

Abstract
WIN antiviral compounds bind human rhinovirus, as well as enterovirus and parechovirus, in an internal cavity located within the viral protein capsid. Access to the buried pocket necessitates deviation from the average viral protein structure identified by crystallography. We investigated the dissociation of WIN 52084 from the pocket in human rhinovirus 14 by using an adiabatic, biased molecular dynamics simulation method. Multiple dissociation trajectories are used to characterize the pathway. WIN 52084 exits between the polypeptide chain near the ends of βC and βH in a series of steps. Small, transient packing defects in the protein are sufficient for dissociation. A number of torsion-angle transitions of the antiviral compound are involved, which suggests that flexibility in antiviral compounds is important for binding. It is interesting to note that dissociation is associated with an increase in the conformational fluctuations of residues never in direct contact with WIN 52084 over the course of dissociation. These residues are N-terminal residues in the viral proteins VP3 and VP4 and are located in the interior of the capsid near the icosahedral 5-fold axis. The observed changes in dynamics may be relevant to structural changes associated with virion uncoating and its inhibition by antiviral compounds.
Keywords: antiviral activity, antiviral binding, ligand binding effects, protein molecular dynamics, picornavirus
Picornaviruses, or small RNA-containing virus, are icosahedral (pseudo T = 3) animal viruses. Sixty copies of the protomer (Fig. 1A), comprising four viral proteins (VP1, VP2, VP3, and VP4), form the viral capsid that surrounds the single-strand RNA. Members of the picornavirus family include human rhinovirus (HRV), poliovirus, coxsackievirus, and hepatitis A virus. These viruses cause a broad spectrum of human and animal diseases (common cold, poliomyelitis, foot-and-mouth disease, and hepatitis), and considerable effort has been focused on the development of antiviral compounds against picornaviruses (1). The most effective compounds are long, hydrophobic compounds with three aromatic rings separated by an aliphatic chain. WIN 52084 is one antiviral compound in a series designed to inhibit HRV and other related viruses. The chemical structure of WIN 52084 (Fig. 1B) is methyloxazoline and phenoxy rings linked by an aliphatic chain of seven carbon atoms to a methylisoxazole group. These antiviral compounds bind to a hydrophobic pocket buried within VP1 (2–5). Binding provides entropic stabilization (6, 7) and prevents viral replication by inhibiting the uncoating process (1, 8, 9).
Fig. 1.

HRV protomer, WIN 52084, and VP1 structure. (A) Ribbon representation of the HRV14 protomer, with WIN 52084 shown as space-filling spheres (magenta). The protomer comprises VP1 (blue), VP2 (green), VP3 (red), and VP4 (yellow). The icosahedral 5-fold axis lies between the VP3 terminus and VP1 in the upper right corner and is ≈10–15° away from perpendicular to the plane of view. (B) Chemical structure of WIN 52084. (C) van der Waals space-filling representation of VP1 (blue) and the C terminus of VP3 (red). WIN 52084 is present but not visible.
VP1, VP2, and VP3 (each ≈30 kDa) fold into the same topology of an eight-stranded, antiparallel β-sandwich of two sheets “CHEF” and “BIDG.” VP4 (≈6 kDa) is largely extended in conformation and located along the internal surface of the capsid (Fig. 1 A). The hydrophobic pocket lies beneath a deep depression that circumscribes the 5-fold rotation axis of the icosahedral capsid known as the “canyon” (2). The bound WIN ligand is inaccessible to solvent and therefore is not visible in a space-filling representation (Fig. 1C). The methyloxazoline group is ≈20 Å from the nearest solvent-accessible surface. There is a region, named the pore, near the end of the CHEF sheet that seems to be the entrance to the pocket. The C terminus of VP3 (shown in red in Fig. 1C) wraps around VP1 along the “floor” of the canyon and across the top of the pore.
The molecular details of how antiviral compounds enter and exit the buried hydrophobic pocket are unknown and not readily investigated by experiment. Internal binding by a ligand to a protein clearly requires motion of the protein and possibly of the ligand, but the necessary conformational rearrangements are not obvious. We took a computational approach and used molecular dynamics (MD) to examine in atomic detail the process of WIN 52084 dissociation from HRV14. Dissociation of diatomic molecules from the internal heme pocket of myoglobin was investigated early on by computational approaches (10–12). Small diatomic molecules readily migrate in the protein matrix such that a variety of paths could be identified directly by simulations and subjected to detailed theoretical analysis. WIN 52084 is a considerably more complex ligand, and a straightforward definition of an appropriate dissociation coordinate is not possible. Accordingly, we used an external force based on a general coordinate related to the distance of WIN 52084 from the hydrophobic pocket to explore dissociation of this flexible ligand from the buried pocket of VP1. The method has been applied to probe surface binding of an antigen (13) and has provided important insights about the dissociation process. The study reported here elucidates the conformational changes and motions required for a large ligand to dissociate from the buried cavity. Effects on residues distant from the pocket are also observed. This observation is of particular interest, because these residues surround the 5-fold axis at which RNA is thought to exit the virus. Features that may aid structure-based research of antiviral compounds are described also.
