

Abstract
As an initial approach to studying the molecular replication mechanisms of hepatitis C virus (HCV), a major causative agent of acute and chronic liver disease, we have recently developed selectable self-replicating RNAs. These replicons lacked the region encoding the structural proteins and instead carried the gene encoding the neomycin phosphotransferase. Although the replication levels of these RNAs within selected cells were high, the number of G418-resistant colonies was reproducibly low. In a search for the reason, we performed a detailed analysis of replicating HCV RNAs and identified several adaptive mutations enhancing the efficiency of colony formation by several orders of magnitude. Adaptive mutations were found in nearly every nonstructural protein but not in the 5′ or 3′ nontranslated regions. The most drastic effect was found with a single-amino-acid substitution in NS5B, increasing the number of colonies ∼500-fold. This mutation was conserved with RNAs isolated from one cell line, in contrast to other amino acid substitutions enhancing the efficiency of colony formation to a much lesser extent. Interestingly, some combinations of these nonconserved mutations with the highly adaptive one reduced the efficiency of colony formation drastically, suggesting that some adaptive mutations are not compatible.
The Hepatitis C virus (HCV) is a distinct member of the family Flaviviridae, comprising a group of enveloped viruses to which the flaviviruses, with the prototype Yellow fever virus, and the animal-pathogenic pestiviruses like Classical swine fever virus and Bovine viral diarrhea virus (BVDV) belong (41). These viruses have in common a single-stranded RNA genome of positive polarity carrying one long open reading frame (ORF) that is flanked at the 5′ and 3′ ends by nontranslated regions (NTRs). In HCV, the genome has a length of ∼9,600 nucleotides and encodes a ∼3,000-amino-acid-long polyprotein carrying the structural proteins in the amino-terminal quarter and the nonstructural (NS) proteins in the remainder (for reviews, see references 4 and 43).
During and after translation, the polyprotein is cleaved in the structural region by host cell enzymes, and the NS proteins are cleaved by two viral proteinases, giving rise to at least 10 different products. These are arranged from the amino to the carboxy terminus as follows: core (C)–envelope protein 1 (E1)–E2–p7–NS2–NS3–NS4A–NS4B–NS5A–NS5B (22, 23, 54). The amino-terminal products C, E1, and E2 are the major constituents of the virus particle, and they are released from the polyprotein precursor by host cell signal peptidases (23). The function of the small hydrophobic polypeptide p7 is unknown. NS2 and the amino-terminal domain of NS3 constitute the NS2-3 proteinase, responsible for polyprotein cleavage at the NS2/3 junction (20, 24). NS3 carries two enzymatic activities residing in two well-defined globular domains (62): a chymotrypsin-like serine proteinase spanning the ∼180 amino-terminal NS3 residues, and nucleoside triphosphatase (NTPase)/helicase activities, residing in the remainder (21, 31, 49, 54). NS4A is a 54-residue-long protein that forms a stable complex with the NS3 proteinase domain and enhances enzymatic activity (3, 14, 36, 52). The function of NS4B currently is not known. NS5A is a highly phosphorylated protein that is released with rather slow kinetics from an NS4B-5A precursor (1, 3, 30, 44, 53). The role of NS5A in replication has not been determined. However, NS5A appears to be involved in resistance against the antiviral activity of alpha interferon (12, 13). NS5A can interact with the interferon-induced double-stranded RNA activated protein kinase PKR that is responsible for a reduction of translation in interferon-treated cells via phosphorylation of eIF2α (17, 18). This interaction inhibits PKR activity, leading to continued protein synthesis in the presence of alpha interferon. NS5B is the RNA-dependent RNA polymerase (RdRp) (6, 37, 59).
Translation of the polyprotein is directed by an internal ribosome entry site (IRES) spanning most of the 5′ NTR and requiring ∼15 nucleotides of the ORF for full activity (27, 45, 55, 57). The first ∼45 nucleotides of the 5′ NTR are dispensable for IRES activity but are probably important for RNA replication. The 3′ NTR has a tripartite structure composed of a variable region following the stop codon of the ORF, a poly(U/UC) tract of variable length, and a highly conserved 98-nucleotide-long sequence designated the X tail (34, 50, 51, 58). Recent in vivo studies demonstrated that a genome lacking the variable region is viable, whereas the X tail is essential for replication (35, 61).
Molecular analyses of HCV replication have so far been hampered by the lack of convenient animal models and efficient cell culture systems. Although infection of primary cells or cell lines with high-titer HCV-containing sera is possible, replication levels in these cultures are too low to allow detailed studies of HCV replication (reviewed in reference 4). Since for numerous positive-strand RNA viruses, including the closely related flavi- and pestiviruses, the transfection of cloned virus genomes into permissive cells allowed productive RNA replication, analogous approaches have been pursued for HCV. However, in spite of the availability of cloned infectious genomes (5, 33, 60), unequivocal demonstration of HCV RNA replication after transfection of cultured cells has so far not been possible (reviewed in reference 4). As an alternative, we have recently developed selectable subgenomic RNAs replicating autonomously to very high levels after transfection into the human hepatoma cell line Huh-7 (38). Selection of these RNAs lacking the complete structural region from C up to p7 or even NS2 was made possible by insertion of the gene encoding neomycin phosphotransferase (neo) downstream of the HCV IRES, whereas translation of the HCV NS proteins was directed by the IRES of the Encephalomyocarditis virus (EMCV). Although replication levels of these RNAs within a selected cell line were high, the number of G418-resistant colonies obtained after selection was very low.
In this study, we analyzed the replicating HCV RNA species in selected cells and provide direct experimental proof that these RNAs carry adaptive mutations. Except for NS4A, adaptive mutations were found in all HCV NS proteins but not in the 5′ or 3′ NTR or in neo or EMCV IRES sequences. The levels of adaptation were different with a single-amino-acid substitution in NS5B, increasing the efficiency of colony formation by almost 1,000-fold. Interestingly, this and two other adaptive mutations in the NS3 helicase affect residues located on the surface of the molecules, suggesting that these sites are important for interaction with viral or host cell factors. Finally, we provide evidence that some of the highly adaptive mutations are only functional when isolated but are inhibitory when combined in a single replicon.
MATERIALS AND METHODS
Cell cultures.
