

Summary
Injury is common in the life of organisms. Because the extent of damage cannot be predicted, injured organisms must determine how much tissue needs to be restored. Although it is known that amputation position affects the regeneration speed of appendages, mechanisms conveying positional information remain unclear. We investigated tissue dynamics in regenerating caudal fins of the African killifish (Nothobranchius furzeri) and found position-specific, differential spatial distribution modulation, persistence, and magnitude of proliferation. Single-cell RNA sequencing revealed a transient regeneration-activated cell state (TRACS) in the basal epidermis that is amplified to match a given amputation position and expresses components and modifiers of the extracellular matrix (ECM). Notably, CRISPR-Cas9-mediated deletion of the ECM modifier sequestosome 1 (sqstm1) increased the regenerative capacity of distal injuries, suggesting that regeneration growth rate can be uncoupled from amputation position. We propose that basal epidermis TRACS transduce positional information to the regenerating blastema by remodeling the ECM.
Subject areas: Biological sciences, Zoology, Molecular biology, Cell biology
Graphical abstract
Highlights
-
•
Amputation position changes tissue-wide proliferation response
-
•
Regeneration deploys transient regeneration-activated cell states
-
•
Sqstm1 slows down regenerative outgrowth in distal injuries
-
•
Prediction: positional information is transduced by ECM changes during regeneration
Biological sciences; Zoology; Molecular biology; Cell biology
Introduction
For many organisms, including humans, the preservation of anatomical form and function depends in great part on the periodic elimination and restoration of cells. This process is referred to as tissue homeostasis. In mammals, the rate of tissue homeostasis varies widely across organs; i.e., self-renewal of the intestinal and lung epithelia has been estimated to take 5 days and up to 6 months, respectively.1,2 As such, specific mechanisms exist in adult tissues responsible for sustaining specific rates of tissue homeostasis to mitigate normal, physiological wear and tear. The intricate balance of tissue homeostasis can be severely disrupted by injury. During their lifespans, all multicellular organisms are likely to experience some kind of injury. Unlike physiologically regulated tissue homeostasis, injuries and the extent of the damage incurred are unpredictable. This creates a challenge for organisms to monitor and deploy an anatomically specific regeneration response proportional to the magnitude of the injury.
During regeneration, a specialized structure known as blastema forms through the rapid proliferation and subsequent differentiation of multiple cell types to restore the missing tissue.3 For instance, it has been shown that osteoblast differentiation is accelerated in regenerating fins. Newly differentiated osteoblasts appear 4 days earlier in regenerated tissue compared to the pre-existing tissue, also referred to as the stump.4 It is not known whether there is a regeneration-specific osteoblast differentiation program or if differentiation trajectories used during tissue homeostasis can be accelerated during regeneration. Regeneration has also been shown to alter rates of tissue growth. In vertebrates, the speed of regeneration differs when different amounts of tissue are lost.5 For example, fish caudal fin amputations close to the base of the appendage, a proximal amputation, display higher growth rates than amputations performed further away from the base of the fin, a distal amputation.6,7
To date, it is not known how injured tissues detect amputation position and what processes may encode positional information during regeneration. Efforts to identify deposited positional information prior to injury led to the identification of transcripts and proteins that are differentially expressed along the proximo/distal (P/D) axis in the intact zebrafish fin.8 However, these differences are lost during regeneration, leading to the hypothesis that positional information must be redefined during regeneration.8 Alternatively, it has been proposed that migratory progenitor cells retain positional identity from their locations prior to injury. This opens up the possibility that progenitors communicate positional information to the new tissue to determine regeneration growth rate.9,10 But the mechanism of positional information retention and potential relay to other cells has not yet been identified.
Furthermore, it has been suggested that positional information is encoded in the tissue within the thickness of the bone at the plane of amputation11,12 and that mechanical distension of the epidermis during wound closure constitutes a direct measurement of the amputation position by the wound epidermis. A wave of mechanical distension in the basal epidermis was shown to propagate to different lengths according to the amputation position.13 To add to the complexity of this process, it was shown that there is a 2-day time window at the beginning of regeneration when positional information is possibly reestablished de novo. If blastemas are impaired in their proliferative ability during this time window, the wrong positional information is encoded in the regenerated appendage, so multiple rounds of regeneration consistently grow abnormal tissue sizes in the absence of any further impairment of proliferation.14 It is conceivable that independent mechanisms of positional information coordinating the regenerative response exist. There may be redundant and complementary means to adequately relay and deposit positional information into the new tissue. This would ensure that form and function are restored following diverse and unpredictable injury.
Here, we deploy spatial and temporal analysis of proliferation and single-cell transcriptomic profiling to measure molecular and cellular changes along the caudal fin P/D axis. We chose the African killifish Nothobranchius furzeri for our studies because of the reduced complexity of differential gene expression and gene regulation compared to the more broadly utilized zebrafish.15 We show that amputation position influences the length of time (persistence) it takes for tissues to progress through regeneration. We report on the discovery of a basal epidermal subpopulation that shows a transient regeneration-activated cell state (TRACS) likely to participate in mediating positional information in the regenerating blastema. Altogether, our study demonstrates that amputations along the P/D axis result in defined spatial and temporal rates of proliferation. We propose that such dynamics likely initiate the proportional changes in tissue architecture that may ultimately define the scale and rate of regeneration of amputated tissues.
Results
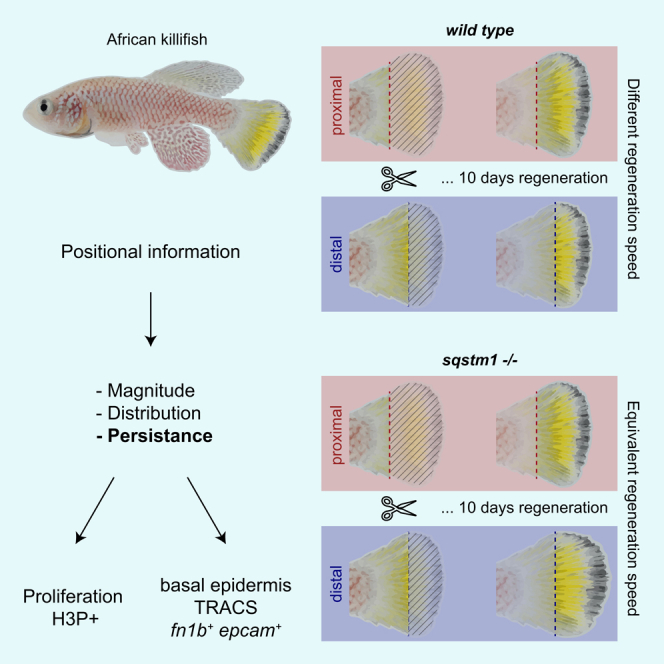
Amputation position influences regeneration growth rate in killifish caudal fins
We investigated whether the killifish N. furzeri displays regeneration growth rate differences at different amputation positions in the caudal fin. We restricted our analyses to male individuals of the same size because the caudal fin has a clear pigmentation pattern that allows for consistent amputation at the same position across multiple individuals (Figures 1A, S1A, S1C, and S1D). Whether there are sex-determined differences that influence regeneration growth rate in killifish remains to be investigated. We first established the effect of amputation position by carrying out regeneration time courses at two different amputation positions along the P/D axis (Figures 1B and S1E). Consistent with other fishes, killifish showed higher regeneration lengths in proximal injuries compared to distal injuries during the first 10 days of regeneration. We built a linear mixed model to test the statistical significance of two fixed effects: (1) time (days post-amputation; dpa) and (2) amputation position (AmpPos, prox, and dist). We observed a highly statistically significant contribution of these components to regeneration growth (Figure 1E). The result is growth rate differences when comparing distinct amputation positions, with proximal injuries displaying a higher growth rate when compared to distal injuries (Figures 1C, 1F, and S1B). We quantified growth rate (mm/day) by measuring the regeneration length and dividing by the time between adjacent time points (Figure 1D). We observed growth rate differences between proximal and distal injuries within 2- and 7-day post-amputation (dpa), with the peak growth rate difference occurring at 3.5 dpa and both injuries reaching and maintaining the same growth rate from 7 dpa onward (Figures 1F and S1F). We also observed significant differences in growth acceleration and deceleration when comparing both proximal and distal injuries (Figure S1G).
Figure 1.

Amputation position triggers differential regenerative outgrowth in killifish
(A) Side view of sexually mature adult male killifish.
(B) Experimental design for longitudinal analysis of regenerative outgrowth.
(C) Representative images of regeneration time course of proximal and distal injuries aligned at the boundary between the fish scales and the caudal fin (orange dotted line); amputation line is displayed in magenta. Scale bar: 1 mm.
(D) Definition of regeneration length in the context of the new tissue, pre-existing tissue, bone, amputation, and fish scales. The difference in regeneration length divided by the time between time points corresponds to the regeneration growth rate expressed in mm/day.
(E) Regeneration growth quantified in proximal and distal injuries over a 10-day time course. Statistical significance is indicated for fixed effects of a linear mixed model. Polygon area represents mean ± SEM. ∗∗∗, p < 0.001 (see STAR Methods).
(F) Regeneration growth rate in proximal and distal injuries. Gray dashed vertical lines correspond to the most different growth rate or the earliest time point when growth rate is the same between proximal and distal injuries, respectively. Colored dashed lines with indicated slopes correspond to growth acceleration and slowing down (n = 50). Polygon area represents mean ± SEM.
See also Figure S1.
We conclude from these experiments that regenerative outgrowths have at least two components that are influenced by amputation position: (1) a magnitude component, as indicated by the higher growth rates of proximal injuries when compared to distal injuries (Figures 1F and S1F) and (2) a time component in which proximal injuries progress to higher growth rates at a sooner time point compared to distal injuries (Figures 1F and S1G). This later observation may not have been detected in prior studies due to the lower temporal resolution reported7,11 (Figure S1B). We reasoned that the first component could be explained by expanding the progenitor pool to match the amputation position as previously observed.7,12 However, the second component suggested an additional layer of regulation that influences the pace at which progenitors become activated to mount a corresponding regenerative response to the amputation position. We hypothesized that amputation position changes both the number of progenitor cells recruited to the regeneration event and the amount of time (persistence) pro-proliferative signals persist in the tissue. We tested this idea by quantifying the cellular proliferation dynamics in proximal and distal injuries during the formation of the initial stages of blastema formation and before the tissue undergoes regenerative outgrowth: 12 to 48 hours post-amputation (hpa).
Amputation position determines the duration of tissue-wide proliferation
We previously reported that between 14 and 24 hpa fins display high levels of proliferation in the pre-existing tissue (more than 1.5 mm away from the plane of amputation, tissue-wide proliferation; Figures S2F and S2G), whereas earlier time points (12 hpa) have proliferation levels similar to unamputated fins (homeostasis proliferation), and later time points display high levels of proliferation localized to the plane of amputation (up to 0.2 mm away from the plane of amputation, blastema local proliferation, Figures S2F–S2G).15 We investigated the effect of amputation position on tissue-wide proliferation over time by whole-mount immunofluorescence against the mitosis marker phospho-Histone H3 (H3P), automatic nuclei segmentation, and cytometry analyses (Figures 2A–2C and S2A). We observed that both proximal and distal injuries displayed tissue-wide proliferation at 18 hpa (Figure 2C; 18 hpa prox dist). However, the distribution of proliferative events diverged after 18 hpa, with distally amputated fins reducing cell division in fin tissue located away from the plane of amputation (−1.5 mm) and proximally amputated fins sustaining such proliferation for an additional 18 h (Figure 2C’, asterisks). In contrast, the tissue adjacent to the plane of amputation (−0.075 mm) in both distally and proximally amputated fins displayed a sustained increase in proliferation (Figure 2C”). We also observed a positive correlation between increased proliferation events in thicker (proximal) than in thinner fin tissues (distal) as previously described7 (Figures S2C–S2E). Thus, amputation position appears to influence the length of time the regenerating tissue undergoes tissue-wide proliferation (Figure 2D). We conclude from these experiments that the number of cells recruited for cell division as well as the persistence of tissue-wide proliferation depend on the location of the amputation plane and that both events likely determine blastema size and regeneration growth rate.
Figure 2.

Amputation position influences temporal and spatial distribution of proliferating cells
(A) Experimental design of regeneration time course at two different amputation positions.
(B) Top: single bone representative image of masked H3P in greyscale with cyan DAPI background. Scale bar: 0.5 mm. Bottom: heatmap representation of H3P density along the P-D axis relative to the amputation plane.