Computational Methods
HRV14–WIN 52084 Simulation System. The simulation system of the HRV14 protomer (PDB ID code 2RS1) with explicit solvation and equilibrium simulations have been described (14). The system has ≈12,500 protein atoms and ≈20,000 water atoms. The MD simulations were performed by using charmm (15, 16), with the effect of the full virus capsid modeled by using icosahedral, rotational symmetry (17). Three initial coordinate sets for the dissociation simulations were taken from a 1.2-ns equilibration simulation at 400-ps intervals and are denoted as I400, I800, and I1,200.
Dissociation Simulations. The dissociation of WIN 52084 from the buried hydrophobic pocket of VP1 was accomplished on a time scale accessible to simulation by using adiabatically biased MD analogous to previous investigations of protein unfolding (18) and ligand dissociation (13). Dissociation was induced by an external biasing potential that penalizes fluctuations that decrease the distance of WIN 52084 atoms from the center of the HRV14 capsid. The dissociation coordinate, ρ, for the biasing potential is defined by the average squared distance, r2i, from the virus center for WIN atoms, i.
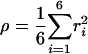 |
[1] |
Here, the summation i is over six heavy atoms from the methyloxazoline ring, the ring that is closest to the solvent-accessible surface (see Fig. 1 A). An increasing value of ρ is achieved by using the external potential, Vbias (18),
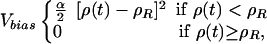 |
[2] |
where α is the force constant for the biasing potential, and ρR is the largest value of ρ over the time 0 to t –Δt. If at the simulation step ρ(t) spontaneously increases to a value >ρR, then the biasing potential is zero, no biasing force is applied, and the value of ρR is updated to ρ(t). The choice for the magnitude of α is a compromise between a large value, which would unphysically alter the natural fluctuations of the system, and a small value, whereby dissociation progresses too slowly to be practical. Other than application of the external potential, the conditions for the dissociation simulations were identical to those for the equilibrium simulations.
Multidimensional Scaling and Principal Component Analysis. Description of the conformational changes of the ligand along the dissociation pathway was facilitated by use of principal component analysis. We use the symmetric matrix A with elements equal to the pairwise RMS coordinate difference between superimposed structures along the trajectory (19):
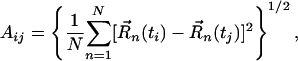 |
[3] |
where Rn(ti) is the Cartesian coordinate for atom n in step i of the trajectory, N is the number of atoms used in the analysis, and the i and j indexes span the length of the trajectory. The RMS coordinate difference is a simple measure of structure similarity and the matrix A allows a representation of the conformational sampling of a dissociation trajectory matrix in terms of a small number of dimensions. In our analysis, the summation is over all heavy atoms of WIN 52084. The RMS deviation matrix was constructed from 560 structures of WIN 52084 taken at 8-ps intervals from the biased simulation calculated with α = 0.0001 kcal/mol·Å4 (1 kcal = 4.18 kJ). Each structure was overlaid by a least-squares fit to the first structure of the trajectory.
A centered matrix C is constructed with elements Clm.
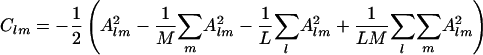 |
[4] |
C is considered equivalently to the correlation matrix of Cartesian coordinates. The matrix of eigenvectors and diagonal matrix of eigenvalues are obtained by diagonalization of C according to the procedure of principal component analysis (19).
Results
Dissociation Simulations. Multiple dissociation trajectories were calculated by using alternative initial conditions and biasing force constants, α (Table 1), to identify common features that are likely to be important for antiviral compounds to exit the pocket. Each trajectory was continued until the nonbonding interaction energy between the antiviral compound and HRV14 was zero, to give a total simulation time of 7.5 ns. The times required for the antiviral compound to dissociate increase with smaller values of α but are comparable for alternative initial configurations. A single trajectory was calculated with α = 0.0001 kcal/mol·Å4 for practical reasons associated with the relatively long dissociation time and the size of the virus-simulation system.
