

Abstract
Fluorescence microscopy is a vital tool in biomedical research but faces considerable challenges in achieving uniform or bright labeling. For instance, fluorescent proteins are limited to model organisms, and antibody conjugates can be inconsistent and difficult to use with thick specimens. To partly address these challenges, we developed a labeling protocol that can rapidly visualize many well-contrasted key features and landmarks on biological specimens in both thin and thick tissues or cultured cells. This approach uses established reactive fluorophores to label a variety of biological specimens for cleared-tissue microscopy or expansion super-resolution microscopy (ExM) and is termed FLARE (Fluorescent Labeling of Abundant Reactive Entities). These fluorophores target chemical groups and reveal their distribution on the specimens; amine-reactive fluorophores such as hydroxysuccinimidyl esters target accessible amines on proteins while hydrazide fluorophores target oxidized carbohydrates. The resulting stains provide signals analogous to traditional general histology stains such as H&E (hematoxylin and eosin) or PAS (periodic acid-Schiff) but use fluorescent probes that are compatible with volumetric imaging. In general, the stains for FLARE are performed in the order of carbohydrates, amine, and DNA, and the incubation time for the stains varies from 1 h to 1 d depending on the combination of stains and the type and thickness of the biological specimens. FLARE is powerful, robust, and easy to implement in labs that already routinely do fluorescence microscopy.
Editorial Summary:
Carbohydrates, proteins, and nucleic acids in cultured cells and thick tissues are fluorescently labeled via FLARE for cleared-tissue or expansion microscopy.
Introduction
Fluorescence microscopy is a technique that is commonly used in the biomedical sciences. It offers the powerful ability to visualize structures or molecules in three dimensions within biological specimens and gives relevant context to the study of the functions of these structures. However, the success of fluorescence microscopy measurements often hinges on the ability of fluorescent labels to detect the objects of interest in a specimen. Immunolabeling of specimens using antibodies is popular due to its high specificity and contrast, but it faces key challenges.1,2 First, many biological molecules or structures of interest lack good commercially-available antibodies that are specific to relevant targets, particularly for specimens that have been heavily fixed or processed. Second, the relatively large size of antibodies (~150,000 Daltons, or ~15 nm, for an immunoglobulin G) can often make staining of samples >100 µm in thickness very time consuming due to slow diffusion through the sample, with sample preparation times that can take weeks. Fluorescent proteins, which are also popular fluorescent probes, require genetic manipulation and are thus limited to a subset of laboratory model organisms.
To partially address these limitations, we developed a fluorescent labeling method that uses reactive probes to target abundant chemical groups on biological samples. We termed the method FLARE for Fluorescent Labeling of Abundant Reactive Entities,3 and have used this method successfully to label cultured cells, mouse tissues, and human tissues.3–5
Development of FLARE
FLARE operates analogously to classic histology stains such as H&E4 that are widely used to reveal overall tissue or cell physiology. However, most histology stains are either nonfluorescent or have poor fluorescence properties, making them ill-suited to volumetric imaging. Additionally, histology stains are typically affinity-based, such that they may be washed out of thick samples during multi-round processing used in some powerful, recently developed techniques in super-resolution microscopy and cleared-tissue microscopy. In contrast, FLARE utilizes covalent stains with good fluorescence properties (e.g., bright and photostable) that are compatible with volumetric imaging, as well as a range of sample fixation and sample processing methods including paraffin embedding, super-resolution expansion microscopy (ExM), and cleared-tissue microscopy. Paraffin-embedding of specimens, in the form of formalin-fixed paraffin-embedded (FFPE) blocks, is commonly used in pathology and research labs due to ease of tissue sectioning and the ability to store tissues for long periods of time, but the process may degrade specimen antigenicity for labeling with antibodies. FLARE provides histology-stain-like contrast in FFPE specimens while offering greater multiplexing capabilities and high-resolution, volumetric imaging capabilities for standard thin (5–10 µm) or moderately thick FFPE sections (10–100 µm, or thicker).
While a variety of innovative super-resolution microscopy methods have been developed to date5–7, ExM uses a unique approach in which swellable polymer hydrogels grown within fixed specimens uniformly enlarge the sample in three dimensions.8–10 This enables features to become resolvable in the enlarged state at ~70 nm resolution or better even when using conventional microscopes that have a physical diffraction-limited resolution of ~250 nm. Importantly, FLARE is able to brightly label many of the major structures within cells and tissues, which is particularly valuable for ExM since bright signals are required for reliable detection of fluorescence after expansion. In order to maximize the amount of protein available for staining via FLARE, we focus on a protein-retention variation of ExM termed MAP (Magnified Analysis of the Proteome)11 that uses detergent and heat denaturation to homogenize specimens prior to expansion, although we do show that FLARE can also reveal major features for enzymatic digestion-based ExM.3 Tissue-clearing utilizes various methods to minimize light scattering for the volumetric study of thick intact specimens (100–1000 µm or more)12,13, but labeling such thick specimens remains a persistent challenge, particularly with large labels like antibodies. Because FLARE labels are small molecules, they can be easily tuned to rapidly penetrate thick specimens to produce uniform stains.
Currently, the chemical reactions used for FLARE to label biological specimens target amine groups, which are abundant on proteins, and oxidized carbohydrates, which are abundant on basement membranes among other structures (Fig. 1). However, FLARE can also target other chemical groups (e.g. thiols, carboxylates, etc.) that are abundant in biological samples. This method can be conveniently performed using a range of fluorescent dyes that are commercially available across the visible and near-infrared ranges in order to suit a specific experiment or instrument. FLARE is generally easy to combine with other forms of stains, if desired, including immunolabeling and DNA fluorescence in situ hybridization (DNA FISH).14
Fig. 1. Overview of FLARE for fluorescent labelling of biological samples.
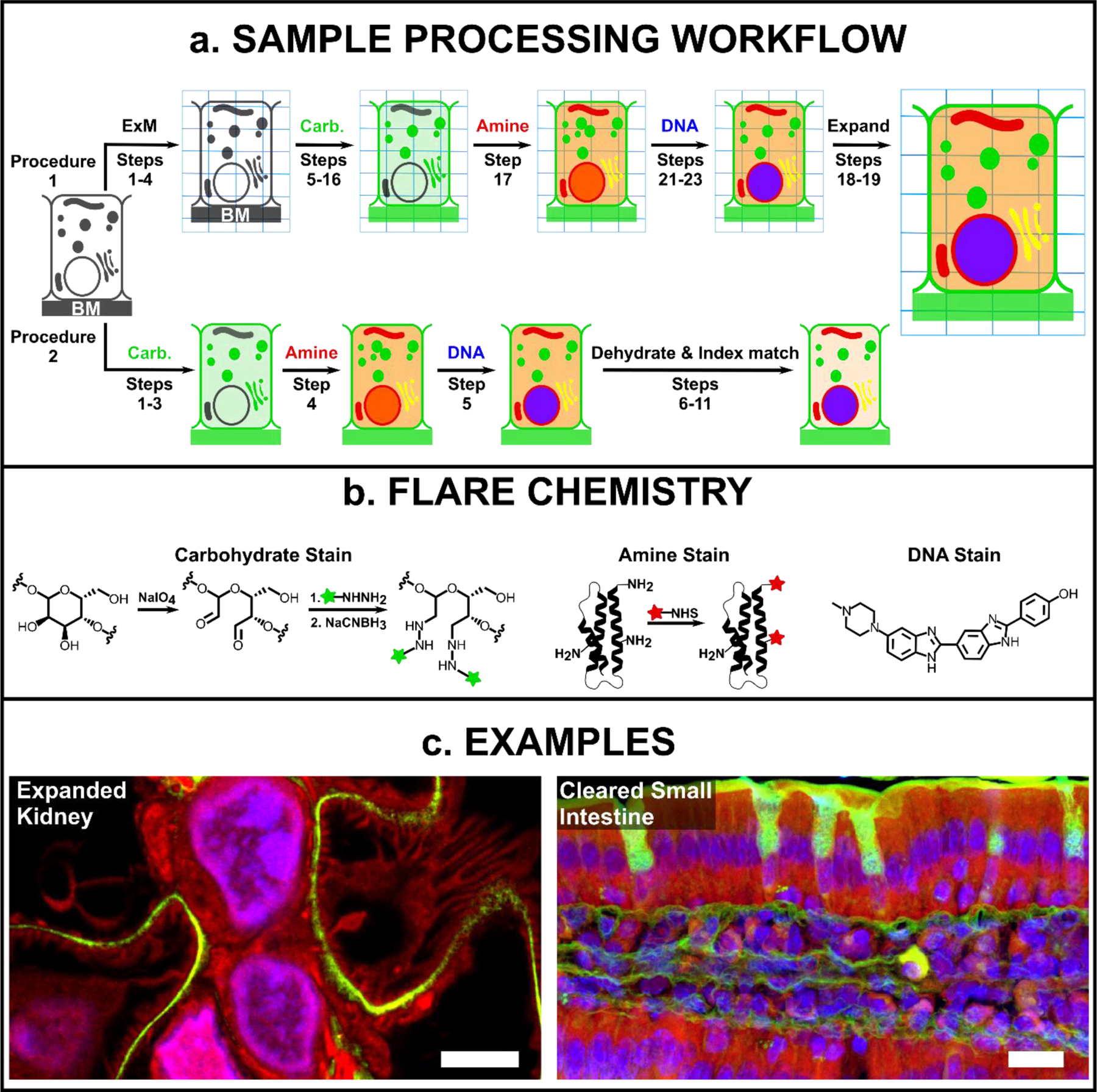
(a) General schematic for FLARE staining and sample processing workflow for ExM (procedure 1) and cleared-tissue microscopy (procedure 2), where BM is the basement membrane. (b) Details of FLARE chemistry. Carbohydrates are oxidized to aldehydes using sodium periodate, coupled to hydrazide-functionalized dyes, and stabilized by reduction with sodium cyanoborohydride. N-hydroxysuccinimide (NHS)-functionalized dyes are used to label amine groups on proteins. DNA is labeled non-covalently using standard DNA-labeling dyes such as Hoechst. (c) Example confocal microscopy images of expanded mouse kidney (100 μm thickness) and optically-cleared mouse small intestine tissues (100 μm thickness) that have been stained by FLARE, where carbohydrates are green, accessible amines (on proteins) are red, and DNA is blue. Scale bars are 3 μm (left, pre-expansion units) and 10 μm (right).
In H&E staining, hematoxylin stains negatively charged components, such as DNA, blue while eosin stains positively charged protein components pink. Inspired by H&E staining, we employed N-hydroxysuccinimide (NHS) functionalized fluorophores to covalently label amines on proteins, and fluorescent DNA-staining dyes such as Hoechst or SYBR Green to stain cell nuclei (Fig. 1b).3 In addition, we adapted the PAS stain to covalently label oxidized carbohydrates by first oxidizing the carbohydrates to aldehydes using sodium periodate and then subsequently reacting the aldehydes with hydrazide-functionalized fluorophores (Fig. 1b).3 The amine and carbohydrate-labeling reactions for FLARE are well-established for protein and carbohydrate bioconjugation, but they are rarely used on intact biological samples for high resolution and high contrast fluorescence imaging purposes. During an early phase of the FLARE protocol development, we demonstrated the use of NHS-functionalized fluorophores to rapidly (<5 min) label the surface of unexpanded and uncleared clinical specimens to enable rapid pathology analysis of breast lumpectomy tissue.15,16 In addition, recently published papers by other groups also incorporated the use of NHS-functionalized fluorophores to study protein distributions on expanded cultured cells, C. elegans, zebrafish, and various mouse organ tissue sections.17–20 Finally, a number of false-coloring algorithms have been developed to computationally render images of tissues, stained with fluorescent analogs of common pathology stains (e.g. H&E), in order to mimic the appearance of those (absorptive) pathology stains as viewed with a transmitted-light microscope.21,22
Comparison with other methods
Labeling of specimens with antibodies is a powerful approach that can be used to visualize specific molecules of interest in biological specimens. However, antibodies face certain drawbacks such as poor penetration through thick complex specimens, lot-to-lot variability, or sometimes inconsistent commercial availability. There can also be labeling difficulties with highly processed biological specimens, such as FFPE tissues, in which antigen targets may be degraded and not bound well by antibodies. In recent years, there has been substantial progress in improving the speed and penetration of antibody labeling for thick specimens, such as the use of sophisticated instruments23, novel sample manipulation24, optimized procedures25, and new tissue permeabilization reagents26, even though several of the other challenges remain. If the labeling of specific molecules is required, we suggest that practitioners use antibodies or small molecule probes that are specific for the desired target. However, if the labeling of general structures is desired, then FLARE offers many advantages, including broad compatibility with other specific labeling methods. For instance, Bertiaux et al. used NHS ester dyes to delineate the general morphological context of malaria parasites while also studying specific proteins of its cytoskeleton.27
Microscopists have, for many decades, stained specimens with a range of general contrast, small molecule labels that are typically examined using transmitted light (typically with thin sections), including H&E and PAS, among others.28 While many of these stains are either nonfluorescent or have poor fluorescent properties (e.g., broad spectra or a tendency to photobleach), some of the stains have been used for fluorescent microscopy, particularly with the eosin component of H&E21,29–32, and less commonly with the PAS stain fuchsin.33,34 These classical histology stains are viable options for fluorescent staining of unexpanded specimens, but care should be taken in some cases. Eosin is an affinity-based stain and can be relatively easy to wash away, so there may be challenges with retaining the stain in multi-step protocols. The classical PAS histology stain, fuchsin, is covalently bound to the sample by means of a rather labile imine bond that may be hydrolyzed over time in solution and, additionally, the fluorescence emission spectrum of fuchsin is broad and can potentially hinder multiplexed imaging possibilities. On its own, eosin or fuchsin (PAS) staining may be sufficient for some experiments, particularly for thin specimens, but they are not easily used in combination. In contrast, the covalent reactions used in FLARE irreversibly bind fluorophores to amines and carbohydrates on the sample and thus, the labels are able to tolerate many harsh processing steps (e.g., for DNA FISH, immunolabeling, tissue clearing, etc.). Additionally, NHS- and hydrazide-functionalized fluorophores are commercially available for a large number of fluorophores with outstanding properties (brightness and resistance to photobleaching) in virtually any spectral band across the visible to near infrared in the range 400–800 nm.
