognosis because of the lack of effective therapies. Mixed-lineage protein kinase 3 (MLK3) is a protein that is often upregulated in TNBC and involved in driving the tumorigenic potential of cancer cells. Here, we present a selective MLK3 degrader, CEP1347-VHL-02, based on the pan-MLK inhibitor CEP1347 and a ligand for E3 ligase von Hippel–Lindau (VHL) by employing proteolysis-targeting chimera (PROTAC) technology. Our compound effectively targeted MLK3 for degradation via the ubiquitin-proteasome system in several cell line models but did not degrade other MLK family members. Furthermore, we showed that CEP1347-VHL-02 robustly degraded MLK3 and inhibited its oncogenic activity in TNBC, measured as a reduction of clonogenic and migratory potential, cell cycle arrest, and the induction of apoptosis in MDA-MB-468 cells. In conclusion, we present CEP1347-VHL-02 as a novel MLK3 degrader that may be a promising new strategy to target MLK3 in TNBC.
Introduction
Mixed-lineage protein kinase 3 (MLK3, MAP3K11) is a member of the MLK family of serine/threonine kinases, which belongs to mitogen-activated protein kinase kinase kinases (MAP3Ks). MLK3 is well-known to activate the c-Jun N-terminal kinase (JNK) and p38 MAPK pathways through phosphorylation and activation of the MAP2Ks MKK4/7 and MKK3/6, respectively.1 MLK3 also activates the extracellular signal-regulated kinase (ERK) MAPK pathway through both direct mitogen-activated protein kinase kinase (MEK) phosphorylation and as a scaffold to promote BRAF activation.2−4 In addition to its role in MAPK signaling, MLK3 regulates the activity of multiple oncogenic proteins and signaling pathways, thereby modulating the proliferation, migration, and invasive potential of cancer cells in a context-dependent manner.5 Previous studies extensively investigated MLK3 as a potential therapeutic target in breast cancer. Chen et al. showed that MLK3 expression is significantly elevated in breast cancer cell lines compared with epithelial nontumorigenic breast cell lines. The authors found that the catalytic activity of MLK3 and its downstream signaling to JNK and activator protein-1 (AP-1) promote the migration of mammary epithelial cells through the upregulation of genes that are associated with the epithelial-to-mesenchymal transition.6 MLK3 signaling through JNK may also phosphorylate the cytoskeletal protein paxillin, which in turn decreases Rho activity to promote focal adhesion turnover and the migration of triple-negative breast cancer (TNBC) cells.7 Additionally, MLK3 was found to promote TNBC spread by the direct phosphorylation and activation of p21-activated kinase 1 (PAK1), resulting in regulation of paxillin and the enhancement of nuclear factor-κB (NF-κB) activity.8,9 Moreover, MLK3 was reported to increase expression of the oncogenic transcription factor FOS-related antigen-1 (FRA-1), accompanied by the elevation of matrix metalloproteinases in TNBC.10 Contrary to its oncogenic functions, MLK3 was shown to promote apoptosis in estrogen receptor (ER)-positive and human epidermal growth factor receptor 2 (HER2)-positive breast cancers.11,12 Thus, MLK3 may have both tumor-promoting and tumor-suppressing functions, depending on the breast cancer subtype. Although MLK3 represents an attractive target for the treatment of human cancers, including TNBC, the pharmacological targeting of this kinase poses many challenges. Several MLK3 inhibitors, such as CEP1347, were pursued as new investigational anticancer drugs, but the utility of these compounds was limited by their low specificity. Furthermore, conventional small-molecule inhibitors do not inhibit kinase-independent and scaffolding functions of MLK3, which were previously shown to drive tumorigenic phenotypes independent of its catalytic activity.2,4,5,13,14
Targeted protein degradation (TPD) is a rapidly growing field that opens new possibilities for the selective targeting of protein kinases in cancer. Proteolysis-targeting chimeras (PROTACs) represent a major TPD technology, which was successfully applied to degrade numerous oncogenic proteins.15−18 PROTACs are heterobifunctional compounds that consist of three elements: a pharmacophore that recognizes a targeted protein of interest (POI), a ligand for recruiting an E3 ligase, and a linker. These compounds simultaneously bind the POI and E3 ligase, thereby forming a ternary complex, subsequently leading to POI ubiquitination and proteasomal degradation.19 There are several examples of small-molecule kinase inhibitors that have been used as pharmacophores for the synthesis of new PROTACs. Compared with primary small-molecule inhibitors, PROTACs allow for the inhibition of both catalytic and noncatalytic functions of targeted kinases and may offer higher target specificity.20,21
In the present study, we present a novel PROTAC compound that targets MLK3 kinase for degradation based on the pan-MLK inhibitor CEP1347 and ligand for E3 ligase von Hippel–Lindau (VHL). We demonstrate that this PROTAC robustly degrades MLK3 but no other MLKs in cancer cell line models. Furthermore, we show that the PROTAC-mediated degradation of MLK3 decreased the tumorigenic potential of TNBC cells.
Results
Development and Characterization of MLK3-Targeting PROTACs
MLK3 was previously reported to increase the tumorigenic potential of cancer cells in both a kinase-dependent and -independent manner. To target it for degradation and eliminate its noncatalytic effect, we designed different PROTACs based on the MLK3 inhibitor CEP1347 linked with different E3-ubiquitin ligase ligands (Figure 1). We synthesized eight PROTACs composed of CEP1347 and ligand for the von Hippel–Lindau E3 ubiquitin ligase (VHL) coupled with various linkers (CEP1347-VHL-01–CEP1347-VHL-08). We also obtained compounds in which CEP1347 was linked with the inhibitor of apoptosis protein (IAP) ligand (CEP1347-IAP-01) and with the cereblon (CRBN) ligand (CEP1347-CRBN-01).
Figure 1.
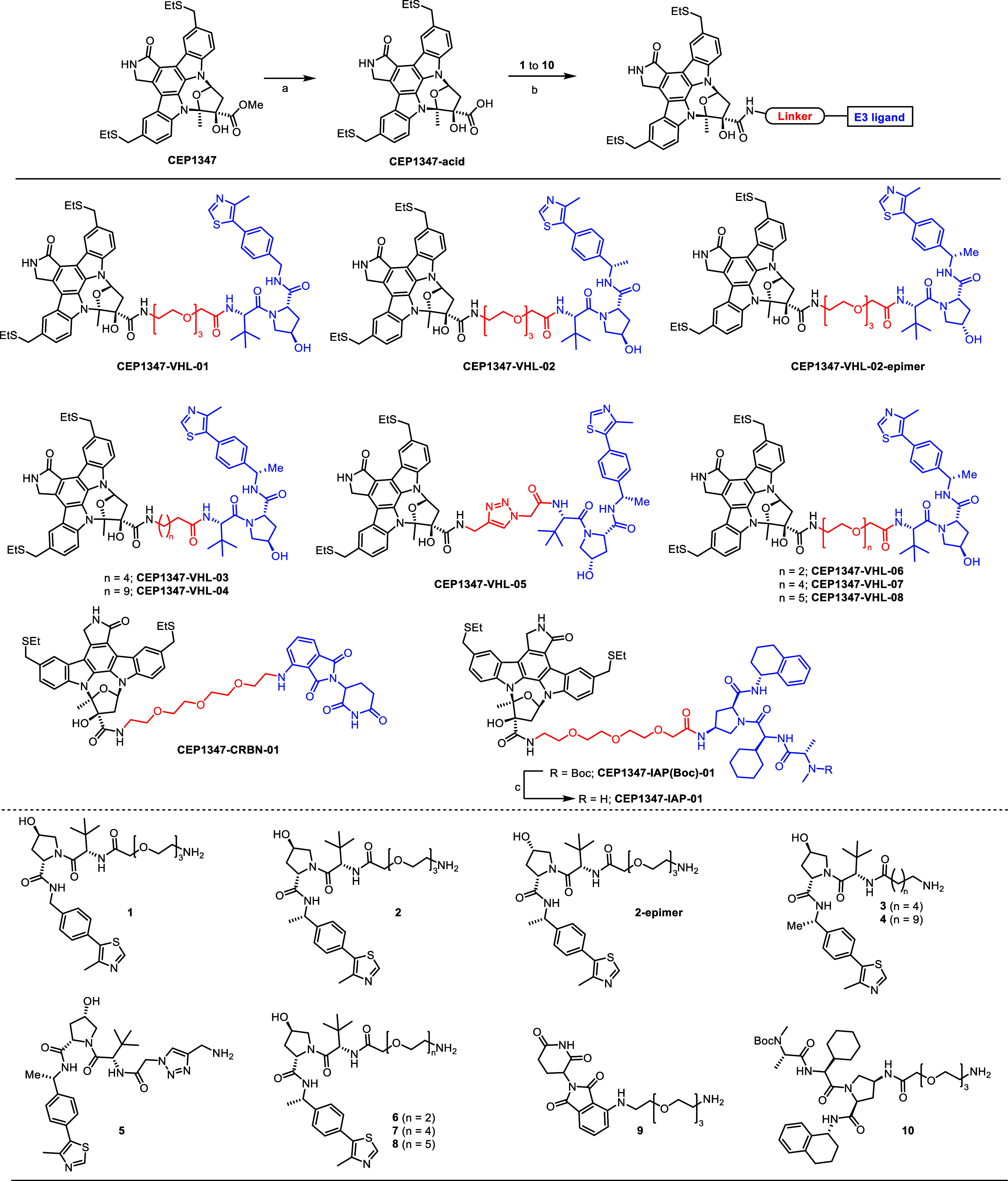
Synthesis of MLK3-targeting PROTACs: reaction conditions (a) aq LiOH, THF; (b) 1 to 10, HATU, DIPEA, DMF; (c) TFA/CH2Cl2.
The efficiency of MLK3 degradation by the synthesized PROTAC compounds was assessed by immunoblotting A375 and HCC1806 cell lines with doxycycline (DOX)-induced MLK3 overexpression (A375_MLK3 and HCC1806_MLK3, respectively). To determine the optimal length of the linker for achieving effective MLK3 degradation, we treated A375_MLK3 cells with eight VHL-based PROTACs (CEP1347-VHL-01, CEP1347-VHL-02, CEP1347-VHL-03, CEP1347-VHL-04, CEP1347-VHL-05, CEP1347-VHL-06, CEP1347-VHL-07, and CEP1347-VHL-08) for 24 h at different concentrations, ranging from 0.1 to 5 μM (Figure 2A). Additionally, we tested three VHL-based PROTACs (CEP1347-VHL-01, CEP1347-VHL-02, and CEP1347-VHL-03) in the HCC1806_MLK3 cell line (Figure 2B). Out of all VHL-based PROTACs tested, CEP1347-VHL-02 degraded MLK3 at the lowest concentrations in both cell lines (Figure 2A,B). We also verified MLK3 degradation by CEP1347-IAP-01 and CEP1347-CRBN-01 PROTAC in the A375_MLK3 cell line (Figure S1). We observed more potent degradation with PROTACs that contained ligands for VHL compared with PROTACs containing the IAP or CRBN ligands. Overall, based on these results, we selected CEP1347-VHL-02 as the most promising compound for further validation.
Figure 2.

MLK3 degradation by CEP1347-VHL-based PROTACs in A375_MLK3 and HCC1806_MLK3 cells. (A,B) Western blot analysis of MLK3 levels in the A375 and HCC1806 cell lines with DOX-inducible MLK3 overexpression after 24 h treatment with different VHL-based PROTACs at a concentration range of 0.1–5 μM. Dimethyl sulfoxide (DMSO) was used as a control. (C) Western blot analysis of MLK3 levels in the HCC1806 cell line with DOX-inducible MLK3 overexpression treated with 0.5 μM CEP1347-VHL-02 PROTAC for the indicated times (2–24 h). DMSO was used as a control. (D) Western blot analysis of MLK3 levels in the HCC1806 cell line with DOX-inducible MLK3 overexpression after 24 h treatment with CEP1347-VHL-02 PROTAC and CEP1347-VHL-02-epimer at a concentration range of 0.25–1 μM. DMSO was used as a control. (E) Western blot analysis of MLK3 levels in the HCC1806 cell line with DOX-inducible MLK3 overexpression pretreated with 1 μM carfilzomib for 1 h, followed by CEP1347-VHL-02 PROTAC treatment at the indicated concentration range of 0.25–1 μM for 7 h. DMSO was used as a control. (F) Western blot analysis of MLK1, MLK2, and MLK4 levels in the A375 cell line with DOX-inducible MLK1, MLK2, and MLK4 overexpression, respectively, after 24 h treatment with CEP1347-VHL-02 PROTAC at the indicated concentration range of 0.1–5 μM. DMSO was used as a control.
CEP1347-VHL-02 Compound Rapidly and Selectively Degrades MLK3 via the Ubiquitin-Proteasome Pathway
Next, we evaluated the dynamics of MLK3 degradation by CEP1347-VHL-02 in the HCC1806_MLK3 cell line. This PROTAC reduced MLK3 levels after 2 h and led to nearly complete degradation after 24 h of treatment (Figure 2C). MLK3 protein levels were not reduced by the inactive cis-epimer of CEP1347-VHL-02 PROTAC, which does not bind VHL, indicating that MLK3 was degraded in a VHL-dependent manner (Figure 2D). Next, we investigated the mechanism of MLK3 degradation by the CEP1347-VHL-02 compound. The treatment of HCC1806_MLK3 cells with the proteasome inhibitor carfilzomib prevented MLK3 degradation by CEP1347-VHL-02, confirming that MLK3 was degraded explicitly through the ubiquitin-proteasome system (Figure 2E). We then evaluated the selectivity of CEP1347-VHL-02. We utilized three cell lines that were generated using A375 melanoma cells with the DOX-inducible overexpression of MLK1, MLK2, and MLK4 (A375_MLK1, A375_MLK2, A375_MLK4 respectively), which are members of MLK family. We found no changes in MLK1, MLK2, or MLK4 levels after CEP1347-VHL-02 treatment, suggesting that CEP1347-VHL-02 PROTAC selectively targets MLK3 over other related kinases (Figure 2F).