Table 1. Simulation times required for dissociation in trajectories calculated with the indicated α values.
| Time, ps
|
|||
|---|---|---|---|
| α, kcal/mol·Å4 | I400 | I800 | I1,200 |
| 0.1 | 72.4 | 78.2 | 77.6 |
| 0.05 | 82.9 | 84.1 | 83.0 |
| 0.01 | 99.5 | 79.6 | 91.2 |
| 0.002 | 162.7 | 153.0 | 147.3 |
| 0.0005 | 509.8 | 404.0 | 314.2 |
| 0.0001 | 5024.0 | ||
Initial coordinates were taken from equilibrium simulation at 400-ps intervals.
Smaller forces are generated by a biasing potential with lower α values (results not shown). It is expected that smaller values of α impose minimal perturbation on the dynamics of the system such that the observed fluctuations most closely approximate the actual unbinding process. As such, discussion of the dissociation pathway analysis considers the seven trajectories calculated with α ≤ 0.002 kcal/mol·Å4 but with the primary focus being on the trajectories with α = 0.0005 and 0.0001 kcal/mol·Å4. These values are slightly smaller than the smallest value found to be sufficient to dissociate a ligand from a surface binding site of an antibody (13).
The nonbonding energy between HRV14 and the antiviral compound was followed over the course of dissociation to help identify HRV14 regions that contribute to the forces involved. WIN 52084 interacts strongly with VP1 residues and the C-terminal residues of VP3. Little or no direct interaction exists with either VP2 or VP4, which is consistent with the packing arrangement within the protomer (Fig. 1).
Perturbations in HRV14 After Unbinding. Dissociation of WIN 52084 from the internal binding pocket of VP1 requires flexibility in the protein structure. Changes in HRV14 conformation were identified by comparing a measure of the positional fluctuations in the biased simulations to fluctuations in the equilibrium simulation where no external perturbation is applied. Any significant structural changes that result from WIN 52084 dissociation contribute toward larger fluctuations in the bias simulations. A large deviation in position, even if transient, should be evident in a difference in positional distributions between the two trajectories. Therefore, we consider the RMS fluctuation, ΔR:
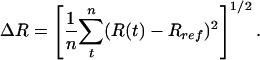 |
[5] |
The atomic fluctuations for the equilibrium simulation, ΔReq, are calculated as usual with Rref equal to the time-averaged position from that equilibrium simulation. For the biased simulations, the fluctuations, ΔR′bias, are calculated relative to the same equilibrium average coordinates. Coordinates, R(t), were taken at 0.2-ps intervals, and the average was taken over the length of the trajectory. We evaluate the difference in these two fluctuation quantities: ΔΔR =ΔR′bias –ΔReq. Values of ΔΔR were averaged from seven trajectories and over side-chain atoms to give 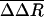 . Only substantial changes in structure caused by the dissociation process, consistent across multiple simulations, are expected to give large values of
. Only substantial changes in structure caused by the dissociation process, consistent across multiple simulations, are expected to give large values of 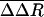 .
.
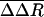 is plotted by residue in Fig. 2. Large
is plotted by residue in Fig. 2. Large 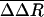 values are observed for several residues in VP1, VP3, and VP4 (shown as spheres in Fig. 3). The VP1 residues are part of either the buried pocket or the exit pore so that the observed effects are readily understood. Residues 1099–1106 (residue numbering for VP1, VP2, VP3, and VP4 starts at 1001, 2001, 3001, and 4001, respectively) are in the βC strand of the CHEF sheet that forms part of the pocket. (WIN 52084 exits between βC and βH.) Residue 1266 forms part of the pore. Residues 1088, 1158, and 1163 line the canyon near the pore exit. Similarly, the large
values are observed for several residues in VP1, VP3, and VP4 (shown as spheres in Fig. 3). The VP1 residues are part of either the buried pocket or the exit pore so that the observed effects are readily understood. Residues 1099–1106 (residue numbering for VP1, VP2, VP3, and VP4 starts at 1001, 2001, 3001, and 4001, respectively) are in the βC strand of the CHEF sheet that forms part of the pocket. (WIN 52084 exits between βC and βH.) Residue 1266 forms part of the pore. Residues 1088, 1158, and 1163 line the canyon near the pore exit. Similarly, the large 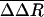 value for the C terminus of VP3, residue E3236, is easily rationalized, because this residue covers the entrance to the pore and interacts with K1103 in βC.
value for the C terminus of VP3, residue E3236, is easily rationalized, because this residue covers the entrance to the pore and interacts with K1103 in βC.