The radically different approach to microscopy used by ExM has created some unique requirements for fluorescent probes compared to microscopy with unexpanded specimens. Expansion tends to dilute signals substantially (e.g., 4-fold linear expansion produces a 64-fold volumetric dilution) and it is challenging to retain affinity-based probes during the procedure due to the extensive washing and somewhat harsh digestion or denaturation procedures. It is therefore important to use bright fluorescent labeling and to retain probes prior to expansion. There has recently been considerable innovation in the development of small-molecule probes for ExM. For example, Shi et al.35, Karagiannis et al.36, Wen et al.37, and Götz et al.38 developed small-molecule ExM probes that contain three chemical groups including (1) a group to target a small molecule (e.g. to a lipid, to the actin cytoskeleton using phalloidin, or to a genetically-encoded reactive protein), (2) a reporting group such as a fluorophore, biotin, or click-chemistry group, and (3) a label-retention group such as an acryloyl group that can be covalently linked to a polymer hydrogel. While these custom probes are very powerful, for instance in their ability to target specific molecules, their synthesis requires substantial chemical expertise beyond the scope of most biomedical researchers interested in using these tools. In contrast, FLARE only offers the ability to label relatively general structures, rather than specific molecules, but it is very bright, uses off-the-shelf reagents, and can be used to label a sample before or after expansion depending on the overall desired workflow (Fig. 2).
Fig. 2. A summary of suggested workflows when combining FLARE with DNA FISH and immunofluorescence for clearing or expansion.
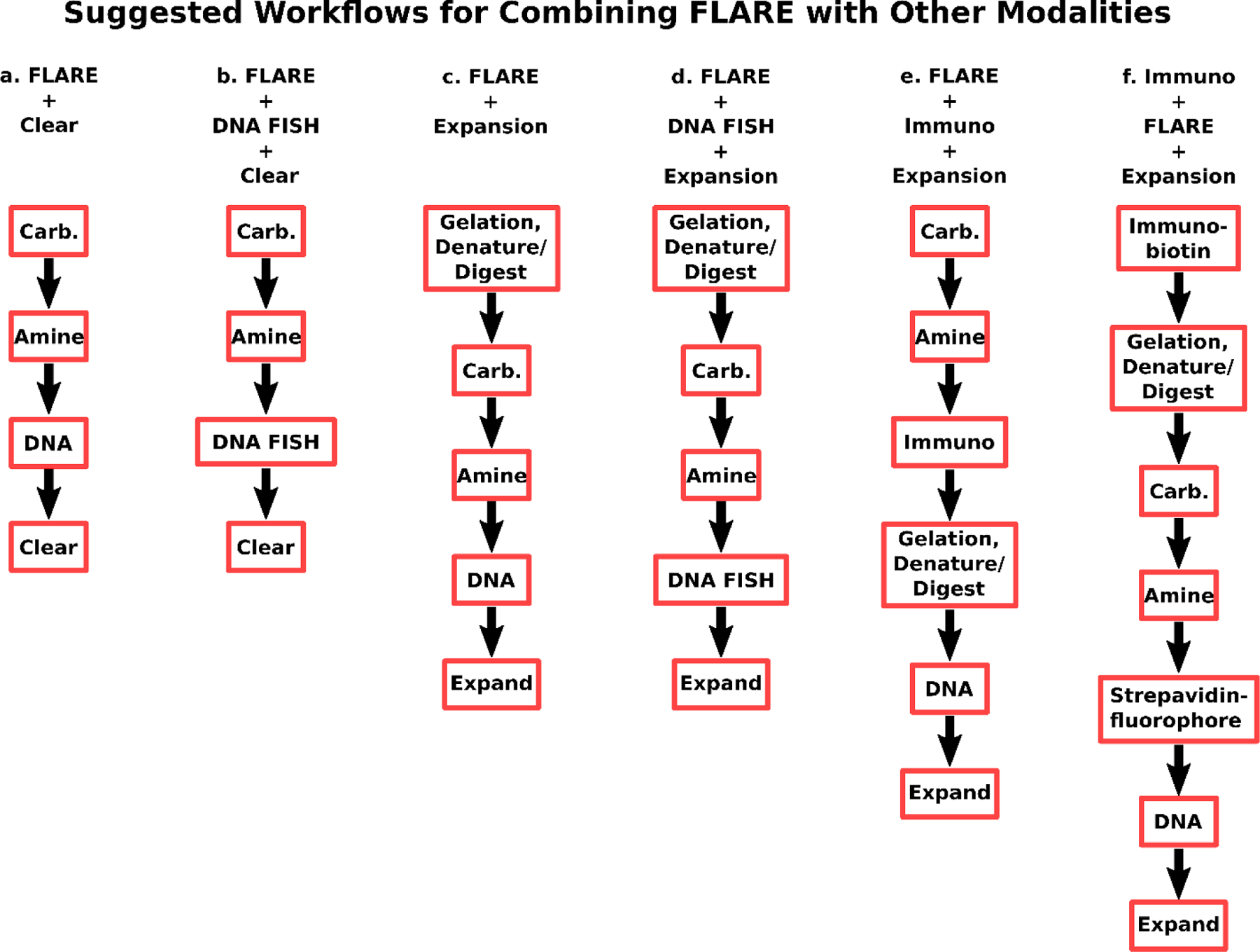
(a) A basic FLARE stain consists of carbohydrate, amine, and DNA labeling and may be followed by tissue clearing for unexpanded specimens. (b) DNA FISH may be added to workflow (a) after FLARE staining, although the noncovalent DNA stain is omitted since the stain is poor after the harsh conditions used for DNA FISH due to possible dehybridization of double-stranded DNA. (c) ExM and FLARE may be combined with gelation and denaturation (or digestion) of a sample, FLARE staining, and expansion by incubation in water. (d) DNA FISH may be added to workflow (c) after FLARE staining, again omitting the nonspecific DNA stain as in (b). (e) FLARE may be combined with immunostaining and ExM by labeling of carbohydrates, labeling of amines, immunolabeling, gelation and denaturation (or digestion), labeling of DNA, and then expansion in water. In case FLARE is found to perturb downstream immunolabeling, it is possible to use alternative protocol (f). “Carb” is carbohydrate stain; “Amine” is amine stain; “DNA” is DNA stain; “Clear” is organically cleared; “Denature” is protein denaturation using sodium dodecyl sulfate; “Digest” is enzymatic protein digestion; “Expand” is expansion in water; “DNA FISH” is DNA fluorescence in situ hybridization; “Immuno” is immunolabeling with standard primary and secondary antibodies; “Immuno-biotin” is immunolabeling with biotin-labeled primary antibodies or a combination of unlabeled primary and biotin-labeled secondary antibodies.
Applications and limitations
FLARE is currently applied only to fixed specimens, but is well suited to a range of specimen types and sample processing techniques. Expanded cultured cells and fresh-fixed tissue sections labeled using FLARE revealed major landmarks and sub-cellular organelles in great detail (Fig. 1c and Fig. 3). During the development of FLARE, we focused on the use of retinal pigment epithelial cultured cells and mouse kidney tissue as model systems with ExM, although many ExM samples can be brightly labeled by FLARE. For instance, other groups have recently used NHS-functionalized dyes to broadly label proteins in a range of expanded specimens including C. elegans, zebrafish, various mouse organ tissue sections, and various cultured cell lines.17–20 The method for C. elegans expansion was able to visualize important features using NHS-functionalized dyes with adequate contrast despite potential protein loss due to enzymatic digestion. M’Saad et al.18 combined the MAP protocol11 with iterative expansion,39 which allowed expansion by ~18-fold, and labeled the resulting gels with NHS-ester functionalized dyes to study whole-cell protein distributions at very high resolution. Damstra et al.19 reported a new single-step ~10-fold expansion protocol that together with NHS-ester dye labeling reveals details at a very high spatial resolution. Unexpanded tissue sections are easily labeled by FLARE either for highly processed FFPE specimens (Fig. 4, Supplementary Code) or specimens that are freshly fixed (Fig. 5) or cryopreserved. Uniform tissue labeling can be achieved for thin sections (e.g., 5 µm thick FFPE sections) or thick ones (1 mm fresh-fixed sections, Fig. 6) although some tuning of the labeling protocol is required as described for thick specimens and, in addition, imaging deep into thick samples generally requires optical clearing.
Fig. 3. Comparison of fixation conditions for expanded RPE cells stained by FLARE.
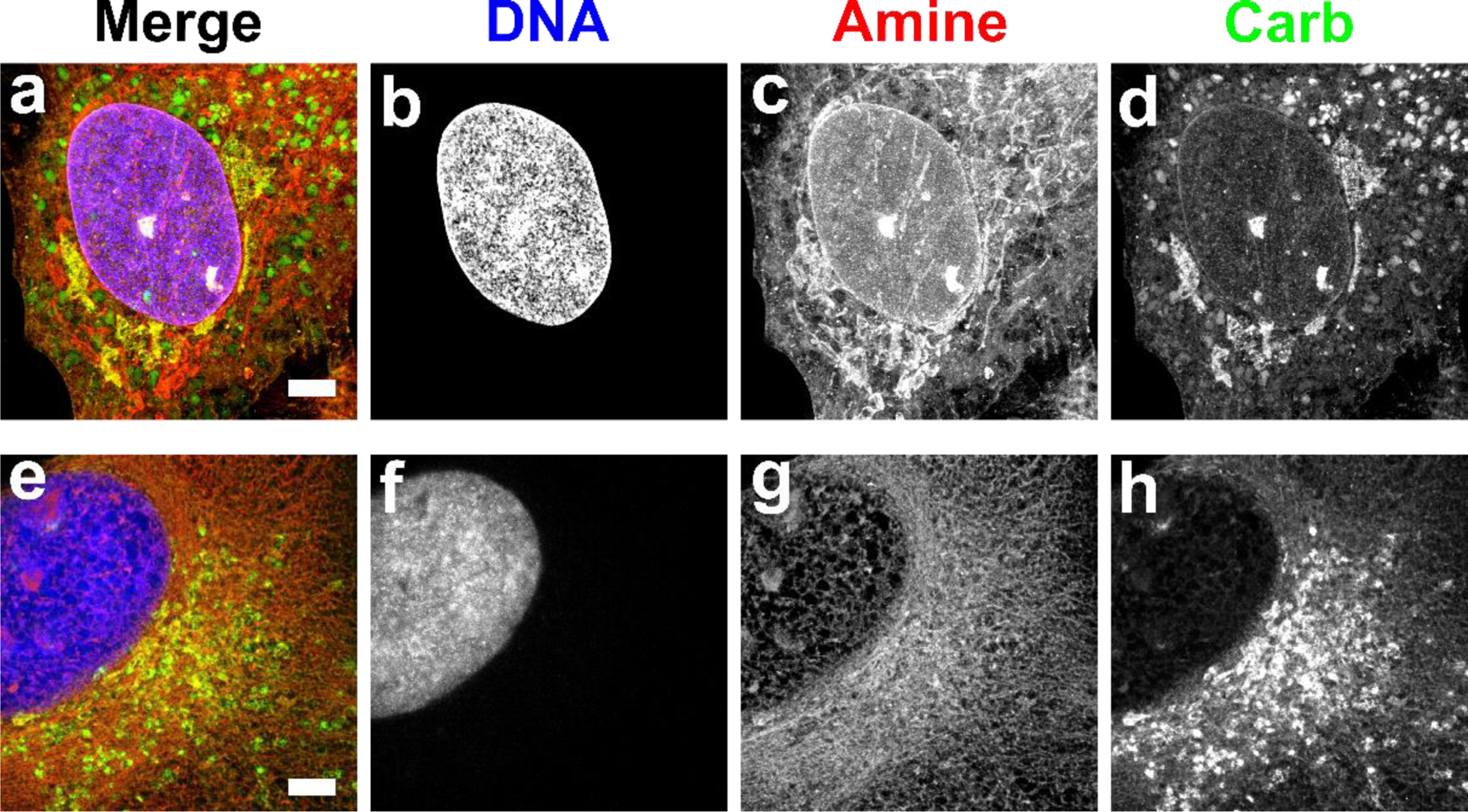
Red corresponds to amine stain, green corresponds to carbohydrate stain, and blue corresponds to nuclear stain. (a-d) Fixation with paraformaldehyde and glutaraldehyde generally preserved membrane-bound organelles and other structures while (e-h) fixation with glyoxal showed substantial perturbations. Scale bar (pre-expansion): 3 µm (a-h). Panels a-d were adapted from Mao et al.3 Science Advances. AAAS Publishing Group.
Fig. 4. FLARE staining of 5 µm thick FFPE human tissue microarrays.
