

ABSTRACT
CD38, a multifunctional enzyme involved in NAD+ catabolism, is hypothesized to act as a metabolic checkpoint for antitumor CD8 T cells. A recent study discovered that, apart from its direct metabolic mechanisms, CD38-mediated RyR2-AKT-TCF1 signaling regulates responsiveness to anti-PD1 cancer therapy at the molecular level. These findings advocate multiprong CD38 targeting to overcome resistance to immune checkpoint blockade therapy.
KEYWORDS: Cancer immunotherapy, CD38, immune checkpoint blockade, metabolism, PD1, T cell exhaustion, TCF1
Cancer immunotherapies address tumor-associated immunosuppression and rejuvenate otherwise dysfunctional antitumor immunological activities. Immune checkpoints, such as programmed cell death 1 (PD1), are the key mediators of cancer-associated immune dysfunction. As such, immune checkpoint blockade (ICB) provides a practical approach to rejuvenate immune dysfunction, especially through the antitumor actions of CD8 T cells. Clinical and preclinical data now clearly identify functionally competent CD8 T cells as a positive predictor of ICB efficacy.
Antitumor CD8 T cell functional competence is governed through complex biological processes and constitutes, among other effector functions, direct cytotoxicity or cytokine-mediated targeting of cancer cells. As these effector processes require energy (e.g., ATP) as well as macromolecules (amino acids, nucleotides), cell metabolism forms the key regulatory framework for CD8 T cell functional competence.1 Indeed, various stages of CD8 T cell biology, including activation, recruitment, cytokine release, and proliferation, are directly governed by metabolic underpinnings. Interestingly, the tumor microenvironment (TME) often hosts a non-conducive, hostile metabolic milieu that dampens CD8 T cell functional competence. Herein, fast-growing cancer cells hijack various metabolic pathways to excessively consume nutrients that are otherwise needed by CD8 T cells and produce metabolites that impart immunosuppression. The TME-associated metabolic landscape represents a formidable obstacle in promoting and sustaining the antitumor functions of CD8 T cells, even after the administration of ICB.
NAD+ metabolite acts as a co-factor within major energy and macromolecule biosynthesis pathways. Not surprisingly, cancers perturb NAD+ metabolism to sustain their metabolic demands. In the context of adaptive immunity, NAD+ regulates T cell development as well as macrophage plasticity and polarity. As such, NAD+ and its precursors (e.g., NMN) hold therapeutic potential to boost the immune system and enhance antitumor immune responses. As NAD+ biosynthesis and catabolism are governed through the enzymatic actions of intracellular and extracellular enzymes, therapeutic strategies often focus on exploiting enzymes involved in NAD+ biology. One such enzyme is CD38.2 CD38 is a multifaceted enzyme that degrades NAD to produce secondary metabolites such as ADPR and cADPR, which contribute to the adenosinergic pathway and calcium signaling. CD38 is often expressed on CD8 T cells, yet its role in CD8 cell functional competence remains poorly understood.
A recent study described how CD38-mediated signaling in CD8 T cells impedes the efficacy of anti-PD1 ICB therapy.3 Herein, authors first found a higher frequency of CD38 positive CD8 T cells in B16 melanoma TME, wherein CD38hi cells showed compromised antitumor activities compared to the CD38lo population. Interestingly, CD38hi CD8 T cells showed an exhausted (Tex) phenotype characterized by high PD1, Tim3, Tox, and lower TCF1 and mitochondrial OXPHOS. Adoptive transfer of CD38hi Tex transgenic melanoma antigen-specific CD8 T cells (Pmel) failed to control tumor growth and showed reduced responsiveness to anti-PD1 ICB. On the contrary, CD38lo Pmel CD8 T cells produced the desired tumor control. Subsequent scRNA analysis on CD38hi CD8 T cells from B16 melanoma TME and publicly available tumor biopsy samples reiterated a negative correlation between CD8 Tex phenotype, Tcf7 (TCF1 encoding gene) expression and ICB refractoriness. This correlation between CD38hi T cell profile and exhaustion state from the murine model was also observed in chronically stimulated human CD8 T cells. Mechanistically, in CD38hi CD8 Tex cells, binding of cADPR to ryanodine receptors (RyRs), specifically RyR2, promoted elevated calcium levels, leading to chronic AKT activation and Tcf7 suppression. From the therapeutic perspective, CD38 inhibition with shRNA or 8Br-cADPR (inhibitor of CD38 cyclase activity) restored antitumor functional competence in chronically stimulated CD8 T cells. Similarly, adoptively transferred transgenic CD38−/− Pmel CD8 cells exhibited high TCF1 expression and improved anticancer functionality. Finally, inhibition of various targets within the RyR2-AKT-TCF1 axis (as listed in Figure 1) restored ICB efficacy. Together, these findings identified multiprong mitigation strategies to overcome CD38-mediated immunosuppression impeding ICB therapy.