(C) Heatmap representation of averaged H3P density in regenerating fins at different amputation positions. Time course is displayed in vertical orientation, n = 12.
(C′) Statistical analysis per time point corresponding to 200 μm segment 1.5 mm away from the amputation plane; polygon area represents mean ± SEM; n = 12. ∗, FDR < 0.05; ∗∗∗, FDR < 0.001, Wilcoxon rank-sum test.
(C'') Statistical analysis per time point corresponding to 200 μm segment adjacent to the amputation plane; polygon area represents mean ± SEM; n = 12. ∗, FDR < 0.05, Wilcoxon rank-sum test.
(D) Regeneration progression model shows proliferation time shift between proximal and distal injuries where tissue-wide proliferation is launched sooner in distal injuries, and it is maintained for longer in proximal injuries.
(E) Experimental design of gradient-cut regeneration dataset where images were collected at 24 hpa.
(F) Blastema size and proliferation peak definition relative to bone length, bone fracture, and regenerating edge.
(G) Linear correlation between proliferation peak distance to the amputation and bone length. Larger bones correspond to distal amputations.
(H) Linear correlation between blastema size and bone length.
(I) Linear scaling of 1.83 μm proliferation peak distance to the amputation per 1 μm of blastema size.
(J) Schematic of length relationship between blastema size and proliferation peak in proximal and distal injuries.
Amputation position predicts blastema size and spatial distribution of proliferation
Interestingly, most dividing cells during tissue-wide proliferation (24 hpa) are located within the epidermis (Figure S2A). We reasoned that amputation position may shape the spatial distribution of tissue-wide proliferation. To test this idea, we generated spatial proliferation profiles in a proximal-distal gradient of amputations. We performed immunostaining against H3P in a cohort of fish amputated at different positions along the P/D axis and quantified the resulting proliferation profiles at 24 hpa (Figure 2E). Previous work has shown that proximal injuries have greater numbers of dividing progenitor cells than distal injuries.7 We reasoned that the spatial distribution of dividing cells may reflect an interplay between how many cells are dividing and for how long they are being deployed tissue-wide. We investigated the possibility of a spatial control mechanism by calculating the distance between the proliferation peak, corresponding to the highest density of proliferating cells, with the plane of amputation (Figure 2F). We observed that bone length, which is a proxy for amputation position, significantly correlates with both the proliferation peak distance to the plane of amputation (p < 0.001, Figure 2G) and the size of the blastema (p < 0.001, Figure 2H). At 24 hpa, blastemas are mainly composed of wound epidermis, which migrates to cover the exposed tissue after amputation.16,17 We observed that proximal injuries have larger blastemas than distal injuries, which is consistent with the displacement of more epidermis to close a larger wound. The scale of epidermis displacement may propagate information further away from the injury into the pre-existing tissue, which is what we observed when correlating blastema size with proliferation peak (Figure 2I). This positive correlation (by a factor of 1.83) suggests a proportional spatial remodeling of the pre-existing tissue that matches the amputation position (Figure 2J). We hypothesize from these findings that a cellular entity may exist that relays such positional information and sets the regeneration event to recruit migrating mesenchyme for a correct blastema size and growth rate. To test this hypothesis, we profiled the pre-existing tissue by single-cell RNA sequencing (scRNA-seq) at 12 hpa, right before the tissue-wide proliferation response is observed.
Single-cell expression analyses reveal transcriptional diversity in intact and regenerating fins
We sought to determine whether cellular and gene expression differences could be identified along the P/D axis of the regenerating caudal fin. We chose to use single-cell transcriptomic profiling because of the inconclusive differences between proximal and distal injuries observed in published bulk measurements.8 We generated three scRNA-seq regeneration datasets using the 10x Genomics CellPlex multiplexed technology that allowed us to both increase the number of cells analyzed and generate biological replicates per sample (Figure S3A, see STAR Methods). First, we sampled regenerating and control fins sharing equivalent anatomical compositions (caudal dataset, Figure 3A). Second, we collected intact dorsal fins from unamputated control fish (dorsal homeostasis, Figure 3E) and intact dorsal fins from fish regenerating their caudal fin for 12 h (dorsal 12 hpa, Figure 3E). And third, we profiled 12 hpa stumps from fins with proximal or distal injuries (Figure 3I). This strategy allowed us to distinguish the transcriptional responses resulting from amputation position, tissue anatomy, and regeneration.
Figure 3.

Amputation induces regeneration transcriptional response within epidermis subpopulation at 12 hpa
(A) Experimental design of 12 hpa and homeostasis tissue collection for scRNA-seq on the caudal fin. Fins were uncut or 50% cut, 12 h later the stump was trimmed to match the corresponding anatomical region between the two samples, then tissues were dissociated and subjected to scRNA-seq.
(B) RRG enrichment analysis on DE genes between 12 hpa and homeostasis caudal cells split by clusters defined in the integrated dataset (656 clusters); Fisher’s exact test, FDR < 0.01.
(C) UMAP distribution of caudal clusters (CCs) within the caudal dataset, cell numbers per sample, and RRG enrichment analysis; Fisher’s exact test, p < 0.001.
(D) Heatmap of DE genes between caudal 12 hpa and caudal homeostasis, and top GO terms associated with the gene list (FDR < 0.05). Cells were randomly sampled to balance both 12 hpa and homeostasis groups.
(E) Experimental design of 12 hpa and homeostasis intact dorsal fin collection for scRNA-seq. Caudal fins were uncut or 50% cut, 12 h later the intact dorsal fin was collected, dissociated, and subjected to scRNA-seq.
(F) RRG enrichment analysis on DE genes between 12 hpa and homeostasis dorsal cells split by clusters defined in the integrated dataset (656 clusters); Fisher’s exact test, FDR < 0.01.
(G) UMAP distribution of dorsal clusters (DC) within the dorsal dataset, cell numbers per sample, and RRG enrichment analysis; Fisher’s exact test, p < 0.001.
(H) Heatmap of DE genes between dorsal 12 hpa and dorsal homeostasis, and top GO terms associated with the gene list (FDR < 0.05). Cells were randomly sampled to balance both 12 hpa and homeostasis groups.
(I) Experimental design of proximal and distal regenerating caudal fins collected for scRNA-seq. Fins were amputated either proximal or distal, 12 h later the stump was trimmed to remove the scales and muscle, then tissues were dissociated and subjected to scRNA-seq.
(J) RRG enrichment analysis on DE genes between proximal and distal cells split by clusters defined in the integrated dataset (656 clusters); Fisher’s exact test, FDR < 0.01.
(K) Heatmap of DE expressed genes between proximal and distal cells within the caudal clusters (CCs). No significant GO terms were found (FDR < 0.05). Cells were randomly sampled to balance both proximal and distal groups.
(L) Heatmap of DE expressed genes between proximal and distal cells within the dorsal clusters (DC), and single GO terms associated with the gene list (FDR < 0.05). Cells were randomly sampled to balance both proximal and distal groups.
We then integrated the obtained multiplexed scRNA-seq datasets above alongside a previously published blastema dataset15 to generate gene expression clusters and a cell identity reference atlas (see methods for integration and quality control). The resulting integrated dataset contained 84,616 cells distributed into 32 cell clusters (cluster resolution: 1), belonging to 11 cell types expected to reside in fin tissues (Figure S3B). We assigned cell clusters to cell types based on both known molecular markers and gene ontology enrichment analysis (Figure S3C). To identify any cell states that may be regeneration-activated in a larger array of potential cell states, we generated a collection of 656 clusters by iteratively changing the cluster resolution in the integrated dataset (cluster resolution: 0.2–2). By doing this, we generated clusters that partially overlapped with each other and represented different degrees of transcriptional variance. This approach allowed us to leverage the transcriptional diversity captured across multiple experiments with cells obtained specifically from cut (regeneration) and uncut (homeostasis) caudal and dorsal fins.
Epidermis subpopulation expresses early regeneration-responsive transcriptional signature
To generate differentially expressed (DE) gene lists, we split the integrated dataset into caudal and dorsal datasets (13,678 and 21,992 cells, respectively) and compared cells from homeostasis and regeneration samples to each other. In the caudal fin, out of 656 clusters, 337 clusters (51%) showed more than 10 DE genes, and 219 clusters (33%) showed more than 50 DE genes. Similarly, in the dorsal fin, 353 clusters (54%) showed more than 10 DE genes, and 279 clusters (42%) showed more than 50 DE genes. We asked whether the DE gene lists obtained for each cluster were significantly enriched for previously reported regeneration responsive genes (RRG).15 We found 8 caudal clusters (CC, Figure 3B), and 23 dorsal clusters (DC, Figure 3F) with statistically significant enrichment for RRG (padj < 0.001). Interestingly, all RRG-enriched clusters belonged to the epidermis. We investigated the degree of overlap between the CC and DC by going back to the integrated dataset and grouping together all cells within each definition. We observed that 86% of cells within the CC and 88% of cells within the DC are not present within the reciprocal definition and concluded that both CC and DC represent different epidermal cell states (Figures 3C and 3G). We considered the possibility that data integration failed and that the high degree of non-overlap between CC and DC were batch effect artifacts. However, we confirmed the presence of dorsal cells within the CC and caudal cells within the DC (Figure S3D). We interpret these data to mean that the identified CC and DC represent epidermal cell states that respond to regeneration in a context-dependent manner.
We next identified DE genes between regeneration and homeostasis in both caudal and dorsal cells (1,450 and 5,259 cells, respectively) within the CC and caudal and dorsal cells (2,558 and 3,832 cells, respectively) within the DC (Figure S3D). Interestingly, half the cells within the CC belong to the proliferating epidermis as indicated by elevated expression levels of proliferation markers (e.g., ccna, mki67, and pcna; Figure S3E). We also confirmed a highly significant overrepresentation of RRG in all four cell groups albeit exclusively within the upregulated component (padj < 0.001, Figure S3F). Additionally, we observed a high degree of overlap (∼40%) of DE genes within all four groups of cells, suggesting that the two epidermal subpopulations appear to be mounting a similar transcriptional response to regeneration (no bias, Figure S3G).
To further investigate the transcriptional signature observed in CC and DC, we performed gene ontology (GO) term analysis on the differentially expressed gene list (Figures 3D, 3H, and S3G; Tables S1 and S2). We found 110 and 115 GO terms in CC and DC, respectively. Within the upregulated genes, top terms (padj < 0.05) were represented by DNA replication (GO:0006260) and ribosome biogenesis (GO:0042254). Conversely, within downregulated genes, the most significant terms were associated with translation (GO:0006412) and ribosome (GO:0005840). Interestingly, we found 144 regeneration responsive genes with a CC bias enriched for unfolded protein binding (GO:0051082), 180 genes with a DC bias enriched for genes involved in DNA replication (GO:0006260) and DNA repair (GO:0006281), and 250 genes with no cluster bias with genes enriched for the proteasome complex (GO:0000502) and protein catabolic process (GO:0030163, Figure S3G). We conclude from our analyses that injury triggers a local and remote RRG transcriptional response in epithelial cells of both cut (caudal) and uncut (dorsal) fins. In uncut dorsal fins, the response is detected in cells already committed to proliferation (dorsal clusters, Figure 3F), whereas in cut fins, the response is found in a subtype of non-proliferative cells (caudal clusters, Figure 3B).