To further validate the selectivity of CEP1347-VHL-02, we performed a shotgun proteomic analysis of protein levels in the total cell extracts from HCC1806_MLK3 cells upon inducing MLK3 expression by DOX and treating with either CEP1347-VHL-02 PROTAC or DMSO (Figure 3A,B and Supplementary Table 1). Our analysis revealed that MLK3 was the only kinase that was significantly degraded by the CEP1347-VHL-02 compound (Figure 3B, black arrow). Beyond MLK3, we observed a reduction in the levels of 11 additional proteins following PROTAC treatment, as marked by the blue boxed area in Figure 3B. Importantly, 10 out of these 11 proteins that showed decreased expression were found to be upregulated in response to DOX-mediated overexpression of MLK3 (highlighted in the yellow boxed area of Figure 3A). This implies that the action of CEP1347-VHL-02 indirectly leads to the diminished expression of proteins dependent on the presence of MLK3 in our cellular model (Figure 3A,C). The efficacy of MLK3 degradation by CEP1347-VHL-02 in these samples was further validated through immunoblot analysis (Figure 3D).
Figure 3.

Shotgun proteomic analysis of total cell protein extracts from HCC1806_MLK3 cell line following the treatment with CEP1347-VHL-02 PROTAC. (A,B) HCC1806_MLK3 cells were treated with DOX to induce MLK3 overexpression. Next, cells were incubated with 0.5 μM CEP1347-VHL-02 PROTAC or DMSO as a control for 24 h. (A) Volcano plot displaying the log2 fold change (Log2 FC, x axis) against the t test-derived–log10 statistical (n = 3) p-value (y axis) for all protein groups detected in the total cell extract from HCC1806_MLK3 cells following DOX induction of MLK3 expression. The abundance changes thresholds of Log2 FC ≥ |1.0| and the significance threshold of −log10 p-value ≥ 2.0 were applied to identify protein groups with levels reproducibly decreased (indicated in blue) or increased (indicated in red) in response to the DOX treatment. MLK3 protein is indicated with a black arrow. (B) Volcano plot displaying the log2 fold change (Log2 FC, x axis) against the t test-derived–log10 statistical (n = 3) p-value (y axis) for all protein groups detected in the total cell extract from HCC1806_MLK3 cells following induction of MLK3 expression and the treatment with CEP1347-VHL-02 PROTAC. The abundance changes thresholds of Log2 FC ≥ |1.0| and the significance threshold of −log10 p-value ≥ 2.0 were applied to identify protein groups with levels reproducibly decreased (indicated in blue) or increased (indicated in red) in response to the PROTAC treatment. MLK3 protein is indicated with a black arrow. (C) Venn diagram of the number of overlapping upregulated proteins from DOX vs NO DOX (yellow boxed area on volcano plot) and downregulated proteins from PROTAC vs DMSO (blue boxed area on volcano plot) MS analysis. MLK3 was not included. (D) Immunoblotting analysis of MLK3 level in HCC1806_MLK3 cells after DOX induction and CEP1347-VHL-02 PROTAC treatment.
CEP1347-VHL-02 PROTAC Degrades Endogenous MLK3 in Breast Cancer Cell Lines
To verify whether CEP1347-VHL-02 degraded endogenous MLK3 in cell lines with high expression of this kinase, we performed additional experiments using breast cancer cell lines MCF-7 (ER+ subtype) and MDA-MB-468 (TNBC subtype). We assessed MLK3 degradation in a time- and concentration-dependent manner. We found that CEP1347-VHL-02 rapidly degraded MLK3 in both cell lines, with PROTAC reaction half-life (t1/2) values of 2.5 and 4 h for MCF-7 and MDA-MB-468, respectively (Figure 4A). CEP1347-VHL-02 also showed a high potency of MLK3 degradation, with a half-maximal degradation concentration (DC50) of 50 nM and maximal levels of degradation (Dmax) of 77% for MCF-7 and DC50 of 300 nM and Dmax of 63% for MDA-MB-468 (Figure 4B). Next, to ensure that the observed lower amounts of MLK3 after PROTAC treatment were due to protein degradation rather than changes in mRNA levels, we analyzed MLK3 gene expression by real-time quantitative polymerase chain reaction (RT-qPCR) in cells that were treated with CEP1347-VHL-02. As expected, MLK3 mRNA levels were unaffected by PROTAC treatment, confirming that the observed MLK3 downregulation by CEP1347-VHL-02 was not attributable to transcriptional effects (Figure S2A). We also confirmed that the degradation of endogenous MLK3 protein by CEP1347-VHL-02 occurred through the ubiquitin-proteasome pathway, as pretreatment with carfilzomib prevented MLK3 downregulation in both cell lines (Figure 4C). We then showed that neither the inactive cis-epimer of CEP1347-VHL-02 nor CEP1347 compound downregulated MLK3 in breast cancer cell lines (Figure S2B,C). Furthermore, CEP1347-VHL-02 did not degrade endogenous MLK1, MLK2, or MLK4 in MCF-7 and MDA-MB-468 cells, confirming the selectivity of this PROTAC for MLK3 (Figure S2D), which is consistent with the results obtained in A375 cells (Figure 2F).
Figure 4.

Degradation of endogenous MLK3 by CEP1347-VHL-02 PROTAC in MCF-7 and MDA-MB-468 cell lines. (A) Western blot analysis of MLK3 levels in MCF-7 and MDA-MB-468 cell lines treated with 1 μM CEP1347-VHL-02 PROTAC for the indicated times (4–48 h). DMSO was used as a control. The percentage of remaining MLK3 protein during the time of treatment was quantified and plotted in the lower panel, and t1/2 of the PROTAC reaction was determined. (B) Western blot analysis of MLK3 levels in MCF-7 and MDA-MB-468 cell lines after 24 h treatment with CEP1347-VHL-02 PROTAC at a concentration range of 0.1–5 μM. DMSO was used as a control. The percentage of remaining MLK3 protein for the indicated concentration range was quantified and plotted in the lower panel, and DC50 and Dmax were determined. (C) Western blot analysis of MLK3 levels in MCF-7 and MDA-MB-468 cell lines pretreated with 1 μM carfilzomib for 1 h, followed by 1 μM CEP1347-VHL-02 PROTAC treatment for 7 h. DMSO was used as a control.
We also tested the efficacy of other VHL-based PROTACs with varying linkers in MDA-MB-468 and MCF-7 breast cancer cell lines. These experiments confirmed that the best degradation potency in both cell lines was achieved by CEP1347-VHL-02 (Figures 4A and S3A), which further corresponds to the results seen in cell lines with MLK3 overexpression. Finally, we also analyzed the efficacy and selectivity of endogenous MLK3 degradation by CEP1347-IAP-01 PROTAC in both cell lines. Similarly to CEP1347-VHL-02, IAP-based PROTAC showed selectivity toward MLK3 as it did not degrade MLK1, MLK2, and MLK4, however, the potency of MLK3 degradation was inferior compared to CEP1347-VHL-02 (Figure S3B,C).
Subsequently, we conducted shotgun proteomic analysis to assess the protein profiles in total cell extracts from MCF-7 and MDA-MB-468 cells following treatment with CEP1347-VHL-02 or vehicle control (DMSO). The mass spectrometry was unable to detect endogenous MLK3 in these samples, thus precluding confirmation of its degradation via this technique, nevertheless, immunoblotting analysis distinctly showed significant degradation of MLK3 following treatment with CEP1347-VHL-02 (Figure S4A,B). Importantly, our analysis revealed no degradation of proteins, including other members of the MLK family, underscoring the specificity of the treatment as no significant changes were observed in the protein landscape (Figure S4 and Supplementary Table 1). Finally, we assessed the activity of two major downstream pathways that are activated by MLK3: MEK/ERK and MKK/JNK pathways. Immunoblotting analysis showed no changes in the activity of the ERK pathway, whereas the JNK pathway was significantly downregulated upon CEP1347-VHL-02 treatment in both MCF-7 and MDA-MB-468 cell lines (Figure S5A,B).
Degradation of Endogenous MLK3 by the CEP1347-VHL-02 Compound Decreases the Tumorigenic Potential of MDA-MB-468 but Not MCF-7 Cells
To determine whether endogenous MLK3 degradation by CEP1347-VHL-02 PROTAC affects cancer cell proliferation and survival, we performed a series of phenotypic assays on MCF-7 and MDA-MB-468 cell lines. We observed a significant reduction of the clonogenic potential of MDA-MB-468 cells after treatment with CEP1347-VHL-02 compared with DMSO-treated cells (Figure 5A, right panel). Interestingly, we did not observe this effect in the MCF-7 cell line (Figure 5A, left panel). To verify this finding with an alternative method, we knocked down MLK3 in MCF-7 and MDA-MB-468 cells (Figure S6A). A colony formation assay that was performed on cells in which MLK3 was silenced confirmed a significant reduction of the clonogenic potential of MDA-MB-468 cells and no changes in MCF-7 cells compared with control cells (Figure S6B). We also tested the effects of CEP1347-VHL-02 treatment under 3D cell culture conditions, which more closely reflect tumor growth in vivo compared to two-dimensional culture. We found that after the treatment with CEP1347-VHL-02, MDA-MB-468 cells grown on hydrogel-coated plates showed compromised formation of spheroids (Figure S7A). Similarly, CEP1347-VHL-02 PROTAC treatment caused a reduction in the growth of spheroids formed by MDA-MB-468 cells grown on low-attachment surface plates (Figure S7B). No changes in spheroid formation or size after the treatment with CEP1347-VHL-02 PROTAC were observed for MCF-7 cells grown in 3D conditions (Figure S7A,B).
Figure 5.

Degradation of endogenous MLK3 by the CEP1347-VHL-02 compound decreased the tumorigenic potential of MDA-MB-468 cells but not MCF-7 cells. (A) Colony formation assay of MCF-7 and MDA-MB-468 cell lines after CEP1347-VHL-02 PROTAC treatment. (Upper panel) Representative photographs of colonies stained with crystal violet formed by MCF-7 and MDA-MB-468 cells after 15 days of 1 μM CEP1347-VHL-02 PROTAC treatment. DMSO was used as a control. (Lower panel) Bar chart that summarizes data from three independent experiments. Each data point represents the mean ± standard deviation (SD) from three independent experiments. The statistical analysis was performed using an unpaired t-test (***p < 0.001) compared with control. (B) Cell cycle analysis in MCF-7 and MDA-MB-468 cell lines after CEP1347-VHL-02 PROTAC treatment. (Upper panel) Representative graphs of cell cycle analysis with propidium iodide staining in MCF-7 and MDA-MB-468 cell lines after 48 h of 1 μM CEP1347-VHL-02 PROTAC treatment. DMSO was used as a control. (Lower panel) Quantified data from two independent experiments. Each data point represents the mean ± SD from three independent experiments. The statistical analysis was performed using one-way ANOVA (***p < 0.001). (C) Cell viability analysis of MCF-7 and MDA-MB-468 cell lines after CEP1347-VHL-02 PROTAC treatment. (Upper panel) Representative graphs of cell viability analysis with annexin V and propidium iodide staining in MCF-7 and MDA-MB-468 cell lines after 48 h of 1 μM CEP1347-VHL-02 PROTAC treatment. DMSO was used as a control. (Lower panel) Quantified data from three independent experiments. Each data point represents the mean ± SD from three independent experiments. The statistical analysis was performed using one-way ANOVA (***p < 0.001). (D) Boyden chamber transwell migration assays of MCF-7 and MDA-MB-468 cell lines after CEP1347-VHL-02 PROTAC treatment. (Left panel) Representative images of a randomly selected area of MCF-7 and MDA-MB-468 cells that migrated through the membrane after 24 h of 1 μM CEP1347-VHL-02 PROTAC treatment. Cells were stained with crystal violet. (Right panel) Quantification of MCF-7 and MDA-MB-468 cells treated with 1 μM CEP1347-VHL-02 PROTAC and DMSO that migrated through the insert. The statistical analysis was performed using an unpaired t-test (**p < 0.01) compared with DMSO as a control. Standard error bars are shown.
Furthermore, our data showed that the reduced proliferation of MDA-MB-468 cells was likely caused by G2/M phase cell cycle arrest after CEP1347-VHL-02 treatment, as indicated by the flow cytometry analysis (Figure 5B). We also observed the induction of apoptosis in MDA-MB-468 cells after PROTAC treatment (Figure 5C). No changes were observed in MCF-7 in cell cycle and there was no significant increase in cell death after CEP1347-VHL-02 treatment (Figure 5B,C). We also assessed the migratory potential of both cell lines upon MLK3 degradation using transwell migration assays. We observed a reduction of MDA-MB-468 cell migration after treatment with CEP1347-VHL-02 PROTAC compared with DMSO-treated cells (Figure 5D, lower panel). On the contrary, CEP1347-VHL-02 treatment did not decrease the migratory potential of MCF-7 cells (Figure 5D, upper panel). Collectively, these results suggested that MLK3 degradation reduced tumorigenic and invasive potential selectively in TNBC cells, where MLK3 plays a protumorigenic role.
Discussion and Conclusions
MLK3 has been recently recognized as a potential therapeutic target in multiple types of malignancies. The functional role of MLK3 and its downstream signaling has also been well-documented in neurodegenerative diseases, including Parkinson’s disease and Alzheimer’s disease.5,22 Despite growing knowledge of the role of MLK3 in cancer and other human diseases, the pharmacological modulation of this kinase remains challenging. In the present study, we developed and characterized the first PROTACs that selectively target MLK3 kinase for degradation. We further characterized our lead compound, CEP1347-VHL-02, in different cell line models and demonstrated that this PROTAC induced potent, rapid, and selective degradation of both overexpressed and endogenous MLK3 protein.