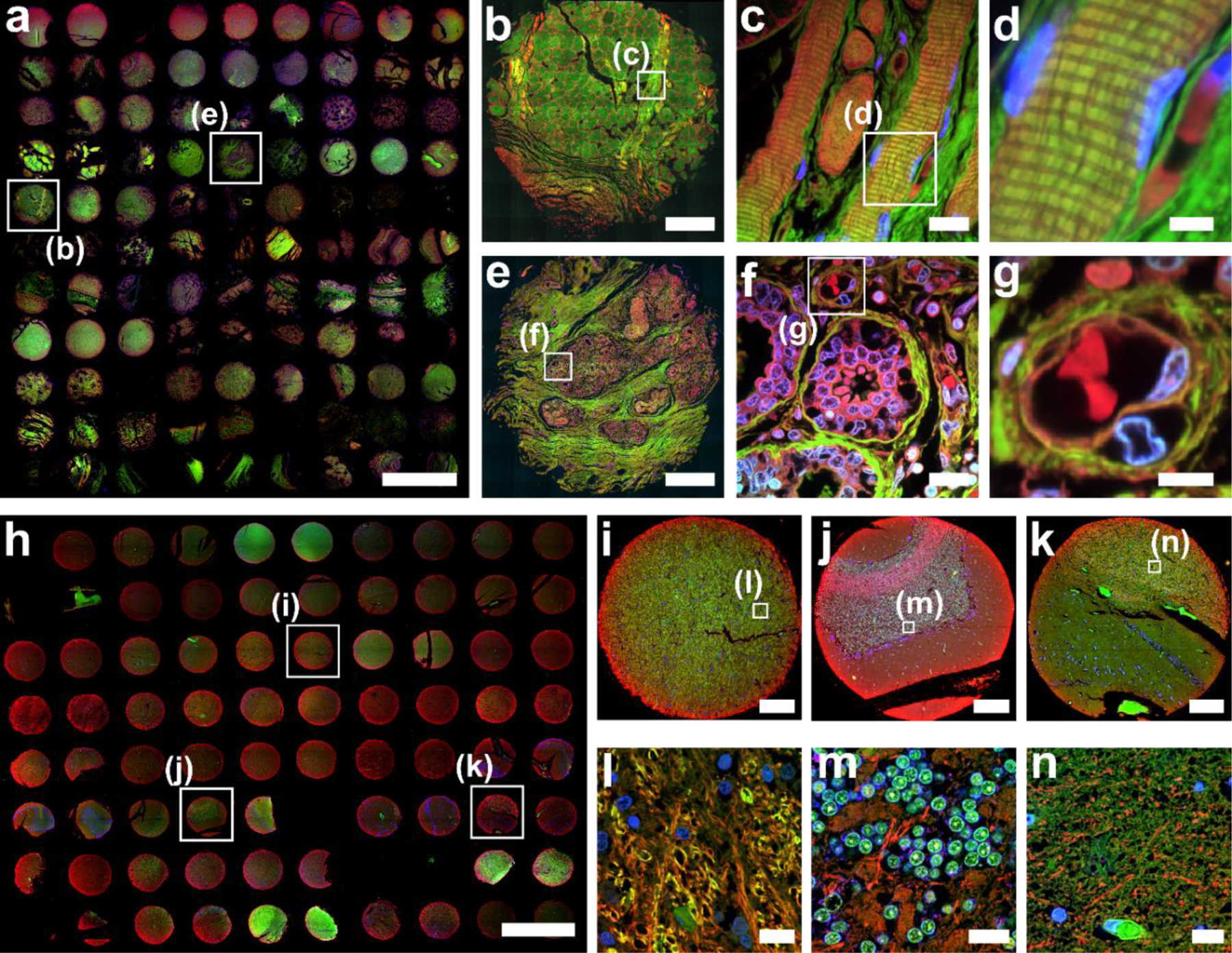
(a) Overview image of 96 human organ tissue cores that were FLARE-stained for amines (red), carbohydrates (green), and DNA (blue), and then tiled using a 4× objective lens on a widefield microscope. See also Supplementary Figures 1-2 for a larger version of a) as well as a diagram of which cores represent which tissues. Zoom-in views of a tonsil core from the boxed region in panel a) were recorded by confocal microscopy with (b) tiling using a 20× objective lens and (c-d) single-region imaging using a 60× objective lens. The cross-striations of skeletal muscle fibers were well-visualized in c-d) and fibrocytes and erythrocytes were distinctively labeled in panel d). Zoom-in views of the breast core from the boxed region in panel a) were recorded by confocal microscopy with (e) tiling using a 20× objective lens and (f-g) single-region imaging using a 60× objective lens. Breast lobules surrounded by carbohydrate-rich connective tissue were well-visualized in e-g), as were details of secretory alveoli and nearby blood vessels. (h) Overview image of 75 human brain tissue cores that were FLARE-stained for amines (red), carbohydrates (green), and DNA (blue), and then tiled using a 4× objective lens on a widefield microscope. See also Supplementary Figures 3-4 for a larger version of h) as well as a diagram of which cores represent which regions of the brain. Cores for (i) midbrain, (j) cerebellum, and (k) hippocampus, selected from panel h), highlight the variable distribution of amines and carbohydrates within the brain. Zoom-in views (l-n) of the regions indicated in boxes of (i-k) were imaged using a 60× objective lens, revealing highly distinct tissue morphologies such as the cell bodies of cerebellar granule cells that are brightly labeled by the carbohydrate stain in (m). Scale bars are 3 mm (a, h), 300 μm (b,e,i-k), 15 μm (c, f, l-n), 6 μm (d, g). The custom Wolfram Mathematica scripts used for stitching tiling images (Fig. 4a and 4h) are available as Supplementary Code.
Fig. 5. Compatibility of FLARE with diverse procedures.
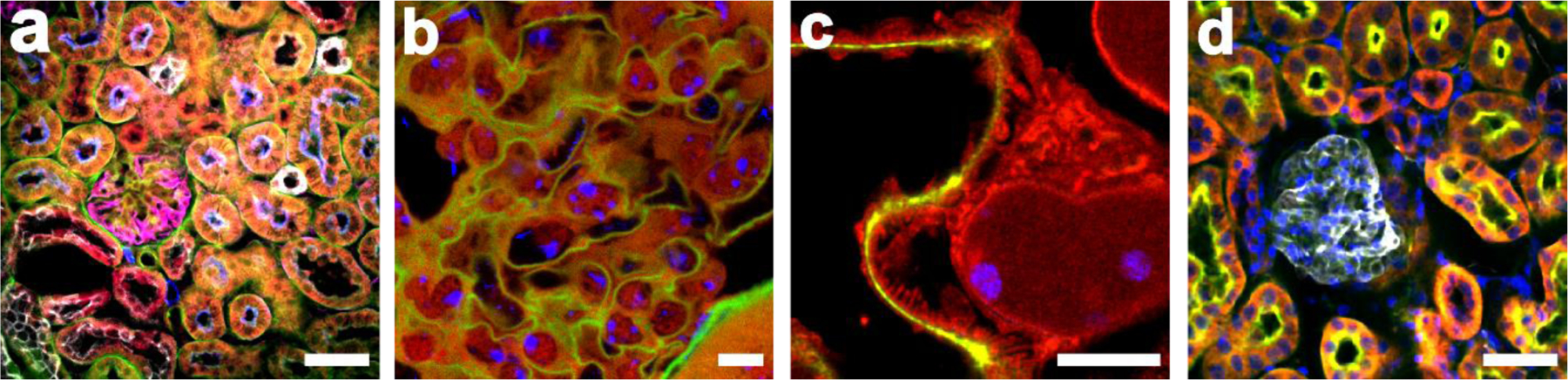
FLARE-stained 100 μm thick mouse kidney sections prepared using various procedures that include two different ExM variants, tissue clearing, and the combination with either immunostaining or DNA FISH. (a) 5-color confocal microscopy image of unexpanded mouse kidney tissue that was stained for carbohydrates (green), amines (red), and then immunostained for podocalyxin (magenta), aquaporin-1 (blue), and cytokeratin 8+18 (grey). (b) Confocal microscopy image of unexpanded cleared mouse kidney tissue that was stained for carbohydrates (green), amines (red), and then DNA FISH (blue) against MaSat DNA. (c) Confocal microscopy image of expanded mouse kidney tissue that was treated by the detergent-based protocol and stained for carbohydrates (green), amines (red), and then DNA FISH (blue) against pericentromeric major satellite (MaSat) DNA. (d) Confocal microscopy image of enzyme digested expanded mouse kidney tissue that was stained for carbohydrates (green), amines (red), DNA (blue) and then immunostained for podocalyxin (grey). Scale bars are 50 μm (a, d), 5 μm (b) and 3 μm (c, pre-expansion units). Panels a-b were adapted from Mao et al.3 Science Advances. AAAS Publishing Group.
Fig. 6. Gelation chamber setup.

(a) Schematic of tissue sample gelation: i. spread a tissue sample on a coverslip (24mm × 50mm) and construct a spacer on each side using a stack of two #1.5 cover glass (~160 μm each); ii. Add a few drops of the freshly prepared gel solution to cover the entire sample area; iii. Place another coverslip (24 mm × 50 mm) on the top; iv. Once the gel has formed, peel off the top glass slide and trim off the excess gel. (b) Step-by-step photos correspond to i-iv in panel (a).
It is important to consider the features being stained in a given sample when using FLARE. For example, in expanded cultured cells, we found that many subcellular organelles were easily recognized, including mitochondria, lysosomes, the Golgi apparatus, nuclear pores, kinetochores, and centromeres. However, other structures, notably the cytoskeleton, were not easily identified despite a relatively high protein abundance.3 Sub-cellular organelles were not generally as easily discerned in relatively more complex 3D tissues, although we were able to observe details of nuclei and mitochondria in expanded mouse renal tubules in addition to details of basement membranes and interdigitated podocyte cells in expanded mouse renal glomeruli. Cell boundaries are brightly outlined in muscle cells in the carbohydrate stain (e.g., smooth muscle cells of the mouse small intestine3) but not in some other specimens, while bands of connective tissue tend to be enriched in both carbohydrates and amines in human prostate tissue.
Taken together, the above observations suggest that the features that are revealed by FLARE depend on the distribution of the reactive groups in the specimen, the relative reactivity of those groups, and overall sample processing procedures (including fixation) which may consume or mask some reactive groups. Not surprisingly, classic histology stains, which were the inspiration for FLARE, also show contrast and features that depend prominently on the sample itself.40 While FLARE will not stain every desired structure in every specimen of interest, published examples can in some cases help practitioners judge the likelihood of success. Additionally, since FLARE is easy and inexpensive, it can be readily attempted on other specimens of interest.
There may be structures or molecules that will simply not be possible to label with high contrast using FLARE alone. In such cases, researchers will need to use traditional specific labels, such as immunolabeling of proteins or labeling of nucleic acids by FISH. If desired, however, FLARE can often be combined with the use of these specific labels (e.g., antibodies or DNA oligonucleotides), and the FLARE dyes can be chosen in virtually any spectral region across the visible or near-infrared to accommodate other probes. In terms of antibody labeling, we found that 14 out of 16 tested antibodies were able to label their targets on cells or tissues after FLARE, and that the carbohydrate oxidation step appeared to be the step that perturbed subsequent antibody labeling for 2 out of the 16 tests (amine labeling did not perturb antibody binding in any of the tests).3 If desired, it is possible to perform immunolabeling before FLARE to avoid oxidation-induced loss of antigenicity,2 and it may be possible to use milder oxidation that preserves antigenicity although we have not yet tested this thoroughly. For the combination of FLARE and DNA FISH, we performed FLARE before DNA FISH and achieved the expected results (Fig. 5b and 5c), showing that FLARE does not substantially perturb DNA FISH.
The detailed procedures for FLARE that we describe here are compatible with a variety of types of specimens, sample formats, and applications, including the examples shown for expanded cells and tissues, cleared thick tissues, and thin uncleared tissue sections. Although FLARE is useful for obtaining relatively general contrast in order to quickly explore the general physiology of specimens, researchers who wish to selectively stain specific structures or molecules are advised to use traditional, specific labels. In those situations, it may nonetheless still be useful to combine FLARE with specific labels in order to gain the best of both worlds.
Experimental Design
When designing FLARE labeling experiments, it is crucial to consider the type of information needed from the sample. First, determine whether FLARE is to be used alone, or in combination with another labeling modality. Second, decide whether expansion is needed in order to visualize the biological component of interest at high spatial resolution. While we focus on denaturation-based MAP in our protocol, we have also had success using enzymatic digestion-based ExM (see section FLARE with other modalities). These expansion methods are well described and validated,8–11,17,18,41,42 and we advise selection of an expansion method suitable for the specific sample. Third, plan control experiments for labeling, especially on new biological specimens that have not already been tested and for experiments that seek to combine various labeling methods. For example, if immunolabeling is required in combination with FLARE, first establish whether or not the chemical stains perturb the desired immunolabel and then determine if a shift in staining workflow is necessary.
Overview of the procedures.
Here we present two FLARE procedures applicable to different specimen types, where Procedure 1 outlines how to apply the method either for expanded cultured cells (option A throughout Procedure 1) or for expanded thin (~100 μm) tissue sections (option B throughout Procedure 1), and Procedure 2 details the method for unexpanded cleared thick tissue sections (option A throughout Procedure 2) and tissue microarray (option B throughout Procedure 2). The main stages of the protocol are shown in Fig. 2 and can be divided into two sections: labeling and sample processing. The labeling is done by FLARE which includes a carbohydrate stain (steps 5–16 of Procedure 1 or steps 1–3 of Procedure 2), an amine stain (step 17 of Procedure 1 or step 4 of Procedure 2), and a DNA stain (steps 21–23 of Procedure 1 or step 5 of Procedure 2). Although FLARE does not have a specific requirement for sample fixation, paraffin-embedded samples or other dehydrated samples should be rehydrated prior to performing FLARE. The sample processing steps will depend on what kind of information the researchers wish to obtain. We recommend the use of ExM sample processing (steps 1–4, 20 of Procedure 1) if super-resolution visualization of nanoscale features is required. On the other hand, we recommend optical clearing sample processing (steps 6–11 of Procedure 2) for relatively thick samples where volumetric information is required. For ExM, FLARE can be performed before or after expansion processing, but when combining FLARE with optical clearing, FLARE must be performed first.
In order to demonstrate FLARE’s general utility, we describe procedures that can be applied to a wide variety of samples (including RPE1 cells, mouse kidney tissues, FFPE human tissue microarrays), freshly fixed or paraffin-embedded, from 4 μm to 1 mm thickness. Although several experimental parameters such as incubation times and dye concentrations vary, suggested parameters have been included for each procedure section. Since FLARE aims to label amines, carbohydrates, and nucleic acids that broadly exist in biological organisms, researchers should be able to label their samples with FLARE. However, to achieve a desired result, we recommend researchers begin with a procedure that most closely matches the thickness of their samples. Based on the outcome and issues encountered, labeling can be improved by considering the specific aspects discussed below, and the troubleshooting guidance.
Sequence of labeling while combining with DNA FISH or Immunolabeling.
In general, the preferred sequence for using FLARE is: carbohydrate, amine, and DNA. It is preferred to label carbohydrates first because the periodate oxidation step (that converts carbohydrates to aldehydes) also tends to chemically bleach fluorophores already present on the specimen. By using this order, the amine-reactive fluorophores are introduced after the oxidation step that could otherwise bleach them (Fig. 2a and 2c).