Amputation position biases differential cell state abundance rather than differential gene expression
In the search for the effects of amputation position on the deployment of regeneration, we profiled 12 hpa stumps from fins with proximal or distal injuries with scRNA-seq (Figure 3I). Because proximal and distal injuries generate stumps with slightly different anatomies, we needed first to understand the transcriptional response to regeneration (see previous section). Similar to what we described above, we looked for DE genes between proximal and distal cells in all 656 clusters defined in the integrated dataset. Within these, we found 577 clusters (88%) with more than 10 DE genes and 552 clusters (84%) with more than 50 DE genes. However, we found zero cluster identities with an enriched regeneration-responsive transcriptional signature when we overlap the DE gene lists with the RRG (Figure 3J). This is not surprising since both samples correspond to the same regeneration time point that is mounting a regenerative response. This is more apparent when we look for the DE genes within the epithelial subpopulations identified above. In the CC, we observed 220 genes upregulated in proximal cells and 41 genes upregulated in distal cells (Figure 3K; Table S3). When we look at the DE genes within the DC, we observe 248 genes upregulated in proximal cells and 28 genes upregulated in distal cells (Figure 3L; Table S4). Surprisingly, we could not find any significant enrichment for GO terms among the DE expressed genes in both cluster groups except for the smallest gene list corresponding to 28 upregulated genes in distal cells of the DC (padj < 0.05). The GO terms correspond to proteosome complex, peptidase complex, endopeptidase complex, catalytic complex (GO:0000502, GO:1905368, GO:1905369, and GO:1902494, respectively), and they were all part of the regeneration transcriptional signature present in both CC (Figure 3B) and DC (Figures 3F and S3G). Furthermore, we investigated whether CC or DC show an enrichment for RRG and found that genes upregulated in distal cells within CC have a statistical enrichment for RRG (padj < 0.001, Figure S3F). The distal bias in regenerative gene expression may be interpreted as a temporal bias, meaning that regeneration is deployed sooner in distal injuries, thus shortening the time window for blastema formation. In accordance with previous observations,8 our data confirm that both proximal and distal injuries launch a transcriptionally equivalent response during regeneration. Hence, we next investigated whether amputation position influenced the relative abundance of cell states to look for a differentiation bias between proximal and distal samples.
An alternative to changes in gene expression is that amputation position may favor a particular cell cluster to numerically expand within the tissue during regeneration, thus resulting in differential abundance of a given cell cluster. This kind of transcriptional output would be the result of amputation position favoring differentiation into specific cellular states. To investigate this possibility, we performed differential abundance analysis using the bioinformatic tool Milo18 that computes differential abundance between two samples using k-nearest neighbor graphs and tests the statistical enrichment of cells for a given sample to partially overlapping neighborhoods (Figure 4A). We observed that some neighborhoods in all major cell types in the fin show statistically significant differential abundance between proximal and distal samples (FDR < 0.05). However, two cellular identities showed a higher proportion of differentially abundant neighborhoods: basal epidermis 2 (bsEp2) and mesenchymal 4 (mes4; Figure 4B). We further investigated the differential abundance bias in our caudal dataset where both homeostasis and regeneration samples correspond to equivalent anatomical regions of the tissue (Figure 3A). We found that bsEp2 is significantly more abundant during regeneration compared to homeostasis with all neighborhoods associated to this cluster identity showing significant abundance enrichment during regeneration (FDR < 0.05; Figure 4C). We reasoned that the overlap between the two differential abundance analyses reflect a cell differentiation bias influenced by both regeneration and amputation position. Moreover, we were able to filter out expected abundance biases such as the one observed for mes4 because there is no neighborhood within this cluster that displays abundance enrichment between regeneration and homeostasis (FDR < 0.05; Figure 4C). We believe the abundance differences captured by the proximal and distal comparison in the mes4 cluster are likely due to intrinsic morphological differences between both injuries that are not present in the regeneration and homeostasis comparison.
Figure 4.

Differential abundance analysis identifies fn1b+epcam+ regeneration-activated basal epidermis subpopulation
(A) Two-dimensional representation of differential abundance (DA) example analysis where two neighborhoods (colored circles) surrounding cells are depicted with black. Note that the algorithm performs the neighborhood analysis in a K-nearest neighborhood (KNN) graph.
(B) DA analysis of proximal and distal samples with statistically significant neighborhoods represented in color; the four most abundant cell types are displayed with the cluster definition from Figure S3. (FDR < 0.05), arrowheads correspond to basal epidermis 2 (bsEp2) and mesenchymal 4 (mes4) clusters.
(C) DA analysis of 12 hpa and homeostasis samples with statistically significant neighborhoods represented in color, displayed cell types, statistical significance, and arrowheads is the same as in (B).
(D) Dimensional reduction UMAP plots of regeneration-activated basal epidermis cell state across proximal, distal, 12 hpa, and homeostasis samples. In color is depicted the bsEp2 cluster on top of a background with the remaining cells of the sample.
(E) Heatmap of marker genes unique to the bsEp2 against all other clusters in the integrated dataset. No GO terms were found to be statistically enriched in this gene list (FDR < 0.05).
(F) Dimensional reduction UMAP distribution of subcluster analysis of bsEp2.
(G) Heatmap of downregulated genes that are common in the potential differentiation trajectory from basal epidermis to epidermis, alongside significant GO terms (FDR < 0.01).
(H) MAX projection of whole-mount HCR in situ hybridization of 1 dpa regenerating tail fin. fn1b is displayed in yellow, epcam in magenta, and DAPI in greyscale. White box corresponds to top image in panel (I). Scale bar: 1 mm.
(I) 10 μm MAX projection of the same sample depicted in (H) imaged at higher resolution. Scale bar: 250 μm. White squares correspond to fn1b+ expression domains within the tissue. Bottom left corresponds to mesenchymal cells housed within the bone plates of the fin. Bottom right is regeneration-activated basal epidermis (bsEp2) coexpressing the markers fn1b and epcam. Scale bar: 50 μm.
We further investigated the role of bsEp2 (Figure 4B, arrow) in transducing information of amputation position into the regenerating tissue. We observed that in the integrated UMAP space (Figure S3A), different from most other cell populations, bsEp2 displays a linear trajectory along the UMAP space in between basal epidermis 1 (bsEp1) on one end and proliferating epidermis 1 (proEp1) and epidermis 5 (ep05) populations on the other end (Figure S3B). We looked at the possibility that the differential abundance present between proximal and distal injuries comes from a delay in differentiation displayed as a bias toward one end of the differentiation trajectory. However, in both injuries, the bsEp2 cells are evenly distributed along the UMAP trajectory and it is only their numbers that differ between proximal and distal samples (Figure 4D). We looked for marker genes that distinguish bsEp2 from the rest of the cell clusters and found secreted molecules and modifiers of the extracellular space: fn1b, mmp13a, serpine2, and hbegfa. We also found genes responsible for cytoskeleton structure or remodeling: tpm4a, dapk3, plek2, lima1a, and myo9b (Figure 4E; Table S5). Altogether, our data indicate that differential abundance of cell states, rather than differential gene expression, is the most detectable change during regeneration at different amputation positions along the P/D axis of the caudal fin.
Regeneration-activated basal epidermis remodels the ECM at the injury site
To better understand the transcriptional output of the regeneration-activated bsEp2 subpopulation, we isolated these cells and redefined the PCA and UMAP embedding configurations. We observed three continuous subtypes of regeneration-activated basal epidermis that we interpret to be a differentiation trajectory between basal epidermis and mature epidermis (Figure 4F). We performed differential expression analysis alongside the differentiation trajectory presuming a fate transition from basal epidermis into mature epidermis. We found that 13% and 24% of DE genes were upregulated, whereas the corresponding 87% and 76% of DE genes were downregulated alongside the differentiation trajectory. The data are consistent with previous observations that regeneration promptly shuts down injury-activated transcriptional programs to shift the microenvironment from a scarring-inducing injury to a regeneration competent event.15,19,20 We further investigated the genes that are being downregulated across the regeneration-activated basal epidermis differentiation trajectory. We observed a significant enrichment for genes that are part of the extracellular matrix (GO:0031012), localize to the extracellular region (GO:0005576), participate during embryonic morphogenesis (GO:0048598), and mediate cell-cell signaling by Wnt (GO:0198738, Figure 4G; Table S6). Animals deficient in interleukin-11 signaling display impaired regeneration because of aberrant deposition of fibronectin in the regenerating hearts and poor mesenchymal migration during fin regeneration.21 Interestingly, we found a significant enrichment of the genes downregulated along the bsEp2 differentiation trajectory within the downregulated orthologues in the il11ra knockout mutant (padj < 0.001). These data predict a deficiency in regeneration-activated basal epidermis differentiation that leads to what the authors defined as mammalian-like fibrosis and failure to regenerate.21 We propose that the regeneration-activated basal epidermis at the plane of amputation undergoes mechanical distension during wound healing and upregulates genes known to remodel the cytoskeleton and ECM. Similar ECM remodeling programs during regeneration have been observed in mammalian skin fibroblasts.22
We used confocal microscopy to visualize the regeneration-activated basal epidermis at 1 dpa using in situ hybridization chain reaction (HCR23) probes targeting epithelial cell adhesion molecule (epcam, ENSNFUG00015018342) and fibronectin 1b (fn1b, ENSNFUG00015003666). We observed a robust upregulation of fn1b adjacent to the plane of amputation (Figure 4H). Interestingly, we observed two domains of fn1b expression that correspond to different cell populations: (1) epcam+ fn1b+ cells localized at the interface between the wound epidermis and the exposed mesenchyme and (2) epcam- fn1b+ cells localized inside of the bone one segment away from the plane of amputation (Figure 4I). This result made us return to look for fn1b+ mesenchymal cells in our integrated dataset; we found 617 cells scattered across all four mesenchymal clusters that accounts for 7% of the mesenchymal compartment. Because they do not separate from the main mesenchymal clusters, we conclude that there are not enough fn1b+ mesenchymal cells captured in our experiments to further investigate an fn1b+ regeneration-activated mesenchymal cell state.
The regeneration-activated fn1b+ basal epidermis is a transient cell state
We investigated the spatial and temporal distribution of fn1b+ basal epithelial cells in proximal and distal amputations by HCR in situ hybridization and antibody stains against E-cadherin (Ecad, a.k.a. Cdh1) and H3P. We observed fn1b expression in epithelial cells as soon as 3 hpa, localized to the amputation plane in cells that accumulate at the wound epidermis. At 3 hpa, both proximal and distal injuries show fn1b expression. Proximal injuries show fn1b− cells intermingling with fn1b+ epithelial cells. In contrast, distal injuries show a homogeneous population of epithelial cells expressing fn1b at the amputation plane (Figure 5A). At 6 hpa, there is an increase in fn1b+-expressing cells, and transcript localization shifted from nuclear to cytoplasmic. Both proximal and distal injuries display strong fn1b expression in multiple cell layers at the wound epidermis (Figure 5B). At 1 dpa, fn1b expression continues to localize to multiple cell layers but distal injuries display a reduced expression domain with sharper boundaries between the underlying mesenchyme and the most superficial fn1b− epidermal cell layers (Figure 5C). At 2 dpa, both proximal and distal injuries show narrow fn1b expression domains unambiguously labeling the boundary between the migrating mesenchymal blastema and the wound epidermis (Figure 5D). Moreover, at 3 and 5 dpa, fn1b expression is shut down (Figures S4A and S4B), indicating that the basal epidermal population that upregulates fn1b upon injury may be a TRACS.
Figure 5.

Regeneration-induced fn1b+ Ecad+ basal epithelial subpopulation is transient and non-proliferative
(A) Early nuclear fn1b expression detected by HCR in situ hybridization in wound epidermis (Ecad+, detected by immunofluorescence) at 3 hpa. Scale bar: 250 μm on the left and 25 μm on the right.
(B) Early nuclear and cytoplasmic fn1b expression detected by HCR in situ hybridization in wound epidermis (Ecad+, detected by immunofluorescence) at 6 hpa.
(C) Late cytoplasmic fn1b expression detected by HCR in situ hybridization in wound epidermis (Ecad+, detected by immunofluorescence) at 1 dpa.
(D) Late cytoplasmic fn1b expression detected by HCR in situ hybridization in restricted domain within wound epidermis (Ecad+, detected by immunofluorescence) at 2 dpa.
(E) Time course analysis of percentage fn1b+ cells in H3P+ Ecad+ population; ∗, FDR < 0.05, Wilcoxon rank-sum test. Polygon area represents mean ± SEM.
(F) Hole punch assay shows fn1b (HCR), Ecad (Ab), and H3P (Ab) stains at 1 dpa in proximal amputation position. Scale bar: 1 mm on the left and 100 μm on the right.
(G) Hole punch assay in distal amputation position showing the same as in (F).
(H) Spatial distribution of H3P and fn1b expression in proximal hole punch. Scale bar: 1 mm.
(I) Spatial distribution of H3P and fn1b expression in distal hole punch.