Cell monolayers of the human hepatoma cell line Huh-7 (42) were routinely grown in Dulbecco's modified minimal essential medium (DMEM; Life Technologies, Karlsruhe, Germany) supplemented with 2 mM l-glutamine, nonessential amino acids, 100 U of penicillin, 100 μg of streptomycin, and 10% fetal calf serum (FCS). In cell lines carrying HCV replicons, 500 to 1,000 μg of G418 (Geneticin; Life Technologies) per ml was added to the medium. It should be noted that given G418 concentrations have not been corrected for the amount of active substance as given by the manufacturer. Naive cells as well as replicon-containing cell lines were passaged twice weekly after treatment with 0.05% trypsin–0.02% EDTA and seeding at a dilution of 1:3 to 1:5.
Plasmid constructions.
All numbers given in parentheses refer to a complete HCV genome cloned by our group (EMBL database accession number AJ238799). The vector used to generate all replicon constructs was pFK, derived from pBR322 by deletion of a StyI-EcoRI fragment spanning the complete tetracycline resistance gene and insertion of a multiple cloning site containing the recognition sequences for the restriction enzymes HindIII, ClaI, NotI, SalI, EcoRI, and SpeI upstream of the promoter for T3 RNA polymerase. The original construct pFK-I377neo/NS3-3′/wt (38), containing a HindIII restriction site and the T7 RNA polymerase promoter upstream and a ScaI restriction site downstream of the replicon sequence, was generated by using the HindIII and an SpeI restriction site introduced downstream of the ScaI site for insertion into vector pFK. Plasmids linearized with ScaI are suitable to generate in vitro transcripts terminating exactly with the 3′ end of our HCV isolate.
To functionally analyze replicon variants from cell line 9-13, NcoI fragments (3420 to 8996) from the PCR products cloned into vector pCR and designated pCR9-13B, -C, -F, -H, and -K were first inserted into a truncated replicon vector lacking the HCV 5′ NTR and neo. PmeI-SpeI fragments from these subclones spanning the EMCV IRES up to the end of the 3′ NTR were then transferred into pFK-I377neo/NS3-3′/wt to obtain plasmids pFK9-13B, -C, -F, -H, and -K/NcoI. Plasmids pFK9-13B, -C, -F, -H, -I, and -K/SfiI were obtained by transferring an SfiI fragment (3622 to 8499) from pCR9-13B, -C, -F, -H, -I, and -K into pFK-I377neo/NS3-3′/wt. The following plasmids were generated by three-factor ligations using pFK-I377neo/NS3-3′/wt as a vector, a second fragment isolated from the same construct, and a fragment covering the mutation of interest isolated from pFK9-13F/SfiI by using the following restriction sites: PmeI, BstXI at 4319, and BstXI at 8012 for pFK1283 Arg→Gly; SfiI at 3622, NsiI at 5286, and SfiI at 8499 for the plasmid harboring all NS3 mutations; and PmeI, BssHII at 5923, and EcoRI at 6699 for pFK1936 Pro→Ser.
Plasmids pFK1383 Glu→Ala, pFK1577 Lys→Arg, and pFK1609 Lys→Glu were generated by site-directed mutagenesis using a PCR-based method (25). PCR fragments were inserted by the same cloning strategy as for the replicon with all NS3 mutations. Plasmids pFK2163 E→G, pFK2330 K→E, and pFK2442 I→V were generated by transferring an EcoRI (6699)-XhoI (7186), and XhoI-BclI (7627), and a BclI-MunI (7996) fragment, respectively, from pFK9-13F/Sfi I into pFK-I377neo/NS3-3′/wt. Plasmid pFK5B/2884 Gly was generated by insertion of an SfiI-SpeI fragment (8499 to 9605) from pFK9-13F/NcoI into pFK-I377neo/NS3-3′/wt together with an SfiI fragment from the same plasmid. Combinations of individual mutations with the conserved NS5B substitution were obtained by insertion of SfiI fragments from plasmids with the corresponding point mutation into pFK5B/2884 Gly. The presence of each mutation was confirmed by sequence analysis. Fragments generated by PCR were completely sequenced after subcloning.
Preparation of total RNA and quantification of HCV RNA by Northern blot.
Total RNA was prepared by a single-step isolation method (10), denatured by treatment with 5.9% glyoxal in 50% dimethyl sulfoxide (DMSO) and 10 mM sodium phosphate buffer (pH 7.0), and analyzed after denaturing agarose gel electrophoresis by Northern blot using standard procedures (2). Prior to hybridization, the membrane was stained with methylene blue and cut 1 cm below the 28S rRNA band. The upper strip containing the HCV replicon RNA was hybridized with a 32P-labeled negative-sense riboprobe complementary to the HCV IRES and neo. The lower strip was hybridized with a β-actin-specific antisense riboprobe and used to correct for total RNA amounts loaded in each lane of the gel. HCV- and β-actin-specific bands were quantified by phosphoimaging using a BAS 2500 scanner (Fuji), and the number of replicon molecules was determined by comparison with a serial dilution of in vitro transcripts loaded in parallel onto the gel.
Amplification of replicon RNA by RT-PCR and cloning of amplified DNA fragments.
Total RNA (1 μg) was mixed with 50 pmol of an appropriate reverse transcription primer in a total volume of 10.5 μl and denatured for 10 min at 65°C. Reverse transcription was performed with Expand-RT (Roche Biochemicals, Mannheim, Germany) as recommended by the manufacturer in a total volume of 20 μl. After 1 h at 42°C, 1/20 of the reaction mixture was used for reverse transcription-PCR (RT-PCR) with the Expand Long Template PCR system (Roche Biochemicals) according to the instructions of the manufacturer. Cycle conditions were 2 min of initial denaturation at 94°C and 40 cycles with 20 s at 94°C, 90 s at 54°C, and 60 s multiplied by the number of kilobase pairs of the amplified fragment at 68°C. After 10 cycles, the extension time was increased 10 s for each additional cycle. Finally, the reaction was incubated for 10 min at 68°C. PCR products were purified by preparative agarose gel electrophoresis and inserted into the vectors given below. For the amplification of almost the entire replicon from nucleotide (nt) 59 of the HCV IRES up to the 3′ end of the ORF, primer A9412 was used for cDNA synthesis and primers S59 and A9386 were used for PCR (Table 1). The PCR product was cloned using a Topo TA cloning kit following the protocol of the manufacturer (Invitrogen, Groningen, Netherlands). The constructs were designated pCR 9-13A-K. The 5′ NTR-neo region was amplified using primer A4919 for cDNA synthesis and primers ST7/Hind1-28 and Aneo3′Pme for PCR. The product was restricted with HindIII and PmeI and inserted into the original replicon construct pFK-I377neo/NS3-3′/wt. The EMCV-to-NS3 region was amplified using primer A4919 for reverse transcription and primers SEMCV-PmeI and A3802 for PCR. The product was restricted with PmeI and SfiI and introduced into the original replicon construct. For functional analysis of the 3′ NTR, primer A9605Sca-Spe was used for the reverse transcription reaction and primers S8467/A9605Sca-Spe were used for PCR. The product was restricted with SfiI and SpeI and inserted into the original replicon construct. The constructs were designated 33 to 48.