Over the past decades, several small-molecule inhibitors have been designed to target MLK3 and other MLK family members.23−26 CEP1347 is a semisynthetic derivative of the indolocarbazole natural product K252a, which was initially found to prevent neuronal cell death through inhibition of the MLK-JNK signaling axis.23,24 These observations moved CEP1347 to clinical trials as a disease-modifying therapeutic for Parkinson’s disease, but the clinical studies failed to demonstrate its effectiveness.27 CEP1347 inhibits the catalytic activity of all core MLK family kinases (MLK1–3), with half-maximal inhibitory concentration (IC50) values of 23–51 nM.23 This compound previously showed antineoplastic activity in different types of cancer cell lines, including pancreatic, breast, glioblastoma, and ovarian cancer cells.9,10,28,29 Here, we used CEP1347 as a pharmacophore for the synthesis of MLK3-targeting PROTACs. Among all tested PROTACs, CEP1347-VHL-02 demonstrated the most potent degradation of MLK3. Strikingly, CEP1347-VHL-02 did not lead to the degradation of other MLK family kinases. Therefore, this compound provides a novel strategy to selectively target MLK3. The ability of CEP1347-VHL-02 to discriminate between closely related kinases is not completely understood. Several recent studies reported isoform-specific degraders of protein kinases that were derived from nonselective inhibitors, such as the multikinase inhibitors Foretinib, TAE-684, and the SGK/S6K1 inhibitor R-308.30−32 One postulation is that the superior degradation selectivity of some PROTACs compared with their native pharmacophores may be driven by the differential cooperativity and stability of ternary complex formation and geometry and orientation of the recruited E3 ligase.32,33 Additional research on the mechanisms that dictate the selectivity of CEP1347-VHL-02 is warranted. A better understanding of this phenomenon may lead to the development of novel selective degraders for other MLKs.
An inherent challenge of traditional small-molecule inhibitors that target MLK3 arises from functions of this protein kinase that are independent of its catalytic activity. Previous studies have shown that MLK3 can activate the RAF/MEK/ERK pathway independent of its kinase activity by acting as a scaffold and crucial component of the B-Raf/Raf-1 complex.2 Schroyer et al. demonstrated that the kinase-independent mechanism of ERK1/2 activation by MLK3 promotes the invasion of colorectal cancer cells in response to oxidative stress.14 In another study, MLK3 was found to promote cell migration in a manner independent of its kinase activity by binding to the Rho activator p63RhoGEF/GEFT.13 MLK3-targeting PROTACs appear to be particularly useful tools for targeting and studying noncatalytic functions of MLK3. CEP1347-VHL-02 degraded endogenous MLK3 within 24 h, with a DC50 < 50 nM and 300 nM for MCF-7 and MDA-MB-468, respectively. Thus, our compound provides a much faster reduction of intracellular protein levels compared with gene silencing methodologies, which may also pose such various challenges as lethality and genetic compensation.34−36 Interestingly, we observed a less potent degradation and an increased rate and extent of target recovery in MDA-MB-468 cells compared to the MCF-7 cell line after PROTAC treatment. These findings can be explained by the intrinsic differences between these two cell lines that may affect degradation kinetics and target recovery, such as endogenous expression of the targeted protein and E3 ligase machinery components, cellular permeability, intracellular compound stability, and rate of compound efflux.37,38 Further studies are required to better characterize the pharmacokinetic properties and stability of CEP1347-VHL-02 and define the MLK3 degradation profile across different cell lines and in vivo models.
TNBC is a subtype of breast cancer that is characterized by the absence of estrogen and progesterone receptors and lack of HER2 overexpression. Among all breast cancer subtypes, TNBC is associated with the least favorable prognosis because of its aggressive clinical course and limited targeted treatment options.39,40 Here, we showed that MLK3 degradation by CEP1347-VHL-02 decreased proliferation and mammosphere formation, induced apoptosis, and decreased migratory potential of the TNBC MDA-MB-468 cell line. These effects were not observed in the ER-positive breast cancer MCF-7 cell line, despite a similar degree of MLK3 degradation by PROTAC in both cell lines. Similarly, MLK3 silencing using small interfering RNA (siRNA) decreased the proliferation of MDA-MBA-468 cells but not MCF-7 cells. These results correspond to recent studies that reported that MLK3 kinase activity is higher in TNBC than in hormone receptor-positive tumors, and it selectively facilitates the survival of TNBC.9−11 Although MLK3 depletion profoundly inhibited oncogenic features of MDA-MB-468 cells, neither CEP1347-VHL-02 at 1 μM nor genetic knock-down of MLK3 was sufficient to completely impair the tumorigenic potential of this cell line. The additional feedback loops involving protein kinases and transcriptional pathways are likely activated after the degradation of MLK3 to compensate for its loss and sustain cancer growth, as previously reported for many kinase targets in TNBC.39 Furthermore, we demonstrated that CEP1347-VHL-02 inhibited the activation of JNK, a major downstream effector of the canonical MLK3 signaling. Because JNK phosphorylation was reduced in both MDA-MB-468 and MCF-7 cells, these results do not explain the different phenotypic changes observed in these cell lines. Therefore, future mechanistic studies of MLK3-targeting PROTACs are warranted, as these compounds represent an attractive tool to investigate distinct functions of MLK3 in TNBC vs ER/PR+ breast cancer. While our results highlight MLK3 degradation as a potential treatment strategy in TNBC, further research aiming to investigate the pharmacokinetics properties, in vivo potency, and safety profile of MLK3-targeting PROTACs is needed to advance the translational applications of these compounds.
In summary, we described first-in-class PROTAC that induces the rapid and selective degradation of MLK3 in cancer cell lines. Our study provides new opportunities for the selective targeting of MLK3 in cancer and paves the way for the development of next-generation degraders against MLK3 and related kinases. Notably, MLK3 degradation by CEP1347-VHL-02 significantly reduced the oncogenic potential of TNBC cells, suggesting that the targeted degradation of this kinase may be a feasible treatment strategy for this subtype of breast cancer.
Experimental Section
General Information for Chemical Synthesis
Starting materials were used as received unless otherwise noted. All moisture sensitive reactions were performed in an inert atmosphere of argon with oven-dried glassware. Reagent grade solvents were used for extractions and flash chromatography. Reaction progress was monitored by LC-MS analysis performed on an Agilent UPLC/MS instrument equipped with a RP-C18 column (Poroshell 120 SB-C18, 4.6 × 50 mm, 2.7 μm or Zorbax 300SB-C18, 4.6 × 50 mm, 3.5 μm), dual atmospheric pressure chemical ionization (APCI)/electrospray (ESI) mass spectrometry detector, and photodiode array detector. Flash chromatography was performed by using a RediSepRf NP-silica (40–63 μm 60 Å) or a Teledyne RediSepRf Gold RP-C18 column (20–40 μm 100 Å) in a Teledyne ISCO CombiFlash Rf 200 purification system unless otherwise specified. 1H NMR spectra were recorded on an Agilent 400 MHz spectrometer and are reported in parts per million (ppm) on the δ scale relative to CDCl3 (δ 7.26) and DMSO-d6 (δ 2.50) as internal standards. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, b = broad, m = multiplet), coupling constants (Hz), and integration. 13C NMR spectra were recorded on an Agilent 100 MHz and are reported in parts per million (ppm) on the δ scale relative to CDCl3 (δ 77.00), CD3OD and DMSO-d6 (δ 39.52).
Synthesis and Characterization Data
The well-known E3 ligase-linker compounds 1–4 and 6–10 were synthesized using the reported procedures and also compounds 1, 2, 4, 9 and 10 were commercially available. Compound 5, CEP1347-VHL-01, CEP1347-VHL-02, CEP1347-VHL-02-epimer, CEP1347-VHL-03, CEP1347-VHL-04, CEP1347-VHL-05, CEP1347-VHL-06, CEP1347-VHL-07, CEP1347-VHL-08, CEP1347-IAP-01, and CEP1347-CRBN-01 were synthesized according to the procedures described below. All compounds were subjected to internal quality control; most demonstrated greater than 95% purity and detection of an expected parent ion by UPLC-MS (ESI) (Figure S8).
LC-MS Methods Used for Analysis
Method A: a 4 min gradient of 50% to 95% methanol in water (containing 5 mM ammonium acetate and 0.2% acetic acid) was used with a 7 min run time at a flow rate of 1 mL/min.
Method B: a 4 min gradient of 50% to 95% acetonitrile (containing 0.05% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 6 min run time at a flow rate of 1 mL/min.
Method C: a 4 min gradient of 5% to 95% acetonitrile (containing 0.05% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 6 min run time at a flow rate of 1 mL/min.
Method D: a 7 min gradient of 5% to 95% acetonitrile (containing 0.05% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 10 min run time at a flow rate of 1 mL/min.
(2S,4S)-1-((S)-2-(2-(4-(Aminomethyl)-1H-1,2,3-triazol-1-yl)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide (5)
(2S,4R)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide dihydrochloride (300 mg, 0.580 mmol) in DMF (4 mL), 2-(4-(((tert-butoxycarbonyl)amino)methyl)-1H-1,2,3-triazol-1-yl)acetic acid (149 mg, 0.580 mmol), HATU (232 mg, 0.609 mmol), and DIPEA (202 μL, 1.16 mmol) were added and stirred for overnight. Water was added and extracted with EtOAc. The organic layer was washed with brine and dried over anhydrous MgSO4. The filtrate was concentrated and used for the next step. The crude material was subjected to DCM/TFA (2 mL: 1 mL) and stirred for 2 h. Solvent was removed and purified by combi flash C18 column using water and acetonitrile (both were buffered with 0.05% of TFA) to provide (2S,4R)-1-((S)-2-(2-(4-(aminomethyl)-1H-1,2,3-triazol-1-yl)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide 5 (217 mg, 64%, white solid) over two steps. 1H NMR (500 MHz, CDCl3) δ 8.67 (s, 1H), 8.15 (d, J = 8.4 Hz, 1H), 8.00–7.71 (m, 2H), 7.35–7.28 (m, 4H), 5.22–5.05 (m, 2H), 5.05–4.94 (m, 1H), 4.52–4.43 (m, 2H), 4.37 (s, 1H), 4.14 (s, 2H), 3.82 (d, J = 11.2 Hz, 1H), 3.58 (dd, J = 11.2, 3.4 Hz, 1H), 2.45 (s, 3H), 2.12 (dd, J = 13.4, 7.7 Hz, 1H), 2.00 (ddd, J = 13.5, 9.7, 4.3 Hz, 1H), 1.43 (d, J = 7.0 Hz, 3H), 1.01 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 170.81, 170.75, 167.84, 166.26, 160.96, 150.84, 147.79, 143.68, 132.05, 130.23, 129.42, 126.33, 69.96, 59.19, 58.85, 57.13, 51.86, 48.78, 37.16, 35.40, 34.49, 26.32, 21.99, 15.55. HRMS (m/z): [M + H]+ calcd. for C28H39N8O4S, 583.2815; found, 583.2806.
Synthesis of (5R,7R,8S)-2,11-Bis((ethylthio)methyl)-7-hydroxy-N-((S)-13-((2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carbonyl)-14,14-dimethyl-11-oxo-3,6,9-trioxa-12-azapentadecyl)-8-methyl-15-oxo-5,6,7,8,14,15-hexahydro-13H-16-oxa-4b,8a,14-triaza-5,8-methanodibenzo[b,h]cycloocta[jkl]cyclopenta[e]-as-indacene-7-carboxamide (CEP1347-VHL-01)
CEP1347 (100 mg, 0.16 mmol) was dissolved in THF (5 mL) and added aqueous LiOH (4.3 mg, 0.18 mmol in 1 mL water) at room temperature. The reaction mixture was stirred for 1 h and quenched with aqueous NH4Cl to precipitate the product. The precipitate was collected and dried to provide the (5R,7R,8S)-2,11-bis((ethylthio)methyl)-7-hydroxy-8-methyl-15-oxo-5,6,7,8,14,15-hexahydro-13H-16-oxa-4b,8a,14-triaza-5,8-methanodibenzo[b,h]cycloocta[jkl]cyclopenta[e]-as-indacene-7-carboxylic acid (CEP1347-acid) (85 mg, 87%, white solid), which was used without further purification. To the solution of CEP1347-acid (7 mg, 0.012 mmol) and (2S,4R)-1-((S)-14-amino-2-(tert-butyl)-4-oxo-6,9,12-trioxa-3-azatetradecanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide 1 (8 mg, 0.012 mmol) in DMF (0.8 mL), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate, N-[(dimethylamino)-1H-1,2,3-triazolo-[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide (HATU) (5 mg, 0.012 mmol) and N,N-Diisopropylethylamine (DIPEA) (4 μL, 0.023 mmol) were added at room temperature. The reaction mixture was stirred overnight and water (6 mL) was added to precipitate the product. The precipitate was collected and purified by ISCO combi flash Redi Sef RF basic alumina column (DCM/MeOH = 9:1). The pure fractions were combined to afford the CEP1347-VHL-01 (3 mg, 20%, white solid). 1H NMR (4 00 MHz, dmso) δ 9.11 (s, 1H), 8.95 (s, 1H), 8.63–8.52 (m, 2H), 8.30 (t, J = 5.7 Hz, 1H), 7.94 (d, J = 4.7 Hz, 2H), 7.78 (d, J = 8.4 Hz, 1H), 7.46–7.37 (m, 6H), 6.99 (d, J = 7.7 Hz, 1H), 6.39 (s, 1H), 5.13 (s, 1H), 4.96 (q, J = 17.6 Hz, 2H), 4.54 (d, J = 9.7 Hz, 1H), 4.45–4.30 (m, 3H), 4.28–4.22 (m, 1H), 3.98–3.90 (m, 6H), 3.70–3.38 (m, 14H), 3.27–3.17 (m, 1H), 2.47–2.39 (m, 7H), 2.11–1.85 (m, 6H), 1.25–1.19 (m, 6H), 0.92 (s, 9H). 13C NMR (101 MHz, dmso) δ 172.66, 172.23, 172.19, 169.58, 169.05, 151.89, 148.20, 139.89, 139.41, 136.28, 133.32, 131.14, 130.15, 129.89, 129.29, 129.12, 128.58, 127.91, 127.04, 126.04, 124.86, 124.62, 122.97, 121.47, 119.84, 115.87, 115.36, 114.77, 109.42, 100.58, 85.76, 85.19, 70.92, 70.33, 70.13, 70.07, 70.01, 69.31, 69.17, 59.18, 57.00, 56.16, 46.01, 42.13, 38.94, 38.36, 36.15, 35.99, 35.31, 26.63, 25.28, 25.11, 22.79, 16.37, 14.91. LC-MS retention time (Method A): 4.17 min, purity: 97%. LRMS (m/z): [M + H]+ calcd. for C62H75N8O11S3, 1203.47; found, 1203.40. HRMS (m/z): [M + H]+ calcd. for C62H75N8O11S3, 1203.4717; found, 1203.4723.