DNA FISH may be performed on FLARE-stained specimens using either tissue-clearing or ExM processing (Fig. 2b and 2d). However, the harsh conditions used in DNA FISH procedures appear to dehybridize double-stranded DNA, leading to unsuccessful conventional DNA stains (e.g., DAPI). For the combination of immunolabeling with FLARE, immunolabeling can simply be performed after FLARE (Fig. 2e). In the event that an antibody does not label a sample well after FLARE, we recommend an alternative labeling sequence consisting of immunolabeling using a biotin-conjugated primary or secondary antibody (biotin is resistant to sodium periodate oxidation), followed by FLARE, and then a final incubation with a fluorescent streptavidin conjugate (Fig. 2f).
Integration of FLARE with ExM requires a few considerations. We generally perform FLARE either before all expansion procedures or after gelation and sample homogenization, but we have not observed clear differences in the staining patterns, making either order a viable choice. However, slightly longer incubation times are required for FLARE stains after gelation and sample homogenization, possibly to give the dyes more time to diffuse through a thicker sample after partial (~two-fold) expansion. Unlike ExM approaches that use enzymatic digestion, MAP enables post-gel immunostaining due to better preservation of protein epitopes11. Although we have not tested immunostaining after FLARE on expanded samples, we have previously tested a range of antibodies against non-expanded FLARE-processed samples and we found that most antibodies successfully bound their targets (14/16)3. We, therefore, hypothesize that FLARE-processed specimens are likely to still be stained well with antibodies after expansion for scenarios where the expansion procedure does not impede antibody labeling.
Fluorophore choice.
In this protocol, we typically use the commercially available reactive fluorophores AT647N-NHS and AT565-NHNH2 to label amines and oxidized carbohydrates, respectively, but these may be easily substituted for other fluorophores that have the correct chemical functionality and non-overlapping spectra. If FLARE is performed before gelation, then it is also important to choose fluorophores that survive the oxidizing environment of the hydrogel polymerization step. AT488, AF488, AT565, AF568, and AT647N are all examples of dyes that survive the oxidizing environment of the typical hydrogel polymerization process. If combining FLARE with immunolabeling or DNA FISH, then it is also important to make sure that the spectra of all channels are isolatable from each other.
For instance, FLARE could be performed with AT647N-NHS labeling of amines (664 nm emission maximum), AT565-hydrazide labeling of oxidized carbohydrates (590 nm emission maximum), Hoechst labeling of DNA (460 nm emission maximum), and an immunostain or DNA FISH stain could be performed using AF488 (525 nm emission maximum). The NHS- or hydrazide-functionalized dyes can be swapped for dyes in other spectral ranges by purchasing other commercially-available dyes with the same reactive chemical groups. For instance, in Fig. 5b and 5c, we selected ATTO-488-NHS to label amines so that we could also use ATTO-647N oligonucleotides for DNA FISH. Intriguingly, Sim et al.20 reported some dependence of the stain specificity with NHS labeling based on the hydrophobicity of different fluorophores.
Tuning of the chemistry for uniform labeling.
Some reaction conditions were tuned to improve FLARE labeling. First, for both expanded and unexpanded samples, we selected a pH of ~6.0 for the AT647N-NHS incubation buffer that promotes stain uniformity, in contrast with typical protein bioconjugation reaction conditions that use a higher pH (~8.4) to promote faster or more complete reactions.43 This is because NHS-esters are less reactive and also hydrolyze (to a non-reactive state) more slowly at a lower pH; under these conditions, fluorophores are able to penetrate more deeply into the sample prior to reaction/hydrolysis.44,45 Second, for expanded samples, due to the negative charge of the hydrogel, we used a relatively high salt concentration (1 M NaCl) in the 100 mM sodium acetate buffer to enable negatively charged molecules to penetrate better through the hydrogel network via electrostatic screening. Third, for unexpanded cleared samples, in addition to lowering the pH, we introduced the organic solvent THF as a co-solvent during the staining incubation in order to facilitate the dye diffusion process. Other organic solvents that are miscible with water and do not interfere with the reaction could also be used. The goal of optimizing these parameters is to increase diffusion relative to the coupling reaction in order to produce a uniform stain, especially in thick specimens. Our procedure should work well for cleared samples that are less than 1 mm thick and for expanded samples that are less than 100 μm thick. Researchers might need to adjust pH, incubation time, or salt concentration (for expanded samples) when working with thicker samples.
Level of expertise needed.
FLARE does not require specific expertise for the implementation. However, researchers who are not familiar with the procedures for ExM are recommended to consult previously published expansion protocols.8–11,17,18,41,42
Materials
Biological materials
- Cell line or tissue of interest. In the procedures we include specific instructions for the use of the following as example applications:
- H-tert Retinal Pigment epithelium (H-tert RPE-1 cells, ATCC, cat. no. CRL-4000, RRID: CVCL_4388, https://scicrunch.org/resolver/RRID:CVCL_4388) !Caution The cell lines used in your research should be regularly checked to ensure they are authentic and frequently be tested for mycoplasma infection.
- Mouse tissue (two-month-old C57BL/6 male mice) !Caution Any experiments using mouse material must conform to relevant Institutional and National regulations. Use of this protocol to generate the results shown here was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Washington (Protocol No.: PROTO20180063). Procedures involving animals were conducted according to the institutional guidelines that are in compliance with National Institutes of Health (NIH) policies.
- Human tissue microarray (US Biomax, Inc. Catalog No.: BNC17011at, FDA999rt) !CAUTION Any experiments using human material must conform to relevant Institutional and National regulations and require patient consent.
- Human kidney tissue (NW Biotrust, Biomaterial ID: 312620). !Caution Any experiments using human material must conform to relevant Institutional and National regulations. Use of this protocol to generate the results shown here was approved by the University of Washington Review Board with deidentification.
Reagents
10× phosphate buffered saline, pH 7.4 (PBS; Fisher Bioreagents, cat. no. L-5400)
Sodium azide (NaN3; Fisher Scientific, cat. no. S227I)
Sodium acetate, anhydrous (NaOAc; Fisher Scientific, cat. no. S209)
Sodium chloride (NaCl; Fisher Scientific, cat. no. 271)
4-Morpholineethanesulfonic acid (MES; Sigma-Aldrich, cat. no. M8250)
Sodium periodate (NaIO4; Sigma-Aldrich, cat. no. 311448)
Sodium cyanoborohydride (NaCNBH3; Sigma-Aldrich, cat. no. 156159) !Caution NaCNBH3 is toxic and releases flammable gas upon contact with water; wear appropriate PPE and handle with care. ▲Critical NaCNBH3 is sensitive to humidity and should be stored in a dry environment.
ATTO 647N NHS ester (AT647N-NHS; Sigma-Aldrich, cat. no. 18373)
ATTO 565 hydrazide (AT565-NHNH2; ATTO-TEC GmbH, cat. no. AD565)
Hoechst 33258 (Sigma-Aldrich, cat. no. B2883)
SYBR Green I (Invitrogen, cat. no. S7563)
Deionized water (DI water)
Ammonium persulfate (APS; Bio-Rad Laboratories, cat. no. 161–0700)
Tetramethylethylenediamine (TEMED; Bio-Rad Laboratories, cat. no. 161–0800) !Caution TEMED is flammable, and a toxic respiratory hazard; handle in small amounts and work in a well-ventilated area.
32% (vol/vol) paraformaldehyde (PFA; Electron Microscopy Sciences, cat. no. RT15714) !Caution PFA is a toxic respiratory hazard; handle in small amounts and work in a well-ventilated area.
50% (vol/vol) glutaraldehyde (GA; Electron Microscopy Sciences, cat. no. 16300) !Caution GA is a toxic respiratory hazard; handle in small amounts and work in a well-ventilated area.
1,4-Piperazinediethanesulfonic acid (PIPES; Sigma-Aldrich, cat. no. P6757)
Triethylene glycol diamine tetraacetic acid (EGTA; Alfa Aesar, cat. no. ALFAA16086)
Magnesium chloride (MgCl2; Sigma-Aldrich, cat. no. M8266)
40% (wt/vol) glyoxal (Sigma-Aldrich, cat. no. 128465)
Sodium borohydride (NaBH4; Fisher Scientific, cat. no. AC189300050) !Caution NaBH4 is toxic and releases flammable gas upon contact with water; wear appropriate PPE and handle it in small amounts. ▲Critical NaBH4 may adversely react with some fluorophores (such as cyanine dyes).
40% (wt/vol) acrylamide (AA; Bio-Rad Laboratories, cat. no. 161–0140) !Caution AA is carcinogenic and may cause nervous system damage; wear appropriate PPE and work in a well-ventilated area.
2% (wt/vol) bis-acrylamide (BA; Bio-Rad Laboratories, cat. no. 161–0142) !Caution BA is carcinogenic and may cause nervous system damage; wear appropriate PPE and work in a well-ventilated area.
Sodium acrylate (SA; Sigma-Aldrich, cat. no. 408220)
VA-044 (Thermo Fisher Scientific, cat. no. NC0471397)
Triton X-100 (Sigma-Aldrich, cat. no. X100)
Poly-lysine (Sigma-Aldrich, cat. no. P8920)
Sodium dodecyl sulfate (SDS; Sigma-Aldrich, cat. no. L3771)
Tris base (Tris; Fisher Scientific, cat. no. BP152–500)
Tetrahydrofuran (THF; Fisher Scientific, cat. no. T425) !Caution THF is volatile and flammable; wear appropriate PPE and work in a fume hood.
Dichloromethane (DCM; Fisher Scientific, cat. no. D37) !Caution DCM is toxic, volatile, and flammable; wear appropriate PPE and work in a fume hood.
Ethyl cinnamate (EC; Sigma-Aldrich, cat. no. 112372)
Anhydrous dimethyl sulfoxide (DMSO, Sigma-Aldrich, cat. no. 276855)
Equipment
Rectangular #1.5 glass coverslip (Fisher Scientific, cat. no. 12544E, 24 × 50 mm)
12 mm round coverslip (Electron Microscopy Sciences, cat. no. 72230–01)
Forceps (Electron Microscopy Sciences, cat. no. 78318–3)
Parafilm M Bemis (VWR, cat. no. 52858)
Razor blade (American Line, cat. no. 66–0089)
12 well-plate (Pipette, cat. no. 712011)
6 well-plate (Pipette, cat. no. 229106)
Paint brush (Black Art Materials, cat. no. 05130–1000)
Scintillation vial, large enough (~28 mm diameter) to hold tissue section (Wheaton, cat. no. 986546)
1.5 mL microcentrifuge tube (VWR, cat. no. 76332–064)
Heat blocks (VRW, Model: MDHB4)
Rocking platform shaker (VWR, Model:100)
Cell incubator (Thermo Fisher Scientific, Model: 370; relative humidity: 90%; CO2 concentration: 5%)
Reagents set-up
1× PBS (pH 7.4).
For 1 L of 1× PBS, add 100 mL 10× PBS to 900 mL DI water. Store at room temperature (20–25°C).
20% (vol/vol) Triton X-100 solution.
For 10 mL of 20% (vol/vol) Triton X-100 solution, add 2 mL pure Triton X-100 to 8mL DI water. CRITICAL Warm the pure Triton X-100 in a 60 °C bath for a few minutes prior to use for easier handling. Store at 4 °C until use, with a recommended storage period of 6 months.
10× PEM buffer.
For 100 mL of 10× PEM buffer, add 30.2 g of PIPES, 0.38 g of EGTA, 95 mg MgCl2to 90 mL of DI water. Adjust pH with aqueous 1 M NaOH or HCl to pH 7 and subsequently adjust total volume to 100 mL with DI water. Once prepared, sore at 4 °C until use, with a recommended storage period of 6 months.
Extraction solution.
Extraction solution contains 100 mM aqueous PIPES buffer pH 7, 1 mM EGTA, 1 mM MgCl2, 0.5% (vol/vol) Triton X-100 solution, and it can be prepared from above 10× PEM buffer and 20% (vol/vol) Triton X-100 solution. For 10 mL of extraction solution, add 1 mL of 10× PEM buffer, 0.25 mL of 20% (vol/vol) Triton X-100 and 8.75 mL of DI water. Prepare fresh each time.
PFA/GA fixation solution.
PFA/GA fixation solution contains 100 mM aqueous PIPES buffer pH 7, 1 mM EGTA, 1 mM MgCl2, 3.2% (vol/vol) PFA, 0.1% (vol/vol) GA, and it can be prepared from 10× PEM buffer, 32% (vol/vol) PFA stock, and 50% (vol/vol) GA stock. For 10 mL of PFA/GA fixation solution, add 1 mL of 10× PEM buffer, 1 mL of 32% (vol/vol) PFA stock, 20 μL of 50% (vol/vol) GA stock, and 7.98 mL of DI water. Prepare fresh each time.
PFA fixation solution.
Use 10× PEM buffer and 32% (vol/vol) PFA stock to prepare a PFA fixation solution that contains 100 mM aqueous PIPES buffer (pH 7), 1 mM EGTA, 1 mM MgCl2, 4% (vol/vol) PFA. For 10 mL of PFA fixation solution, add 1 mL of 10× PEM buffer, 1.25 mL of 32% (vol/vol) PFA stock, and 7.75 mL of DI water. Prepare fresh each time.
3% (vol/vol) Glyoxal mixture solution (pH 5).
The glyoxal mixture solution contains 0.75% (vol/vol) glacial acetic acid, 3% (vol/vol) glyoxal, 19.9% (vol/vol) absolute ethanol. For 10 mL of glyoxal mixture solution, add 75 µL of glacial acetic acid, 0.3 mL of glyoxal, 1.99 mL of ethanol, and 5 mL of DI water. The pH is adjusted to 5 using 1M NaOH before adjusting the total volume to 10 mL with DI water. Prepare fresh each time.
NaOAc buffer.
For 25 mL of NaOAc buffer, add 246 mg NaOAc powder to 25 mL of DI water. Adjust pH to 5 using 1M HCl. The buffer can be stored at room temperature, and should be stable for at least two weeks.
NaOAc/NaCl buffer.