We next sought to define the distribution of fn1b+ basal epidermis on the orthogonal plane. We observed that throughout the time course all fn1b+ cells always localize to the wound epidermis where presumably basement membrane was lost after amputation (Figures S5A and S5B). Also, there are more fn1b+ cells in the inter-ray domain compared to the corresponding orthogonal plane along the fin ray, consistent with previous observations in zebrafish24 (Figures S5A–S5F). Furthermore, we observed a shift from an earlier time point when fn1b+ cells represent a multi-layer cell compartment to a single-layer compartment over time that delineates the growing tip of the mesenchyme (Figure 5D). We interrogated the proliferative nature of the regeneration-activated fn1b+ basal epidermis with H3P staining and observed that few fn1b+ are positive for H3P. We segmented H3P+ cells and classified them into Ecad+ fn1b+, Ecad+ fn1b−, Ecad− fn1b+, or Ecad− fn1b− cells using cytometry analysis (Figures 5E, 5F, and S4C–S4E). We quantified the percentage of fn1b+ cells within the H3P+ Ecad+ population and found that proximal injuries maintain fn1b expression within the proliferating epidermis for longer time compared to distal injuries (Figure 5E). This is not the case if we look at the Ecad− compartment (mesenchyme) where we see that the fn1b+ dividing cells quickly plummet over time independent of amputation position (Figure S4F). We measured the spatial distribution of the fn1b+ Ecad+ H3P+ cells and found that distal injuries maintain the same spatial distribution across 1 and 2 dpa (Figure S4H). However, proximal injuries display a narrower distribution for these cells at 2 dpa (Figure S4H). Lastly, we investigated the possibility of interaction between adjacent bones following injury. We performed punch amputation using a 1.5-mm diameter biopsy punch at proximal or distal positions and analyzed fn1b expression and H3P at 1 dpa. We observed robust fn1b expression all around the circumference of the punch in both proximal and distal injuries, with much higher expression at the interface between the wound epidermis and the bone fractures. However, fn1b expression was solely localized to fractured bones, which also displayed higher proliferation levels in contrast with adjacent bones that were not amputated (Figures 5F and 5G). Interestingly, we observed fn1b expression in a stab injury in the absence of exposed mesenchyme, but the expression domain was smaller and weaker compared to punch amputated fins (Figure S4G). We quantified the spatial distribution of proliferation as well as fn1b expression in the biopsy punch. We found in both proximal and distal injuries that the anterior side of the punch displays a clear separation between the fn1b expression domain and the proliferation domain (Figures 5H and 5I). Yet, the posterior side of the punch displays overlapping fn1b expression and proliferation. This suggests that the nature of the injuries and the formation of a blastema is different between the anterior and the posterior side of the punch as shown previously in zebrafish.25 Altogether, we observe that fn1b+ basal epidermal cells represent a TRACS with the potential to transduce positional information to the regenerating blastema. Once a blastema is formed (2 dpa), the basal epidermis TRACS loses fn1b expression, returning to a homeostatic basal epidermis cell state.
sqstm1KO decouples regeneration growth rate from amputation position
We sought to functionally test the ability of basal epidermis TRACS to transduce positional information at their corresponding amputation position. We identified the candidate gene sequestosome 1 (sqstm1, ENSNFUG00015002500) as a bsEp2 marker in our scRNA-seq experiments (Figure 6A). We deleted 524-bp fragment spanning the first exon of sqstm1 by CRISPR-Cas9-mediated transgenesis and successfully generated heterozygote and homozygote sqstm1 mutants (Figures 6B and 6C). All mutant fish developed normally, and we were able to breed both sexes. The sqstm1 mutant allele segregated with mendelian ratios, and diapause embryos responded to temperature in the same fashion than wild-type siblings. We observed sqstm1 expression as early as 6 h post-amputation localized to the wound epidermis with only a fraction of fn1b+ bsEp2 TRACS expressing sqstm1 (Figure 6D). Proximal injuries displayed a higher number of expressing sqstm1 cells, and at 24 h post-amputation, most sqstm1 expression had shut down, with few cells maintaining sqstm1 and fn1b expression localized to the interphase between the fn1b expression domain (bsEp2) and the wound epidermis (Figure 6E). We next quantified the regeneration growth in wild-type, heterozygote, and homozygote mutants (Figures 6F and 6G). We found that both heterozygote and homozygote sqstm1 mutants displayed an increase in regeneration growth exclusively in distal injuries (Figures 6H–6J).
Figure 6.

sqstm1 KO displays proximal phenotype in all amputation positions
(A) Feature plot of sqstm1 (ENSNFUG00015002500) with bsEp2 highlighted in green.
(B) Transgenic strategy to delete the first exon of sqstm1 locus. ChIP-seq tracks displaying normalized H3K4me3 and H3K27ac signal from published dataset.15
(C) Genotyping gel to identify wild-type (+/+), heterozygote (+/−), and homozygote (−/−) sqstm1 mutants; 1 kb Plus DNA ladder was run on the first well.
(D) Wild-type expression of sqstm1 (magenta) and fn1b (yellow) in proximal and distal injuries detected by HCR in situ hybridization at 6 hpa. Nuclear dye POPO-1 was used for counterstaining. Scale bar: 250 μm on the left and 25 μm on the right.
(E) Wild-type expression of sqstm1 (magenta) and fn1b (yellow) in proximal and distal injuries detected by HCR in situ hybridization at 24 hpa. Nuclear dye POPO-1 was used for counterstaining. Scale bar: 250 μm on the left and 25 μm on the right.
(F) Representative images of heterozygote sqstm1 mutant regeneration time course of proximal and distal injuries aligned at the amputation plane (magenta dotted line). The boundary between the fish scales and the caudal fin is indicated in orange dotted line. Scale bar: 1 mm.
(G) Representative images of homozygote sqstm1 mutant regeneration time course of proximal and distal injuries aligned at the amputation plane (magenta dotted line). The boundary between the fish scales and the caudal fin is indicated in orange dotted line. Scale bar: 1 mm.
(H) Regeneration growth measurements in wild-type siblings. Statistical significance is indicated for fixed effects of a linear mixed model. Polygon area represents mean ± SEM. ∗∗∗, p < 0.001 (see STAR Methods).
(I) Regeneration growth measurements in heterozygote sqstm1 mutant siblings. Statistical significance is indicated for fixed effects of a linear mixed model. Polygon area represents mean ± SEM. ∗∗∗, p < 0.001 (see STAR Methods).
(J) Regeneration growth measurements in homozygote sqstm1 mutant siblings. Statistical significance is indicated for fixed effects of a linear mixed model. Polygon area represents mean ± SEM. ∗∗∗, p < 0.001 (see STAR Methods).
Discussion
Amputation position controls at least three regeneration components
The position of amputation along the proximo/distal (P/D) axis of a limb has been shown to influence the magnitude (number of cells) and volume of pre-existing tissue that participate in the regenerative response. For instance, amputations at different P/D positions lead to differences in growth rate, number of proliferating cells, or number of cells expressing a regeneration-induced gene.7,11,26 It is well documented that gene expression domains expand in relation to the amputation position, which in turn has been associated with the magnitude of gene expression: the more cells mounting a regeneration response, the larger the expression domain.7,12,27 Careful quantitative analyses, however, have provided an alternative interpretation where independent of the number of cells that participate during regeneration, amputation position influences the spatial distribution of these cells. It was shown in axolotls that amputation position determines how far away into the pre-existing tissue progenitor cells will migrate to contribute to the regenerating outgrowth, supporting the idea that positional information defines the distance from which cells contribute to regeneration.28
Our results are consistent with amputation position affecting the distribution and persistence of transient tissue changes during regeneration. We found that proliferation profiles are defined by amputation position (Figure 2G). The location of the highest mitotic density (peak proliferation) is proportional to the amputation position, and the size of blastema at 1 dpa also scales linearly with amputation position (Figures 2G–2I). We believe both magnitude (i.e., number of dividing cells or number of cells expressing fgf20) and location (i.e., peak proliferation at 150 μm from the amputation or cell migration to the blastema) represent two dimensions of regeneration that are influenced by positional information.
Duration of regeneration processes, on the other hand, has been more challenging to address because of the temporal resolution needed to capture regeneration transitions. It has been shown that osteoblast differentiation is accelerated during regeneration4 and that cell-cycle length can also be altered during regeneration.17,29 In our study, we captured temporal shifts in regenerative outgrowth acceleration (Figure 1G), tissue-wide proliferation shut-down (Figure 2C), and gene expression (Figure 5G). We observed that these processes last different amounts of time relative to amputation position. The current model considers magnitude and distribution to be the effectors of positional information during regeneration, but here we present persistence as a third component that enables regeneration to respond to positional information. To our knowledge it has not been unambiguously shown that amputation position defines not only the magnitude and extent of tissue involved but also the duration of the biological processes launched during regeneration. Whether there are different mechanisms behind each component or they share a common regulator remains to be experimentally tested.
Injury context defines epidermal regeneration cell states
A growing body of evidence indicates that transitional cell states arise following amputation to orchestrate the necessary steps for successful regeneration.30,31,32 Injury-induced cellular states have been shown to lead to a spatial compartmentalization of cellular behavior both near and far from the place of insult.33,34,35 For instance, it has been shown in planarians that Erk signaling propagates tissue-wide during regeneration and that regeneration ability is lost when its propagation is impaired.35 Furthermore, in mammalian skin, the cycling characteristics of epidermal progenitor clones are differentially regulated in areas surrounding or away from growing hair follicles, suggesting that location influences spatiotemporal control of cell potencies.36 We found that upon amputation, fin epidermis transitions into a cell state characterized by a transcriptomic signature enriched in DNA synthesis and ribosome biogenesis genes, irrespective of whether the epidermis was adjacent to an amputation or resided in an uncut fin (Figures 3D and 3H). Our data show that amputation triggers a cell state transition in the epidermis in very distant tissues that were never cut. Interestingly, such cell states were defined by different epidermal subclusters in cut and uncut fins, with the cell state shift of the epidermis in a distant uncut organ restricted to cells that were already committed to undergo cell division (e.g., pcna+ and ccna+ cells). We propose that regeneration induces a cell state that primes the epidermis to prepare for rapid proliferation, and the presence of a wound within the same organ pushes the tissue to further recruit epidermal cells that do not show a proliferative phenotype. Whether distant transcriptional responses captured in our study contribute to a feedback mechanism to integrate regenerative signals remains to be investigated.
Basal epidermis deploys cell states that are dependent on amputation position
We detected small transcriptional changes when comparing scRNA-seq data from distal and proximal amputations (Figures 3K and 3L). However, when we compared cell-type abundance between distal and proximal amputations, we identified large transcriptional changes in one of the two basal epidermal clusters (Figures 4B and 4C). Because the cells shared cell-type identity with basal epidermis but differed in gene expression enrichment depending on whether the fins were cut proximally or distally, we concluded that these differences likely corresponded to different cell states of the same cell type. Moreover, we failed to detect this basal epidermal cell state during fin homeostasis (Figure 4D), indicating that the identified cell state exists only during regeneration.
The identified basal epidermal cell state was characterized by significant enrichment of ECM modifiers and components such as fibronectin (fn1b), Wnt ligands, and genes associated with embryonic morphogenesis. Genes encoding ECM components have been shown to be upregulated during regenerative outgrowth,37 and fn1b has been previously identified as a regenerating basal epidermis marker gene.38 Moreover, disruption of basement membrane components is known to reduce regenerative ability,39 and TRACSs have been shown to upregulate gene expression of matrix metalloproteinases that is required for whole-body regeneration.30 Finally, we followed the basal epidermal cell state over time and found it to be transient (Figures 5A–5D, S5A, and S5B). Interestingly, the fn1b+ basal epidermal TRACS uncovered in this study may be orthologous to one of the two fn1b+ epidermis populations observed previously in zebrafish, where early fn1b+ epidermis was transient and later fn1b+ epidermis contributed to resident epithelial cells.24 Our data suggest, therefore, a previously unsuspected role for TRACS in vertebrate regeneration.
Regeneration growth rate can be decoupled from amputation position
We report that CRISPR-Cas9-mediated deletion of the first exon of sqstm1 increases the ability of distal injuries to regenerate (Figures 6F–6J). We conclude that amputation position controls regeneration growth rate by an inhibitory mechanism that represses regeneration in distal amputations. Mutants defective in sqstm1 display high regeneration growth rates independent of amputation position, which suggests that regeneration growth rate is saturated in the mutants, and it can be uncoupled from positional information. Interestingly, fin bone patterning has also been shown to be uncoupled from regenerative outgrowth,40 suggesting that positional information encompasses a complex relationship of modular regeneration programs. Furthermore, we observed sqstm1 expression at early time points not only in fn1b+ basal epidermis TRACS but also in fn1b− suprabasal epidermal cells that localize to the wound epidermis (Figures 6D and 6E). The compartmentalization of fn1b and sqstm1 expression in partially overlapping epidermal cells suggests a differentiation trajectory where fn1b+ basal epidermis TRACS are destined to die by apoptosis through an autophagy mechanism mediated by Sqstm1, as suggested by others in zebrafish.24 Interestingly, SQSTM1-dependent autophagy has been shown to promote FN1 degradation in human cancer cells.41 We hypothesize that the sqstm1KO mutant fails to eliminate fn1b+ basal epidermis TRACS, thus maintaining a more persistent ECM remodeling phenotype and forming blastemas with saturated regeneration growth rate. Yet, the role of ECM remodeling in blastema formation, the existence of a saturated regeneration growth rate, and the function of Sqstm1 mediating apoptosis of fn1b+ basal epidermis TRACS remains to be functionally tested.