Synthesis of (5R,7R,8S)-2,11-Bis((ethylthio)methyl)-7-hydroxy-N-((S)-13-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidine-1-carbonyl)-14,14-dimethyl-11-oxo-3,6,9-trioxa-12-azapentadecyl)-8-methyl-15-oxo-5,6,7,8,14,15-hexahydro-13H-16-oxa-4b,8a,14-triaza-5,8-methanodibenzo[b,h]cycloocta[jkl]cyclopenta[e]-as-indacene-7-carboxamide (CEP1347-VHL-02)
To the solution of CEP1347-acid (25 mg, 0.042 mmol) and (2S,4R)-1-((S)-14-amino-2-(tert-butyl)-4-oxo-6,9,12-trioxa-3-azatetradecanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide 2 (29 mg, 0.046 mmol) in DMF (1.5 mL), HATU (17 mg, 0.044 mmol) and DIPEA (14 μL, 0.083 mmol) were added at room temperature and stirred for overnight. Water (6 mL) was added to precipitate the product. The precipitate was collected and purified by ISCO combi flash Redi Sef RF silica gel column chromatography (DCM/MeOH = 9:1) to afford the CEP1347-VHL-02 (28 mg, 55%, white solid). 1H NMR (400 MHz, CDCl3) δ 8.73 (s, 1H), 8.59 (s, 1H), 8.42 (s, 1H), 7.82 (d, J = 8.6 Hz, 1H), 7.72–7.65 (m, 2H), 7.43 (d, J = 7.3 Hz, 1H), 7.39–7.31 (m, 3H), 7.25–7.20 (m, 2H), 7.11 (dd, J = 8.4, 1.7 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.72 (dd, J = 7.7, 4.7 Hz, 1H), 6.28 (s, 1H), 5.18–5.06 (m, 1H), 4.85 (q, J = 9.9 Hz, 2H), 4.55–4.24 (m, 3H), 4.09–3.87 (m, 8H), 3.84–3.55 (m, 12H), 3.52–3.41 (m, 1H), 3.17–3.09 (m, 1H), 2.85 (q, J = 7.4 Hz, 2H), 2.40 (q, J = 7.5 Hz, 2H), 2.37 (s, 3H), 2.11 (s, 3H), 2.10–2.05 (m, 2H), 1.65–1.41 (m, 6H), 1.19 (t, J = 7.4 Hz, 3H), 1.15 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 172.86, 172.58, 170.81, 170.46, 170.32, 150.22, 144.11, 139.88, 135.99, 131.86, 130.19, 129.54, 129.46, 128.46, 126.76, 126.33, 126.07, 124.92, 124.24, 121.22, 120.03, 117.07, 115.72, 115.42, 113.25, 107.65, 100.85, 85.93, 85.51, 77.23, 71.80, 71.64, 70.57, 70.28, 70.05, 69.51, 59.20, 58.13, 56.85, 53.43, 48.78, 46.13, 42.80, 39.66, 36.67, 36.47, 35.99, 29.71, 26.55, 25.91, 22.96, 22.52, 15.72, 14.82, 14.52. LC-MS retention time: 4.30 min (Method A), 2.90 (Method B), 4.45 min (Method C) purity: 97%. LRMS (m/z): [M + H]+ calcd. for C63H77N8O11S3, 1217.49; found, 1217.20. HRMS (m/z): [M + H]+ calcd. for C63H77N8O11S3, 1217.4874; found, 1217.4877.
Synthesis of (5R,7R,8S)-2,11-Bis((ethylthio)methyl)-7-hydroxy-N-((S)-13-((2S,4S)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidine-1-carbonyl)-14,14-dimethyl-11-oxo-3,6,9-trioxa-12-azapentadecyl)-8-methyl-15-oxo-5,6,7,8,14,15-hexahydro-13H-16-oxa-4b,8a,14-triaza-5,8-methanodibenzo[b,h]cycloocta[jkl]cyclopenta[e]-as-indacene-7-carboxamide (CEP1347-VHL-02-epimer)
CEP1347-VHL-02-epimer was synthesized using the same reaction procedure as CEP1347-VHL-02. Starting materials CEP1347-acid (14 mg, 0.022 mmol), (2S,4S)-1-((S)-14-amino-2-(tert-butyl)-4-oxo-6,9,12-trioxa-3-azatetradecanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide 2-epimer (13 mg, 0.022 mmol, white solid) and HATU (9 mg, 0.023 mmol), DIPEA (0.044 mmol, 44 μL) were used. CEP1347-VHL-02-epimer (6 mg, 20%). 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 8.51 (s, 1H), 7.95 (t, J = 5.6 Hz, 1H), 7.89–7.79 (m, 2H), 7.53–7.39 (m, 2H), 7.38–7.28 (m, 4H), 7.25–7.19 (m, 2H), 7.11 (d, J = 8.4 Hz, 1H), 6.83–6.71 (m, 1H), 6.61 (s, 1H), 5.39–5.28 (m, 2H), 5.01 (p, J = 7.1 Hz, 1H), 4.63–4.51 (m, 2H), 4.50–4.36 (m, 2H), 4.32 (s, 1H), 4.05–3.92 (m, 4H), 3.90–3.68 (m, 13H), 3.52 (dt, J = 15.9, 8.0 Hz, 1H), 3.00–2.93 (m, 1H), 2.65 (q, J = 7.1 Hz, 2H), 2.52–2.46 (m, 2H), 2.45 (s, 3H), 2.18 (d, J = 15.7 Hz, 3H), 2.14 (s, 3H), 2.00 (td, J = 9.2, 4.5 Hz, 1H), 1.47–1.38 (m, 6H), 1.35–1.19 (m, 5H), 1.01 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 171.80, 171.60, 170.65, 170.49, 169.08, 141.41, 138.53, 135.02, 131.23, 129.97, 128.71, 128.59, 125.76, 125.39, 124.83, 124.12, 123.39, 120.81, 119.51, 115.13, 113.86, 112.69, 106.69, 99.65, 84.45, 84.41, 76.20, 70.12, 69.91, 69.80, 69.39, 69.34, 68.68, 58.83, 57.51, 55.50, 52.41, 48.19, 41.80, 38.31, 35.51, 34.25, 33.94, 30.89, 28.68, 28.30, 26.20, 25.36, 25.22, 24.97, 24.70, 24.49, 21.67, 21.32, 20.83, 13.54, 13.10. LC-MS retention time (Method B): 3.08 min, purity: 99%. LRMS (m/z): [M + H]+ calcd. for C63H77N8O11S3, 1217.49; found, 1217.20. HRMS (m/z): [M + H]+ calcd. for C63H77N8O11S3, 1217.4874; found, 1217.4860.
Synthesis of (5R,7R,8S)-2,11-Bis((ethylthio)methyl)-7-hydroxy-N-(6-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-6-oxohexyl)-8-methyl-15-oxo-5,6,7,8,14,15-hexahydro-13H-16-oxa-4b,8a,14-triaza-5,8-methanodibenzo[b,h]cycloocta[jkl]cyclopenta[e]-as-indacene-7-carboxamide (CEP1347-VHL-03)
CEP1347-VHL-03 was synthesized using the same reaction procedure as CEP1347-VHL-02. Starting materials CEP1347-acid (11 mg, 0.018 mmol), (2S,4R)-1-((S)-2-(6-aminohexanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide 3 (10 mg, 0.018 mmol), HATU (7 mg, 0.019 mmol), and DIPEA (6 μL, 0.036 mmol) were used. The reaction mixture was stirred for overnight and the water was added to the reaction mixture. Workup with EtOAc (5 × 5 mL) and the combined layers were dried over anhydrous Na2SO4. The concentrated crude material was purified by an ISCO combi flash Redi Sef RF basic alumina column chromatography (CH2Cl2/MeOH) to give the CEP1347-VHL-03 (7 mg, 30%, white solid). 1H NMR (400 MHz, dmso) δ 9.11 (d, J = 1.7 Hz, 1H), 8.96 (s, 1H), 8.59 (s, 1H), 8.37–8.31 (m, 2H), 8.00–7.91 (m, 2H), 7.82–7.75 (m, 2H), 7.44–7.34 (m, 5H), 6.99 (dd, J = 7.3, 4.9 Hz, 1H), 6.34 (s, 1H), 5.08 (d, J = 3.5 Hz, 1H), 5.04–4.85 (m, 4H), 4.53 (d, J = 9.3 Hz, 1H), 4.42 (t, J = 8.0 Hz, 1H), 4.27 (d, J = 5.7 Hz, 1H), 4.01–3.91 (m, 5H), 3.60 (d, J = 3.3 Hz, 2H), 3.29–3.17 (m, 3H), 2.47–2.42 (m, 6H), 2.31–2.22 (m, 1H), 2.19–2.14 (m, 1H), 2.09 (s, 3H), 2.04–1.94 (m, 2H), 1.78 (ddd, J = 12.8, 8.5, 4.6 Hz, 1H), 1.59–1.48 (m, 4H), 1.37–1.19 (m, 11H), 0.94 (s, 9H). 13C NMR (101 MHz, dmso) δ 172.48, 172.37, 172.23, 171.07, 170.08, 151.91, 148.21, 145.09, 139.44, 136.28, 133.33, 131.55, 131.14, 130.14, 129.88, 129.29, 129.27, 127.04, 126.83, 126.36, 126.03, 124.87, 124.61, 122.96, 121.45, 119.83, 115.87, 115.52, 114.78, 109.41, 100.52, 85.75, 85.21, 69.23, 59.00, 56.82, 56.72, 48.14, 46.01, 42.54, 39.11, 38.18, 36.00, 35.67, 35.39, 35.32, 29.28, 26.93, 26.72, 25.69, 25.13, 25.10, 22.86, 22.81, 16.43, 14.91. LC-MS retention time: 4.36 min (Method A), 3.10 min (Method B), purity: 92%. LRMS (m/z): [M + H]+ calcd. for C61H73N8O8S3, 1141.47; found, 1141.20. HRMS (m/z): [M + H]+ calcd. for C61H73N8O8S3, 1141.4713; found, 1141.4706.
(5R,7R,8S)-2,11-Bis((ethylthio)methyl)-7-hydroxy-N-(11-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-11-oxoundecyl)-8-methyl-15-oxo-5,6,7,8,14,15-hexahydro-13H-16-oxa-4b,8a,14-triaza-5,8-methanodibenzo[b,h]cycloocta[jkl]cyclopenta[e]-as-indacene-7-carboxamide (CEP1347-VHL-04)
CEP1347-VHL-04 was synthesized using the same reaction procedure as CEP1347-VHL-02. Starting materials CEP1347-acid (10 mg, 0.017 mmol), trifluoroacetate salt of (2S,4R)-1-((S)-2-(11-aminoundecanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide 4 (13 mg, 0.018 mmol, white solid), HATU (7 mg, 0.017 mmol) and DIPEA (6 μL, 0.036 mmol) were used to provide CEP1347-VHL-04 (8.8 mg, 44%). 1H NMR (500 MHz, CDCl3) δ 8.60 (s, 1H), 8.37 (s, 1H), 7.86 (d, J = 8.4 Hz, 1H), 7.80 (s, 1H), 7.65 (t, J = 5.9 Hz, 1H), 7.49–7.38 (m, 2H), 7.38–7.29 (m, 4H), 7.19–7.13 (m, 1H), 7.10–7.02 (m, 1H), 6.78–6.70 (m, 2H), 6.17 (d, J = 8.7 Hz, 1H), 5.14 (s, 1H), 5.04 (p, J = 7.1 Hz, 1H), 4.70 (t, J = 7.9 Hz, 1H), 4.53–4.42 (m, 3H), 4.37–4.30 (m, 1H), 4.09–3.95 (m, 3H), 3.79 (s, 2H), 3.58–3.44 (m, 2H), 3.44–3.33 (m, 1H), 3.09–3.03 (m, 1H), 2.68 (q, J = 7.4 Hz, 2H), 2.57–2.37 (m, 6H), 2.16 (t, J = 7.6 Hz, 2H), 2.11–1.96 (m, 4H), 1.72–1.65 (m, 2H), 1.62–1.51 (m, 2H), 1.45–1.17 (m, 18H), 1.01 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 173.87, 172.83, 172.72, 172.17, 169.82, 150.45, 148.39, 143.34, 139.75, 136.09, 132.20, 131.76, 131.08, 130.86, 129.85, 129.65, 128.89, 126.84, 126.52, 126.01, 125.30, 125.13, 124.39, 121.52, 120.45, 116.88, 115.95, 115.16, 113.45, 107.87, 100.94, 85.62, 85.55, 70.11, 58.63, 57.58, 56.84, 48.95, 46.24, 42.94, 39.54, 36.71, 36.61, 36.41, 35.65, 35.24, 29.64, 29.35, 29.20, 29.16, 27.00, 26.63, 26.14, 25.99, 25.63, 22.44, 22.34, 16.12, 14.90, 14.73. LC-MS retention time (Method B): 4.10 min, purity: 95%. LRMS (m/z): [M + H]+ calcd. for C66H83N8O8S3, 1211.55; found, 1211.20. HRMS (m/z): [M + H]+ calcd. for C66H83N8O8S3, 1211.5496; found, 1211.5529.