TABLE 1.
Oligonucleotides used for amplification of HCV RNAs from cell line 9-13
| Name | Sequence |
|---|---|
| 3′-Ligaa | ACT AGT GAC TAC GAC GTG ATA GTG CCA GTC TGA CGA AGC TTG ACG ATG |
| 1/3′ | CAT CGT CAA GCT TCG TCA GAC TGG |
| 2/3′ | CAC TAT CAC GTC GTA GTC ACT AGT |
| 5′-Ligab | GGG CGA AUU GGG UAC CGG GCC CCC CCU CGA GA CUA GUG ACU ACG ACG UGA UAG UGC CAG UCU GAC GAA GCU UGA CGA UGU CUA G |
| S-Liga1 | ACT AGT GAC TAC GAC GTG ATA GTG C |
| S-Liga2 | GCC AGT CTG ACG AAG CTT GAC GAT G |
| Aneo3′Pme | ACA AGA GTT TAA ACT CAG AAG AAC TCG TCA AGA AG |
| A220 | AGG CAT TGA GCG GGT TGA TCC |
| A325 | GAC CTC CCG GGG CAC TCG CAA GCA CC |
| A349 | GTG CTC ATG GTG CAC GGT CTA CGA |
| A3802 | GAG AGT AGG CTC CCC CTG CTG TC |
| A4919 | AGC ACA GCC CGC GTC ATA GCA CTC G |
| A9386 | TTA GCT CCC CGT TCA TCG GTT GG |
| A9412 | CAG GAT GGC CTA TTG GCC TGG AG |
| A9605Sca-Spe | TGC ACT AGT AGT ACT TGA TCT GCA GAG AGG CCA GTA TC |
| SEMCV-PmeI | ACA AGA GTT TAA ACA GAC CAC AAC GGT TTC CCT CTA GC |
| ST7/Hind1-28 | GCT AAG CTT CGT AAT ACG ACT CAC TAT AGC CAG CCC CCG ATT GGG GGC GAC ACT CC |
| S59 | TGT CTT CAC GCA GAA AGC GTC TAG |
| S8467 | GTA ATA CCC TCA CAT GTT ACT TGA |
| S9392 | CCA GGC CAA TAG GCC ATC CTG |
| S9503 | CCT TTG GTG GCT CCA TCT TAG |
3′ end blocked with an amino link.
RNA synthesized by in vitro transcription.
Cloning of 3′ and 5′ ends of replicon RNA.
To determine the 5′ and 3′ termini of HCV RNAs replicating in selected cell lines, replicon RNA was isolated from total RNA by hybridization to a biotinylated oligonucleotide, and captured RNA was ligated with synthetic oligonucleotides that could be used for hybridization with primers for RT-PCR. To isolate HCV replicon RNA from total RNA of cell line 9-13, 200 μg of total RNA was dissolved in 30 μl of hybridization buffer (80% deionized formamide, 40 mM HEPES [pH 7.5], 1 mM EDTA, 400 mM NaCl), mixed with 10 pmol of the 5′ biotinylated oligonucleotide A349 (see Table 1), heated for 1 min to 95°C, and incubated overnight at 40°C. After addition of 400 μl of TEN100 (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 100 mM NaCl) and 1 mg of streptavidin-coated paramagnetic beads (Roche Biochemicals) equilibrated with binding buffer, the mixture was incubated for 30 min at room temperature with bottom-over-top shaking. After three washes with TEN1000 (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 1 M NaCl), beads were equilibrated with ligation buffer (10 mM Tris-Cl [pH 7.5], 1 mM hexamine-cobalt-chloride, 10 mM MgCl2, 5 mM dithiothreitol [DTT], 1 mM ATP, 30% DMSO) (34). From 150 to 200 pmol of an appropriate oligonucleotide (see below) was ligated to the captured RNA in ligation buffer supplemented with 2 U of RNasin (Promega, Mannheim, Germany) and 30 U of T4 RNA ligase (Gibco Life Technologies) in a total volume of 100 μl for 6 h at 19°C. Beads were washed three times with TEN100, and RNA was eluted two times with 30 μl of RNase-free water for 3 min at 95°C. For the 3′ end, 200 pmol of the synthetic oligonucleotide 3′-Liga (Table 1) phosphorylated at its 5′ end using T4 polynucleotide kinase (Roche Biochemicals) was added to the reaction, and 5 μl of the eluted RNA was subjected to reverse transcription using primer 1/3′ and 100 U of reverse transcriptase from Moloney murine leukemia virus (Life Technologies) in a total volume of 10 μl following the instructions of the manufacturer. After 1 h at 48°C, the reaction was adjusted to 55 mM KCl, 17 mM Tris-HCl (pH 8.4), 2.3 mM MgCl2, 0.24% Tween 20, 50 pmol each of primers S9392 and 1/3′, and 2.5 U of Taq DNA polymerase (Gibco) in a final volume of 50 μl, and used for PCR with the following cycle conditions: 3 min at 95°C for initial denaturation, 35 cycles with 30 s at 95°C, 1 min at 59°C, and 1 min at 72°C, and a final 5-min incubation at 72°C. Then 5 μl of the reaction was used for a second PCR with primers S9503 and 2/3′ using the same cycle conditions. PCR products were purified by preparative agarose gel electrophoresis and inserted into the pCR vector using a Topo TA cloning kit according to the instructions of the manufacturer (Invitrogen). Determination of the 5′ ends of replicon RNA was done in the analogous way using an artificial in vitro transcript for ligation (5′Liga; see Table 3). Prior to RNA ligation, the in vitro transcript was dephosphorylated and purified by preparative denaturing polyacrylamide gel electrophoresis. Primer A349 was used for reverse transcription, and primers S-Liga1 and A325 were used for first PCR. Second PCR was done with primers S-Liga2 and A220.