For 25 mL of NaOAc/NaCl buffer, add 246 mg NaOAc powder and 1.75 g NaCl to 25 mL of DI water. Adjust pH to 5 using 1M HCl. The buffer can be stored at room temperature, and should be stable for at least two weeks.
MES buffer.
For 25 mL of MES buffer, add 586 mg MES and 263 mg NaCl to 25 mL of DI water. Adjust pH to 6 using 1M NaOH. The buffer can be stored at room temperature, and should be stable for at least two weeks.
AT565-NHNH2 master stock.
Add 200 µL pf anhydrous DMSO to 1 mg AT565-NHNH2 to give a concentration of 5 mg/mL (6.65 mM). Store at −20 °C until use, with recommended maximum storage of 6 months.
AT647N-NHS ester master stock.
Add 200 µL of anhydrous DMSO to 1 mg of AT647N-NHS to give a concentration of 5 mg/mL (5.93 mM). Store at −20 °C until use, with recommended maximum storage of 2 months. ▲Critical Conditions should be kept anhydrous as NHS-ester is prone to hydrolysis.
SYBR Green master stock.
Prepare a master stock of SYBR Green with a concentration of 10 mg/mL (19.6 mM) using anhydrous DMSO. Add 1 mL of anhydrous DMSO to 10 mg of SYBR Green. Store at −20 °C until use, with recommended storage of 12 months.
Monomer solution (cells).
A monomer solution for cells contains 20% (wt/vol) AA, 10% (wt/vol) SA, 0.05% (wt/vol) BA, 4% (wt/vol) PFA, and 0.67% (wt/vol) TEMED. For 4 mL of cell monomer solution, add 2 mL of 40% (wt/vol) AA, 400 mg SA, 0.1 mL BA, 0.5 mL of 32% (vol/vol) PFA and 26.8 µL of TEMED to 1273.2 µL of 1× PBS. ▲Critical The monomer solution can be stored at 4 °C for maximum 1 week, protected from light. The monomer solution may degrade during this period, therefore it is recommended to prepare fresh each time if possible.
Monomer solution (tissues).
A monomer solution for tissues contains 20% (wt/vol) AA, 10% (wt/vol) SA, 0.05% (wt/vol) BA, and 4% (wt/vol) PFA. Store at 4 °C for up to 1 week, protected from light. For 4 mL of tissue monomer solution, add 2 mL of 40% (wt/vol) AA, 400 mg SA, 0.1 mL BA, and 0.5 mL of 32% (vol/vol) PFA to 1300 µL of 1× PBS. ▲Critical The monomer solution can be stored at 4 °C for maximum 1 week, protected from light. The monomer solution may degrade during this period; therefore, it is recommended to prepare fresh each time if possible.
Denaturing solution.
Denaturing solution contains 200 mM SDS, 200 mM NaCl, and 50 mM Tris. For 20 mL of denaturing solution, add 1.73 g SDS, 350 mg NaCl, 182 mg Tris base to 20 mL of water. Adjust pH to 9.3 using 1M HCl. Store at 37 °C until use. ▲Critical Warm the mixture to dissolve the solids faster prior to pH adjustments.
PBST solution.
PBST solution contains 0.1% Triton X-100 and 0.02% (wt/vol) NaN3 in 1× PBS. For 1 L of PBST solution, add 13 mg of NaN3 and 1 mL of Triton X-100 to 999 mL of 1× PBS. Store at room temperature until use, with recommended storage period of 6 months.
Hoechst 33258 master stock.
Prepare a Hoechst 33258 master stock with a concentration of 10 mg/mL (23.4 mM) using anhydrous DMSO by adding 1 mL of anhydrous DMSO to 10 mg of Hoechst 33258. Store at −20 °C until use.
THF/NaOAc solution.
For 2 mL of solution, add 1mL of THF to 1 mL of NaOAc buffer. Prepare fresh each time.
THF/MES solution.
For 2 mL of solution, add 1 mL of THF to 1 mL of MES buffer. Prepare fresh each time.
THF/PBS solution.
For 2 mL of solution, add 1 mL of THF to 1 mL of 1× PBS buffer. Prepare fresh each time.
Tissue extraction.
We typically obtain mouse tissue specimens following cardiac perfusion with 1× PBS for 3 min followed by 4% PFA (vol/vol) solution in 1× PBS for 5 min (7 mL/min). Organs of interest were then collected and post-fixed in 4% (vol/vol) PFA solution in 1× PBS. Depending on the size of the organ, the post-fixation period varies from 1 hour (for kidney) to 6 hours (for liver). Afterwards, organs were washed with a 1× PBS solution three times and sliced by a vibratome to 100-μm thickness.
Procedure 1
FLARE for Expanded Specimens
CRITICAL FLARE labeling of fixed and expanded specimens is described here either for routine cultured cells (indicated as option A throughout the Procedure, where our model specimen was H-tert Retinal Pigment Epithelium cells) or for thin (~100 μm) tissue sections (option B, where our model specimen was mouse kidney tissue sections). The FLARE labeling procedure is quite general and should work well with many tissues (brain, muscle, kidney, etc.); however, expansion-based methods may require optimization to ensure uniform expansion with different specimens or thicker/thinner specimens, and researchers encountering difficulties should consult the primary literature for examples similar to their specimen. Other researchers have recently applied NHS labeling to a range of expanded specimens including Yu et al.17 who labeled whole expanded C. elegans organisms, Damstra et al.19 who labeled other expanded cultured cells and expanded mouse brain, and Sim et al.20 who labeled expanded zebrafish larvae and various expanded mouse tissue sections. The expansion method used in this protocol was adapted and modified from Ku et al.11 The bold numbers in brackets denote the quantities used during the experiment. As a general start, we would suggest following the processing and staining procedures as written for either the cell or tissue section. After which, optimize the label, if needed, using the troubleshooting section.
-
1To prepare cell culture samples follow option A, and for tissue samples follow option B.
- Cell culture pre-processing and reduction •Timing ~12 h
- Grow cells to ~70–90% confluency on #1.5 round coverslips. For H-tert RPE-1 cells, this can be achieved by seeding ~30,000–70,000 cells/well on #1.5 round coverslips in 24-well culture plates and incubate for 12 h at 37 °C.
- Fix the cells using 0.125mL of PFA/GA fixation solution for 10 min at room temperature (~20 °C). Store fixed cells in 1× PBS with 3 mM sodium azide at 4 °C prior to use. ■Pause Point Fixed cells can be stored for up to 6 months at 4 °C.
- OPTIONAL Prepare a fresh, aqueous 10 mM NaBH4 solution [4 mg NaBH4 in 10 mL DI H2O]. CRITICAL STEP Steps 1A iii-v Reduction steps are optional, but are advised in order to quench remaining aldehydes from the fixation process and to lower the amount of GA-induced fluorescence.
- OPTIONAL Incubate the fixed cells with the NaBH4 solution prepared in Step 1Aiii for 10 min [~0.5 mL per well, 24-well plate].
- OPTIONAL Wash cells with 1× PBS [0.5 mL per well, repeat three times]. ■Pause Point Store up to 6 months in 1× PBS with 3 mM sodium azide, depending on your fixation conditions and cell sample stability.
- Tissue pre-processing •Timing ~1–6 h
- After removal of the organ of interest, fix the tissue in 4% (vol/vol) PFA at 4 °C. The length of time required for fixation is dependent on the thickness of the tissue (e.g., adult mouse kidney is immersion-fixed in 4% (vol/vol) PFA for 1 h, while adult mouse liver is fixed for 6 h).
- Slice the tissue on a vibratome into ~100 µm sections and store in a multiwell plate in 1× PBS buffer with 3 mM sodium azide at 4 °C until ready for further processing. ■Pause Point Fixed tissue slices can be stored for up to 6 months at 4 °C.
Monomer infusion •Timing 24 h
CRITICAL For the relatively thin (~100 µm) tissue specimens focused on here, immersion of the sample within the monomer solution is sufficient for the production of uniform gels46, whereas the original MAP procedure11 used cardiac perfusion to enhance monomer infusion throughout relatively thick whole mouse organs. In any event, the FLARE labeling procedure is well-suited to either variation of the gelation procedure.
-
2
Incubate cell samples or tissue samples with cell monomer solution or tissue monomer solution, respectively. If using cells, add the cell monomer solution to each well of a 24-well plate and incubate at room temperature (RT) for 24 h [0.5 mL per well]. If using tissue, incubate the sample in tissue monomer solution overnight at 4 °C [1 mL per well]. Protect samples from light during incubation.
Gelation
-
3For gelation of cell samples follow option A, for tissue samples follow option B.
- Cell sample gelation •Timing 30 min
- Prepare a fresh, aqueous 10% (wt/vol) APS solution [10 mg APS in 100 μL DI H2O]. Keep the solution on ice until use.
- Pipette 98 μL of monomer stock solution into a microcentrifuge tube.
- Pipette 2 μL of 10% (wt/vol) APS solution into the microcentrifuge tube, quickly mix, and add 70 μL of mixed solution onto a piece of Parafilm (~5 cm × 5 cm).
- Use the forceps to carefully take out the round coverslip with fixed cells from the well, and carefully place the coverslip with cells facing down onto the solution prepared from the previous step. ▲Critical Step Make sure the gelation is done on the side of the coverslip that is covered with cells. ?Troubleshooting
- Wait 30 min for the solution to polymerize at RT. Protect the solution from light during polymerization. Note that any solution left in the microcentrifuge tube can be used to judge the progress of the gelation process.
- Use a ruler to measure the gel size to obtain the pre-expansion size for calculation of expansion factor later in Step 19.
- Tissue sample gelation •Timing 2 h for mouse kidney tissue
- Add one drop of monomer solution [99 μL of fresh monomer solution + 1 μL of 10% (w/v) VA-44] on the tissue surface, and place another rectangular #1.5 coverslip on top to create a tissue gelation chamber. The drop of monomer solution should cover the entire sample area (up to ~ 1 cm2) (Fig. 6b-c and 6f-g).
- Put the sample in a glass chamber and purge with nitrogen before shutting the airtight lid. Incubate the chamber at 45 °C for 2 h.
Denaturation
-
4For denaturation of cell samples follow option A, for tissue samples follow option B.
- Denaturation of cell samples •Timing 60 min
- Use a razor blade to carefully peel off the gel from the Parafilm. Avoid peeling the gel off the coverslip at this point as this could damage the cells.
- Transfer the gel attached to the round coverslip into a 20 mL scintillation vial containing denaturing solution [10 mL solution for each sample].
- Denature the cell sample for 60 min at 95 °C. Note that the hydrogel will typically expand ~2-fold during this process. The heating process can be done using heat blocks on an orbital shaker. ?Troubleshooting
- Transfer the gel to a large volume of PBST solution to wash out denaturing reagents. This wash step should be repeated three times for 30 minutes each time. Next, use 1× PBS to wash away detergents prior to chemical staining. ■Pause Point Store up to a week in 1× PBS with 3 mM sodium azide at 4 °C.
- Denaturation of tissue samples •Timing 48 h for mouse kidney tissue
- Denature the gelled samples for the subsequent FLARE staining and expansion steps. For mouse kidney tissue demonstrated here, incubate in denaturing solution at 70 °C for 24 h followed by an additional 24 h at 90 °C.42 Thinner or more easily denatured tissues such as brain can be incubated for briefer periods while thicker or less easily denatured tissues may require longer incubations, as described by Ku et al.11. Based on the sample stiffness, harsher or milder temperatures might be needed. Researchers should determine the desired conditions for their samples. However, regardless of the success of the denaturation, the gelled samples can still be labeled using FLARE.
Carbohydrate stain of FLARE •Timing 5 h 45 min
-
5
Oxidation of carbohydrates to aldehydes. Prepare a fresh, aqueous 20 mM NaIO4 solution [43 mg NaIO4 in 10 mL NaOAc/NaCl buffer].
-
6
Cut a piece of either the expanded cell-based hydrogel (from Step 4A) or the expanded tissue-based hydrogel (from Step 4B) to a suitable size (e.g., 1×1 cm2) and transfer both the hydrogel and the 20 mM NaIO4 solution [10 mL] into a 6-well plate. We refer to both gel systems as “hydrogel” in subsequent steps. (Fig. 7a-b)
-
7
Let the oxidation reaction run for 1 h while gently agitating the container on an incubator shaker at 37 °C. Protect the solution from light during the reaction.
-
8
Remove the solution from Step 7 and wash the sample with NaOAc buffer at room temperature for 10 min each with gentle agitation [10 mL per wash, repeat three times]. ▲Critical Step Excess NaIO4 not removed by washing may oxidize the dyes in the subsequent coupling step, reducing fluorescence signal.
-
9
Transfer the hydrogel sample into a well of a 12-well plate.
-
10
Hydrazone formation to couple hydrazide dye to oxidized carbohydrate aldehydes. Dilute AT565-NHNH2 master stock in NaOAc buffer to final concentration 5 μg/mL and add to the well containing sample [1 μL dye in 1 mL NaOAc buffer, for 6.66 μM final concentration]. ?Troubleshooting
-
11
Let the reaction run for 3 h with gentle agitation on an orbital shaker.
-
12
Reduction to form a stable bond between the oxidized carbohydrates and fluorophore. Prepare a fresh 100 μL of 5 M NaCNBH3 solution [31.4 mg NaCNBH3 in 100 μL NaOAc buffer]..
-
13
Add 20 μL of the NaCNBH3 solution directly to the sample well of Step 10.
-
14
Allow the sample to react for 30 min with gentle agitation.
-
15
Wash the sample with the NaOAc buffer for 15 min [2 mL each, repeated twice]. Then allow the sample to remain in the NaOAc buffer for a longer period of time [1 h or overnight depending on the sample] in order to remove excess unconjugated hydrazide dye. The removal of dyes can be observed by the increasing lack of color in the hydrogel over time during washing (Fig. 7c).
-
16
Wash the sample with 1× PBS solution for 15 min. [2 mL]
Fig. 7. A step-by-step illustration of the FLARE procedure for the expanded sample.