(5R,7R,8S)-2,11-Bis((ethylthio)methyl)-7-hydroxy-N-((1-(2-(((S)-1-((2S,4S)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-2-oxoethyl)-1H-1,2,3-triazol-4-yl)methyl)-8-methyl-15-oxo-5,6,7,8,14,15-hexahydro-13H-16-oxa-4b,8a,14-triaza-5,8-methanodibenzo[b,h]cycloocta[jkl]cyclopenta[e]-as-indacene-7-carboxamide (CEP1347-VHL-05)
CEP1347-VHL-05 was synthesized using the same reaction procedure as CEP1347-VHL-02. Starting materials CEP1347-acid (15 mg, 0.025 mmol), (2S,4S)-1-((S)-2-(2-(4-(aminomethyl)-1H-1,2,3-triazol-1-yl)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide 5 (17 mg, 0.030 mmol), HATU (10 mg, 0.026 mmol) and DIPEA (9 μL, 0.050 mmol) were used to afford CEP1347-VHL-05 (9 mg, 30%, white solid). 1H NMR (500 MHz, DMSO) δ 9.05 (d, J = 1.7 Hz, 1H), 8.91 (s, 1H), 8.81 (t, J = 6.0 Hz, 1H), 8.61–8.50 (m, 1H), 8.46–8.30 (m, 2H), 7.94–7.86 (m, 3H), 7.74 (d, J = 8.5 Hz, 1H), 7.37–7.35 (m, 4H), 7.32–7.28 (m, 2H), 6.98–6.92 (m, 1H), 6.39 (s, 1H), 5.34–5.10 (m, 2H), 5.08–4.76 (m, 4H), 4.56–4.31 (m, 4H), 4.19 (s, 1H), 3.99–3.79 (m, 4H), 3.63–3.41 (m, 2H), 3.22–3.15 (m, 1H), 2.42–2.35 (m, 8H), 2.11 (d, J = 3.8 Hz, 1H), 2.01 (s, 3H), 1.98–1.91 (m, 1H), 1.70 (ddd, J = 12.9, 8.7, 4.5 Hz, 1H), 1.30 (d, J = 7.0 Hz, 3H), 1.16 (t, J = 7.3 Hz, 6H), 0.90 (s, 9H). 13C NMR (126 MHz, DMSO) δ 172.74, 172.24, 170.99, 169.38, 165.79, 151.96, 148.23, 145.09, 145.03, 139.46, 136.30, 133.37, 131.57, 131.17, 130.17, 129.93, 129.30, 129.26, 127.08, 126.85, 126.38, 126.06, 125.08, 124.85, 124.61, 122.98, 121.48, 119.86, 115.89, 115.39, 114.80, 100.59, 85.29, 69.24, 59.10, 57.36, 56.91, 51.79, 48.18, 46.03, 44.05, 42.54, 38.19, 36.03, 36.00, 35.32, 26.82, 25.13, 22.92, 22.89, 16.46, 14.94. LC-MS retention time (Method C): 4.23 min, purity: 96%. LRMS (m/z): [M + H]+ calcd. for C60H68N11O8S3, 1166.44; found, 1166.20. HRMS (m/z): [M + H]+ calcd. for C60H68N11O8S3, 1166.4414; found, 1166.4440.
(5R,7R,8S)-2,11-Bis((ethylthio)methyl)-7-hydroxy-N-(2-(2-(2-(((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-2-oxoethoxy)ethoxy)ethyl)-8-methyl-15-oxo-5,6,7,8,14,15-hexahydro-13H-16-oxa-4b,8a,14-triaza-5,8-methanodibenzo[b,h]cycloocta[jkl]cyclopenta[e]-as-indacene-7-carboxamide (CEP1347-VHL-06)
CEP1347-VHL-06 was synthesized using the same reaction procedure as CEP1347-VHL-02. Starting materials CEP1347-acid (20 mg, 0.033 mmol), (2S,4R)-1-((S)-2-(2-(2-(2-aminoethoxy)ethoxy)acetamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide 6 (22 mg, 0.037), HATU (13 mg, 0.035 mmol) and DIPEA (12 μL, 0.066 mmol) were used. After the consumption of CEP1347-acid standard workup was performed and purified by silica gel column chromatography (DCM/MeOH = 9:1) to afford CEP1347-VHL-06 (19 mg, 49%, white solid). 1H NMR (400 MHz, dmso) δ 9.11 (d, J = 1.8 Hz, 1H), 8.95 (s, 1H), 8.59 (s, 1H), 8.42 (t, J = 5.9 Hz, 1H), 8.30 (d, J = 7.7 Hz, 1H), 7.96–7.88 (m, 2H), 7.79 (d, J = 8.5 Hz, 1H), 7.51–7.36 (m, 5H), 7.31 (d, J = 8.3 Hz, 2H), 7.04–6.96 (m, 1H), 6.38 (s, 1H), 5.15–4.78 (m, 4H), 4.57 (d, J = 9.6 Hz, 1H), 4.39 (t, J = 8.1 Hz, 1H), 4.27 (s, 1H), 4.03–3.84 (m, 6H), 3.70–3.36 (m, 10H), 3.28–3.19 (m, 1H), 2.47–2.42 (m, 4H), 2.41 (s, 3H), 2.10 (s, 3H), 2.04–1.92 (m, 2H), 1.75 (ddd, J = 12.9, 8.5, 4.7 Hz, 1H), 1.31 (d, J = 7.0 Hz, 3H), 1.25–1.17 (m, 6H), 0.94 (s, 9H). 13C NMR (101 MHz, dmso) δ 172.66, 172.23, 170.83, 169.53, 169.05, 151.90, 148.19, 145.04, 139.41, 136.29, 133.33, 131.53, 131.16, 130.14, 129.89, 129.33, 129.26, 127.04, 126.76, 126.71, 126.35, 126.04, 124.84, 124.62, 122.96, 121.46, 119.84, 115.87, 115.35, 114.78, 109.44, 100.57, 85.75, 85.23, 70.95, 70.06, 69.81, 69.32, 69.18, 59.03, 56.89, 56.18, 48.19, 46.01, 44.21, 42.44, 38.96, 38.07, 36.25, 35.99, 35.31, 26.74, 25.12, 22.81, 16.41, 14.91. LC-MS retention time (Method B): 3.15 min, purity: 99%. LRMS (m/z): [M + H]+ calcd. for C61H73N8O10S3, 1173.46; found, 1173.20. HRMS (m/z): [M + H]+ calcd. for C60H68N11O8S3, 1173.4612; found, 1173.4637.
(5R,7R,8S)-2,11-Bis((ethylthio)methyl)-7-hydroxy-N-((S)-16-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidine-1-carbonyl)-17,17-dimethyl-14-oxo-3,6,9,12-tetraoxa-15-azaoctadecyl)-8-methyl-15-oxo-5,6,7,8,14,15-Hexahydro-13H-16-oxa-4b,8a,14-triaza-5,8-methanodibenzo[b,h]cycloocta[jkl]cyclopenta[e]-as-indacene-7-carboxamide (CEP1347-VHL-07)
CEP1347-VHL-07 was synthesized using the same reaction procedure as CEP1347-VHL-02. Starting materials CEP1347-acid (10 mg, 0.017 mmol), (2S,4R)-1-((S)-17-amino-2-(tert-butyl)-4-oxo-6,9,12,15-tetraoxa-3-azaheptadecanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide dihydrochloride 7 (25 mg, 0.033), HATU (13 mg, 0.033 mmol) and DIPEA (14 μL, 0.083 mmol) were used. After the consumption of CEP1347-acid standard workup was performed and purified by silica gel column chromatography (DCM/MeOH = 9:1) to afford CEP1347-VHL-07 (6.4 mg, 31%, white solid). 1H NMR (500 MHz, CDCl3) δ 8.66 (s, 1H), 8.45 (s, 1H), 8.23 (s, 1H), 7.89 (d, J = 8.6 Hz, 1H), 7.73 (s, 1H), 7.53–7.27 (m, 5H), 7.26 (s, 5H), 6.87–6.53 (m, 1H), 5.88 (s, 1H), 5.11–5.00 (m, 1H), 4.82–4.64 (m, 1H), 4.62–4.24 (m, 4H), 4.05–3.54 (m, 21H), 3.01–2.89 (m, 1H), 2.71–2.63 (m, 2H), 2.44 (d, J = 12.9 Hz, 5H), 2.26 (s, 1H), 2.14 (s, 3H), 2.08–2.07 (m, 1H), 1.47–1.42 (m, 6H), 1.24 (d, J = 7.3 Hz, 3H), 1.02 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 172.98, 172.73, 171.19, 170.37, 170.01, 150.46, 143.70, 139.67, 136.06, 132.24, 130.64, 130.15, 129.73, 129.56, 129.44, 129.01, 126.73, 126.50, 126.37, 126.03, 125.25, 125.06, 124.41, 121.81, 120.42, 117.35, 116.05, 115.12, 113.67, 107.78, 100.64, 85.60, 85.54, 70.99, 70.77, 70.72, 70.61, 70.44, 70.36, 70.05, 69.75, 58.75, 57.13, 53.44, 48.79, 46.12, 42.84, 39.21, 36.48, 36.33, 36.10, 35.34, 26.49, 26.44, 26.08, 25.75, 22.42, 15.67, 14.73, 14.57. LC-MS retention time (Method A): 4.44 min, purity: 99%. LRMS (m/z): [M + H]+ calcd. for C65H81N8O12S3, 1261.51; found, 1261.20. HRMS (m/z): [M + H]+ calcd. for C65H81N8O12S3, 1261.5136; found, 1261.5160.
(5R,7R,8S)-2,11-Bis((ethylthio)methyl)-7-hydroxy-N-((S)-19-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidine-1-carbonyl)-20,20-dimethyl-17-oxo-3,6,9,12,15-pentaoxa-18-azahenicosyl)-8-methyl-15-oxo-5,6,7,8,14,15-hexahydro-13H-16-oxa-4b,8a,14-triaza-5,8-methanodibenzo[b,h]cycloocta[jkl]cyclopenta[e]-as-indacene-7-carboxamide (CEP1347-VHL-08)
CEP1347-VHL-08 was synthesized using the same reaction procedure as CEP1347-VHL-02. Starting materials CEP1347-acid (10 mg, 0.017 mmol), (2S,4R)-1-((S)-20-amino-2-(tert-butyl)-4-oxo-6,9,12,15,18-pentaoxa-3-azaicosanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide dihydrochloride 8 (16 mg, 0.020), HATU (8 mg, 0.020 mmol) and DIPEA (14 μL, 0.083 mmol) were used. After the consumption of CEP1347-acid standard workup was performed and purified by silica gel column chromatography (DCM/MeOH = 9:1) to afford CEP1347-VHL-08 (12 mg, 55%, white solid). 1H NMR (500 MHz, DMSO) δ 9.06 (d, J = 1.8 Hz, 1H), 8.90 (s, 1H), 8.56 (s, 1H), 8.36 (d, J = 7.7 Hz, 1H), 8.27 (t, J = 5.9 Hz, 1H), 7.90–7.87 (m, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.41–7.24 (m, 7H), 6.95 (dd, J = 7.4, 4.8 Hz, 1H), 6.34 (s, 1H), 5.04 (s, 1H), 5.00–4.75 (m, 3H), 4.47 (d, J = 9.5 Hz, 1H), 4.37 (t, J = 8.3 Hz, 1H), 4.20 (s, 1H), 3.93–3.81 (m, 5H), 3.57–3.39 (m, 19H), 3.37–3.12 (m, 8H), 2.42–2.36 (m, 6H), 2.05 (s, 3H), 2.01–1.91 (m, 2H), 1.70 (ddd, J = 13.1, 8.7, 4.5 Hz, 1H), 1.29 (d, J = 7.0 Hz, 3H), 1.19–1.14 (m, 6H), 0.86 (s, 9H). 13C NMR (126 MHz, DMSO) δ 172.69, 172.26, 170.93, 169.47, 168.98, 148.22, 145.19, 139.44, 136.31, 133.35, 131.58, 131.16, 130.15, 129.91, 129.29, 127.07, 126.79, 126.38, 126.06, 124.89, 122.99, 121.51, 119.86, 115.90, 115.41, 114.79, 100.62, 85.22, 70.90, 70.30, 70.25, 70.06, 70.02, 69.24, 69.19, 59.03, 56.98, 56.16, 48.21, 46.03, 42.46, 38.96, 38.20, 36.20, 36.00, 35.32, 26.69, 25.13, 22.93, 22.84, 16.45, 14.93. LC-MS retention time (Method A): 4.43 min, purity: 97%. LRMS (m/z): [M + H]+ calcd. for C67H85N8O13S3, 1305.54; found, 1305.20. HRMS (m/z): [M + H]+ calcd. for C67H85N8O13S3, 1305.5398; found, 1305.5416.