Fig. 2.

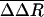 for each residue of viral proteins: ΔΔR = ΔRbias′ – ΔReq, where ΔRbias′ and ΔReq are from Eq. 5, and
for each residue of viral proteins: ΔΔR = ΔRbias′ – ΔReq, where ΔRbias′ and ΔReq are from Eq. 5, and 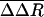 is the average of seven biased simulations with biasing force constants of 0.002, 0.0005, and 0.0001 kcal/mol·Å4.
is the average of seven biased simulations with biasing force constants of 0.002, 0.0005, and 0.0001 kcal/mol·Å4.
Fig. 3.

Capsid structure at the 5-fold axis. (Left) Ribbon drawing of VP1 (blue) and part of VP3 (red) and VP4 (yellow), with residues with large 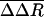 shown as spheres. These residues are VP1 residues 99–109, 221, and 226; VP3 N-terminal residues 1–10 and C-terminal residues 233–236; and VP4 N-terminal residues 29–42. WIN 52084 is colored green. (Right) N-terminal residues of VP3 and VP4 from five protomers around the 5-fold rotation axis. Colors are as described for Left.
shown as spheres. These residues are VP1 residues 99–109, 221, and 226; VP3 N-terminal residues 1–10 and C-terminal residues 233–236; and VP4 N-terminal residues 29–42. WIN 52084 is colored green. (Right) N-terminal residues of VP3 and VP4 from five protomers around the 5-fold rotation axis. Colors are as described for Left.
Relatively large conformational fluctuations are also observed for residues never in contact with WIN 52084 over the entire dissociation pathway, including 10 residues at the N terminus of VP3 and the N-terminal 14 residues of VP4. The VP3 N terminus contacts the N terminus of VP4 from a different protomer. Together, these termini wrap around the 5-fold axis to form an annulus on the inner side of the capsid (Fig. 3). We observe that the fluctuations around the 5-fold annulus increase as the antiviral compound moves out of the buried pocket. This observation for unbinding is particularly interesting, because externalization of RNA in viral uncoating is thought to occur through the opening at the 5-fold axis (20, 21). The increased fluctuations are consistent with effects associated with the presence of antiviral compounds reported from crystallographic studies on HRV16 (21) and equilibrium MD simulations on HRV14 (14). For residues at the N terminus of VP3, the B-factors are larger in the crystallographic analysis of HRV16 from the native virus structure than in complex with a WIN compound. Additionally, the picosecond-to-nanosecond time-scale fluctuations are comparatively large for N-terminal residues of VP3 and VP4 near the 5-fold axis but are smaller in the presence of antiviral compounds. Thus, the large 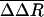 as a result of dissociation from residues surrounding the 5-fold axis likely reflects the higher-magnitude fluctuations exhibited by these residues in the absence of the antiviral compound.
as a result of dissociation from residues surrounding the 5-fold axis likely reflects the higher-magnitude fluctuations exhibited by these residues in the absence of the antiviral compound.
Conformational Flexibility of WIN 52084. The size of antiviral compounds and the buried location of the hydrophobic pocket raises the question of whether flexibility is required for binding (22). WIN 52084 has a seven-carbon, fully reduced alkyl chain and is correspondingly conformationally flexible. Additionally, bound WIN 52084 is mobile within the pocket (14, 23). To examine this question, torsional rotations of WIN 52084 were enumerated for the dissociation trajectories. A number of rotations are observed along the entire alkyl chain as the antiviral compound moves through the pocket and escapes into solvent. The frequency of dihedral transitions is high, with approximately five transitions for each torsion angle during dissociation. Similarly, high numbers of dihedral transitions are observed in all dissociation simulations. Moreover, the torsional rotations lead to nonlinear conformations of the antiviral agent (illustrated below). Together, these features indicate that flexibility facilitates access to the pocket.