(a) The gel embedded tissue section shown in Figure 6 partially expanded after the denaturation and became transparent. (b) The tissue section was cut into three pieces. One piece (star) was used for FLARE staining and the other two were saved for other purposes. (c) The tissue section was stained pink after using ATTO 565 hydrazide for the carbohydrate stain. (d) Subsequently, the amine stain performed by using ATTO 647N-NHS ester made the tissue section light violet. (e) The tissue section was slightly colored even after the full expansion, demonstrating the characteristic of intense labeling of FLARE. The gel was laying down on a coverslip (24 mm × 50 mm) while taking step-by-step photos.
Amine stain of FLARE
-
17To label amines on cell samples, follow option A, for amine labelling of tissue samples, follow option B.
- Cell sample amine labeling •Timing 1 h
- Prepare 2.5 μg/mL AT647N-NHS from master stock [add 1 μL dye to 2 mL 1× PBS for 2.97 μM final concentration]. ?Troubleshooting
- Incubate sample (from step 16) with AT647N-NHS solution for 1 h with gentle agitation and while covered to protect from light.
- Wash the sample with 1× PBS for 30 min [2mL each, repeated twice]. Store in 1× PBS for further use.
- Tissue sample amine labeling •Timing 6 h for mouse kidney tissue
- Incubate tissue sample (from step 16) with a final concentration of 5 μg/mL (5.9 μM) AT647N-NHS in MES. We incubate for 6 h for 100 μm thick expanded kidney tissue sections to help ensure a uniform stain (Fig. 7d). Faster stains can be achieved with a higher concentration of dye.
Expansion •Timing 1 h
-
18
Transfer the sample to a 100 mm Petri Dish and cover the dish with an excess volume of DI H2O (~30 mL) and let the sample expand for at least 1 h. Frequent DI H2O change speeds up the expansion process. Note that the sample should be completely submerged in water.
-
19
Carefully remove as much water as possible from the edge of the Petri Dish using a plastic pipette leaving behind the expanded hydrogel sample. Use a ruler to measure the post-expansion size and divide the value by the pre-expansion size (found in Step 3) to obtain the approximate expansion factor (4.5–5-fold). ▲Critical Step This is the macroscopic expansion factor and should be the same as the microscopic expansion factor when the ExM procedure is done properly. However, one should keep an eye out for possible discrepancies between the two expansion factors, especially in specimens that have not been previously validated for expansion (Fig. 7e).
-
20
OPTIONAL If you wish to undertake DNA fluorescence in situ hybridization (FISH), follow the general protocol as described in the Supplementary Method 1 before moving on to the next step in this procedure.
DNA stain •Timing 35 min
▲Critical While conventional DNA staining is a great tool for most samples, it might not work as well after DNA FISH. This is due to the harsh conditions used in the DNA FISH protocol that can dehybridize the double-stranded DNA leading to poor staining by DNA dyes applied later.
-
21
Prepare a 10 μg/mL SYBR Green solution from the master stock [add 1 μL master stock dye to 1 mL DI H2O]. ?Troubleshooting
-
22
Apply the solution directly to the surface of the gel side that contains the cells or tissues and incubate for 20 min. ?Troubleshooting
-
23
Rinse the sample three times with an excess of DI H2O to remove unbound SYBR Green. Note that SYBR Green is a turn-on fluorescent dye with minimal background fluorescence. However, DNA dyes such as Hoechst require thorough washing of the hydrogel to minimize background fluorescence.
Sample mounting •Timing ~5–15 min
-
24
Temporarily transfer the gel to a clean untreated coverslip and remove excess liquid by wicking the edges with tissue paper. Excess water beneath the gel may hinder the adhesion with poly-lysine later (Step 25) and add unnecessary imaging depth. ▲Critical Step Be careful while handling the gel as it is prone to sticking to the tissue paper.
-
25
To eliminate drift during imaging, carefully slide the gel from the untreated coverslip onto a poly-lysine coated coverslip to create strong adhesion between the gel and the poly-lysine surface. It is best to image the sample immediately. ▲Critical Step The poly-lysine coated coverslips for imaging may be prepared ahead of time and are stable for weeks. Evenly spread 2–3 μL of a 1 mg/mL poly-lysine solution on the surface of a hydrophilic coverslip and allow it to dry. Exposing the coverslip to an air plasma in a plasma cleaner for ~1 min is the preferred way to render the slides hydrophilic prior to spreading the poly-lysine solution, but not required. ?Troubleshooting
Imaging and Image analysis •Timing Variable (about 3 hours for doing high-resolution imaging on a 100 µm thick tissue)
-
26
Select an appropriate imaging technique for your sample type (e.g. widefield, confocal, or light-sheet microscopy). See Supplementary Table 1 for a list of which of the three modalities was used for all data sets shown in this paper. For expanded samples, we often use a widefield microscope to check sample usability, and we typically use confocal microscopy for routine experiments since confocal microscopy offers straightforward high-resolution 3D imaging with good optical sectioning. For high-resolution imaging of expanded specimens whose index of refraction is approximately the same as that of pure water, we use a water-immersion lens. It is important to match the index of refraction of the specimen with the immersion medium of the objective lens to avoid serious optical aberrations. Ensure that the objective lens has a large enough working distance to image into the specimen at the desired depth. ?Troubleshooting
-
27
For all data sets from expanded samples, use the free software package ImageJ for image visualization and processing, such as channel merge, image registration, and image smooth. Details for the processing we applied for each figure can be found in Supplementary Table 1.
Procedure 2
FLARE for Unexpanded Cleared Tissue Sections
CRITICAL The following procedure is designed for tissue sections, where we demonstrate FLARE using specific examples of 100 µm mouse kidney tissue (option A throughout Procedure 2) and 4 µm thick FFPE human tissue microarrays (option B throughout Procedure 2). The bold numbers included in brackets are the quantities used during the experiment. To adapt FLARE for thicker samples we advise using the processing and staining procedure for 100 μm thick tissue sections as a general starting point, and then further optimize the labeling using the troubleshooting section. For instance, due to the need for dyes to travel a longer distance through thicker tissues, we recommend a longer incubation time.
Carbohydrate stain of FLARE •Timing 4 h
-
1Oxidation of carbohydrates to aldehydes can be performed on either tissue section samples (option A, see also Fig. 8 for an example of FLARE labeling of a ~ 1 mm thick tissue sample) or tissue microarray samples (option B).
- Oxidation of tissue section samples
- Prepare a fresh, aqueous 100 mM NaIO4 in pH 5.0 NaOAc buffer [42.8 mg NaIO4 in 2 mL NaOAc buffer].
- Use a paint brush to gently transfer a tissue section into the above solution in a scintillation vial. Note that having the tissue section float, initially, at the surface of the solution in the well plate makes the transfer to a scintillation vial easier and helps avoid damage to the tissue section.
- Let the reaction run for 30 min while gently agitating the sample on an orbital shaker or rocker. Ensure that the tissue is in constant motion within the solution. Protect the solution from light during the reaction.
- Oxidation of tissue microarray samples
- Since the tissue cores are tightly mounted on the glass slide, the scintillation vial used for tissue sections is too small for the glass slide. Therefore, the oxidation solution is directly applied on the TMA. To be more specific, prepare fresh 100 mM NaIO4 solution and drop 1 mL of the solution onto the tissue cores mounted on the glass slide before lightly covering the tissue cores using Parafilm M. Let the reaction run for 30 min.
-
2Hydrazone formation to couple hydrazide dye to oxidized carbohydrate aldehydes can be performed on either tissue section samples (option A) or tissue microarray samples (option B).
- Hydrazone formation for tissue section samples
- Remove the solution in Step 1Aiii and wash the sample with NaOAc buffer [10 mL per wash, 10 min each wash, repeat three times].▲Critical Step Excess NaIO4 not removed by washing may oxidize the dyes in the subsequent coupling step, reducing fluorescence signal.
- Dilute AT565-NHNH2 master stock in fresh THF/NaOAc solution to final concentration 2.5 μg/mL (Fig. 9e) [1 μL of master stock dye in 2 mL THF/NaOAc solution, for 3.33 μM final concentration]. ?Troubleshooting
- Remove NaOAc washing buffer from Step 2Ai and add the diluted dye solution from Step 2Aii. Incubate the sample on the orbital shaker for 2 h. Ensure that the tissue is in constant motion within the solution. Protect the solution from light during the reaction.
- Hydrazone formation for TMA samples
- Prepare AT565-NHNH2 solution by diluting AT565-NHNH2 master stock in NaOAc solution to final concentration 2.5 μg/mL.
- Wash TMA with NaOAc solution 3 times [1 mL each]
- Drop 1 mL of dye solution onto the washed tissue cores on the glass slide.
- Cover the tissue cores on the glass slide with Parafilm M, sandwiching the dye solution in between the glass slide and the parafilm, and let the reaction run for 2 h.
-
3Reduction to form a more stable amine bond can be performed on either tissue section samples (option A) or tissue microarray samples (option B).
- Reduction of tissue section samples
- Prepare 100 μL of fresh 5 M NaCNBH3 solution [31.4 mg NaCNBH3 in 100 μL NaOAc buffer]. ▲Critical Step NaCNBH3 should be stored in an inert condition. NaCNBH3 is a milder reductant than NaBH4. NaBH4 may react with certain fluorophores (such as many cyanine dyes).
- Add 10 μL of NaCHBH3 solution directly into the reaction mixture in Step 2Aiii.
- Allow the sample to react for 30 min on an orbital shaker.
- Wash the sample with fresh THF/NaOAc solution [10 mL per wash, 10 min per wash, repeat two times].
- Wash the sample with fresh THF/MES solution (Fig. 9f) [10 mL, 10 min]. ■Pause Point Sample can be stored overnight in THF/MES solution at the room temperature.
- Reduction of tissue microarray samples
- Slowly remove the Parafilm M and then add 10 μL of prepared 5M NaCNBH3 solution.
- Cover the TMA sample with Parafilm M again. Let the reaction run for 30 min.
- Wash the sample with fresh NaOAc solution [1 mL, 5 min per wash, repeat two times].
Fig. 8. FLARE labeling of ~1 mm thick kidney tissue.
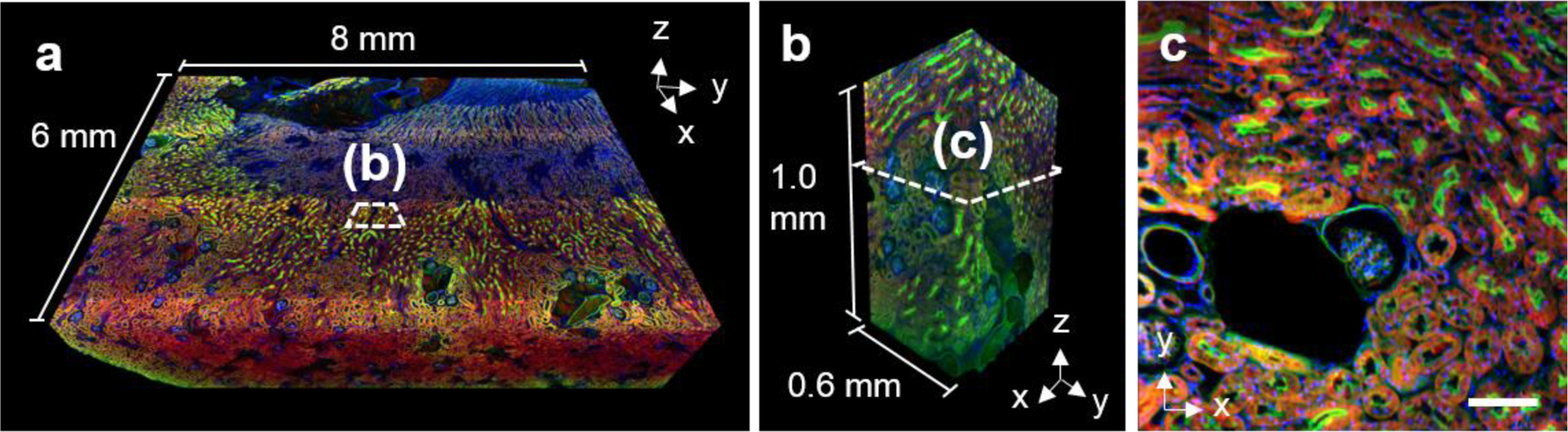
(a) Volumetric imaging of a large mouse kidney section (8 mm × 6 mm × 1 mm) imaged with open-top light-sheet microscopy. Amines (red), carbohydrates (green), and DNA (blue) reveal unique structures throughout the tissue. (b) The zoom-in view from the boxed region in panel a) shows that there is uniform labeling of structures through the full ~1 mm tissue thickness. (c) A single optical section from the boxed region in panel b) demonstrates highly specific amine, carbohydrate, and DNA staining of kidney tissue microstructure. Scale bar, 100 μm (c).
Fig 9. A step-by-step illustration of the FLARE procedure for the optically cleared sample.

(a) A flat, 200 μm mouse kidney section was placed on a #1.5 coverslip. (b-c) The same section was performed with FLARE, optically cleared, and incubated in ethyl cinnamate and sandwiched between two #1.5 coverslips. (d) The mounted sample (yellow box) was directly placed on a widefield microscope. (e-h) A scintillation vial was used to perform the FLARE procedure, including (e) carbohydrate stain, (f) wash, (g) amine stain, (h) DNA stain. The grid lines are separated by ~2 mm. The amine labeling was not uniform on purpose to demonstrate the result without using MES/THF co-solvent mixture.
Amine Stain •Timing 2 h (100 µm thick specimen)
-
4The amine stain can be performed on either tissue section samples (option A) or TMA samples (option B).
- Amine stain for tissue section samples
- Prepare 2.5 μg/mL AT647N-NHS from master stock [add 1 μL dye stock to 2 mL THF/MES solution, for 2.97 μM final concentration]. ?Troubleshooting
- Incubate sample with AT647N-NHS solution from Step 3A for 2 hours with gentle rocking. Ensure that the tissue is in constant motion within the solution (Fig. 9g). Protect the solution from light during the reaction.
- Wash the sample with fresh THF/MES solution [10 mL per wash, repeat two times].
- Wash the sample with fresh THF/PBS solution (Fig. 9h) [10 mL, 10 min]. ■Pause Point Sample can be stored overnight in THF/PBS solution at the room temperature.