TABLE 3.
CFU/μg of HCV RNA obtained with the parental replicon (wild type) and with RNAs carrying given amino acid substitutions in the HCV polyproteina
| HCV protein(s) | Amino acid substitution | Mean CFU/μg of RNA ± SD |
|---|---|---|
| None | None | 40 ± 14 |
| NS3 | 1283 Arg→Gly | 285 ± 60 |
| NS3 | 1383 Glu→Ala | 70 ± 25 |
| NS3 | 1577 Lys→Arg | 20 ± 5 |
| NS3 | 1609 Lys→Glu | 150 ± 65 |
| NS3 | All NS3 mutations | 385 ± 25 |
| NS4B | 1936 Pro→Ser | 500 ± 210 |
| NS5A | 2163 Glu→Gly | 2,650 ± 700 |
| NS5A | 2330 Lys→Glu | 35 ± 5 |
| NS5B | 2442 Ile→Val | 20 ± 10 |
| NS3-5B′ | 9-13F (SfiI fragment) | 4,250 ± 650 |
| NS5B | 2884 Arg→Gly | 23,000 ± 3,000 |
| NS3 + NS5B | 1283 Arg→Gly + 1383 Glu→Ala + 1577 Lys→Arg + 1609 Lys→Glu + 2884 Arg→Gly | 51,000 ± 6,000 |
| NS4B + NS5B | 1936 Pro→Ser + 2884 Arg→Gly | 750 ± 400 |
| NS5A + NS5B | 2163 Glu→Gly + 2884 Arg→Gly | n.a. |
| NS5A + NS5B | 2330 Lys→Glu + 2884 Arg→Gly | 19,000 ± 3,000 |
| NS5B + NS5B | 2442 Ile→Val + 2884 Arg→Gly | 9,000 ± 2,000 |
Numbers refer to the amino acid positions of the polyprotein of the HCV consensus genome, which can be retrieved from the EMBL nucleotide sequence database under accession number AJ238799. Values were derived from several independent transfection experiments using serial dilutions of each RNA construct. n.a., not applicable because no or only one colony was obtained in several independent experiments.
Sequence analysis.
Thermo Sequenase fluorescent-labeled primer cycle sequencing kit with 7-deaza-dGTP (Amersham-Pharmacia Biotech, Freiburg, Germany) was used for sequencing reactions with IRD-41-labeled primers (MWG-Biotech, Ebersberg, Germany) following the instructions of the manufacturer. Reactions were analyzed on a Licor DNA sequencer 4000 (MWG-Biotech).
In vitro transcription.
To generate run-off transcripts of HCV replicons, plasmid DNA was first linearized with AseI and then incubated with ScaI (New England Biolabs, Bad Schwalbach/Taunus, Germany). After extraction with phenol-chloroform and ethanol precipitation, DNA was dissolved in RNase-free deionized water and used for in vitro transcription reactions containing 80 mM HEPES (pH 7.5), 12 mM MgCl2, 2 mM spermidine, 40 mM DTT, 3.125 mM each NTP, plus 1 U of RNasin (Promega), 0.05 μg of restricted plasmid DNA, and 1 U of T7 RNA polymerase (Promega) per μl. After 2 h at 37°C, an additional 0.5 U of T7 RNA polymerase was added, and the reaction was incubated for another 2 h. Transcription was terminated by the addition of 2 U of RNase-free DNase (Promega) per μg of plasmid DNA and 1 h of incubation at 37°C. For purification of transcripts, the reaction was adjusted to a total volume of 600 μl by the addition of 60 μl of 2 M sodium acetate (pH 4.5) and water and extracted once each with acidic phenol and chloroform. After isopropanol precipitation, RNA was dissolved in RNase-free water, and the concentration was determined by measurement of the optical density at 260 nm. Integrity of the RNA was checked by denaturing agarose gel electrophoresis.
Electroporation and selection of G418-resistant cell lines.
Total RNA (10 μg) isolated from cell lines and corresponding to 1 to 10 ng of HCV replicon RNA or 1 to 1,000 ng of in vitro transcripts adjusted with total RNA from naive Huh-7 cells to a final amount of 10 μg were used for electroporation. To determine the efficiency of electroporation, 500 ng of a plasmid carrying the firefly luciferase gene under the control of the human cytomegalovirus immediate-early promoter/enhancer complex was included in every transfection. Subconfluent monolayers of Huh-7 cells were detached from the culture dish by trypsin treatment, washed twice with phosphate-buffered saline, and adjusted to a concentration of 107 cells per ml in Cytomix (56) containing 1.25% DMSO (39). Then 400 μl of the cell suspension was mixed with RNA by gentle pipetting, transferred to an electroporation cuvette (0.4-cm gap width; Bio-Rad, Munich, Germany), and subjected to an electric pulse at 960 μF and 270 V using a Gene pulser system (Bio-Rad). Cells were immediately transferred to 8 ml of DMEM containing 10% FCS and 1.25% DMSO, and 7 ml was seeded in a 10-cm-diameter cell culture dish. The remainder was seeded in a 35-mm-diameter dish, and at 24 h posttransfection, these cells were lysed and luciferase activity was determined using standard procedures (2). Unless otherwise stated, the medium in the 10-cm dish was replaced 24 h after electroporation with DMEM supplemented with 10% FCS and G418 (500 μg/ml) (Life Technologies). Medium was changed once a week, and 3 to 4 weeks after electroporation, colonies were stained with Coomassie brilliant blue (0.6 g/liter in 50% methanol–10% acetic acid). We note that under the conditions given above, the transfection efficiency (number of RNA-positive cells) was 20 to 50% as determined with a Semliki forest virus replicon RNA allowing the expression of beta-galactosidase and counting of blue-stained cells.
RESULTS
Experimental approach.
We have recently described the construction of subgenomic HCV RNAs replicating autonomously after transfection into the human hepatoma cell line Huh-7 (38). These RNAs were composed of the following elements (Fig. 1): the HCV 5′ NTR, directing translation of neomycin phosphotransferase; the EMCV IRES, allowing translation of the HCV NS3-5B polyprotein; and the authentic 3′ NTR. After linearization at an engineered ScaI restriction site, run-off transcripts were generated using T7 RNA polymerase and purified RNA was used for electroporation of Huh-7 cells. After 24 h, cells were subjected to selection with G418, allowing the isolation of cells that supported continuous replication of HCV RNAs. These cells grew and formed colonies that could be isolated and expanded for further analysis. Alternatively, colonies were fixed on the cell culture dish, stained, and counted. The efficiency of colony formation (ECF) is reflected in the number of colonies obtained in a given transfection. It is expressed as colony-forming units per microgram of transfected HCV RNA (CFU/μg), and these terms will be used throughout this report.