Dissociation Pathway. Dissociation proceeds in a series of steps illustrated by the time profiles shown in Fig. 4 for trajectories with biasing force constants α = 0.0001 and 0.0005 kcal/mol·Å4. Jumps occur at similar values of the dissociation coordinate ρ0.5 in all simulations but are delineated most clearly with α = 0.0001 kcal/mol·Å4. In the region, S2, ρ0.5 increases slowly from 137 to 140 Å for most trajectories and is sampled for nearly 3 ns in the α = 0.0001 kcal/mol·Å4 simulation. An abrupt transition occurs between S2 and the region S3. In S3, the antiviral compound moves continuously over a larger range of ρ0.5 between 145 and 150 Å but for a shorter time period relative to S2. From S3 and ρ0.5 ≈ 150 Å, the antiviral compound has reached the surface of VP1 and starts to become solvated. Dissociation proceeds more rapidly through a series of shorter-lived steps most perceptible in the profile for α = 0.0001 kcal/mol·Å4 (Fig. 4, Inset). WIN 52084 is fully dissociated at ρ0.5 = 170 Å. The discrete jumps in the time profiles for transition between S2 and S3 and after S3 suggest that dissociation of the antiviral compound from these regions of the pore involves crossing an energy barrier (discussed below).
Fig. 4.

The time profile of dissociation coordinate (ρ0.5) for the biased simulations, with α = 0.0001 kcal/mol·Å4 (Left) and 0.0005 kcal/mol·Å4 (Right). S2 is the region of simulation for ρ0.5 from 137 to 140 Å and corresponding time from 0.3 to 3.4 ns, and S3 is the region of simulation for ρ0.5 from 144 to 150 Å and corresponding time from 3.5 to 4.1 ns. (Inset) Enlarged part of Left between 3.4 and 4.3 ps.
How the long-chain WIN compound adjusts conformationally while exiting the deeply buried pocket was evaluated by a principal component analysis of coordinate differences (19) along the dissociation simulation calculated with α = 0.0001 kcal/mol·Å4. The analysis allows visualization of the conformational change in a reduced dimensionality of RMS deviations between WIN structures. The two largest eigenvalues account for >75% of the total; thus, much of the information is represented by a two-dimensional projection of the corresponding eigenvectors PC1 and PC2 (Fig. 5). Similar to a cluster analysis, the points from the two vectors indicate the path followed by the antiviral compound on a surface that approximates distances among the subsets of structures (19). The antiviral compound is observed to cluster in four major regions of this surface directly related to the dissociation profile. Two clusters correspond to S2 of the dissociation profile (gold), one to S3 (green), and one to the extended conformation as WIN 52084 enters solvent (blue). The structure of WIN 52084 moves away from original bound conformation (red) as it travels to one end of the pocket (S2) and to the pore opening (S3). As it exits into solvent, the structure closely resembles the original conformation.
Fig. 5.

Principal component analysis of matrix C (Eq. 4) calculated from 560 structures of WIN 52084 from the biased simulation with α = 0.0001 kcal/mol·Å4. The plot shows components of the two eigenvectors (PC1 and PC2) with the largest eigenvalues. The stars are colored according to simulation time: red, 0–64 ps; yellow, 64–3,480 ps; green, 3,480–4,032 ps; blue, 4,032–4,480 ps.
The structural characteristics of WIN 52084 associated with the substates identified in Figs. 4 and 5 are illustrated by trajectory snapshots in Fig. 6. Structures of WIN 52084 and nearby HRV14 residues from the α = 0.0001 kcal/mol·Å4 trajectory are shown for the initial equilibrium state, the plateau regions S2 and S3, and close to fully dissociated. In the equilibrium bound state, WIN 52084 is approximately linear and deep in the pocket. In S2 (2 ns), the oxazoline ring end of the antiviral compound has moved radially outward, with a small transverse displacement in the alkyl chain and isoxazole ring. The configuration is bent near the ether oxygen. Over the S3 region (3.5–3.8 ns), WIN 52084 moves radially outward from ρ0.5 = 145 to 150 Å, the bend angle increases and its position travels down the alkyl chain, and the oxazoline ring begins to pierce the protein surface. WIN 52084 is observed to exit between polypeptide chains at the end of βC and βH. Side-chain rearrangements were observed for residues 1101–1106, 1221, and 1266.
Fig. 6.

WIN 52084 dissociation pathway. (A) The pathway illustrated with overlaid snapshots of WIN 52084 and a single copy of VP1 (blue) and VP3 (red). (B) Individual snapshots illustrating dissociation for α = 0.0001 kcal/mol·Å4. Each panel is labeled with the dissociation coordinate ρ0.5 (Å) and simulation time (ps) corresponding to the profile in Fig. 4 Left. Blue, VP1 residues 98–107, 220–222, and 265–267; red, VP3 residues 232–235; yellow, WIN 52064.