Figure 1.
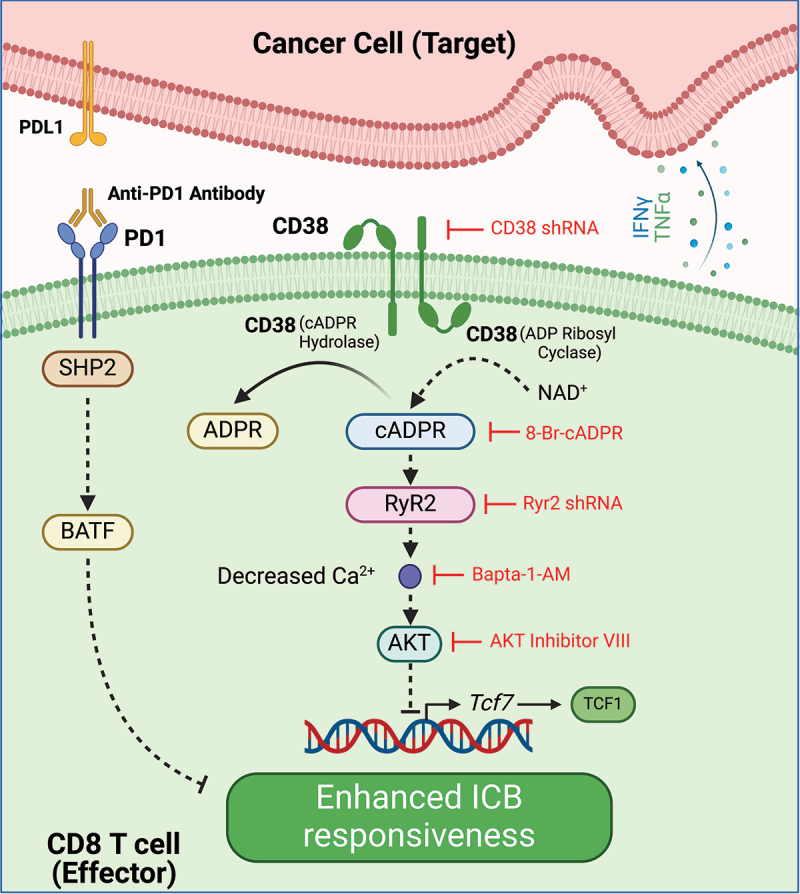
Multiprong strategies to overcome CD38-mediated CD8 T cell dysfunction to enhance ICB responsiveness: through its hydrolase/NADase activity, CD38 degrades NAD to form ADPR/cADPR, a secondary messenger in cell signaling pathways. In CD38hi CD8 T cells, cADPR regulates Ryr2 calcium channel activation, which elevates intracellular Ca2+ levels resulting in chronic activation of AKT. Such CD38-mediated RyR2-AKT activation causes downregulation of TCF1 and promotes terminal differentiation of CD8 T cells leading to the unresponsiveness to anti-PD1 therapy. Genetic or pharmacological inhibition of various targets within CD38- RyR2-AKT-TCF1 axis, restores antitumor activities of CD38hi CD8 T cells and improves responsiveness to ICB. Created with BioRender.com.