FIG. 1.

Experimental approach used to establish cell lines carrying self-replicating HCV RNAs. neo, neomycin phosphotransferase gene; EI, EMCV IRES; T7, promoter of the bacteriophage T7 RNA polymerase. Since for optimal HCV IRES activity ∼36 nucleotides of the core open reading frame are required, 12 amino acid residues of the core protein are fused to the amino terminus of the neomycin phosphotransferase. For details, see the text.
Evidence for adaptive mutations in HCV replicons.
Although within a selected cell line replication levels were high, this system was limited by the fact that the number of colonies obtained after transfection with replicon RNAs was very low. On average, only 20 to 40 colonies were obtained per microgram of in vitro transcript (Fig. 1). Two possibilities could account for this effect. First, only a few cells in the transfection reaction were permissive and allowed persistent replication, or, second, during selection, adaptive mutations were generated within the replicon which enhanced replication levels to an extent sufficient to establish a G418-resistant cell colony. In the first case, all cells in a cell line obtained after selection should have been permissive and therefore supported high-level HCV replication. To examine this possibility, the well-characterized cell line 9-13 (38) obtained after transfection with an NS3-5B replicon, as shown in Fig. 1, was used for supertransfection with an HCV RNA in which the neo sequence was replaced by a reporter gene, allowing discrimination between the parental neo replicon and the supertransfected HCV RNA. For comparison, naive Huh-7 cells were transfected with the reporter replicon in parallel. However, in several independent experiments, no difference in reporter activity was observed between cell lines carrying selectable replicons and naive cells (data not shown), indicating that the small number of colonies was not due to a small number of permissive cells.
To analyze the second possibility, total RNA was isolated from cell line 9-13. If adaptive mutations had occurred during selection of these cells, the ECF of HCV RNA contained in total RNA of these cells should be significantly higher than the ECF of the parental in vitro transcript. Therefore, serial dilutions of this total RNA were analyzed by Northern blot, and the amount of HCV-specific RNA was quantitated by phosphoimaging. Total RNA (10 μg) (depending on replication levels, corresponding to 1 to 10 ng of HCV RNA) was transfected into naive Huh-7 cells, and 24 h later cells were subjected to G418 selection. For comparison, 10 to 1,000 ng of replicon RNA generated by in vitro transcription and adjusted with RNA from naive Huh-7 cells to a total amount of 10 μg was transfected in parallel. In the initial set of experiments, cells in both transfections were routinely subjected to selection with G418 at a concentration of 1 mg/ml as described recently (38). Under these conditions, ∼20 colonies were obtained per microgram of in vitro transcript (Table 2). With the total RNA isolated from cell line 9-13, so few colonies were obtained that no conclusion about the ECF could be drawn. However, when we reduced the G418 concentration to 500 or 250 μg/ml, a clear difference became visible. While the ECF of the original replicon was only moderately affected under reduced G418 concentration, the ECF obtained with HCV RNA from cell line 9-13 increased tremendously and was several orders of magnitude higher compared to the in vitro transcript (Table 2).
TABLE 2.
Influence of G418 selection on the CFU/μg of HCV replicon RNA obtained from different sourcesa
| RNA | CFU/μg at G418 concn (μg/ml):
|
||
|---|---|---|---|
| 1,000 | 500 | 250 | |
| Total 9-13 RNA | n.a. | 85,000 ± 20,000 | 480,000 ± 120,000 |
| In vitro transcript | 16 ± 2 | 28 ± 9 | 74 ± 10 |
Values are means ± standard errors. n.a., not applicable.
Sequence analysis of cell culture-adapted HCV RNAs and functional testing.
The most likely explanation for the observed differences in the ECF was that HCV RNA replicating in cell line 9-13 contained adaptive mutations. Therefore, nearly the complete replicon from nt 59 in the HCV IRES up to nt 9386 in the variable region of the 3′ NTR was amplified by long-distance RT-PCR, and the sequences of the HCV ORFs of several independent clones were determined (Fig. 2). Sequences at the 5′ and 3′ ends were analyzed after RNA ligation and nested RT-PCR as described in Materials and Methods. Within the 5′-terminal portion (nt 1 to 208) of the 5′ NTR (nt 1 to 341), only a few nonconserved nucleotide changes were found, and no consistent extra sequences or deletions at the very 5′ end could be observed. Within the analyzed 3′-terminal region (nt 9503 to 9605) of the 3′ NTR (nt 9375 to 9605), only two nucleotide changes were found with all eight clones sequenced, and there were no deletions or insertions of nucleotides at the very 3′ end. In contrast to this high conservation, several amino acid changes were found with each of the eight analyzed NS3-5B sequences. There was no clustering of mutations with respect to a particular NS protein, and the number of mutations varied significantly between the individual clones. While clones 9-13C and 9-13K contained only seven amino acid substitutions, clone 9-13A carried 11. Stop codons around positions 4100 and 8900 were found with clones 9-13A and 9-13I, respectively, whereas a single nucleotide deletion in NS3 with clone 9-13G and a single nucleotide insertion in NS5B with clone 9-13I led to frameshift mutations. There were no conserved mutations with the exception of a glycine substitution for arginine within NS5B at amino acid position 2884 that was found in all polyprotein sequences from cell line 9-13 (Fig. 2). However, this mutation was not conserved with RNAs isolated from another cell line (5-15) (38) carrying the analogous HCV replicon (data not shown).
FIG. 2.

Sequence analysis of HCV polyproteins recloned from cell line 9-13 and result of functional testing of different HCV fragments. The parental replicon construct is shown at the top, with numbers below referring to nucleotide positions of the HCV genome (for details, see the legend to Fig. 1). HCV polyproteins from eight independent clones are shown as open bars. The positions of nucleotide and amino acid sequence deviations from the original consensus sequence are indicated by vertical lines and labeled as follows: black, amino acid substitution; gray, silent nucleotide change; ↓, nonsense mutation; 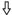 , frameshift mutation. The single conserved amino acid substitution in NS5B is marked with a diamond. The positions of the recognition sequences for the restriction enzymes SfiI and NcoI used for subcloning into the parental construct are indicated with dotted lines. The names of the corresponding constructs are given at the left, and the CFU/μg in vitro transcript obtained with each of the subcloned NcoI or SfiI fragments after selection with G418 (500 μg/ml) is given on the right. n.d., not determined.