We attempted to identify structural features of HRV14 or interactions with the antiviral compound that could be the molecular basis for the energy barriers implied from the profiles shown in Fig. 4, but our analysis of the trajectories found no consistent structural changes of the protein correlated in time with the transitions. One HRV14 interaction that seems necessary to disrupt in order for the antiviral compound to exit is a solvent-exposed ion pair: the C-terminal end of VP3 wraps around VP1 such that the last residue, E3236, lies in the canyon region blocking the pore of the pocket and forms an ion pair with K1103 (Fig. 6B Left). The ion pair is observed in the crystallographic structure and is a strong interaction in equilibrium simulations. For WIN 52084 to exit the pocket, this interaction must be broken. Still, the loss of the ion pair does not correlate in time with any of the transitions observed in the dissociation time profile. Additionally, alternative ionic interactions with R1094 or solvent compete for E3236 such that the E3236–K1103 interaction can be disrupted independent of WIN 52084 dissociation. Indeed, in the trajectory illustrated in Fig. 6, the salt bridge is disrupted early on and independent of dissociation, unlike other simulations in which this salt bridge is disrupted by WIN moving out of the pocket. For these reasons, the ion pair is not likely to be an energy barrier for dissociation of ligands from the internal cavity of VP1.
Discussion
In all dissociation simulations the antiviral compound exits the VP1 cavity following the same path; the initial displacement is a small displacement of the oxazoline ring to a position between residues 1103–1108 and 1215–1221 and residue 1266 at the end of the CHEF sheet (Fig. 6, region S2). Flexibility of the antiviral compound is a key feature of dissociation, because the alkyl chain undergoes a number of dihedral transitions during dissociation that allow WIN 52084 to twist its way through the pocket. Only localized, transient structural changes occur in VP1 during dissociation. A recent study suggested that addition of antiviral compounds during assembly might improve access to the pocket (24); however, the absence of large-amplitude motions of VP1 in the dissociation trajectories indicates that the hydrophobic pocket is fully accessible in the assembled virus particle. Dissociation follows a sequence of jumps within the VP1 cavity so that WIN 52084 stalls in two regions, S2 and S3, implying the existence of at least two energy barriers. Escape from the S2 and S3 regions is by transient packing defects in the protein in correspondence with the configuration of WIN 52084 appropriate for snaking out of the pocket and through the pore region. We were unable to identify any specific structural feature of HRV14 or any HRV14–WIN 52084 interaction that consistently correlated with transitions between S2 and S3, or out of S3, although deviations from equilibrium fluctuations were observed for several residues over the course of multiple dissociation trajectories (Fig. 2). The dissociation process for a hydrophobic compound seems to be restricted only by the overall shape of the pore and without steric blocks from specific protein residues while being facilitated by torsional rotations in the WIN compound. The pocket provides a hydrophobic compound seclusion from solvent.
Antiviral Drug Design. The dissociation of WIN 52084 from HRV14 occurs with a large number of dihedral rotations in the WIN compound and the development of a near 90° bend angle in the alkyl chain (Fig. 6). It is reasonable therefore that the efficacy of antiviral inhibitors depends on torsional rigidity, because the kinetics of binding, as well as affinity, can affect antiviral activity. The potential requirement for flexibility was recognized some time ago (22), and the results reported here now provide evidence for that proposal. Preliminary calculations using a rigid form of WIN 52084 reinforce the importance of flexibility.
The pattern of dihedral rotations differs somewhat for multiple dissociation simulations. Rotations at alternative positions facilitate dissociation, and not all of the alkyl bonds rotate. As such, it is not inconsistent that WIN compounds with a double bond have antiviral activity (22). In this regard, it is of interest to compare the antiviral compounds WIN 55865 and WIN 56278, which have one double bond in a five-carbon alkyl chain. These compounds are identical except in the configuration of the double bond; the configuration is cis in WIN 55865 and trans in WIN 56278. Nevertheless, WIN 55865 is less efficacious by an order of magnitude, and it is speculated that the cis bond cannot adopt as easily to travel the nonlinear pathway through the pore and into the hydrophobic pocket. Our results suggest that further decreasing the saturation of the alkyl chain would be disadvantageous, because making a highly restrained ligand is likely to generate a large kinetic barrier for binding and thus diminish drug efficacy.