Basal epidermis reads and writes positional information during blastema formation
We present a model for the role of the fn1b+ basal epidermis TRACSs on reading and writing positional information (Figures 7A–7C). In this model, basal epidermis cells adjacent to the amputation plane transition to a TRACS following loss of cell polarity by the absence of basement membrane and mechanical distension caused by wound closure.13 We propose that the degree of force applied to the epidermis is proportional to the thickness of the bone at the plane of amputation, along with the corresponding surface area between exposed mesenchyme and wound epidermis, leading to a proportional number of basal epidermal cells to acquire a basal epidermis TRACS. The transient cell state in turn remodels the extracellular space at the plane of amputation to recruit a correct number of migrating mesenchyme, resulting in the formation of a concomitantly scaled regeneration blastema. We propose, therefore, that the differences in persistence of proliferation (Figure 2C), the spatial distribution of dividing cell in the tissue (Figure 2G), and the resulting number of fn1b+ basal epidermis TRACS along the P/D axis, all play a role in codifying the corresponding positional information that results in properly scaled regeneration growth rates. The use of time to impart positional information during embryonic development has been proposed by theorists to influence positional precision.42 Previously, it has been suggested that the epidermis does not play a role in conveying positional information during regeneration.43 However, our study highlights that transient epidermis cell states are part of a complex multicellular process (blastema formation) that regulates the speed of regeneration relative to amputation position. Altogether, our findings have revealed a role for position of amputation in regulating the duration of a proliferative state in injured tissues as well as transient cell states that may relay positional information in animal regeneration.
Figure 7.

Model for positional information relay during regeneration by basal epidermis
(A) Overview of the fin and proximal and distal definition in this study relative to pigment pattern. Thickness of the fin drawn to scale is showed at the bottom of the image. Scale bar: 2 mm.
(B) Color definition for the model presented in (C) and bone thickness comparison between proximal and distal injuries.
(C) Model for de novo deposition of positional information during regeneration by basal epidermis transient regeneration-activated cell state.
Limitations of the study
Our study uncovered new dynamics of proliferative activities in injured tissues and the generation of a well-defined cell state of basal epidermis cells in response to injury. However, these reproducible and measurable responses await mechanistic analyses to determine whether they are causal to or correlated with regeneration. Still needed are genetic gain- or loss-of-function studies. In our efforts to genetically manipulate the TRACS, we failed to generate mutants of genes expressed in TRACS that were not embryonic lethal except for sqstm1, suggesting the need for conditional mutant alleles for the study of regeneration in adults. Additionally, our study was solely conducted in males; therefore, further experimentation is needed to test any sex possible differences.
Resource availability
Lead contact
Requests for reagents and resources should be directed to and will be fulfilled by the lead contact, Alejandro Sánchez Alvarado (asa@stowers.org).
Materials availability
This study did not generate new unique reagents, however reagents presented in this study are available from the lead contact with a completed materials transfer agreement and with reasonable compensation by requestor for its processing and shipping.
Data and code availability
-
•
Original data underlying this manuscript is publicly available and can be accessed from the Stowers Original Data Repository at http://www.stowers.org/research/publications/libpb-2453. Single cell RNA-seq data have been deposited at GEO and are publicly available under accession number GSE260629 at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE260629.
-
•
All original code has been deposited at Zenodo and is publicly available under https://doi.org/10.5281/zenodo.10725332.
-
•
Any additional information required to reanalyze the data reported in this work is available from the lead contact, Alejandro Sánchez Alvarado (asa@stowers.org).
Acknowledgments
We thank T. Piotrowski, R. Barajas Azpeleta, C. Barradas Chacón, A. Accorsi, V. Doddihal, F. G. Mann, and A. Karabulut for critical reading of the manuscript; current and past members of the Sánchez Alvarado lab at SIMR; T. Piotrowski, R. Krumlauf, and N. Rohner for insightful scientific discussion; and current and past members of the Technology Centers at SIMR who provided technical support. Vector graphics from Ibrandify (Freepik). We thank J. Toyoshiba for support with professional photography of the killifish. We thank the Behrensen and Guzmán Palma families for cultivating scientific excellence and immense kindness in our dear friends Camila and Pablo. We thank the KUMC Genomics Core for generating data on the Illumina NovaSeq 6000 System. The KUMC Genomics Core is supported by the Kansas Intellectual and Developmental Disabilities Research Center (NIH U54 HD 090216), the Molecular Regulation of Cell Development and Differentiation—COBRE (P30 GM122731-03), and the NIH S10 High-End Instrumentation Grant (NIH S10OD021743). This project was funded by the Stowers Institute for Medical Research and the Howard Hughes Medical Institute. Original data underlying this manuscript can be accessed from the Stowers Original Data Repository at: http://www.stowers.org/research/publications/libpb-2453.
Author contributions
Conceptualization, A.O.G., W.W., and A.S.A.; methodology, A.O.G., D.Z., R.R.S., A.R.S., J.R., K.F., J.A.M., and B.Y.R.; software, A.O.G., J.R., C.E.B., E.J.R., D.A.A., and B.Y.R.; validation, A.O.G.; formal analysis, A.O.G. and N.Z.; investigation, A.O.G., D.Z., R.R.S., A.R.S., A.S., J.R., D.A.A., K.F., B.Y.R., and W.W.; resources, D.Z., R.R.S., C.E.B., E.J.R., D.A.A., N.Z., J.A.M., A.G.P., and A.S.A.; data curation, A.O.G. and E.J.R.; writing—original draft, A.O.G. and A.S.A.; writing—review & editing, A.O.G. and A.S.A.; visualization, A.O.G.; supervision, A.O.G., R.R.S., A.G.P., W.W., and A.S.A.; project administration, A.O.G., R.R.S., A.G.P., and A.S.A.; funding acquisition, A.G.P. and A.S.A.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-Histone H3 (Ser10) (D2C8) XP Rabbit mAb #3377 | Cell Signaling Technology | RRID#AB_1549592 |
| Purified Mouse Anti-E-Cadherin | BD Transduction Laboratories | RRID#AB_397581 |
| F(ab')2-Goat anti-Mouse IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | Cat#A48286 |
| F(ab')2-Goat anti-Mouse IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 555 | Thermo Fisher Scientific | Cat#A48287 |
| Chemicals, peptides, and recombinant proteins | ||
| MS-222 | Sigma-Aldrich | Cat#E10521 |
| PFA | Electron Microscopy Sciences | Cat#15710 |
| Ethyl alcohol | Sigma-Aldrich | Cat#E7023 |
| H2O2 | Sigma-Aldrich | Cat#H1009 |
| Formamide | Sigma-Aldrich | Cat#S4117 |
| Goat Serum | Thermo Fisher Scientific | Cat#16210072 |
| Western Blocking Reagent | Sigma-Aldrich | Cat#11921673001 |
| Horse Serum | Thermo Fisher Scientific | Cat#26050070 |
| DMSO | Sigma-Aldrich | Cat#472301 |
| NaN3 | Sigma-Aldrich | Cat#S2002 |
| FBS | Thermo Fisher Scientific | Cat#A5256801 |
| DAPI | Thermo Fisher Scientific | Cat#62248 |
| YOYO-1 Iodide | Thermo Fisher Scientific | Cat#Y3601 |
| EasyIndex | LifeCanvas Technologies | N/A |
| Probe Hybridization Buffer | Molecular Instruments | N/A |
| Probe Wash Buffer | Molecular Instruments | N/A |
| Amplification Buffer | Molecular Instruments | N/A |
| Bovine Serum Albumin | Fisher Scientific | Cat#BP9706100 |
| Collagenase type 2 | Worthington Biochemical | Cat#LS004174 |
| Draq5 | Biostatus | Cat#DR50200 |
| Poloxamer 188 | Sigma-Aldrich | Cat#P5556 |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 3′ Reagent Kits v3.1 | 10x Genomics | Cat#PN-1000128 |
| 3ʹ CellPlex Kit Set A | 10x Genomics | Cat#PN-1000261 |
| Chromium Next GEM Single Cell 3′ Reagent Kits v3.1 with Feature Barcode technology for Cell Multiplexing | 10x Genomics | Cat#PN-1000269 |
| Deposited data | ||
| Original data | Stowers Original Data Repository | http://www.stowers.org/research/publications/libpb-2453 |
| Single cell RNA-seq | GEO GSE260629 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE260629 |
| Code | Zenodo | https://doi.org/10.5281/zenodo.10725332 |
| Experimental models: organisms/strains | ||
| African turquoise killifish ZMZ-1002 wild type strain | Sánchez Alvarado Lab | N/A |
| Oligonucleotides | ||
| gRNA-1: sqstm1: TGGACTTGATGTTTGAGTTATGG | This paper | N/A |
| gRNA-2: sqstm1: CGGCAATGGTCCCTCATTAGCGG | This paper | N/A |
| genotyping primer Fw: sqstm1: TTGATGAGGGAGCCCGAAAG | This paper | N/A |
| genotyping primer Rv: sqstm1: ATGACTAAGCGGCTTTCCCAA | This paper | N/A |
| Software and algorithms | ||
| MicroManager | https://micro-manager.org/ | RRID#SCR_016865 |
| Linear Stack Alignment with SIFT | https://imagej.net/plugins/linear-stack-alignment-with-sift | N/A |
| Fiji | https://fiji.sc/ | RRID#SCR_002285 |
| Grid/Collection stitching Plugin | https://imagej.net/plugins/grid-collection-stitching | N/A |
| H3P segmentation | Bio-protocol 13(24): e4908 | https://doi.org/10.21769/BioProtoc.4908 |
| Proliferation profile | This paper | N/A |
| 10x Genomics Cell Ranger v6.0.1 | https://www.10xgenomics.com/support/software/cell-ranger/latest/release-notes/cr-release-notes | N/A |
| R v4.2.3 | https://www.r-project.org/ | RRID#SCR_001905 |
| Seurat v4.3.0 | https://satijalab.org/seurat/articles/get_started.html | RRID#SCR_016341 |
| clusterProfiler v4.6.2 | http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html | RRID#SCR_016884 |
| miloR v0.1.0 | https://marionilab.github.io/miloR/index.html | N/A |
Experimental model and study participant details
Animals
African killifish Nothobranchius furzeri were reared at the Stowers Institute and all animal procedures were performed with IACUC and IBC approval (Protocol ID: 2022-137, IBC Registration: IBC-2015-01-asa). The aquatic animal program meets all federal regulations and has been fully accredited by AAALAC International since 2005. Killifish were single housed in polycarbonate tanks (1.4 L), with a 14:10 h light:dark photoperiod. All experiments were performed with the ZMZ1002 wild type line. Only males 2 to 3 months-old were used for our study because of pigment pattern and body size.
Method details
Fin amputation
Experimental fish were anesthetized using 200 mg/L MS-222 (Sigma-Aldrich, Cat. E10521) for 3 min and placed on top of a plastic Petri dish lid (VWR, Cat. 25384-302). Fins were amputated using a disposable razor blade (VWR, Cat.55411-050) or a rapid core punch (World Precision Instruments, Cat. 504647) at the experimental plane of amputation (Figure S1A). After amputation the fish was placed back in its housing tank and observed for recovery within 5 min.
Longitudinal analysis of growth rate, imaging, and quantification
Experimental fish were anesthetized using 200 mg/L MS-222 for 3 min and placed on their right side on top of a plastic Petri dish lid. Glass coverslip (Epredia, Cat. 152222) was placed on top of the tail to avoid water reflection and room temperature system water was added to fully immerse the tail in water. Images were collected every 12 h for the first 5 days of regeneration and every 24 h for the remaining of the experiment. Images were acquired with a INFINITY3-6URC (Lumenera) camera on a Leica M205 FCA microscope with a Leica PlanApo 0.63X objective controlled by MicroManager (RRID: SCR_016865),44,45 white was balanced with a clean kimwipe (Kimberly Clark, Cat. 34155) and the background was set dark with reflective light coming from both the bottom and the top. After image acquisition the fish was placed back in its housing tank and observed for recovery within 5 min.