, frameshift mutation. The single conserved amino acid substitution in NS5B is marked with a diamond. The positions of the recognition sequences for the restriction enzymes SfiI and NcoI used for subcloning into the parental construct are indicated with dotted lines. The names of the corresponding constructs are given at the left, and the CFU/μg in vitro transcript obtained with each of the subcloned NcoI or SfiI fragments after selection with G418 (500 μg/ml) is given on the right. n.d., not determined.
To analyze whether these mutations conferred an adaptive phenotype, we decided to introduce nearly the complete polyprotein coding region into the parental replicon by using NcoI restriction sites (Fig. 2). Transferred fragments contained almost all amino acid substitutions found with each clone, including the conserved one within NS5B. Since clones 9-13A, -G, and -I carried stop codons or frameshift mutations within the NcoI fragment, they were not included in this analysis. In several independent experiments, no colonies were obtained after transfection of naive Huh-7 cells and selection at various G418 concentrations. This result suggested that all subcloned fragments contained inactivating mutations that might have been introduced either by the HCV polymerase during replication or the RT-PCR process. One potential candidate was the conserved mutation within NS5B. Therefore, SfiI fragments that lacked this particular substitution were introduced into the parental replicon (Fig. 2). Owing to a stop codon or a frameshift mutation with clones 9-13A and 9-13G, respectively, they were excluded, whereas the deleterious mutations contained in clone 9-13I were downstream of the SfiI fragment, and therefore this clone was included in this analysis. After selection of transfected naive Huh-7 cells, three clones still gave no viable colonies (clones 9-13B, -H, and -K), whereas the ECF of the three other clones was reproducibly higher compared with the parental replicon. Clones 9-13C and 9-13I had ∼500 CFU/μg of RNA, whereas that of clone 9-13F was increased a further 10-fold (Fig. 2). These results demonstrated that the polyprotein sequences of these three clones contained adaptive mutations, whereas inactivating mutations must have been present in the sequences of the other clones (9-13B, -H, and -K). Furthermore, since the NcoI fragment of clone 9-13F differed from the SfiI fragment only by the conserved amino acid substitution in NS5B at position 2884, these results suggested that this particular mutation was inactivating.
Identification of several adaptive amino acid substitutions in the HCV polyprotein.
Owing to its having the highest ECF, we focused our subsequent analyses on clone 9-13F. Within the SfiI fragment of this clone, eight amino acid changes were found: four within NS3, one in NS4B, two within NS5A, and one close to the amino terminus of NS5B, whereas NS4A was unaltered. To determine which of these mutations was responsible for the adaptation, each substitution was introduced individually into the parental replicon construct, and the CFU/μg of in vitro transcript was determined for each construct in parallel with RNA from clone 9-13F. To analyze for a possible synergism or additive effect of adaptive mutations, an additional construct was generated carrying all NS3 substitutions found with clone 9-13F. The results of this analysis are shown in Fig. 3 and summarized in Table 3. Two mutations within NS3 at amino acid positions 1383 and 1577 of the polyprotein did not increase the number of colonies obtained. The same was true with the substitution close to the carboxy terminus of NS5A and the one at the amino terminus of NS5B (amino acid positions 2330 and 2442, respectively). In contrast, the two other amino acid changes that were both located in the helicase domain increased the ECF ∼5-fold (Fig. 3 and Table 3). Interestingly, this value was further increased ∼2-fold with the construct carrying all NS3 mutations, indicating that the two mutations in the helicase domain were additive.
FIG. 3.

Single-amino-acid substitution in NS5A increases ECF almost 100-fold. The basic construct with the amino acid substitutions found in the SfiI fragment of clone 9-13F is shown in the center. Results obtained with single-amino-acid substitutions are given below. Colonies shown on each cell culture dish were obtained after transfection of Huh-7 cells with 500 ng of each mutant or construct 9-13F, shown at the top. The individual amino acid substitutions are written below each stained culture dish, with the arrow pointing to the substituting residue. Numbers refer to the amino acid position of the HCV polyprotein. A summary of these results is given in Table 3.
A peculiar phenotype was found with the NS4B mutation. About 2 weeks posttransfection, a large number of small colonies were reproducibly found in these transfections, but during continued selection most cells died. Yet the overall number of colonies obtained with 1 μg of in vitro transcript of this mutant was significantly higher compared with the parental replicon (∼10-fold; Table 3). The most drastic effect was found with the glycine substitution for glutamic acid at amino acid position 2163, which is approximately in the middle of NS5A. This particular substitution increased the ECF ∼100-fold. In fact, the CFU/μg of RNA of this mutant was similar to that obtained with the SfiI fragment carrying all mutations. Thus, we had identified the major determinant of adaptation in the SfiI fragment corresponding to amino acids 1094 to 2720 of the polyprotein. Furthermore, the results demonstrated that adaptation could be achieved by mutations in several HCV NS proteins, albeit with different efficiencies.
Cell culture adaptation of the HCV replicon is mainly achieved by a single-amino-acid substitution in NS5B.
Although we had identified a major determinant of adaptation, the amount of HCV RNA contained in cell line 9-13 was still ∼30-fold higher compared to the replicon with the adaptive NS5A mutation (85,000 versus ∼3,000 at 500 μg of G418 per ml) (Table 2 and 3). The most likely explanation was that additional adaptive mutations outside of the sequence analyzed were present in HCV RNAs replicating in cell line 9-13. Therefore, fragments corresponding to the 5′ NTR up to the 3′ end of neo, the EMCV IRES up to the amino-terminal region of NS3, and the carboxy-terminal half of NS5B up to the 3′ end were amplified from total RNA of cell line 9-13 and inserted into the parental replicon construct (Fig. 4). In vitro transcripts from 16 to 18 clones of each type were generated and analyzed after transfection into naive Huh-7 cells for their ECF. As summarized in Fig. 4, the majority of the 5′-end fragments were inactive, and the CFU of 5 of 18 clones was comparable to the parental nonadapted replicon. In the EMCV NS3 fragment, the majority of fragments were active but no adaptation was observed. A completely different picture was found with the 3′-end fragment. Five of 16 tested fragments were inactive, three were comparable to the wild type, and eight had a higher CFU. Most interestingly, five of these clones had a CFU that was ∼1,000-fold higher compared with the nonadapted replicon and almost reached the level of HCV RNA isolated from cell line 9-13.