Viral Uncoating. The dissociation of WIN 52084 was observed to affect the dynamics of HRV14 residues near the icosahedral 5-fold axis. These changes are of particular interest in view of the inhibition of viral uncoating by WIN compounds. Cell attachment and uncoating to release viral RNA are associated with large conformational transitions in the capsid that transform the overall structure of the particle (25) and perturb interactions at the 5-fold axis (26, 27). In addition to release of viral RNA, the N termini of VP1 and VP4 are externalized from their internal location in the capsid core. Although the atomic details of the uncoating pathway are unknown, it has been suggested that RNA and possibly VP4 exit through the channel at the 5-fold axis of the capsid (20, 21). A focus on the potential role of the 5-fold axis is also reinforced by an elastic network normal-mode analysis (28). One factor in the mechanism of the WIN antiviral activity predicted from computational studies (6) and subsequently supported by experiment (7) is entropic stabilization by the antiviral compound. Enthalpic stabilization is implicated in the case of receptor-mediated uncoating (29). Another mechanism proposed for WIN antiviral activity is that WIN raises the free energy of the barrier to uncoating by interfering with conformational changes in the virus particle needed to release RNA (2, 3, 5). Antibody neutralization and limited proteolysis studies find evidence consistent with large-scale conformational fluctuations that transiently expose VP4 on the viral surface at equilibrium and independent of cell attachment (30, 31). These fluctuations occur in the absence of WIN 52084 but are severely depressed in its presence (30). The dynamic properties observed computationally here and previously (14) seem to reflect features that could underlie the larger-amplitude “breathing” motions and the effect of WIN compounds occupying the VP1 pocket. The physical basis for smaller atomic fluctuations in these annulus-forming residues far from the pocket may be the cause for inhibition of a conformational transition for uncoating.
Conclusions
Dissociation of a buried antiviral compound was studied with MD simulations of HRV14 capsid by applying an external force counteracting fluctuations that moved the compound radially inward toward the capsid center. The biasing force allowed investigation on a time scale accessible to simulation methods. Large deviations from equilibrium fluctuation magnitudes or atomic positions were avoided by use of a small force constant. It is important to note that similar protein and ligand conformational effects and reaction profiles were observed for multiple dissociation simulations. Based on the minimal perturbation imposed by the external force and the consistent behavior for multiple simulations, it is concluded that the simulation results provide reasonable models for dissociation that show important features of the exit pathway. Dissociation proceeds in a series of steps. Two steps have longer lifetimes and correspond to displacement of the antiviral compound to a position near the end of the pocket and then moving through the pore region. Once the compound penetrates the surface and enters the solvent region, interaction with the virus diminishes and dissociation proceeds more rapidly. Transient packing defects from rotations in side chains of different HRV14 residues seem to be the only protein motion needed for unbinding. No large conformational changes are required. Conformational flexibility of the antiviral compound seems to be important for accessing the internal hydrophobic pocket in VP1, although the specific requirements for rotation of a given bond position cannot be determined from this study. It is particularly notable that dissociation is observed to occur with an increase in the fluctuations of N-terminal residues in VP3 and VP4. These residues surround the 5-fold axis of the capsid where RNA (20, 21) and possibly VP4 (21) are thought to exit during uncoating. Larger thermal fluctuations may reflect a greater conformational flexibility important for viral uncoating. Future simulation studies should provide the level of atomic detail needed to understand the physical basis for these long-range effects of antiviral compounds.
Acknowledgments
We thank Jeff Evansek for helpful discussion on the interpretation of matrix C. This work was supported by National Institutes of Health Grant AI39639 (to C.B.P.) and a Purdue University reinvestment grant to support the Markey Center of Structural Biology.
Author contributions: C.B.P. designed research; Y.L. and Z.Z. performed research; Y.L., Z.Z., and C.B.P. analyzed data; and Y.L., Z.Z., and C.B.P. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HRV, human rhinovirus; MD, molecular dynamics.
References
- 1.Diana, G. D., McKinlay, M. A. & Treasurywala, A. (1997) in Structure Biology of Viruses, eds. Chiu, W., Burnett, R. & Garcea, R. (Oxford Univ. Press, New York), pp. 432–450.