For image quantification we first integrated the image sequence into a stack that was registered using Linear Stack Alignment with SIFT.46 The bones to quantify and the base of the fin where the scales end was outlined by hand with the "Straight" and the "Oval" tools in Fiji (RRID: SCR_002285).47 The edge of the fin was outlined by traditional thresholding of the images to create a mask, the mask was then subtracted from a one pixel dilate of itself to create a new image with the Edge location as a separate channel on the stack. For each bone the intersection between the oval (fish scales) and the straight ROIs was centered and the image was rotated to align the bone with the x axis. A crop of the preexisting bone tissue was used to register the movie using StackReg48 and the registration matrix was propagated to the rest of the stack. The distance between the scales and the bone fracture is defined as the "bone length" and the distance between the bone fracture and the edge of the tissue is defined as the "regeneration length". Growth rate measurements were done by subtracting the average regeneration length on one timepoint from the antecedent timepoint and dividing the result by the length of time between the two timepoints, growth rate is expressed in millimeters per day. Changes in growth rate over time is defined as "growth acceleration", and it was calculated using a linear regression of at least three timepoints to measure the slope of the linear model.
Sample collection and bleaching for IF, HCR or HCR-IF
Fish were euthanized for 5 min with 500 mg/L MS-222 followed by hypothermic shock in 4°C cold system water for 30 min, in between the MS-222 treatment and the hypothermic shock, fin of interest was collected and fixed with 4% PFA (Electron Microscopy Sciences, Cat. 15710) 0.1% Tween 20 1X PBS for 4 h at room temperature. The fins were dehydrated in 25%–50%-75%–100% EtOH (Sigma-Aldrich, Cat. E7023) train in PBST (0.1% Tween 20 1X PBS) for at least 4 h per step. Fins were left for at least one night in 100% EtOH (200 proof), this step is very important for enabling reagent penetration and successful bleaching. A first bleaching step was done with 6% H2O2 (Sigma-Aldrich, Cat. H1009) 80% EtOH under an LED lamp (3W) for 12 h at room temperature, then rehydration was done on the reverse train 50%-25%-EtOH-PBST for at least 4 h per step. A second bleaching step was done with 3% H2O2 5% formamide (Sigma-Aldrich, Cat. S4117) 0.1% Tween 20 in 1X SSC under an LED lamp (3W) for 1 h at room temperature. Two 5-min washes were done with PBST before proceeding with staining.
IF staining
Following bleaching, samples were blocked overnight with 10% Goat Serum (Thermo Fisher, Cat. 16210072), 5% Western Blocking Reagent solution (Sigma-Aldrich, Cat. 11921673001), 2.5% Horse Serum (Thermo Fisher, Cat. 26050070), 5% DMSO (Sigma-Aldrich, Cat. 472301), 123 mM NaN3 (Sigma-Aldrich, Cat. S2002) in PBST. Two 5-min washes with PBST were done after blocking and the samples were incubated with primary antibody solution: anti-H3P at 1:400 dilution (Cell Signaling Technology, catalog number: 3377S, RRID: AB_1549592), anti-Ecad at 1:200 dilution (BD Biosciences, catalog number: 610182, RRID: AB_397581) diluted in 10% FBS (Thermo Fisher, Cat. A5256801), 5% DMSO, 123 mM NaN3 in PBST for 24 h at room temperature in roller shaker (Cole-Parmer, Cat. UX-51901-23). Four 20-min washes with PBST at room temperature were done in between primary and secondary antibody incubation. Secondary antibody solution: anti-rabbit AF488 at 1:400 dilution (Thermo Fisher, Cat. A48286), anti-mouse AF555 at 1:500 dilution (Thermo Fisher, Cat. A48287) diluted in 10% FBS, 5% DMSO, 123 mM NaN3 in PBST for 24 h at room temperature in roller shaker protected from ambient light. Six 20-min washes with PBST at room temperature were done afterward. Nuclear staining was done with 1 μg/mL DAPI (Thermo Fisher, Cat. 62248), or 1nM YOYO-1 Iodide (Thermo Fisher, Cat. Y3601) in PBST overnight at room temperature in roller shaker, and two 20-min washes with PBST at room temperature were done prior to clearing. Overnight tissue clearing was done with 1mL of EasyIndex (LifeCanvas Technologies) and the sample was mounted on round coverslip bottom petri dishes (Fisher Scientific, Cat. 50-305-805) in the same medium that was used for clearing, an additional glass coverslip was placed on top of the sample and volume was minimized to get the sample to lay flat at the bottom of the Petri dish.
HCRv3-IF staining
Following bleaching, samples were pre-hybridized with 500μL probe hybridization buffer (Molecular Instruments) preheated at 37°C for 30 min. In the meantime, probe solution was prepared by adding 4μL of each probe set to 500 μL hybridization buffer preheated at 37°C. Hybridization was done for 24 h at 37°C in rocking shaker (Fisher Scientific, Cat. 88-861-025) with the tubes oriented vertically on a rack so the tissue is not damaged. Samples were washed 4 times for 15 min with probe wash buffer (Molecular Instruments) at 37°C in rocking shaker, followed by 2 washes for 15 min each with SSCT at room temperature in roller shaker. Pre-amplification was done with 500 μL of room temperature amplification buffer (Molecular Instruments) for 30 min in roller shaker. In the meantime, snap cooling of h1 and h2 harpins corresponding to AF647 and AF546, were incubated at 95°C for 90 s and then cooled at room temperature for 30 min in the dark. Amplification solution was prepared by adding h1 and h2 harpins in 500 μL amplification buffer at room temperature. Samples were incubated in amplification solution for 24 h at room temperature in roller shaker, washed 5 times with 0.01% Tween 20 1X SSC, and washed once with PBST for 15 min per wash at room temperature in roller shaker. Samples were fixed with 4% PFA 0.1% tween PBST for 1 h at room temperature and washed once with PBST for 15 min at room temperature in roller shaker. For HCR-only samples we counterstained with nuclear stain, cleared and mounted the same way than IF (see above). For immunofluorescence following HCR we skipped the blocking step and went straight to primary antibody incubation and followed the IF staining protocol (see above).
Confocal imaging
Images were acquired with an Orca Flash 4.0 sCMOS 100fps at full resolution on a Nikon Eclipse Ti microscope equipped with a Yokagawa CSU W1 10,000 rpm Spinning Disk Confocal with 50 μm pinholes. Samples were illuminated with 405nm(5.73mw), 488nm(6.75mw), 561nm(5.84mw) and 640nm(6.62mw) lasers (LUNV 6-line Laser Launch) with nominal power measures at the objective focal plane. This spinning disk confocal is equipped with a quad dichroic filter for excitation with 405/488/561/640nm. Emissions filters used to acquire this image were 430–480 nm for DAPI or YOYO-1, 507–543 nm for AF488, 570–640 nm for AF555 or AF546 and 662.5–737.5 nm for AF647.
For Figure 2, we used a Nikon Plan Apochromat Lambda 10x objective lens, N.A. 0.45, 0.78 μm/px objective with 200 ms exposure for IF channels, and 100 ms for DAPI channel at 3 μm z resolution. For Figures 4, 5, S2, S4, and S5 high magnification images we used a Nikon Plan Apochromat Lambda LWD 40× objective lens, N.A 1.15, 0.283 μm/px with 300 ms exposure for HCR channels and 100 ms for DAPI channel at 3 μm z resolution. For Figure 5 low magnification images we used a Nikon Plan Apochromat Lambda 4× objective lens, N.A. 0.2, 1.735 μm/px with 200 ms exposure for HCR channels and 50 ms for YOYO-1 channel at 25 μm z resolution.
H3P segmentation and cytometry analysis
Tiled images were stitched using the Grid/Collection stitching Plugin49 from Fiji, and X, Y, and Z coordinates were identified for H3P positive nuclei segmented following the pipeline published elsewhere.50 Quadrants in Figure S4C (cytometry analysis) was done using fluorescence minus one (FMO) negative fn1b (HCR) and Ecad (IF) controls.
Proliferation profile build
The analysis starts with segmentation of the tail into regions—each region contains exactly one ray and number of regions is equal to number of rays. The ray is determined by slope and intercept of the line determining the bone in the Cartesian coordinates. The i-th region boundaries (for all regions except the first and the last) are computed as a straight line representing a bisector cutting the angle between the i-th and (i+1)-th rays. The upper boundary of the first (upper) region and the lower boundary of the last (lowest) region are computed to make the rays inside the region to be its bisector. For each region we find all cells with coordinates X, Y belonging to this part of the plane using the data from the segmentation step (see above).
As region boundaries are not parallel lines, we can determine their intersection point that is served as the region center. Using this point we convert all X, Y coordinates of each point of interest (cells, scales, edge points, amputation points etc.) into two other numbers r and ‘Y to bone’ (the last one is ready for the output). The radial coordinate r is determined as a distance between the center and the given point, while the ‘Y to bone’ coordinate is measured as distance from the point to the bone along the normal to the bone. We use the ray line to compute the distance between two points along this line (Figure S2B).
Note that there are two types of regions – the central regions that have amputation points and the side regions free of such points. The regions of both types are characterized by the sequence of edge points. These special points are used for a special (scaled) measure of the cell position. Specifically, when the region has amputation points, we find the average radial distance between the region center and all amputation points. When we use the edge points, we generate a polynomial approximation R(ϕ) of the curve corresponding to the ordered sequence of the edge points in the region boundary. The function R(ϕ) determines the dependence of the edge point radial distance from the region center as a function of the angular polar coordinate ϕ.
Then the following distances along the ray are computed: from the cell to the edge, from the cell to the averaged amputation position, these values allow to produce ‘r bone Amp’ and ‘r bone Edge’. We also find the distance from the scale center to the edge, from the bifurcation point to the edge, from the amputation point to the edge to be added to corresponding output files.
Once we generated distance measurements of each given H3P positive nuclei to the amputation plane parallel to the bone, we bin the bone axis every 50 μm, and we counted the H3P positive nuclei within each bin to generate heatmaps of proliferation distribution (Figure 2C). Next, we iteratively slide the 50 μm bin for 1 μm at a time along the bone axis to generate a proliferation density profile along the bone dimension, the peak proliferation (Figure 2G), corresponds to the highest value of the proliferation density profile after a median blur (kernel = 3).
Single cell dissociation, CellPlex labeling and flow cytometry
Fish were euthanized at the expressed time point and tissues of interest were rinsed in cold 0.1% BSA (Fisher Scientific Cat. BP9706100) 1X PBS (PBS-BSA). The tissue was minced using a fine blade and single cell suspension was achieved by dissociation in 10mL 1 mg/mL collagenase type 2 (Worthington Biochemical, Cat. LS004174) for 5 mins at 37dC followed by 70 μm filtration (Fisherbrand, Cat. 22-363-548). The cells were rinsed by adding 25mL 0.1% BSA 1X PBS (PBS-BSA) and centrifuged for 3 min at 500 g at 4C with slow deacceleration (slow deacceleration will help keep the pellet intact).
A small aliquot of the cells was counted using 500 nM Draq5 (Biostatus, Cat. DR50200) solution in an EC800 flow cytometer. For regular 10x run, cells were stained with 1 μg/mL DAPI, 500 nM Draq5 in PBS-BSA at 4e6 cells/mL and sorted as described below. For CellPlex 10x run, cells were labeled with 3ʹ CellPlex Kit Set A (10x Genomics, PN-1000261) according to the manufacturer’s directions. A small aliquot was counted one more time using 500 nM Draq5 solution in an EC800 flow cytometer, and different samples were pooled together to balance the cell pool equally across samples. Then cells were stained with DAPI-Draq5 (same as above) at 4e6 cells/mL and sorted as described below. We employed CellPlex to collect biological replicates of all our conditions as well as prevent batch effects between samples within a multiplex experiment.