FIG. 4.

Identification of adaptive mutations in the 3′-proximal region of the HCV replicon. The basic construct is shown at the top, with the restriction sites used for subcloning indicated above. Three fragments corresponding to the 5′ NTR up to the 3′ end of neo, the EMCV IRES up to the amino-terminal region of NS3, and the carboxy-terminal half of NS5B up to the end of the 3′ NTR were amplified by RT-PCR from cell line 9-13 and inserted into the parental replicon construct. From 16 to 18 clones from each fragment (indicated with the pictograms at the top of the table) were isolated, and 300 ng of the corresponding in vitro transcripts was used for transfection into naive Huh-7 cells. The CFU/μg of in vitro transcript generated from each replicon construct is given at the left, and the number of clones in each category is shown on the right. For the 3′ fragment, five clones were obtained with >10,000 CFU/μg of RNA. Note that for the nonadapted parental replicon, the CFU/μg of RNA is 1 to 100 (indicated by shading).
To identify the mutation(s) responsible for this phenotype, sequences of the five highly and three intermediately adapted clones as well as of one inactive clone were analyzed. To our great surprise, all fragments carried the same glycine-for-arginine substitution in NS5B at amino acid position 2884 of the polyprotein that we had found in the HCV replicons recloned from cell line 9-13 and that appeared to be inactivating (Fig. 5). In all highly adapted replicons, this was the only amino acid change in the HCV polyprotein. Three of these replicons (34, 42, and 47; Fig. 5) carried an additional nonconserved nucleotide substitution in the variable region of the 3′ NTR. Since this region is not essential for replication in vivo (61), it appears to be rather tolerant of nucleotide substitutions. Apart from some minor rearrangements in the poly(U/UC)-tract, the 3′ NTRs of the highly adapted replicons were well conserved. In case of the X tail, only clone 37 had one nucleotide substitution. This uridine→cytosine transition at position 9518 was located in the loop region of the first stem-loop structure of the X tail (7), recently identified as part of a binding site for polypyrimidine tract-binding protein (28). In the three intermediately adapted replicons (35, 43, and 39), in addition to the highly adaptive mutation in NS5B, one or two additional amino acid substitutions were found in the NS5B fragment. Apart from one nucleotide substitution in the variable region of clone 43, the 3′ NTRs were highly conserved. Although we have not yet examined these additional NS5B mutations in detail, they probably counteract the highly adaptive substitution leading to an intermediate level of adaptation. In case of the inactive replicon (number 36), two other NS5B mutations were found that apparently destroyed the replication competence of this RNA.
FIG. 5.

Sequence analysis of replicons carrying various 3′-terminal fragments that were isolated from cell line 9-13. The region subcloned into the parental construct and composed of the 3′-terminal region of NS5B (beginning at nucleotide position 8499) and the tripartite 3′ NTR is given on the top. The amino acid substitution in NS5B found in each subclone is indicated with a black vertical line and labeled with a diamond; the positions of all other amino acid changes are depicted with gray lines. Nucleotide substitutions in the 3′ NTR are indicated with gaps and labeled with a star. The CFU/μg of in vitro transcript obtained with each clone is given on the right. The tripartite structure of the 3′ NTR composed of the variable region (VR), the poly (U/UC)n tract, and the highly conserved X tail is indicated. Numbers to the left refer to the individual clone. Note that all cloned fragments carried some minor rearrangements in the poly (U/UC)n tract.
Since all subcloned 3′ fragments carried, in addition to the conserved NS5B mutations, some minor rearrangements in the poly(U/UC) tract or single-nucleotide substitutions at other positions in the 3′ NTR, we could not rule out the possibility that these alterations contributed to the adaptation. Therefore, the conserved NS5B substitution was introduced into the parental replicon, and two independent clones were analyzed as described above. As summarized in Table 3, this replicon, designated rep5B/2884Gly, was as efficient as the highly adapted replicons carrying the complete 3′ fragments. In fact, the ECF of this cloned replicon was comparable to that obtained with HCV RNA isolated from cell line 9-13. Thus, we had identified the most important adaptive mutation.
Evidence for incompatibility of adaptive mutations in NS5A and NS5B.
In the experiments summarized in Fig. 2, we had found that a replicon carrying the SfiI fragment from clone 9-13F was replication competent, whereas the analogous replicon carrying in addition the conserved NS5B substitution at position 2884 was inactive. This latter replicon, designated 9-13F/NcoI, was constructed in three different ways to exclude the possibility that inactivating mutations had been introduced inadvertently into the replicon sequence, and in independent experiments, all three RNAs did not give rise to G418-resistant colonies. On the other hand, the results described above clearly showed that this particular NS5B mutation contributed most to adaptation. At least two possibilities could account for this discrepancy. First, the combination of mutations contained in the SfiI fragment with this NS5B substitution increased HCV RNA replication to a level that was cytotoxic. In this case, all cells supporting high-level replication would be lost and no colonies could be formed. Second, the mutation in NS5B was incompatible with those contained in the SfiI fragment.
To analyze for potential cytopathogenicity, several experiments were performed. First, naive Huh-7 cells were cotransfected with the 9-13F/NcoI replicon and a plasmid directing the expression of firefly luciferase under control of the human cytomegalovirus promoter. For comparison, Huh-7 cells were transfected only with the reporter construct. In case of cytopathogenicity of the replicon, transfected cells should be eliminated, leading to a reduction in expression of the reporter gene relative to cells transfected only with the reporter construct. However, no difference was found between these transfected cells (data not shown). Second, naive Huh-7 cells were cotransfected with 10 ng of rep5B/2884Gly and increasing amounts of the potentially cytopathogenic 9-13F/NcoI-RNA (Fig. 6). Since large amounts of transfected input RNA can lead to a reduction in the number of colonies, a cotransfection of rep5B/2884Gly with an inactive 9-13F/NcoI RNA was performed as a control in parallel. This replicon carried a deletion of 10 amino acids in the active site of the NS5B RdRp and therefore, by definition, could not replicate. In case of cytopathogenicity of the parental 9-13F/NcoI replicon, the number of colonies obtained in this cotransfection should be much lower compared to the control reaction with the inactive replicon. However, no difference in the number of G418-resistant colonies was found (Fig. 6). In summary, these results indicated that the 9-13F/NcoI replicon was not cytopathogenic and implied that the highly adaptive NS5B mutation was incompatible with one or several mutations contained in the SfiI fragment of clone 9-13F.