- 2.Smith, T. J., Kremer, M. J., Luo, M., Vriend, G., Arnold, E., Kamer, G., Rossmann, M. G., McKinlay, M. A., Diana, G. D. & Otto, M. J. (1986) Science 233, 1286–1293. [DOI] [PubMed] [Google Scholar]
- 3.Grant, R. A., Hiremath, C. N., Filman, D. J., Syed, R., Andries, K. & Hogle, J. M. (1994) Curr. Biol. 4, 784–797. [DOI] [PubMed] [Google Scholar]
- 4.Muckelbauer, J. K., Kremer, M., Minor, I., Diana, G., Dutko, F. J., Groarke, J., Pevear, D. C. & Rossmann, M. G. (1995) Structure (London) 3, 653–667. [DOI] [PubMed] [Google Scholar]
- 5.Hiremath, C. N., Filman, D. J., Grant, R. A. & Hogle, J. M. (1997) Acta Crystallogr. D 53, 558–570. [DOI] [PubMed] [Google Scholar]
- 6.Phelps, D. K. & Post, C. B. (1995) J. Mol. Biol. 254, 544–551. [DOI] [PubMed] [Google Scholar]
- 7.Tsang, S. K., Danthi, P., Chow, M. & Hogle, J. M. (2000) J. Mol. Biol. 296, 335–340. [DOI] [PubMed] [Google Scholar]
- 8.Fox, M. P., Otto, M. J. & McKinlay, M. A. (1986) Antimicrob. Agents Chemother. 30, 110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diana, G. D., McKinlay, M. A., Otto, M. J., Akullian, V. & Oglesby, C. (1985) J. Med. Chem. 28, 1906–1910. [DOI] [PubMed] [Google Scholar]
- 10.Straub, J. E. & Karplus, M. (1991) Chem. Phys. 158, 221–248. [Google Scholar]
- 11.Case, D. A. & Karplus, M. (1979) J. Mol. Biol. 132, 343–368. [DOI] [PubMed] [Google Scholar]
- 12.Kottalam, J. & Case, D. A. (1988) J. Am. Chem. Soc. 110, 7690–7697. [Google Scholar]
- 13.Paci, E., Caflisch, A., Pluckthun, A. & Karplus, M. (2001) J. Mol. Biol. 314, 589–605. [DOI] [PubMed] [Google Scholar]
- 14.Speelman, B., Brooks, B. R. & Post, C. B. (2001) Biophys. J. 80, 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S. & Karplus, M. (1983) J. Comput. Chem. 4, 187–217. [Google Scholar]
- 16.MacKerell, A. D. J., Bashford, D., Bellott, M., Dunbrack, R. L., Evanseck, J. D., Field, M. J., Fischer, S., Gao, J., Guo, H., Ha, S., et al. (1998) J. Phys. Chem. 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- 17.Cagin, T., Holder, M. & Pettitt, B. M. (1991) J. Comput. Chem. 12, 627–634. [Google Scholar]
- 18.Paci, E. & Karplus, M. (1999) J. Mol. Biol. 288, 441–459. [DOI] [PubMed] [Google Scholar]
- 19.Troyer, J. M. & Cohen, F. E. (1995) Proteins Struct. Funct. Genet. 23, 97–110. [DOI] [PubMed] [Google Scholar]
- 20.Hogle, J. M. (2002) Annu. Rev. Microbiol. 56, 677–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hadfield, A. T., Lee, W. M., Zhao, R., Oliveira, M. A., Minor, I., Rueckert, R. R. & Rossmann, M. G. (1997) Structure (London) 5, 427–441. [DOI] [PubMed] [Google Scholar]
- 22.Mallamo, J. P., Diana, G. D., Pevear, D. C., Dutko, F. J., Chapman, M. S., Kim, K. H., Minor, I., Oliveira, M. & Rossmann, M. G. (1992) J. Med. Chem. 35, 4690–4695. [DOI] [PubMed] [Google Scholar]
- 23.Phelps, D. K. & Post, C. B. (1999) Protein Sci. 8, 2281–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang, Y., Simpson, A. A., Ledford, R. M., Bator, C. M., Chakravarty, S., Skochko, G. A., Demenczuk, T. M., Watanyar, A., Pevear, D. C. & Rossmann, M. G. (2004) J. Virol. 78, 11061–11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curry, S., Chow, M. & Hogle, J. M. (1996) J. Virol. 70, 7125–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basavappa, R., Syed, R., Flore, O., Icenogle, J. P., Filman, D. J. & Hogle, J. M. (1994) Protein Sci. 3, 1651–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hewat, E. A. & Blaas, D. (2004) J. Virol. 78, 2935–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tama, F. & Brooks, C. L. (2002) J. Mol. Biol. 318, 733–747. [DOI] [PubMed] [Google Scholar]
- 29.Tsang, S. K., McDermott, B. M., Racaniello, V. R. & Hogle, J. M. (2001) J. Virol. 75, 4984–4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis, J. K., Bothner, B., Smith, T. J. & Siuzdak, G. (1998) Proc. Natl. Acad. Sci. USA 95, 6774–6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li, Q., Yafal, A. G., Lee, Y. M. H., Hogle, J. & Chow, M. (1994) J. Virol. 68, 3965–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]