For cell sorting, the sample buffer and the cell sorter sheath fluid contained 0.1% Poloxamer 188 (Sigma-Aldrich, Cat. P5556) to reduce shear stress and improve post-sort cell viability. Sorting of live cells was performed on a 6-laser BD S6 FACSymphony equipped with a 100-μm nozzle, and chilled to 4 °C at all times. Because the Draq5-DAPI staining produced no spillover into the target detectors on this cell sorter, no compensation was performed. Due to the heterogeneous nature of the sample, an FSC/SSC scatter gate was not used to avoid biasing the sorted sample for specific cell types, instead we triggered acquisition with Draq5 to capture all nucleated events. The Draq5-DAPI staining pattern displayed minor shifts over the course of the sort, which was accounted for by adjustments of the sort gate. To ascertain sample quality, a post-sort viability assay was performed on the BD S6 FACSymphony prior to further processing of each sample. The sorter was flushed with sample buffer (cold 0.1% BSA 1X PBS) for 1 min between samples to remove leftover nuclear stain and prevent secondary staining. Samples with post-sort viability >96% were used for library preparation and sequencing.
scRNAseq library preparation and sequencing
For regular 10X
One proximal and one distal single cell library were generated using conventional methods. Dissociated, sorted cells were assessed for concentration and viability via Luna-FL cell counter (Logos Biosystems). Cells deemed to be at least 97% viable were loaded on a Chromium Single Cell Controller (10x Genomics), based on live cell concentration. Libraries were prepared using the Chromium Next GEM Single Cell 3′ Reagent Kits v3.1 (10x Genomics) according to manufacturer’s directions. Resulting cDNA and short fragment libraries were checked for quality and quantity using a 2100 Bioanalyzer (Agilent Technologies) and Qubit Fluorometer (Thermo Fisher Scientific). With cells captured estimated at ∼8,000–9,000 cells per sample, libraries were pooled and sequenced to a depth necessary to achieve at least 42,000 mean reads per cell on an Illumina NovaSeq 6000 instrument utilizing RTA and instrument software versions current at the time of processing with the following paired read lengths: 28∗10∗10∗90bp.
For CellPlex 10x
Three multiplexed libraries were generated encompassing: 1) regeneration and homeostasis samples, 2) dorsal samples, and 3) proximal and distal samples. The CellPlex labeled, pooled cell samples were assessed for concentration and viability via Luna-FL cell counter (Logos Biosystems). Samples with cells deemed to be at least 97% viable were loaded on a Chromium Single Cell Controller (10x Genomics), based on live cell concentration targeting 20,000, 30,000 or 50,000 cells. Libraries were prepared using the Chromium Next GEM Single Cell 3′ Reagent Kits v3.1 with Feature Barcode technology for Cell Multiplexing (10x Genomics) according to manufacturer’s directions. Resulting cDNA, short fragment libraries, and CMO libraries were checked for quality and quantity using a 2100 Bioanalyzer (Agilent Technologies) and Qubit Fluorometer (Thermo Fisher Scientific). Multiplexed gene expression and CMO libraries were pooled, as specified by manufacturer, and sequenced to a depth necessary to achieve at least 26,000 mean reads per cell on an Illumina NovaSeq 6000 instrument utilizing RTA and instrument software versions current at the time of processing with the following paired read lengths: 28∗10∗10∗90bp.
scRNAseq alignment primary analysis
Fastqs were demultiplexed using cellranger mkfastq (10x Genomics Cell Ranger51 6.0.1) with default settings. Cellranger multi (10x Genomics Cell Ranger 6.0.1) was used to align the fastqs against Ensembl52 104 Nfu_20140520 and filter, count barcodes and UMIs. Feature-barcode matrices were analyzed as described below.
scRNAseq quality control, integration, differential expression analysis and GO enrichment
Demultiplexed sample feature count matrices were loaded into R using Seurat53 (Seurat 4.3.0), mitochondrial percentages were calculated using the PercentageFeatureSet function and a median + 2 sd threshold was use to filter out cells with high mitochondrial gene expression, the resulting percent.mt distribution in the integrated object had a median of 3.9% and the highest value was 21.5%. Low nFeature_RNA count was filtered with median - 1.2 sd and high nFeature_RNA count was filtered with median + 4 sd, the resulting nFeature_RNA distribution in the integrated object had a median of 1536 and the highest value was 5259 nFeature_RNA. We decided to apply relative statistical thresholds to compensate between batch effects and sequencing depth between samples considering doublets were removed during demultiplexing on the previous step. After quality control, all samples were normalized individually using the SCTransform function regressing percent.mt from the model. All the samples for integration were put on a list and anchor features were identified with SelectIntegrationFeatures (nfeatures = 8000), samples were prepared for integration using the function PrepSCTIntegration and the anchor features, and integration anchors were identified with the function FindIntegrationAnchors using the list containing the samples, SCT as the normalization method and the anchor features. Finally, all samples were integrated using the function IntegrateData with the integration anchors and SCT as the normalization method. Once integrated, RunPCA, RunUMAP and FindNeighbors functions were run on the integrated assay, and clusters were computed with the FindClusters function by iteratively changing the cluster resolution from 0.2 to 2 in 0.1 intervals. Cell markers of differential expression analysis was done using the FindMarkers function and selecting the subset of cells to compare to each other. GO enrichment was performed using the enrichGO function from the clusterProfiler package54 (clusterProfiler 4.6.2) and a custom killifish database built with the makeOrgPackage function from the AnnotationForge package.55 We used a corresponding table that connects Ensembl IDs to their corresponding Gene Ontology terms pulled down from Ensembl’s BioMart tool56 (Ensembl 104).
Differential abundance analysis using milo
To understand the enrichment of certain cell types between conditions, differential abundance between the cell neighborhoods for a given condition were tested using miloR (v0.1.0).18 We tested for differential abundance between the conditions (proximal vs. distal, regenerated vs. homeostasis). Using Seurat generated graph, a K-nearest neighborhood (KNN) graph was precomputed which assigned the cells to a neighborhood with the parameters (k = 20, d = 30, prop = 0.1). Cells within each neighborhood for a given sample were then counted and tested for differential abundance using a generalized linear design framework while accounting for multiple comparison testing using the spatial FDR in miloR. We then annotated the neighborhoods with the cell clusters and neighborhoods with a fraction less than 0.7 were annotated to be “mixed”.
Quantification and statistical analysis
We used R57 (version 4.2.3) to do statistical analysis. All numbers of individuals used for the experiments and the statistical test method used can be found in the corresponding figure legends. Generally, we assigned the significance level based on p value in following manner: ∗p < 0.05; ∗∗p < 0.01, ∗∗∗p < 0.001.
For statistical modeling of time-series regeneration data, we built linear mixed model using the lme4 package (version 1.1–34) in R (version 4.2.3) for the regeneration length. We built a linear mixed model using data from all time points, excluding 30 out of 1260 measurements in our statistical models that correspond to one individual that died early during the experiment. We included the time factor (dpa) and the amputation position factor (prox and dist) as fixed effects, and random intercept and slope for the time factor within each fish individual as random effects.
CRISPR-Cas9 mediated transgenesis
We injected single cell embryos with injection mix using two gRNAs synthesized by IDT (Alt-R CRISPR-Cas9 crRNA): gRNA-1 TGGACTTGATGTTTGAGTTATGG and gRNA-2 CGGCAATGGTCCCTCATTAGCGG. We resuspended 2nmol of each crRNA in 20μL of IDT Nuclease-Free Duplex Buffer (IDT, Cat. 11-05-01-03). Both crRNAs were mixed with Alt-R CRISPR-Cas9 tracrRNA-ATTO 550 (IDT, Cat. 1075928) at a final concentration of 50μM, they were heated up at 95°C for 5 min and allowed to cool down to room temperature to assemble the gRNA. RNP complex was built by incubating the gRNA with Alt-R S.p. HiFi Cas9 Nuclease V3 (IDT, Cat. 1081060) at a final concentration of 27.6μM for 20 min at room temperature. Finally, a 7.5μM solution of RNP diluted in Nuclease-Free water was injected using a microINJECTOR (Tritech Research, Cat. MINJ-D). Genotyping was done using NEB Phusion High-Fidelity DNA Polymerase (NEB, Cat. M0530S) PCR with the oligonucleotides TTGATGAGGGAGCCCGAAAG and ATGACTAAGCGGCTTTCCCAA.
Published: August 21, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.110737.
Supplemental information
Left side of the spreadsheet shows differentially expressed genes, and right side of the spreadsheet shows statistically enriched GO terms (padj < 0.05). Data correspond to heatmap in Figure 3D
Left side of the spreadsheet shows differentially expressed genes, and right side of the spreadsheet shows statistically enriched GO terms (padj < 0.05). Data correspond to heatmap in Figure 3H
Left side of the spreadsheet shows differentially expressed genes, and right side of the spreadsheet shows statistically enriched GO terms (padj < 0.05). Data correspond to heatmap in Figure 3K
Left side of the spreadsheet shows differentially expressed genes, and right side of the spreadsheet shows statistically enriched GO terms (padj < 0.05). Data correspond to heatmap in Figure 3L
Gene list displayed on heatmap in Figure 4E. No GO terms were found to be statistically enriched in this gene list (FDR < 0.05).