- Amine stain for TMA samples
- Prepare AT647N-NHS solution by diluting AT647N-NHS master stock in PBS solution to a final concentration 2.5 μg/mL.
- Wash TMA with PBS solution 3 times [1 mL each]
- Drop 1 mL of the dye solution onto the washed tissue cores on the glass slide. Cover the TMA sample with Parafilm M. Let the reaction run for 30 min.
- Wash the sample with PBS solution [1 mL, 5 min per wash, repeat two times].
DNA Stain •Timing 30 min (100 µm thick specimen)
-
5The DNA stain can be applied to tissue section samples (option A) or TMA samples (option B).
- DNA stain for tissue section samples
- Prepare 5 μg/mL Hoechst 33258 solution [add 1 μL dye stock to 2 mL THF/PBS solution].
- Incubate the sample in the solution prepared in Step 4A for 30 minutes with gentle rocking. Ensure that the tissue is in constant motion within the solution. Protect the solution from light during incubation.
- Briefly wash the sample with fresh THF/PBS solution with gentle rocking [10 mL per wash, repeat twice, 5 min each].
- DNA stain for TMA samples
- Prepare 5 μg/mL Hoechst 33258 solution in PBS solution.
- Drop 1 mL of the dye solution onto the washed TMA sample. Cover the TMA sample with Parafilm M. Let the reaction run for 30 min.
Dehydration and Refractive Index Matching for tissue section samples •Timing ~3.5 h (100 µm thick specimen)
CRITICAL Sample dehydration should only be performed on tissue sections (100 µm) and not on TMA samples (5 µm) because they are very thin. TMA samples are imaged with an air and/or water-immersion objective lens directly. For TMA samples, please proceed to Step 16.
-
6
Transfer the tissue section into fresh 50% (vol/vol) THF in DI water [4 mL].
-
7
Incubate the sample for 30 min with gentle rocking. Protect the solution from light during incubation.
-
8
Incubate the sample for 30 min in 80% THF (vol/vol) in DI water [3.2 mL THF and 0.8 mL DI water]. Protect the solution from light during incubation.
-
9
Incubate the sample for 30 min in 100% THF [4 mL THF]. Protect the solution from light during incubation.
-
10
Soak the sample in DCM until it sinks [4 mL DCM].
-
11
Incubate the sample with EC to match the refractive index [1 mL EC]. The duration of the dehydration and index matching step will depend on the type of tissue and its thickness, but ~2 h is a good starting point. It may be necessary to refresh the EC solution multiple times with longer incubation times for thicker samples to ensure good index-matching. The clarity of the cleared tissue is easy to judge by the unaided eye (Fig. 9b). ■Pause Point It is advisable to image as soon as possible but we have successfully imaged following storage in EC at room temperature for a few days. ?Troubleshooting
Sample mounting (for 100 µm thick specimen only) •Timing 5 min
-
12
Use a paint brush to transfer the tissue sample onto a rectangular #1.5 coverslip. Note that at this point, the tissue sample is now rigid due to dehydration and thus would not entangle with the brush and can be easily transferred.
-
13
Attach a piece of double-sided tape to the surface of each of the two short edges of the coverslip.
-
14
Add one drop of EC to fully cover the sample.
-
15
Press a second rectangular #1.5 coverslip onto the first to form a sandwich structure held together by the double-sided tape. Make sure that the sample is sitting very closely to the surface of the first #1.5 coverslip in the sandwich structure and that there are no air bubbles on the specimen surfaces (Fig. 9c).
Imaging and Image Analysis •Timing Variable
-
16
Image as required for study. There are some considerations in sample imaging that are similar to those mentioned in Step 26 of Procedure 1. A widefield microscope is typically used first to check for general sample quality and a confocal microscope is then used for routine experiments (Fig. 9d). For imaging of very thin (<10 μm) unexpanded specimens, we typically use a widefield microscope with a 4× objective lens. For thicker unexpanded cleared specimens (10–200 μm), we typically use a confocal microscope with an oil-immersion objective lens to avoid optical aberrations when imaging deeper into the EC-cleared specimens. For even thicker cleared specimens (>0.1 mm) we typically use light sheet microscopy46–48. ?Troubleshooting
-
17
Analyze data. As described in Step 27 of Procedure 1, the free software package ImageJ is sufficient to analyze most data sets. However the large mouse kidney data set (Fig. 8) which was acquired by open-top light-sheet microscopy32 was volumetrically rendered using Imaris. Details regarding the data processing we used for each figure can be found in Supplementary Table 1.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1.
Troubleshooting Table
| Step | Problem | Possible reason | Solutions |
|---|---|---|---|
| Procedure 1 step 4. |
Poor or uneven expansion, sample tearing | Inadequate denaturation or digestion of the sample may cause sample tearing or under-expanded regions in the nuclei distribution (see Fig. 10). | Extend the denaturation period and/or repeat the denaturation step at a higher temperature. In the case of enzymatic digestion, an addition of another category of enzyme (such as elastase) may help with digestion49,50 (see Fig. 10). |
| Procedure 1 steps 3, 25, 26 OR Procedure 2 step 16 |
No signal | Lack of signal can arise due to several reasons: | |
| Inappropriate microscope settings | Check that the microscope settings are appropriate to the fluorophores with a suitable positive control. | ||
| Issues with sample gelation | Make sure that the hydrogel polymerization is done on the coverslip side coated with cells as it can be easy to lose track of which side of a coverslip the cells are adhered to (Fig. 11b). Make sure that the sample is positioned as close to the surface of the gel as possible in order to be within the working distance of the objective lens. | ||
| Error with sample mounting | It is possible that the gel is upside down, and the surface of the hydrogel containing the sample is outside the working distance of the objective lens (Fig. 11c). Test this by imaging the other surface of the gel. | ||
| Fluorophore bleaching | The NaIO4 oxidation step will chemically bleach many fluorophores, and adequate removal of NaIO4 is required prior to the hydrazide dye incubation. Repeat the procedure with more thorough washing. Importantly, the hydrazide labeling step should be performed before the NHS-dye coupling reaction, or any other fluorescent labeling reactions. | ||
| Procedure 1 steps 10, 12, 17, 25, 26 OR Procedure 2 steps 2, 3, 4, 16 |
Dim Signal | Dim signals can arise for several reasons: | |
| Reagent degradation can result in an inefficient reaction. | If the carbohydrate stain is dim, it could indicate that NaCNBH3, which can become deactivated with prolonged exposure to moist air, has been hydrolyzed, leaving the hydrazide-dye conjugated to the specimen by a relatively labile hydrazone bond. If the amine channel is dim, it could indicate that the NHS groups on the NHS-dye have been hydrolyzed and thus the dyes were not efficiently coupled to the sample. In either of these cases, researchers can repeat the procedure using fresh aliquots or newly purchased reagents. In addition, proper storage can help slow down the degradation of these reagents (refer to the Reagents set-up section). | ||
| Insufficient dye concentration | The sample staining intensity can be tuned to produce brighter signals by increasing the concentration of the reactive fluorophores used in the labeling reactions. | ||
| Lack of sample exposure to chemicals | With respect to the hydrogel-embedded samples, make sure that the hydrogel surface containing the sample is placed face-up in a container during the labeling reaction, or the dyes may have a harder time accessing the sample resulting in dim signals (Fig. 11a). | ||
| Incorrect objective lens | It is essential to carefully choose the correct objective lens for a given type of sample: water-immersion objective lenses are well-suited to imaging specimens in hydrogels, whereas oil-immersion lenses are well-suited to imaging organic cleared oil refractive index matched EC specimens. The use of a mismatched immersion lens may lead to optical aberrations and loss of signal at substantial depths. | ||
| Inner filter effect | See advice given for an uneven Signal. | ||
| Procedure 1 step 26 OR Procedure 2 step 16 |
Uneven Signal | Uneven signals mostly occur following labeling of thick samples and could be a result of excess dye during a labeling reaction. A thick sample can be labeled so strongly at the surface that the excitation light in the microscope is substantially attenuated within the interior of the sample (“inner filter effect”), producing a very bright surface but an apparent dimmer interior, even if the sample itself is in fact labeled uniformly. | Section the hydrogel or the thick sample and image the cross-section to assess the sample uniformity without this complication (see Fig. 12). If the stain is indeed uniform, stain a new sample with a more dilute dye to produce a dimmer stain that avoids this inner filter effect (Fig. 12). If the stain is in fact enriched at the surface, consider preparing the NHS-dye stock in ~100 mM MES buffer at pH 6.0 to slow down the hydrolysis and coupling reactions while also providing more time for the labeling reaction and dye penetration. In addition, to aid in the penetration of the dye through the expanded samples which are embedded in negatively charged hydrogel, consider using NaOAc/NaCl buffer solution to help allow charged reactants to enter the negatively charged hydrogel gel. Also, make sure that the bottom surface of the tissue sample is not attached to the surface of the container during incubation (Fig. 11a) and to ensure continuous sample motion during gentle agitation. Using a lower concentration of dye may also help improve the uniformity of labeling, but possibly at the cost of decreasing the brightness of the stain. |
| Procedure 1 steps 15, 17 OR Procedure 2 Steps 2, 4 |
Overly Bright Signals | Crosstalk from an overly bright signal in a channel could swamp a weaker signal from a different spectral channel. In addition, an overly bright signal can produce inner filter effect. (see Uneven Signal) Overly bright signals can arise due to several reasons: | |
| Insufficient washing could prevent unconjugated dyes from being removed from the sample resulting in very bright signals. | Washing the sample extensively can fix this problem and improve contrast. | ||
| If the dye concentration is too high, an overly bright sample may be produced. | Lowering the concentration of dye(s) in the coupling reaction(s) can help tune the signal. | ||
| Procedure 2 step 7–11 |
Poor Transparency in Refractive Index Matched Samples | Improper or insufficient tissue clearing can arise for several reasons: | |
| Inadequate dehydration prior to incubation with ethyl cinnamate (EC) or other common refractive index oil. | Thorough dehydration is crucial to achieve a uniform refractive index match throughout the entire specimen thickness and extending the dehydration periods at each stage may help improve the clarity. | ||
| Insufficient incubation with EC | Refresh the EC a couple of times, followed by waiting ~30–60 min or more (depending on the size of the tissue) for the tissue to become fully cleared and index-matched. |
Timing
Procedure 1, FLARE for Expanded Specimens
Step 1, specimen pre-processing: ~12 h (cells), ~1–6 h (mouse kidney tissue)
Steps 2–3, monomer infusion and gelation: ~24.5 h (cells), ~26 h (mouse kidney tissue)
Steps 4, denaturation: ~1 h (cells), ~48 h (mouse kidney tissue)
Steps 5–16, carbohydrate stain: ~6 h
Steps 17, amine stain: ~1–6 h
Steps 18–20, expand: ~1 h
Steps 21–23, DNA stain: ~35 min
Steps 24–25, sample mounting: ~15 min
Steps 26–27, imaging and image analysis: variable
Procedure 2, FLARE for Unexpanded Cleared Tissue Sections
Steps 1–3, carbohydrate stain: ~4 h
Steps 4, amine stain: ~2 h
Steps 5, DNA stain: ~30 min
Steps 6–11, Dehydration and Refractive Index Matching: ~3.5 h
Steps 12–15, sample mounting: ~5 min
Steps 16–17, imaging and image analysis: variable
Anticipated Results
We have applied FLARE to a wide variety of samples including cultured cells, mouse organs, human specimens, as well as different sample processing procedures such as fresh fixation, FFPE preparation, clearing, or expansion. Here we present typical results seen following FLARE labeling of a range of samples (Fig. 3–5, Fig. 8). The 1 mm thick mouse kidney tissue shown in Fig. 8 (the procedure for which is described in Supplementary Table 1) provides an example of longer dye incubation times and other optimizations for labeling thicker samples. We also include figures and data to illustrate possible issues associated with FLARE labeling (Fig. 10–12).
Fig. 10. Demonstration of expansion uniformity using a MAP-based protocol.

Epifluorescence images showing nuclei (DNA) stained by Sybr Green in expanded 100 μm thick mouse kidney tissue sections that were homogenized using a MAP-based protocol at three time points. (a) Incomplete homogenization leads to abundant tears (empty areas) and under-expanded regions (high-density clusters) exhibiting relatively small or abnormally stretched nuclei. (b) Extending the homogenization period (~5 times longer than in panel a) leads to uniform sample expansion that lacks obvious distortions in the expanded state. (c) A further extension of the homogenization period (~25 times longer than in panel a) does not cause a larger expansion factor or damage DNA. Scale bars 200 μm (a,b,c) in pre-expansion units.
Fig. 12. Demonstration of the inner filter effect for densely-labeled samples.
Confocal images of densely labeled 500 µm sections of human nephrectomy kidney tissue (deidentified and obtained from NW Biotrust) stained with AT647N-NHS ester in MES/THF co-solvent showing prominent inner filter effect, with a figure showing imaging procedure. The inner filter effect results from substantial absorption of the excitation light through the depth of a strongly labeled specimen, giving the impression that labeling is stronger at the surface of a specimen. (a) Cross-sectional view of kidney slice z-stack showing strong signal close to the coverslip with decaying intensity at depth (increasing z distance). The kidney slice was then cut, flipped on edge, and (b) reimaged to show the newly revealed surface. The revealed face showed that the stain was uniform across the sample depth (~500 µm), confirming that the artifact in a indeed resulted from the Inner Filter Effect. See Troubleshooting section. Scale bar: 100 µm (a,b).
Effects of fixatives
Fixation is a critical part of many imaging experiments. Because FLARE is able to show the general physiology of samples extremely well, it can be a useful tool for revealing potential perturbations in fixation or other sample processing steps. For example, we compared fixation using paraformaldehyde and glutaraldehyde (Fig. 3a-d) with fixation using glyoxal51 (Fig. 3e-h) in expanded RPE cells. We found that cells fixed with paraformaldehyde and glutaraldehyde revealed mitochondria, lysosomes, the Golgi apparatus, nuclear pores and portions of the cytoskeleton (Fig. 3a-d) while glyoxal-fixed cells showed major perturbations to many of these structures (Fig. 3e-h).