Of note, cancer immunotherapy strategies reprogramming cell metabolism warrant careful considerations. A central metabolite such as NAD+ often sustains metabolic pathways and cell signaling events required for the growth and proliferation of both cancer and immune cells. Not surprisingly, a ‘face-off’ between NAD+ metabolism rate-limiting enzymes nicotinamide phosphoribosyltransferase (NAMPT) and nicotinamide N-methyltransferase (NNMT) in macrophages and cancer-associated fibroblasts (CAFs), respectively, define the anti-PD-1 ICB efficacy in gastric cancer.4 Next, NAD+ itself can induce cell death in CD8 T cells (NAD+ induced cell death; NICD).5,6 In non-small cell lung cancer cells expressing ART1 (adenosine 5′-diphosphate (ADP) – ribosyltransferase-1), ribosyl groups from NAD+ can be transferred to the P2X7R receptor in T cells, potentially leading to NICD. In these ART1-expressing cancers, CD38 inhibition may not favor ICB but rather increase NICD. Indeed, personalized and context-dependent precision approaches hold the key toward exploiting metabolism-based targets within cancer immunotherapy strategies.
In summary, cell metabolism holds the key to functional competence of immune cells and provides therapeutic opportunities for immune rejuvenation.7 As we advance, novel combinatorial strategies in line with patient-specific metabolic landscapes promise to create avenues promoting the efficacy of ICBs and other emerging cancer immunotherapies.
Funding Statement
S.G. is supported by grants from the Canadian Institutes of Health Research (CIHR), Canadian Cancer Society (CCS), Canada Foundation for Innovation- John R. Evans Leaders Fund (CFI-JELF), and Research Nova Scotia (RNS). PN is the Linnea Veinotte Scholar trainee in the Cancer Research Training Program of the Beatrice Hunter Cancer Research Institute, supported by GIVETOLIVE.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Møller SH, Hsueh P-C, Yu Y-R, Zhang L, Ho P-C.. Metabolic programs tailor T cell immunity in viral infection, cancer, and aging. Cell Metab. 2022;34(3):378–3. doi: 10.1016/j.cmet.2022.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy BE, Sadek M, Elnenaei MO, Reiman A, Gujar SA. Targeting NAD+ synthesis to potentiate CD38-based immunotherapy of multiple myeloma. Trends Cancer. 2020;6(1):9–12. doi: 10.1016/j.trecan.2019.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Kar A, Ghosh P, Gautam A, Chowdhury S, Basak D, Sarkar I, Bhoumik A, Barman S, Chakraborty P, Mukhopadhyay A, et al. CD38–RyR2 axis–mediated signaling impedes CD8+ T cell response to anti-PD1 therapy in cancer. Proc Natl Acad Sci USA. 2024;121(11):e2315989121. doi: 10.1073/pnas.2315989121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang Y, Wang Y, Chen G, Sun F, Wu Q, Huang Q, Zeng D, Qiu W, Wang J, Yao Z, et al. Nicotinamide metabolism face-off between macrophages and fibroblasts manipulates the microenvironment in gastric cancer. Cell Metab. 2024;36(8):1806–1822.e11. doi: 10.1016/j.cmet.2024.05.013. [DOI] [PubMed] [Google Scholar]
- 5.Seman M, Adriouch S, Scheuplein F, Krebs C, Freese D, Glowacki G, Deterre P, Haag F, Koch-Nolte F. Nad-induced T cell death: ADP-ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity. 2003;19(4):571–582. doi: 10.1016/S1074-7613(03)00266-8. [DOI] [PubMed] [Google Scholar]
- 6.Stark R, Wesselink TH, Behr FM, Kragten NAM, Arens R, Koch-Nolte F, van Gisbergen KPJM, van Lier RAW. T RM maintenance is regulated by tissue damage via P2RX7. Sci Immunol. 2018;3(30):eaau1022. doi: 10.1126/sciimmunol.aau1022. [DOI] [PubMed] [Google Scholar]
- 7.Al-Habsi M, Chamoto K, Matsumoto K, Nomura N, Zhang B, Sugiura Y, Sonomura K, Maharani A, Nakajima Y, Wu Y, et al. Spermidine activates mitochondrial trifunctional protein and improves antitumor immunity in mice. Science. 2022;378(6618):eabj3510. doi: 10.1126/science.abj3510. [DOI] [PubMed] [Google Scholar]