Synthesis of (5R,7R,8S)-N-(2-(2-(2-(2-((2-(2,6-Dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)amino)ethoxy)ethoxy)ethoxy)ethyl)-2,11-bis((ethylthio)methyl)-7-hydroxy-8-methyl-15-oxo-5,6,7,8,14,15-hexahydro-13H-16-oxa-4b,8a,14-triaza-5,8-methanodibenzo[b,h]cycloocta[jkl]cyclopenta[e]-as-indacene-7-carboxamide (CEP1347-CRBN-01)
To the solution of CEP1347-acid (10 mg, 0.017 mmol) and 4-((2-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)ethyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione 9 (8.3 mg, 0.017 mmol) in DMF (1 mL), HATU (8 mg, 0.02 mmol) and DIPEA (0.051 mmol, 9 μL) were added. The reaction mixture was than stirred for overnight and the crude material was purified by a preparatory HPLC with a XBridge BEH C18 OBD Prep Column, 130 Å, 5 μm, 30 mm × 150 mm reversed-phase column as the stationary phase. Water (buffered with 0.05% trifluoroacetic acid) and MeCN (buffered with 0.05% trifluoroacetic acid) were used as the mobile phase and HPLC conditions: UV collection 254 nm, flow rate 30 mL/min, 30% MeCN as linear gradient for 5 min and 30% → 65% MeCN for 5 to 35 min (HPLC retention time: 27.00 min). The HPLC fractions were combined and lyophilized to yield CEP1347-CRBN-01 (7 mg, 40%, yellow solid). 1H NMR (400 MHz, dmso) δ 11.06 (s, 1H), 9.11 (s, 1H), 8.66–8.57 (m, 1H), 8.33–8.27 (m, 1H), 8.01–7.90 (m, 2H), 7.78 (d, J = 8.5 Hz, 1H), 7.57–7.49 (m, 1H), 7.44–7.38 (m, 2H), 7.13–7.05 (m, 1H), 7.04–6.96 (m, 2H), 6.56 (s, 1H), 6.38 (s, 1H), 5.07–4.88 (m, 4H), 3.98–3.90 (m, 4H), 3.73–3.61 (m, 12H), 3.50–3.18 (m, 5H), 2.90–2.84 (m, 1H), 2.58–2.53 (m, 1H), 2.47–2.39 (m, 5H), 2.10 (s, 3H), 2.02–1.97 (m, 2H), 1.23–1.19 (m, 6H). 13C NMR (101 MHz, dmso) δ 173.28, 172.70, 172.26, 170.50, 169.37, 167.72, 146.81, 139.39, 136.64, 136.26, 133.34, 132.47, 131.15, 129.92, 129.28, 127.06, 126.36, 126.02, 124.85, 124.59, 122.94, 121.45, 119.77, 117.83, 115.86, 115.34, 114.75, 111.11, 109.65, 100.61, 85.75, 85.17, 70.35, 70.28, 70.26, 69.98, 69.31, 69.12, 48.99, 46.02, 44.09, 42.43, 42.11, 38.94, 35.97, 35.30, 31.40, 25.12, 22.80, 22.57, 14.90, 14.88. LC-MS retention time (Method D): 6.33 min, purity: 99%. LRMS (m/z): [M + Na]+ calcd. for C53H58N7O11S2, 1054.3; found, 1054.3. LRMS (m/z): [M + H]+ calcd. for C53H58N7O11S2, 1032.3636; found, 1032.3630.
Synthesis of (5R,7R,8S)-N-(2-(2-(2-(2-(((3S,5S)-1-((S)-2-Cyclohexyl-2-((S)-2-(methylamino)propanamido)acetyl)-5-(((R)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamoyl)pyrrolidin-3-yl)amino)-2-oxoethoxy)ethoxy)ethoxy)ethyl)-2,11-Bis((ethylthio)methyl)-7-hydroxy-8-methyl-15-oxo-5,6,7,8,14,15-hexahydro-13H-16-oxa-4b,8a,14-triaza-5,8-methanodibenzo[b,h]cycloocta[jkl]cyclopenta[e]-as-indacene-7-carboxamide (CEP1347-IAP-01)
To the solution of CEP1347-acid (10.8 mg, 0.018 mmol) and tert-butyl ((S)-1-(((S)-2-((2S,4S)-4-(2-(2-(2-(2-aminoethoxy)ethoxy)ethoxy)acetamido)-2-(((R)-1,2,3,4-tetrahydronaphthalen-1-yl)carbamoyl)pyrrolidin-1-yl)-1-cyclohexyl-2-oxoethyl)amino)-1-oxopropan-2-yl)(methyl)carbamate 10 (14 mg, 0.018 mmol) in DMF (1.5 mL), HATU (7.51 mg, 0.019 mmol) and DIPEA (6 μL, 0.036 mmol) were added at room temperature. The reaction mixture stirred for 1 h and water was added to precipitate the product. The precipitate was collected with the aid of water and was used for the next step without further purification. The dried precipitate was dissolved in CH2Cl2 (5 mL) and then degassed by bubbling with argon. TFA (1 mL) was added at 0 °C under argon bubbling. The reaction mixture was stirred for about 45 min (monitored by LC-MS) under argon at 0 °C. The solvent and TFA were removed using rotary evaporator and purified by a preparatory HPLC with a XBridge BEH C18 OBD Prep Column, 130 Å, 5 μm, 30 mm × 150 mm reversed-phase column as the stationary phase. Water (buffered with 0.05% trifluoroacetic acid) and MeCN (buffered with 0.05% trifluoroacetic acid) were used as the mobile phase and HPLC conditions: UV collection 254 nm, flow rate 30 mL/min, 30% MeCN as linear gradient for 5 min and 30% → 60% MeCN for 5 to 40 min (HPLC retention time: 26.60 min). The HPLC fractions were combined and lyophilized to yield CEP1347-IAP-01 (4 mg, 18%, over 2 steps, white solid). 1H NMR (400 MHz, dmso) δ 9.11 (s, 1H), 8.73 (d, J = 7.9 Hz, 1H), 8.59 (s, 1H), 8.43 (d, J = 8.6 Hz, 1H), 8.36–8.29 (m, 2H), 7.96–7.91 (m, 2H), 7.78 (d, J = 8.4 Hz, 1H), 7.46–7.39 (m, 2H), 7.30–7.26 (m, 2H), 7.15–7.00 (m, 5H), 6.38 (s, 1H), 5.04–4.83 (m, 3H), 4.45–4.26 (m, 3H), 3.99–3.80 (m, 8H), 3.68–3.51 (m, 10H), 3.46–3.30 (m, 2H), 3.27–3.17 (m, 1H), 2.73–2.63 (m, 3H), 2.48–2.42 (m, 7H), 2.38–2.30 (m, 1H), 2.09 (s, 3H), 2.04–1.94 (m, 1H), 1.89–1.53 (m, 11H), 1.29 (d, J = 6.9 Hz, 3H), 1.24–1.19 (m, 7H), 1.13–0.88 (m, 3H). 13C NMR (101 MHz, dmso) δ 172.65, 172.23, 171.38, 169.77, 169.59, 169.03, 139.41, 137.71, 137.41, 136.27, 133.33, 131.14, 129.89, 129.29, 129.00, 128.78, 127.12, 126.06, 124.86, 124.61, 122.96, 121.49, 119.83, 115.87, 114.77, 109.41, 100.58, 85.75, 85.19, 70.89, 70.42, 70.32, 70.12, 69.93, 69.16, 58.97, 56.24, 55.90, 53.33, 47.79, 47.16, 46.00, 42.41, 38.91, 35.98, 35.31, 34.65, 31.18, 30.19, 29.16, 28.88, 28.61, 26.19, 25.95, 25.12, 25.10, 22.78, 20.64, 16.11, 14.91, 14.90. LC-MS retention time: 2.63 min (Method B), 4.22 min (Method C), and purity: 98%. LRMS (m/z): [M + H]+ calcd. for C67H86N9O11S2, 1256.59; found, 1256.40. HRMS (m/z): [M + H]+ calcd. for C67H86N9O11S2, 1256.5888; found, 1256.5887.
Cell Lines and Culture Conditions
The MDA-MB-468 cell line was purchased from German Collection of Microorganisms and Cell Cultures (DSMZ; catalog no. ACC 738). The A375, HCC1806, and MCF-7 cell lines were a kind gift from CRUK Manchester Institute (UK). MCF-7 and MDA-MB-468 cells were cultured in high-glucose Dulbecco’s modified Eagle medium (DMEM; GIBCO, catalog no. 11995–065) supplemented with 10% fetal bovine serum (FBS; Sigma, catalog no. F7524) and 1% antibiotics: penicillin (100 U/ml) and streptomycin (100 μg/mL; Sigma-Aldrich, catalog no. P4333). A375 and HCC1806 cells were cultured in Roswell Park Memorial Institute 1640 (RPMI-1640) medium (GIBCO, catalog no. 21875-034) supplemented with 10% FBS and 1% antibiotics: penicillin (100 U/ml) and streptomycin (100 μg/mL). All cell lines were grown at 37 °C in a humidified 5% CO2/95% air atmosphere. The number of living cells was calculated by Trypan Blue staining using a Countess 3 Automated Cell Counter (Invitrogen). Cell lines were regularly tested for mycoplasma contamination using a PCR-based method.41
Generation of DOX-Inducible Cell Lines
A375 and HCC1806 were used to generate cell lines with the DOX-inducible expression of MLK1–4 as described previously.3,42 Briefly, MLK1–4 plasmids (cloned into pLenti/TO/V5-DEST vector) and pLenti3.3/TR were transfected into HEK293T cells to generate the lentiviral stock. Parental A375 and HCC1806 cells were transduced with lentiviral stocks and transduced cells were subjected to antibiotic selection (Blasticidin [Invitrogen] and Geneticin [Gibco]). Doxycycline (Invitrogen) was used to induce MLK expression.
Protein Isolation and Immunoblotting Analysis
A375 and HCC1806 cells with DOX-inducible expression of MLK1–4 were seeded on a six-well plate at a density of 100 000 cells/well. The next day, MLK overexpression was induced by 1 μg/mL DOX. 24 h after DOX induction, the cells were treated with PROTACs for the indicated times and concentrations. For rescue experiments, cells were pretreated with 1 μM of the proteasome inhibitor carfilzomib for 1 h before CEP1347-VHL-02 PROTAC treatment.
To assess degradation of endogenous MLK in breast cancer cells, MCF-7 and MDA-MB-468 cells were seeded on a six-well plate at a density of 100 000 cells/well. The next day, the cells were treated with PROTACs for the indicated times and concentrations. For rescue experiments, cells were pretreated with 1 μM of the proteasome inhibitor carfilzomib for 1 h before CEP1347-VHL-02 PROTAC treatment.
After the experimental procedures, cells were washed with phosphate-buffered saline (PBS) and scraped on ice in RIPA buffer (ThermoScientific, catalog no. 89901) supplemented with 1% protease inhibitor (Merck, catalog no. 11836170001). Proteins were separated by electrophoresis in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and then transferred to nitrocellulose membranes by wet electrophoretic transfer. Membranes were blocked by incubation with 5% nonfat milk in TBST (0.1% Tween-20) for 1 h at room temperature and then incubated overnight at 4 °C with primary antibodies. The next day, the membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies. Signals were visualized by enhanced chemiluminescence detection (Clarity Max Western ECL Substrate or Clarity Western ECL Substrate, Bio-Rad). Membranes were developed on an Amersham ImageQuant 800 (Cytiva). As a loading control, membranes were incubated with a monoclonal anti-GAPDH or anti-β-actin antibody. Primary antibodies from Santa Cruz Biotechnology: MLK3 (#SC-166639), MLK-2 (#SC-393675), ubiquitin (#SC-8017). Primary antibodies from Cell Signaling Technology: actin (#3700S), MLK1 (#5029), ERK1/2 (#4696S), p-ERK (#4377S), JNK (#9252S), p-JNK (#4671S), GAPDH (#2118). Primary antibody for MLK4 (#A302–610A) was purchased from Bethyl Laboratories
Cell Cycle Analysis
MDA-MB-468 and MCF-7 cells were seeded on a six-well plate at a density of 50 000 cells/well and subsequently treated with 1 μM CEP1347-VHL-02 PROTAC or DMSO as a control. After 48 h of treatment, cells were trypsinized, washed with PBS, fixed in ice-cold methanol, and stored at −20 °C. The next day, cells were stained with FxCycle PI/RNase Staining Solution (Invitrogen, catalog no. F10797). The stained cells were analyzed by flow cytometry (NovoCyte, Agilent). Experiments were repeated two times. The quantitative analysis was performed using ImageJ software.
Annexin V Apoptosis Assay
Apoptotic cells were detected using the Alexa Fluor488 Annexin V/Dead cell Apoptosis Kit (Invitrogen, catalog no. 3 V13245). MDA-MB-468 and MCF-7 cells were seeded on a six-well plate at a density of 50 000 cells/well and treated with 1 μM CEP1347-VHL-02 PROTAC or DMSO as a control. After 48 h of treatment, the cell suspension together with the supernatant was washed and stained with Annexin V for 30 min at room temperature in the dark. Propidium iodide was added 5 min before the analysis. The stained cells were analyzed by flow cytometry (NovoCyte, Agilent). The experiments were repeated at least three times.
Colony Formation Assay
MDA-MB-468 and MCF-7 cells were seeded at a density of 5000 cells/well in a 6 cm plate in triplicate and treated with 1 μM CEP1347-VHL-02 PROTAC or DMSO as a control. The cell medium was exchanged every 5 days. Colonies were visualized after 14 days of culture after staining with crystal violet. The experiments were repeated at least three times.
3D Cell Culture
4000 cells were seeded on the top of hydrogels (LifeGel for 3D Celll Culture, Real Research) and were left to grow with 1 μM CEP1347-VHL-02 PROTAC or DMSO for 10 days. As an alternative, cells were grown in low-attachment surface Nunclon Sphera plates (Thermo Fisher Scientific) in medium with 1 μM CEP1347-VHL-02 PROTAC or DMSO as a control for 10 days.