FLARE on tissue microarrays
FLARE’s simplicity and robustness make the staining method appealing for use with diverse types of samples, such as in the fluorescent staining of FFPE sections in tissue microarrays that have been embedded and stored for ~13 months (Fig. 4a-g and Supplementary Figures 1-2) or 5 years (Fig. 4h-n and Supplementary Figures 3-4) prior to staining. FLARE is able to present the general physiology of these tissues similar to H&E (Fig. 4b, 4e, and 4i-k) with an additional benefit of showing abundant multicolor details with high contrast (Fig. 4c-d, 4f-g, and 4l-n). Since FLARE reveals the accessible amine and carbohydrate groups, extensive fixation or different processing procedures or storage conditions could in principle alter the staining pattern of FFPE sections.
FLARE with other modalities
If desired, FLARE can be optimized for combination with other modalities such as immunolabeling on fresh mouse kidney tissue (Fig. 5a, see Supplementary Table 2 for a summary), DNA FISH on FLARE-stained cleared unexpanded mouse kidney (Fig 5b) and expanded mouse kidney (Fig. 5c). It is prudent to keep in mind that while the labeling order in combination with such modalities may require optimization, FLARE is flexible and can often be applied at different steps in a protocol (Fig. 2). FLARE also reveals major features in combination with enzymatic digestion-based ExM (Fig. 5d), however, digestion-induced protein loss may alter the labeling to some extent.
FLARE on thick tissue
We used FLARE to label a relatively thick (~1 mm) kidney tissue specimen and we were able to uniformly stain the specimen by tuning of buffer conditions (Fig. 8 and Supplementary Video 1). Bioconjugation with NHS-esters tends to be very quick, especially at a reaction-optimized buffer pH of ~8–9. However, this may result in most of the reactions happening on the tissue surface thus creating uneven labeling because hydrolysis is also rapid at this pH and competes with bioconjugation. By using a lower buffer pH of ~6 and a mixed aqueous and organic solvent, we were able to slow down the bioconjugation reaction while also increasing the rate of dye diffusion, thereby allowing the dye to infiltrate the sample fully before bioconjugation could happen, resulting in a more uniform stain. An advantage of the FLARE stain for such a thick specimen is that the labeling can be done in < 24 h, while the use of immunolabels could take days or weeks.
In conclusion, FLARE is a valuable addition to the fluorescent labeling toolkit. It produces bright, well-contrasted, and reliable stains using commercially available reagents. The fluorescence spectra of the FLARE probes can be easily tuned to meet experimental constraints or preferences by choice of commercial fluorophore, as demonstrated by the many examples we provide here and can be found in previous publications3 spanning the entire visible spectrum and into the near-infrared region.
Supplementary Material
Fig. 11. Proper sample orientation during staining and image acquisition.
(a) If the cell sample is facing down (left), the dyes often have lower accessibility to the sample (blue arrows), resulting in dimmer stains. If the cell sample is facing up (right), the dyes have higher accessibility to the sample (blue arrows), resulting in brighter stains. (b) Hydrogel polymerization on the side coated with cells (left) vs polymerization on the incorrect side (right). If polymerization is performed on the side without the cells, then the subsequent hydrogel would not have any cells embedded, resulting in no signal under the microscope. (c) Expanded hydrogel mounted on poly-lysine coated coverslip, where cell sample is within the working distance of the objective (left) and the cell sample on the opposite side of coverslip (right). If the hydrogel is mounted as seen on the right, the cell sample might not be within the working distance of the objective lens.
Key references using this protocol:
Feature-rich covalent stains for super-resolution and cleared tissue fluorescence microscopy (DOI: 10.1126/sciadv.aba4542);
Rapid pathology of lumpectomy margins with open-top light-sheet (OTLS) microscopy (DOI: 10.1364/BOE.10.001257).
Acknowledgments
This work was supported by the University of Washington, NIDDK Diabetic Complications Consortium grants DK076169 and DK115255 (J.C.V.), NIH grants R01 MH115767 (J.C.V.), R01 CA244170 (J.T.C.L.), and K99 CA240681 (A.K.G.), and DoD PCRP grant W81XWH-18-10358 (J.T.C.L.). NW BioTrust, a core service for patient consenting, and NWBioSpecimen, a core service for procurement and annotation of research biospecimens, are supported by National Cancer Institute grant P30 CA015704 (G. Gilliland, principal investigator [PI]), Institute of Translational Health Sciences grant UL1 TR000423 (M. Disis, PI), the University of Washington School of Medicine and Department of Pathology, and Fred Hutchinson Cancer Research Center.
Footnotes
Code Availability
The custom Wolfram Mathematica scripts used for stitching tiling images (Fig. 4a and 4h) are available as Supplementary Code.
Competing Interests
J.T.C.L and A.K.G are co-founders and hold equity in Lightspeed Microscopy Inc., of which J.T.C.L is also a board member. The other authors declare no competing interests.
Data Availability
Fig. 1c, Fig. 3a-d, and Fig. 5a and 5c are reproduced from Mao et al. (DOI: 10.1126/sciadv.aba4542).3 The data in Fig. 8 and Supplementary Video 1 were previously shown in Mao et al. (DOI: 10.1126/sciadv.aba4542)3, but are now presented in an alternative format. The data shown in Fig. 3e-h, Fig. 4, Fig. 5b and 5d, Fig. 10, Fig. 12 has not been published previously. Additional examples of data are available from the corresponding author upon request.
References
- 1.Bradbury A & Plückthun A Reproducibility: Standardize antibodies used in research. Nature 518, 27–29 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Schnell U, Dijk F, Sjollema KA & Giepmans BNG Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods 9, 152–158 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Mao C et al. Feature-rich covalent stains for super-resolution and cleared tissue fluorescence microscopy. Sci. Adv 6, eaba4542 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Titford M Progress in the Development of Microscopical Techniques for Diagnostic Pathology. J. Histotechnol 32, 9–19 (2009). [Google Scholar]
- 5.Sigal YM, Zhou R & Zhuang X Visualizing and discovering cellular structures with super-resolution microscopy. Science 361, 880–887 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schermelleh L et al. Super-resolution microscopy demystified. Nat. Cell Biol 21, 72–84 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Sahl SJ, Hell SW & Jakobs S Fluorescence nanoscopy in cell biology. Nat. Rev. Mol. Cell Biol 18, 685–701 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Chen F, Tillberg PW & Boyden ES Expansion microscopy. Science 347, 543–548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chozinski TJ et al. Expansion microscopy with conventional antibodies and fluorescent proteins. Nat. Methods 13, 485–488 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tillberg PW et al. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nat. Biotechnol 34, 987–992 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ku T et al. Multiplexed and scalable super-resolution imaging of three-dimensional protein localization in size-adjustable tissues. Nat. Biotechnol 34, 973–981 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richardson DS & Lichtman JW Clarifying Tissue Clearing. Cell 162, 246–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tainaka K, Kuno A, Kubota SI, Murakami T & Ueda HR Chemical Principles in Tissue Clearing and Staining Protocols for Whole-Body Cell Profiling. Annu. Rev. Cell Dev. Biol 32, 713–741 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Beliveau BJ et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc. Natl. Acad. Sci 109, 21301–21306 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y et al. Rapid pathology of lumpectomy margins with open-top light-sheet (OTLS) microscopy. Biomed. Opt. Express 10, 1257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie W et al. Microscopy with ultraviolet surface excitation for wide-area pathology of breast surgical margins. J. Biomed. Opt 24, 12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu C.-C. (Jay) et al. Expansion microscopy of C. elegans. eLife 9, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.M’Saad O & Bewersdorf J Light microscopy of proteins in their ultrastructural context. Nat. Commun 11, 3850 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Damstra HGJ et al. Visualizing cellular and tissue ultrastructure using Ten-fold Robust Expansion Microscopy (TREx). http://biorxiv.org/lookup/doi/10.1101/2021.02.03.428837 (2021) doi: 10.1101/2021.02.03.428837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sim J et al. Whole-ExM: Expansion microscopy imaging of all anatomical structures of whole larval zebrafish. http://biorxiv.org/lookup/doi/10.1101/2021.05.18.443629 (2021) doi: 10.1101/2021.05.18.443629. [DOI] [Google Scholar]
- 21.Elfer KN et al. DRAQ5 and Eosin (‘D&E’) as an Analog to Hematoxylin and Eosin for Rapid Fluorescence Histology of Fresh Tissues. PLOS ONE 11, e0165530 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serafin R, Xie W, Glaser AK & Liu JTC FalseColor-Python: A rapid intensity-leveling and digital-staining package for fluorescence-based slide-free digital pathology. PLOS ONE 15, e0233198 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yun DH et al. Ultrafast immunostaining of organ-scale tissues for scalable proteomic phenotyping. http://biorxiv.org/lookup/doi/10.1101/660373 (2019) doi: 10.1101/660373. [DOI] [Google Scholar]
- 24.Ku T et al. Elasticizing tissues for reversible shape transformation and accelerated molecular labeling. Nat. Methods 17, 609–613 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Susaki EA et al. Versatile whole-organ/body staining and imaging based on electrolyte-gel properties of biological tissues. Nat. Commun 11, 1982 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao S et al. Cellular and Molecular Probing of Intact Human Organs. Cell 180, 796–812.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bertiaux E et al. Expansion microscopy provides new insights into the cytoskeleton of malaria parasites including the conservation of a conoid. PLOS Biol 19, e3001020 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Theory and practice of histological techniques. (Elsevier Churchill Livingston, 2013). [Google Scholar]
- 29.Apgar et al. Fluorescence microscopy of rat embryo sections stained with haematoxylin-eosin and Masson’s trichrome method: FLUORESCENCE MICROSCOPY USING HAEMATOXYLIN-EOSIN AND MASSON’S TRICHROME. J. Microsc 191, 20–27 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Giacomelli MG et al. Virtual Hematoxylin and Eosin Transillumination Microscopy Using Epi-Fluorescence Imaging. PLOS ONE 11, e0159337 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glaser AK et al. Light-sheet microscopy for slide-free non-destructive pathology of large clinical specimens. Nat. Biomed. Eng 1, 0084 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glaser AK et al. Multi-immersion open-top light-sheet microscope for high-throughput imaging of cleared tissues. Nat. Commun 10, 2781 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasten FH, Burton V & Glover P Fluorescent Schiff-Type Reagents for Cytochemical Detection of Polyaldehyde Moieties in Sections and Smears. Nature 184, 1797–1798 (1959). [DOI] [PubMed] [Google Scholar]
- 34.Li Z et al. Application of periodic acid-Schiff fluorescence emission for immunohistochemistry of living mouse renal glomeruli by an ‘in vivo cryotechnique’. Arch. Histol. Cytol 69, 147–161 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Shi X et al. Label-retention expansion microscopy. http://biorxiv.org/lookup/doi/10.1101/687954 (2019) doi: 10.1101/687954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karagiannis ED et al. Expansion Microscopy of Lipid Membranes. http://biorxiv.org/lookup/doi/10.1101/829903 (2019) doi: 10.1101/829903. [DOI] [Google Scholar]
- 37.Wen G et al. Evaluation of Direct Grafting Strategies via Trivalent Anchoring for Enabling Lipid Membrane and Cytoskeleton Staining in Expansion Microscopy. ACS Nano 14, 7860–7867 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Götz R et al. Nanoscale imaging of bacterial infections by sphingolipid expansion microscopy. http://biorxiv.org/lookup/doi/10.1101/2020.05.06.080663 (2020) doi: 10.1101/2020.05.06.080663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang J-B et al. Iterative expansion microscopy. Nat. Methods 14, 593–599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ross MH Histology: a text and atlas with correlated cell and molecular biology. (Lippincott Williams & Wil, 2006). [Google Scholar]
- 41.Bucur O et al. Nanoscale imaging of clinical specimens using conventional and rapid-expansion pathology. Nat. Protoc (2020) doi: 10.1038/s41596-020-0300-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Unnersjö-Jess D et al. Confocal super-resolution imaging of the glomeruluar filtration barrier enables by tissue expansion. Kidney Int (2017) doi: 10.1016/j.kint.2017.09.019. [DOI] [PubMed] [Google Scholar]
- 43.Hermanson Greg. Bioconjugate Techniques, 2nd Edition. [Google Scholar]
- 44.Mujumdar RB, Ernst LA, Mujumdar SR, Lewis CJ & Waggoner AS Cyanine dye labeling reagents: sulfoindocyanine succinimidyl esters. Bioconjug. Chem 4, 105–111 (1993). [DOI] [PubMed] [Google Scholar]
- 45.Murray E et al. Simple, Scalable Proteomic Imaging for High-Dimensional Profiling of Intact Systems. Cell 163, 1500–1514 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Power RM & Huisken J A guide to light-sheet fluorescence microscopy for multiscale imaging. Nat. Methods 14, 360–373 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Santi PA Light Sheet Fluorescence Microscopy: A Review. J. Histochem. Cytochem 59, 129–138 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu JTC et al. Harnessing non-destructive 3D pathology. Nat. Biomed. Eng (2021) doi: 10.1038/s41551-020-00681-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chozinski TJ et al. Volumetric, Nanoscale Optical Imaging of Mouse and Human Kidney via Expansion Microscopy. Sci. Rep 8, (2018). 10.1038/s41598-018-28694-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang N et al. Superresolution imaging of Drosophila tissues using expansion microscopy. Mol. Biol. Cell 29, 1413–1421 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richter KN et al. Glyoxal as an alternative fixative to formaldehyde in immunostaining and super‐resolution microscopy. EMBO J e201695709 (2017) doi: 10.15252/embj.201695709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Fig. 1c, Fig. 3a-d, and Fig. 5a and 5c are reproduced from Mao et al. (DOI: 10.1126/sciadv.aba4542).3 The data in Fig. 8 and Supplementary Video 1 were previously shown in Mao et al. (DOI: 10.1126/sciadv.aba4542)3, but are now presented in an alternative format. The data shown in Fig. 3e-h, Fig. 4, Fig. 5b and 5d, Fig. 10, Fig. 12 has not been published previously. Additional examples of data are available from the corresponding author upon request.