Left side of the spreadsheet shows differentially expressed genes, and right side of the spreadsheet shows statistically enriched GO terms (padj < 0.05). Data correspond to heatmap in Figure 4G
References
- 1.Watson J.K., Rulands S., Wilkinson A.C., Wuidart A., Ousset M., Van Keymeulen A., Göttgens B., Blanpain C., Simons B.D., Rawlins E.L. Clonal Dynamics Reveal Two Distinct Populations of Basal Cells in Slow-Turnover Airway Epithelium. Cell Rep. 2015;12:90–101. doi: 10.1016/j.celrep.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014;15:19–33. doi: 10.1038/nrm3721. [DOI] [PubMed] [Google Scholar]
- 3.Seifert A.W., Muneoka K. The blastema and epimorphic regeneration in mammals. Dev. Biol. 2018;433:190–199. doi: 10.1016/j.ydbio.2017.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh S.P., Holdway J.E., Poss K.D. Regeneration of amputated zebrafish fin rays from de novo osteoblasts. Dev. Cell. 2012;22:879–886. doi: 10.1016/j.devcel.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan T.H. Regeneration in teleosts. Archiv für Entwicklungsmechanik der Organismen. 1900;10:120–134. doi: 10.1007/bf02156348. [DOI] [Google Scholar]
- 6.Morgan T.H. Further experiments on the regeneration of the tail of fishes. Archiv für Entwicklungsmechanik der Organismen. 1902;14:539–561. doi: 10.1007/bf02188503. [DOI] [Google Scholar]
- 7.Lee Y., Grill S., Sanchez A., Murphy-Ryan M., Poss K.D. Fgf signaling instructs position-dependent growth rate during zebrafish fin regeneration. Development. 2005;132:5173–5183. doi: 10.1242/dev.02101. [DOI] [PubMed] [Google Scholar]
- 8.Rabinowitz J.S., Robitaille A.M., Wang Y., Ray C.A., Thummel R., Gu H., Djukovic D., Raftery D., Berndt J.D., Moon R.T. Transcriptomic, proteomic, and metabolomic landscape of positional memory in the caudal fin of zebrafish. Proc. Natl. Acad. Sci. USA. 2017;114:E717–E726. doi: 10.1073/pnas.1620755114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kragl M., Knapp D., Nacu E., Khattak S., Maden M., Epperlein H.H., Tanaka E.M. Cells keep a memory of their tissue origin during axolotl limb regeneration. Nature. 2009;460:60–65. doi: 10.1038/nature08152. [DOI] [PubMed] [Google Scholar]
- 10.Autumn M., Hu Y., Zeng J., McMenamin S.K. Growth patterns of caudal fin rays are informed by both external signals from the regenerating organ and remembered identity autonomous to the local tissue. bioRxiv. 2024 doi: 10.1101/2024.03.29.586899. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uemoto T., Abe G., Tamura K. Regrowth of zebrafish caudal fin regeneration is determined by the amputated length. Sci. Rep. 2020;10:649. doi: 10.1038/s41598-020-57533-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stewart S., Yette G.A., Le Bleu H.K., Henner A.L., Braunstein J.A., Chehab J.W., Harms M.J., Stankunas K. Skeletal geometry and niche transitions restore organ size and shape during zebrafish fin regeneration. bioRxiv. 2019;1:1. doi: 10.1101/606970. [DOI] [Google Scholar]
- 13.De Leon M.P., Wen F.-L., Paylaga G.J., Wang Y.-T., Roan H.-Y., Wang C.-H., Hsiao C.-D., Lin K.-H., Chen C.-H. Mechanical waves identify the amputation position during wound healing in the amputated zebrafish tailfin. Nat. Phys. 2023;19:1362–1370. doi: 10.1038/s41567-023-02103-6. [DOI] [Google Scholar]
- 14.Wang Y.T., Tseng T.L., Kuo Y.C., Yu J.K., Su Y.H., Poss K.D., Chen C.H. Genetic Reprogramming of Positional Memory in a Regenerating Appendage. Curr. Biol. 2019;29:4193–4207.e4. doi: 10.1016/j.cub.2019.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W., Hu C.K., Zeng A., Alegre D., Hu D., Gotting K., Ortega Granillo A., Wang Y., Robb S., Schnittker R., et al. Changes in regeneration-responsive enhancers shape regenerative capacities in vertebrates. Science. 2020;369 doi: 10.1126/science.aaz3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poleo G., Brown C.W., Laforest L., Akimenko M.A. Cell proliferation and movement during early fin regeneration in zebrafish. Dev. Dyn. 2001;221:380–390. doi: 10.1002/dvdy.1152. [DOI] [PubMed] [Google Scholar]
- 17.Nechiporuk A., Keating M.T. A proliferation gradient between proximal and msxb-expressing distal blastema directs zebrafish fin regeneration. Development. 2002;129:2607–2617. doi: 10.1242/dev.129.11.2607. [DOI] [PubMed] [Google Scholar]
- 18.Dann E., Henderson N.C., Teichmann S.A., Morgan M.D., Marioni J.C. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol. 2022;40:245–253. doi: 10.1038/s41587-021-01033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gawriluk T.R., Simkin J., Thompson K.L., Biswas S.K., Clare-Salzler Z., Kimani J.M., Kiama S.G., Smith J.J., Ezenwa V.O., Seifert A.W. Comparative analysis of ear-hole closure identifies epimorphic regeneration as a discrete trait in mammals. Nat. Commun. 2016;7 doi: 10.1038/ncomms11164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brant J.O., Boatwright J.L., Davenport R., Sandoval A.G.W., Maden M., Barbazuk W.B. Comparative transcriptomic analysis of dermal wound healing reveals de novo skeletal muscle regeneration in Acomys cahirinus. PLoS One. 2019;14 doi: 10.1371/journal.pone.0216228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allanki S., Strilic B., Scheinberger L., Onderwater Y.L., Marks A., Günther S., Preussner J., Kikhi K., Looso M., Stainier D.Y.R., Reischauer S. Interleukin-11 signaling promotes cellular reprogramming and limits fibrotic scarring during tissue regeneration. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abg6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomes R.N., Manuel F., Nascimento D.S. The bright side of fibroblasts: molecular signature and regenerative cues in major organs. NPJ Regen. Med. 2021;6:43. doi: 10.1038/s41536-021-00153-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi H.M.T., Schwarzkopf M., Fornace M.E., Acharya A., Artavanis G., Stegmaier J., Cunha A., Pierce N.A. Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development. 2018;145 doi: 10.1242/dev.165753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shibata E., Ando K., Murase E., Kawakami A. Heterogeneous fates and dynamic rearrangement of regenerative epidermis-derived cells during zebrafish fin regeneration. Development. 2018;145 doi: 10.1242/dev.162016. [DOI] [PubMed] [Google Scholar]
- 25.Cao Z., Meng Y., Gong F., Xu Z., Liu F., Fang M., Zou L., Liao X., Wang X., Luo L., et al. Calcineurin controls proximodistal blastema polarity in zebrafish fin regeneration. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2009539118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goss R.J. Academic Press, Inc.; 1969. Principles of Regeneration. [DOI] [Google Scholar]
- 27.Akimenko M.-A., Johnson S.L., Westerfield M., Ekker M. Differential induction of four msx homeobox genes during fin development and regeneration in zebrafish. Development. 1995;121:347–357. doi: 10.1242/dev.121.2.347. [DOI] [PubMed] [Google Scholar]
- 28.Currie J.D., Kawaguchi A., Traspas R.M., Schuez M., Chara O., Tanaka E.M. Live Imaging of Axolotl Digit Regeneration Reveals Spatiotemporal Choreography of Diverse Connective Tissue Progenitor Pools. Dev. Cell. 2016;39:411–423. doi: 10.1016/j.devcel.2016.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cura Costa E., Otsuki L., Rodrigo Albors A., Tanaka E.M., Chara O. Spatiotemporal control of cell cycle acceleration during axolotl spinal cord regeneration. Elife. 2021;10 doi: 10.7554/eLife.55665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benham-Pyle B.W., Brewster C.E., Kent A.M., Mann F.G., Chen S., Scott A.R., Box A.C., Sánchez Alvarado A. Identification of rare, transient post-mitotic cell states that are induced by injury and required for whole-body regeneration in Schmidtea mediterranea. Nat. Cell Biol. 2021;23:939–952. doi: 10.1038/s41556-021-00734-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eroglu E., Yen C.Y.T., Tsoi Y.L., Witman N., Elewa A., Joven Araus A., Wang H., Szattler T., Umeano C.H., Sohlmér J., et al. Epicardium-derived cells organize through tight junctions to replenish cardiac muscle in salamanders. Nat. Cell Biol. 2022;24:645–658. doi: 10.1038/s41556-022-00902-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu B., Lelek S., Spanjaard B., El-Sammak H., Simões M.G., Mintcheva J., Aliee H., Schäfer R., Meyer A.M., Theis F., et al. Origin and function of activated fibroblast states during zebrafish heart regeneration. Nat. Genet. 2022;54:1227–1237. doi: 10.1038/s41588-022-01129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jaiswal J., Egert J., Engesser R., Peyrotón A.A., Nogay L., Weichselberger V., Crucianelli C., Grass I., Kreutz C., Timmer J., Classen A.K. Mutual repression between JNK/AP-1 and JAK/STAT stratifies senescent and proliferative cell behaviors during tissue regeneration. PLoS Biol. 2023;21 doi: 10.1371/journal.pbio.3001665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan Y., Chai C., Li P., Zou X., Ferrell J.E., Jr., Wang B. Ultrafast distant wound response is essential for whole-body regeneration. Cell. 2023;186:3606–3618.e16. doi: 10.1016/j.cell.2023.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Simone A., Evanitsky M.N., Hayden L., Cox B.D., Wang J., Tornini V.A., Ou J., Chao A., Poss K.D., Di Talia S. Control of osteoblast regeneration by a train of Erk activity waves. Nature. 2021;590:129–133. doi: 10.1038/s41586-020-03085-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roy E., Neufeld Z., Cerone L., Wong H.Y., Hodgson S., Livet J., Khosrotehrani K. Bimodal behaviour of interfollicular epidermal progenitors regulated by hair follicle position and cycling. EMBO J. 2016;35:2658–2670. doi: 10.15252/embj.201693806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nauroy P., Guiraud A., Chlasta J., Malbouyres M., Gillet B., Hughes S., Lambert E., Ruggiero F. Gene profile of zebrafish fin regeneration offers clues to kinetics, organization and biomechanics of basement membrane. Matrix Biol. 2019;75–76:82–101. doi: 10.1016/j.matbio.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 38.Hou Y., Lee H.J., Chen Y., Ge J., Osman F.O.I., McAdow A.R., Mokalled M.H., Johnson S.L., Zhao G., Wang T. Cellular diversity of the regenerating caudal fin. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aba2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen C.H., Merriman A.F., Savage J., Willer J., Wahlig T., Katsanis N., Yin V.P., Poss K.D. Transient laminin beta 1a Induction Defines the Wound Epidermis during Zebrafish Fin Regeneration. PLoS Genet. 2015;11 doi: 10.1371/journal.pgen.1005437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harper M., Hu Y., Donahue J., Acosta B., Dievenich Braes F., Nguyen S., Zeng J., Barbaro J., Lee H., Bui H., McMenamin S.K. Thyroid hormone regulates proximodistal patterning in fin rays. Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2219770120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu X., Meng L., Li X., Li D., Liu Q., Chen Y., Li X., Bu W., Sun H. Regulation of FN1 degradation by the p62/SQSTM1-dependent autophagy–lysosome pathway in HNSCC. Int. J. Oral Sci. 2020;12 doi: 10.1038/s41368-020-00101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marković A., Briscoe J., Page K.M. Dynamics of positional information in the vertebrate neural tube. bioRxiv. 2024;1:1. doi: 10.1101/2024.07.01.601514. [DOI] [Google Scholar]
- 43.Shibata E., Liu Z., Kawasaki T., Sakai N., Kawakami A. Robust and local positional information within a fin ray directs fin length during zebrafish regeneration. Dev. Growth Differ. 2018;60:354–364. doi: 10.1111/dgd.12558. [DOI] [PubMed] [Google Scholar]
- 44.Edelstein A., Amodaj N., Hoover K., Vale R., Stuurman N. Computer control of microscopes using microManager. Curr. Protoc. Mol. Biol. 2010;14:20. doi: 10.1002/0471142727.mb1420s92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edelstein A.D., Tsuchida M.A., Amodaj N., Pinkard H., Vale R.D., Stuurman N. Advanced methods of microscope control using muManager software. J. Biol. Methods. 2014;1 doi: 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lowe D.G. Distinctive Image Features from Scale-Invariant Keypoints. Int. J. Comput. Vis. 2004;60:91–110. doi: 10.1023/b:Visi.0000029664.99615.94. [DOI] [Google Scholar]
- 47.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thevenaz P., Ruttimann U.E., Unser M. A pyramid approach to subpixel. IEEE Trans. Image Process. 1998;7:27–41. doi: 10.1109/83.650848. [DOI] [PubMed] [Google Scholar]
- 49.Preibisch S., Saalfeld S., Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 2009;25:1463–1465. doi: 10.1093/bioinformatics/btp184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Granillo A.O., Schnittker R.R., Wang W., Sánchez Alvarado A. Quantifying Cell Proliferation Through Immunofluorescence on Whole-Mount and Cryosectioned Regenerating Caudal Fins in African Killifish. Bio. Protoc. 2023;13 doi: 10.21769/BioProtoc.4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8 doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martin F.J., Amode M.R., Aneja A., Austine-Orimoloye O., Azov A.G., Barnes I., Becker A., Bennett R., Berry A., Bhai J., et al. Ensembl 2023. Nucleic Acids Res. 2023;51:D933–D941. doi: 10.1093/nar/gkac958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carlson M., Pagès H. 2023. AnnotationForge: Tools for Building SQLite-Based Annotation Data Packages. R Package Version 1.44.0. [DOI] [Google Scholar]
- 56.Kinsella R.J., Kähäri A., Haider S., Zamora J., Proctor G., Spudich G., Almeida-King J., Staines D., Derwent P., Kerhornou A., et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database. 2011;2011 doi: 10.1093/database/bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.R_Core_Team . R Foundation for Statistical Computing; 2023. R: A Language and Environment for Statistical Computing.https://www.R-project.org/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Left side of the spreadsheet shows differentially expressed genes, and right side of the spreadsheet shows statistically enriched GO terms (padj < 0.05). Data correspond to heatmap in Figure 3D
Left side of the spreadsheet shows differentially expressed genes, and right side of the spreadsheet shows statistically enriched GO terms (padj < 0.05). Data correspond to heatmap in Figure 3H
Left side of the spreadsheet shows differentially expressed genes, and right side of the spreadsheet shows statistically enriched GO terms (padj < 0.05). Data correspond to heatmap in Figure 3K
Left side of the spreadsheet shows differentially expressed genes, and right side of the spreadsheet shows statistically enriched GO terms (padj < 0.05). Data correspond to heatmap in Figure 3L
Gene list displayed on heatmap in Figure 4E. No GO terms were found to be statistically enriched in this gene list (FDR < 0.05).
Left side of the spreadsheet shows differentially expressed genes, and right side of the spreadsheet shows statistically enriched GO terms (padj < 0.05). Data correspond to heatmap in Figure 4G
Data Availability Statement
-
•
Original data underlying this manuscript is publicly available and can be accessed from the Stowers Original Data Repository at http://www.stowers.org/research/publications/libpb-2453. Single cell RNA-seq data have been deposited at GEO and are publicly available under accession number GSE260629 at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE260629.
-
•
All original code has been deposited at Zenodo and is publicly available under https://doi.org/10.5281/zenodo.10725332.
-
•
Any additional information required to reanalyze the data reported in this work is available from the lead contact, Alejandro Sánchez Alvarado (asa@stowers.org).