Transwell Migration Assay
Cell migration was evaluated using Boyden chamber inserts (8-μm pore size; Corning, catalog no. 353182) in a 12-well format. A total of 200 000 cells, resuspended in serum-free DMEM with the addition of 1 μM CEP1347-VHL-02 PROTAC or DMSO as a control, were seeded inside each insert and incubated for 24 h at 37 °C in a humidified 5% CO2 atmosphere. Outside the insert, DMEM supplemented with 20% FBS was used as the chemoattractant. Cells that migrated through the membrane of the chamber were fixed and stained with crystal violet. The quantitative analysis was performed using ImageJ software. The experiments were repeated at least three times.
Mass Spectrometry Analysis
Experimental Settings
For the MS analysis, HCC1806_MLK3 cells were seeded on a six-well plate at a density of 150 000 cells/well. The next day, cells were treated with DOX for 24 h, followed by treatment with 0.5 μM CEP1347-VHL-02 PROTAC or DMSO as a control. After 24 h, cells were lysed in lysis buffer (Cell Signaling Technology, catalog no. 9803S) and subjected to sample preparation and MS analysis.
MCF-7 cells were seeded on a six-well plate at a density of 100 000 cells/well. The next day, cells were treated with 1 μM CEP1347-VHL-02 PROTAC or DMSO as a control. After 24 h, cells were lysed in RIPA buffer (Sigma-Aldrich, catalog no. R0278) and subjected to sample preparation and MS analysis.
MDA-MB-468 cells were seeded on a six-well plate at a density of 150 000 cells/well. The next day, cells were treated with 1 μM CEP1347-VHL-02 PROTAC or DMSO as a control. After 24 h, cells were lysed in urea lysis buffer (8 M urea in 100 mM Tris, pH 8.5) and after 6x dilution in dilution buffer (100 mM HEPES, pH 8.0) subjected to sample preparation and MS analysis.
Sample Preparation
Samples were subjected to chloroform/methanol precipitation, and the resulting protein pellets were washed twice with methanol. The protein pellets were air-dried for ∼10 min and resuspended in 100 mM HEPES (pH 8) by vigorous vortexing and sonication. Proteins were then reduced and alkylated with 10 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP)/40 mM chloroacetic acid (CAA) and digested with trypsin in a 1:50 (w/w) enzyme-to-protein ratio at 37 °C overnight. Digestion was terminated by the addition of trifluoroacetic acid (TFA) to a 1% final concentration. The resulting peptides were labeled using an on-column TMT labeling protocol.43 TMT-labeled samples were compiled into a single TMT sample and concentrated using a SpeedVac concentrator. Peptides in the compiled sample were separated into eight fractions by off-line basic reversed-phase using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo Fisher Scientific).
Liquid Chromatography–Mass Spectrometry Measurement
Prior to the liquid chromatography (LC)-MS measurement, the peptide fractions were resuspended in 0.1% TFA and 2% acetonitrile in water. Chromatographic separation was performed on an Easy-Spray Acclaim PepMap column (15 cm length × 75 μm inner diameter or 50 cm length × 75 μm inner diameter; Thermo Fisher Scientific) at 35 or 55 °C by applying 105–180 min acetonitrile gradients in 0.1% aqueous formic acid at a flow rate of 300 nL/min. An UltiMate 3000 nano-LC system was coupled to a Q Exactive HF-X mass spectrometer via an easy-spray source (all Thermo Fisher Scientific). The Q Exactive HF-X was operated in TMT mode with survey scans acquired at a resolution of 60 000 at m/z 200. Up to 15 or 18 of the most abundant isotope patterns with charges 2–5 from the survey scan were selected with an isolation window of 0.7 m/z and fragmented by higher-energy collision dissociation with normalized collision energies of 32, while the dynamic exclusion was set to 35 s. The maximum ion injection times for the survey scan and dual MS (MS/MS) scans (acquired with a resolution of 45 000 at m/z 200) were 50 and 96 or 150 ms, respectively. The ion target value for MS was set to 3e6 and for MS/MS was set to 1e5, and the minimum AGC target was set to 1e3.
The data were processed with MaxQuant v. 1.6.17.044 and the peptides were identified from the MS/MS spectra searched against Uniprot human reference proteome (UP000005640) using the built-in Andromeda search engine. Reporter ion MS2-based quantification was applied with reporter mass tolerance = 0.003 Da and min reporter PIF = 0.75. Cysteine carbamidomethylation was set as a fixed modification and methionine oxidation, glutamine/asparagine deamination, as well as protein N-terminal acetylation, were set as variable modifications. For in silico digests of the reference proteome, cleavages of arginine or lysine followed by any amino acid were allowed (trypsin/P), and up to two missed cleavages were allowed. The false discovery rate (FDR) was set to 0.01 for peptides, proteins, and sites. A match between runs was enabled. Other parameters were used as preset in the software. Unique and razor peptides were used for quantification enabling protein grouping (razor peptides are the peptides uniquely assigned to protein groups and not to individual proteins). Reporter intensity corrected values for protein groups were loaded into Perseus v. 1.6.10.0.45 Standard filtering steps were applied to clean up each data set: reverse (matched to decoy database), only identified by site, and potential contaminant (from a list of commonly occurring contaminants included in MaxQuant) protein groups were removed. Reporter intensity corrected values were Log2 transformed. Protein groups with all values and at least 2 unique+razor peptides were kept. Reporter intensity values were then normalized by median subtraction within TMT channels. Student’s t tests were performed on these values for groups of samples constituting a given data set. The abundance changes thresholds of Log2 fold change (Log2 FC) ≥ |1.0| and the significance threshold of −Log10 p-value ≥ 2.0 were applied to deliver protein groups with levels deemed reproducibly decreased or increased between the sample groups investigated. This data set has been deposited to the ProteomeXchange Consortium46 via the PRIDE partner repository47 with the data set identifier PXD050260.
RNA Isolation, cDNA Synthesis, and RT-qPCR
Total RNA was isolated from MCF-7 and MDA-MB-468 cells 24 h after PROTAC treatment using the GeneMATRIX Universal RNA Purification Kit (EURX, catalog no. E3598) according to the manufacturer’s instructions. The concentration and purity of RNA samples were determined using a DeNovix DS-11 Spectrophotometer. cDNA was synthesized from 1 μg of total RNA using the HighCapacity cDNA Reverse Transcription Kit (Applied Biosystems, catalog no. 4368814) according to the manufacturer’s protocol. The level of MLK3 gene expression was quantified by RT-qPCR. Each reaction mixture contained 1 μL of 10× diluted cDNA, 5 μL of iTaq Universal SYBR Green Supermix(2x) (Bio-Rad, catalog no. 1725121), and 0.5 μM specific oligonucleotide MLK3 or GAPDH primers in a total volume of 10 μL. Reactions were performed in triplicate on the LightCycler 480 II (Roche) under the following conditions: 5 s at 95 °C, followed by 45 amplification cycles (95 °C for 10 s, 60 °C for 10 s, 72 °C for 10 s). MLK3 expression was normalized to the expression of GAPDH. Sequences of the primers were the following: MLK3 (forward, 5′-GGCGAGCGTATCAGCATG-3′; reverse, 5′-GGGAAAGGTGGGCGAATC-3′), GAPDH (forward, 5′-CCCTTCATTGACCTCAACTAC-3′; reverse, 5′-CCTGCTTCACCACCTTCTTGA-3′).
SiRNA Transient Transfection and Colony Formation Assay
MCF-7 and MDA-MB-468 cells were seeded on a six-well plate at a density of 10 000 cells/well. The next day, cells were transfected with specific siRNAs that targeted MLK3 gene (ON-TARGETplus Human MAP3K11 (4296) SMARTpool siRNA (Dharmacon, catalog no. J-003577)) or with universal negative nontargeting control siRNA (Dharmacon, catalog no. D-001210–05–20) using JetPRIME Transfection Reagent (Polypus, catalog no. 101000046) according to the manufacturer’s instructions. Cells were stained with crystal violet solution 7 days after transfection. The efficiency of MLK3 silencing was validated by immunoblotting.
Statistical Analysis
All experiments were performed at least three times. Statistical analyses were performed using GraphPad Prism 7 software. Statistical significance was determined using Student’s t-test (comparison between two groups) or one-way ANOVA (comparison between more than two groups). The data are expressed as the mean ± SD. Values of p < 0.05 were considered statistically significant.
Acknowledgments
This research was funded by the National Science Centre, Poland (2021/42/E/NZ5/00227 grant to AAM and 2021/41/N/NZ3/02997 to DM). DM was supported by grants from the Polish National Agency for Academic Exchange (PPN/WAL/2020/1/00018) and Fulbright Foundation. This research was also supported by the National Cancer Institute, grant number ZIA BC 011691 to JB. Additionally, this research was supported by the NIH Intramural Research Program through an NCI FLEX award to JB. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. PT-A is supported by NIH grant R03 DE033064, a Cancer Center Support Grant to Fox Chase Cancer Center (P30 CA006927), and Temple University Start up package. For the purpose of Open Access, the author has applied a CC-BY public copyright license to any Author Accepted Manuscript (AAM) version arising from this submission. The graphs were created using GraphPad Prism software. Some pictures were created using BioRender.
Glossary
Abbreviations
- ANOVA
analysis of variance
- DC50
half-maximal degradation concentration
- Dmax
maximal levels of degradation
- DMEM
Dulbecco’s modified Eagle medium
- DMSO
dimethyl sulfoxide
- DOX
doxycycline
- ER
estrogen receptor; ERK, extracellular signal-regulated kinase
- FBS
fetal bovine serum
- FRA-1
FOS-related antigen-1
- HER2
human epidermal growth factor receptor 2
- IAP
inhibitor of apoptosis protein
- IC50
half-maximal inhibitory concentration
- JNK
c-Jun N-terminal kinase
- LC
liquid chromatography
- MAP3K
mitogen-activated protein kinase kinase kinase
- MEK
mitogen-activated protein kinase kinase
- MLK
mixed-lineage kinase
- MS
mass spectrometry
- NF-κB
nuclear factor-κB
- PAK1
p21-activated kinase 1
- PBS
phosphate-buffered saline
- POI
protein of interest
- PROTAC
proteolysis targeting chimera
- RT-qPCR
real-time quantitative polymerase chain reaction
- RPMI
Roswell Park Memorial Institute
- SD
standard deviation
- siRNA
small interfering RNA
- t1/2
half-life of a reaction
- TFA
trifluoroacetic acid
- TNBC
triple-negative breast cancer
- TPD
targeted protein degradation
- VHL
von Hippel–Lindau
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c00577.
Evaluation of IAP- and CRBN-PROTACs (Figure S1); specificity of endogenous MLK3 degradation by CEP1347-VHL-02 PROTAC in MCF-7 and MDA-MB-468 cell lines (Figure S2); evaluation of different PROTACs by Western Blot analysis in MCF-7 and MDA-MB-468 cell lines (Figure S3); shotgun proteomic analysis of total cell protein extracts from MCF-7 and MDA-MB-468 cell lines (Figure S4); analysis of activation of JNK and ERK pathways in MCF-7 and MDA-MB-468 cell lines (Figure S5); knock-down of MLK3 decreases the colony formation potential of MDA-MB-468, but not the MCF-7 cell line (Figure S6); degradation of MLK3 by CEP1347-VHL-02 PROTAC treatment impaired the formation of MDA-MB-468 spheroids in 3D cell culture (Figure S7); NMR spectra and HPLC traces of newly synthesized PROTACs (Figure S8) (PDF)
Shotgun proteomic analysis of protein levels in the total cell extracts from HCC1806_MLK3, MCF-7 and MDA-MB-468 (XLSX)
SMILES data of compounds (CSV)
Author Contributions
# K.K. and D.M. contributed equally. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Gallo K. A.; Johnson G. L. Mixed-Lineage Kinase Control of JNK and P38 MAPK Pathways. Nat. Rev. Mol. Cell Biol. 2002, 3 (9), 663–672. 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- Chadee D. N.; Kyriakis J. M. MLK3 is Required for Mitogen Activation of B-Raf, ERK and Cell Proliferation. Nat. Cell Biol. 2004, 6 (8), 770–776. 10.1038/ncb1152. [DOI] [PubMed] [Google Scholar]
- Marusiak A. A.; Edwards Z. C.; Hugo W.; Trotter E. W.; Girotti M. R.; Stephenson N. L.; Kong X.; Gartside M. G.; Fawdar S.; Hudson A.; et al. Mixed Lineage Kinases Activate MEK Independently of RAF to Mediate Resistance to RAF Inhibitors. Nat. Commun. 2014, 5 (1), 3901. 10.1038/ncomms4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadee D. N.; Xu D.; Hung G.; Andalibi A.; Lim D. J.; Luo Z.; Gutmann D. H.; Kyriakis J. M. Mixed-Lineage Kinase 3 Regulates B-Raf through Maintenance of the B-Raf/Raf-1 Complex and Inhibition by the NF2 Tumor Suppressor Protein. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (12), 4463–4468. 10.1073/pnas.0510651103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandraiah K.; Thylur Puttalingaiah R. The Role of Mixed Lineage Kinase 3 (MLK3) in Cancers. Pharmacol. Ther. 2022, 238, 108269. 10.1016/j.pharmthera.2022.108269. [DOI] [PubMed] [Google Scholar]
- Chen J.; Miller E. M.; Gallo K. A. MLK3 Is Critical for Breast Cancer Cell Migration and Promotes a Malignant Phenotype in Mammary Epithelial Cells. Oncogene 2010, 29 (31), 4399–4411. 10.1038/onc.2010.198. [DOI] [PubMed] [Google Scholar]
- Chen J.; Gallo K. A. MLK3 Regulates Paxillin Phosphorylation in Chemokine-Mediated Breast Cancer Cell Migration and Invasion to Drive Metastasis. Cancer Res. 2012, 72 (16), 4130–4140. 10.1158/0008-5472.CAN-12-0655. [DOI] [PubMed] [Google Scholar]
- Das S.; Nair R. S.; Mishra R.; Sondarva G.; Viswakarma N.; Abdelkarim H.; Gaponenko V.; Rana B.; Rana A. Mixed Lineage Kinase 3 Promotes Breast Tumorigenesis via Phosphorylation and Activation of P21-Activated Kinase 1. Oncogene 2019, 38 (19), 3569–3584. 10.1038/s41388-019-0690-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair R. S.; Kumar S.; Das S.; Singh S. K.; Srivastava P.; Sondarva G.; Rao A.; Sinha S. C.; Xiong R.; Bloem L.; et al. TrkA Expression Directs the Anti-Neoplastic Activity of MLK3 Inhibitors in Triple-Negative Breast Cancer. Oncogene 2023, 42 (14), 1132–1143. 10.1038/s41388-023-02633-6. [DOI] [PubMed] [Google Scholar]
- Rattanasinchai C.; Llewellyn B. J.; Conrad S. E.; Gallo K. A. MLK3 Regulates FRA-1 and MMPs to Drive Invasion and Transendothelial Migration in Triple-Negative Breast Cancer Cells. Oncogenesis 2017, 6 (6), e345–e345 10.1038/oncsis.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangasamy V.; Mishra R.; Mehrotra S.; Sondarva G.; Ray R. S.; Rao A.; Chatterjee M.; Rana B.; Rana A. Estrogen Suppresses MLK3-Mediated Apoptosis Sensitivity in ER+ Breast Cancer Cells. Cancer Res. 2010, 70 (4), 1731–1740. 10.1158/0008-5472.CAN-09-3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S.; Sondarva G.; Viswakarma N.; Nair R. S.; Osipo C.; Tzivion G.; Rana B.; Rana A. Human Epidermal Growth Factor Receptor 2 (HER2) Impedes MLK3 Kinase Activity to Support Breast Cancer Cell Survival. J. Biol. Chem. 2015, 290 (35), 21705–21712. 10.1074/jbc.M115.655563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson-Fields K. I.; Sandquist J. C.; Rossol-Allison J.; Blat I. C.; Wennerberg K.; Burridge K.; Means A. R. MLK3 Limits Activated Gαq Signaling to Rho by Binding to p63RhoGEF. Mol. Cell 2008, 32 (1), 43–56. 10.1016/j.molcel.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroyer A. L.; Stimes N. W.; Abi Saab W. F.; Chadee D. N. MLK3 Phosphorylation by ERK1/2 Is Required for Oxidative Stress-Induced Invasion of Colorectal Cancer Cells. Oncogene 2018, 37 (8), 1031–1040. 10.1038/onc.2017.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekaran P.; Hwang Y. S.; Lee G.-H.; Park J.; Kim J. G.; La Y. K.; Park N. Y.; Kothandaraman R.; Yim M. S.; Choi J.; et al. Degradation of Polo-like Kinase 1 by the Novel Poly-Arginine N-Degron Pathway PROTAC Regulates Tumor Growth in Nonsmall Cell Lung Cancer. J. Med. Chem. 2024, 67, 3307–3320. 10.1021/acs.jmedchem.3c01493. [DOI] [PubMed] [Google Scholar]
- Mao W.; Vandecan N. M.; Bingham C. R.; Tsang P. K.; Ulintz P.; Sexton R.; Bochar D. A.; Merajver S. D.; Soellner M. B. Selective and Potent PROTAC Degraders of C-Src Kinase. ACS Chem. Biol. 2024, 19 (1), 110–116. 10.1021/acschembio.3c00548. [DOI] [PubMed] [Google Scholar]
- Zhou C.; Fan Z.; Gu Y.; Ge Z.; Tao Z.; Cui R.; Li Y.; Zhou G.; Huo R.; Gao M.; et al. Design Synthesis, and Biological Evaluation of Potent and Selective PROTAC Degraders of Oncogenic KRAS G12D. J. Med. Chem. 2024, 67 (2), 1147–1167. 10.1021/acs.jmedchem.3c01622. [DOI] [PubMed] [Google Scholar]
- Xiao B.; Shi Z.; Liu J.; Huang Q.; Shu K.; Liu F.; Zhi C.; Zhang D.; Wu L.; Yang S.; et al. Design Synthesis, and Evaluation of VHL-Based EZH2 Degraders for Breast Cancer. Bioorg. Chem. 2024, 143, 107078. 10.1016/j.bioorg.2023.107078. [DOI] [PubMed] [Google Scholar]
- Békés M.; Langley D. R.; Crews C. M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discovery 2022, 21 (3), 181–200. 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigglestone C. E.; Yeung K.-S. Degradation of Protein Kinases: Ternary Complex, Cooperativity, and Selectivity. ACS Med. Chem. Lett. 2021, 12 (11), 1629–1632. 10.1021/acsmedchemlett.1c00543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Ayuso P.; Brognard J. Degraders: The Ultimate Weapon Against Amplified Driver Kinases in Cancer. Mol. Pharmacol. 2022, 101 (4), 191–200. 10.1124/molpharm.121.000306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno R.; Recio J.; Barber S.; Gil C.; Martinez A. The Emerging Role of Mixed Lineage Kinase 3 (MLK3) and Its Potential as a Target for Neurodegenerative Diseases Therapies. Eur. J. Med. Chem. 2023, 257, 115511. 10.1016/j.ejmech.2023.115511. [DOI] [PubMed] [Google Scholar]
- Maroney A. C.; Finn J. P.; Connors T. J.; Durkin J. T.; Angeles T.; Gessner G.; Xu Z.; Meyer S. L.; Savage M. J.; Greene L. A.; et al. CEP-1347 (KT7515), a Semisynthetic Inhibitor of the Mixed Lineage Kinase Family. J. Biol. Chem. 2001, 276 (27), 25302–25308. 10.1074/jbc.M011601200. [DOI] [PubMed] [Google Scholar]
- Roux P. P.; Dorval G.; Boudreau M.; Angers-Loustau A.; Morris S. J.; Makkerh J.; Barker P. A. K252a and CEP1347 Are Neuroprotective Compounds That Inhibit Mixed-Lineage Kinase-3 and Induce Activation of Akt and ERK. J. Biol. Chem. 2002, 277 (51), 49473–49480. 10.1074/jbc.M203428200. [DOI] [PubMed] [Google Scholar]
- Goodfellow V. S.; Loweth C. J.; Ravula S. B.; Wiemann T.; Nguyen T.; Xu Y.; Todd D. E.; Sheppard D.; Pollack S.; Polesskaya O.; et al. Discovery Synthesis, and Characterization of an Orally Bioavailable, Brain Penetrant Inhibitor of Mixed Lineage Kinase 3. J. Med. Chem. 2013, 56 (20), 8032–8048. 10.1021/jm401094t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline E. M.; Butkovich L. M.; Bradner J. M.; Chang J.; Gelbard H.; Goodfellow V.; Caudle W. M.; Tansey M. G. The Second Generation Mixed Lineage Kinase-3 (MLK3) Inhibitor CLFB-1134 Protects against Neurotoxin-Induced Nigral Dopaminergic Neuron Loss. Exp. Neurol. 2019, 318, 157–164. 10.1016/j.expneurol.2019.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mixed Lineage Kinase Inhibitor CEP-1347 Fails to Delay Disability in Early Parkinson Disease. Neurology 2007, 69 (15), 1480–1490. 10.1212/01.wnl.0000277648.63931.c0. [DOI] [PubMed] [Google Scholar]
- Okada M.; Takeda H.; Sakaki H.; Kuramoto K.; Suzuki S.; Sanomachi T.; Togashi K.; Seino S.; Kitanaka C. Repositioning CEP-1347, a Chemical Agent Originally Developed for the Treatment of Parkinson’s Disease, as an Anti-Cancer Stem Cell Drug. Oncotarget 2017, 8 (55), 94872–94882. 10.18632/oncotarget.22033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswakarma N.; Sondarva G.; Principe D. R.; Nair R. S.; Kumar S.; Singh S. K.; Das S.; Sinha S. C.; Grippo P. J.; Grimaldo S.; et al. Mixed Lineage Kinase 3 Phosphorylates Prolyl-Isomerase PIN1 and Potentiates GLI1 Signaling in Pancreatic Cancer Development. Cancer Lett. 2021, 515, 1–13. 10.1016/j.canlet.2021.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.-T.; Dobrovolsky D.; Paulk J.; Yang G.; Weisberg E. L.; Doctor Z. M.; Buckley D. L.; Cho J.-H.; Ko E.; Jang J.; et al. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-Kinase Degrader. Cell Chem. Biol. 2018, 25 (1), 88–99.e6. 10.1016/j.chembiol.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovell H.; Testa A.; Zhou H.; Shpiro N.; Crafter C.; Ciulli A.; Alessi D. R. Design and Characterization of SGK3-PROTAC1, an Isoform Specific SGK3 Kinase PROTAC Degrader. ACS Chem. Biol. 2019, 14 (9), 2024–2034. 10.1021/acschembio.9b00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. E.; Wang S. L.; Jaime-Figueroa S.; Harbin A.; Wang J.; Hamman B. D.; Crews C. M. Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 2019, 10 (1), 131. 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Smith B. E.; Burslem G. M.; Buhimschi A. D.; Hines J.; Jaime-Figueroa S.; Wang J.; Hamman B. D.; Ishchenko A.; Crews C. M. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem. Biol. 2018, 25 (1), 78–87.e5. 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H.; Sun X.; Rao Y. PROTAC Technology: Opportunities and Challenges. ACS Med. Chem. Lett. 2020, 11 (3), 237–240. 10.1021/acsmedchemlett.9b00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Brolosy M. A.; Stainier D. Y. R. Genetic Compensation: A Phenomenon in Search of Mechanisms. PLoS Genet. 2017, 13 (7), e1006780 10.1371/journal.pgen.1006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz D. M.; Cassiani P. J.; Li L.; Billy E.; Korn J. M.; Jones M. D.; Golji J.; Ruddy D. A.; Yu K.; McAllister G.; et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discovery 2016, 6 (8), 900–913. 10.1158/2159-8290.CD-16-0178. [DOI] [PubMed] [Google Scholar]
- Riching K. M.; Mahan S.; Corona C. R.; McDougall M.; Vasta J. D.; Robers M. B.; Urh M.; Daniels D. L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13 (9), 2758–2770. 10.1021/acschembio.8b00692. [DOI] [PubMed] [Google Scholar]
- Riching K. M.; Caine E. A.; Urh M.; Daniels D. L. The Importance of Cellular Degradation Kinetics for Understanding Mechanisms in Targeted Protein Degradation. Chem. Soc. Rev. 2022, 51 (14), 6210–6221. 10.1039/D2CS00339B. [DOI] [PubMed] [Google Scholar]
- Mehlich D.; Marusiak A. A. Kinase Inhibitors for Precision Therapy of Triple-Negative Breast Cancer: Progress, Challenges, and New Perspectives on Targeting This Heterogeneous Disease. Cancer Lett. 2022, 547, 215775. 10.1016/j.canlet.2022.215775. [DOI] [PubMed] [Google Scholar]
- Bianchini G.; De Angelis C.; Licata L.; Gianni L. Treatment Landscape of Triple-Negative Breast Cancer — Expanded Options, Evolving Needs. Nat. Rev. Clin. Oncol. 2022, 19 (2), 91–113. 10.1038/s41571-021-00565-2. [DOI] [PubMed] [Google Scholar]
- Young L.; Sung J.; Stacey G.; Masters J. R. Detection of Mycoplasma in Cell Cultures. Nat. Protoc. 2010, 5 (5), 929–934. 10.1038/nprot.2010.43. [DOI] [PubMed] [Google Scholar]
- Mehlich D.; Łomiak M.; Sobiborowicz A.; Mazan A.; Dymerska D.; Szewczyk Ł. M.; Mehlich A.; Borowiec A.; Prełowska M. K.; Gorczyński A.; et al. MLK4 Regulates DNA Damage Response and Promotes Triple-Negative Breast Cancer Chemoresistance. Cell Death Dis. 2021, 12 (12), 1111. 10.1038/s41419-021-04405-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S. A.; Rhoads A.; Cocco A. R.; Peckner R.; Haber A. L.; Schweitzer L. D.; Krug K.; Mani D. R.; Clauser K. R.; Rozenblatt-Rosen O.; et al. Streamlined Protocol for Deep Proteomic Profiling of FAC-Sorted Cells and Its Application to Freshly Isolated Murine Immune Cells*. Mol. Cell. Proteomics 2019, 18 (5), 995–1009. 10.1074/mcp.RA118.001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S.; Temu T.; Cox J. The MaxQuant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat. Protoc. 2016, 11 (12), 2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- Tyanova S.; Temu T.; Sinitcyn P.; Carlson A.; Hein M. Y.; Geiger T.; Mann M.; Cox J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13 (9), 731–740. 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Deutsch E. W.; Csordas A.; Sun Z.; Jarnuczak A.; Perez-Riverol Y.; Ternent T.; Campbell D. S.; Bernal-Llinares M.; Okuda S.; Kawano S.; et al. The ProteomeXchange Consortium in 2017: Supporting the Cultural Change in Proteomics Public Data Deposition. Nucleic Acids Res. 2017, 45 (D1), D1100–D1106. 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Bai J.; Bandla C.; García-Seisdedos D.; Hewapathirana S.; Kamatchinathan S.; Kundu D. J.; Prakash A.; Frericks-Zipper A.; Eisenacher M.; et al. The PRIDE Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences. Nucleic Acids Res. 2022, 50 (D1), D543–D552. 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.