FIG. 6.

Evidence that replicon 9-13F/NcoI is not cytopathogenic. Replicon rep5B/Gly2884 (10 ng) was transfected into naive Huh-7 cells either alone or together with 10, 100, or 1,000 ng of the potentially cytopathogenic replicon rep9-13F/NcoI. As a control, cotransfections were performed with rep5B/Gly2884 and given amounts of an inactive rep9-13F/NcoI mutant carrying a deletion of 10 amino acid residues spanning the active site of the NS5B RNA polymerase. About 4 weeks posttransfection, cells were fixed and stained with Coomassie brilliant blue. Colonies obtained after transfection of rep5B/Gly2884 and selection with G418 are shown on the top; results obtained in cotransfections are given below. Representative plates are shown.
To define incompatible mutations, replicons were generated carrying, in addition to the conserved NS5B substitution at position 2884, either all four NS3 mutations, the NS4B substitution, each of the two NS5A mutations, or the second NS5B substitution at position 2442 (Fig. 7). Each of these RNAs was transfected into naive Huh-7 cells, and the ECF was determined in comparison to the parental replicon rep5B/2884Gly. In the combination of the four NS3 mutations with the adaptive NS5B substitution, a ∼2-fold increase in the number of colonies was found, indicating an additive effect of these particular mutations (Fig. 7 and Table 3). The nonadaptive substitution in NS5A at position 2330 did not affect the ECF, but a reduction was found with the other combinations. While the nonadaptive NS5B mutation at position 2442 reduced the number of colonies ∼2-fold, a ∼20-fold reduction was found in case of the combination with the NS4B substitution (Fig. 7 and Table 3). The most drastic effect could be seen with the replicon carrying both the highly adaptive NS5B and the NS5A mutation. In several independent experiments, no or only one colony was obtained with this replicon, indicating that these two mutations are incompatible.
FIG. 7.

Evidence for the incompatibility of adaptive mutations. Replicons were generated carrying both the highly adaptive NS5B mutation at position 2884 and either all four mutations in NS3 or one of the given mutations in NS4B, NS5A, or NS5B found in the SfiI fragment of clone 9-13F. About 4 weeks after transfection into naive Huh-7 cells, they were fixed and stained with Coomassie brilliant blue. The result obtained after transfection of the parental replicon rep5B/Gly2884 is shown on the top, and results obtained with the combination mutants are shown below. Note that in several independent experiments, no or only one colony was obtained with the combination of the highly adaptive NS5B and NS5A mutations (2884 R→G and 2163 E→G). A summary of these results is given in Table 3.
DISCUSSION
For numerous positive-strand RNA viruses, cloned infectious full-length genomes have been used as a valuable tool for studying various aspects of the viral life cycles (reviewed in reference 8). These molecules can be generated in large quantities and, after transfection of permissive cells, direct the production of infectious virus progeny. For HCV, although cloned virus genomes replicating in experimentally inoculated chimpanzees have been described (5, 33, 60), the development of cell culture systems with these RNAs has so far not been possible. The recent development of selectable subgenomic HCV replicons has opened new avenues to the study of HCV replication, persistence, and pathogenesis in cultured cells. However, owing to the small number of HCV RNA-containing cell colonies routinely obtained, the system was of limited use for studies using reverse genetics methodologies. In a search for the reasons for this low efficiency, we found that HCV RNAs must first acquire adaptive mutations that, as shown by the highly adaptive mutation within NS5B, can increase the ECF by up to 3 orders of magnitude. Surprisingly, adaptation can be achieved by many different mutations in different HCV NS proteins, although the level of adaptation can vary drastically.
While adaptive mutations are the major determinant of colony formation, during these studies we found that several other parameters must be considered. Of utmost importance is the G418 concentration. In our initial experiments, selections were performed at a concentration of 1 mg/ml. As shown with total RNA isolated from a selected cell line, very few colonies were obtained under these conditions but when the G418 concentration was lowered, the number of colonies increased tremendously. It was this effect that in our initial studies masked the higher ECF of HCV RNA replicating in selected cell lines compared to the parental replicon (38). The importance of G418 concentration (and drug quality that varies with individual batches) can be explained by the complex interplay between the selective pressure and the replication of HCV RNA. In principle, the level of G418 resistance is determined by the amount of HCV RNA within a cell, which in turn is determined by the replication level. When the amount of transfected HCV RNA is small, as is the case with total RNA, a rather long time may be required for amplification of HCV RNA and development of resistance. In this case, cells subjected to high selective pressure (high G418 concentration) right after transfection will die, because the time required to develop the necessary level of resistance is too long. Consequently, when using total RNA, colonies could only form at low G418 concentrations. However, when cells are transfected with large amounts of in vitro transcripts, a sufficient amount of neomycin phosphotransferase is translated from the input RNA, and since this protein has a very long half-life, cells have a high level of resistance right after transfection and can survive even when the input RNA replicates only at a low level. In fact, even cells in which the RNA does not replicate can grow to a certain extent (until the majority of neomycin phosphotransferase is degraded or diluted after cell division), and this may lead to different efficiencies or even a loss of selection when cell cultures become too dense. This happens, e.g., when the amount of transfected RNA exceeds about 1 μg.
The observation of adaptive mutations provides an explanation for the small number of colonies. Although direct experimental proof is missing, the most likely explanation is that adaptive mutations are required to enhance RNA replication. Therefore, a nonadapted replicon would replicate at a level too low to confer continuous G418 resistance. However, owing to the high error rate of NS5B RdRp, mutations are introduced into the replicon. The majority of mutations might be deleterious or without effect and only in few instances be adaptive. Since only cells carrying adapted replicons develop G418 resistance, the number of colonies would be small after transfection of the parental replicon but large in the case of adapted RNA. According to this assumption, adaptive mutations must be introduced at an early time point after transfection and become fixed in all RNA progeny. In agreement with this hypothesis, we found that the most adaptive mutation in NS5B is conserved with all replicons isolated from cell line 9-13. However, it should be noted that this mutation was not found in other cell lines, suggesting that in these cases adaptive mutations were introduced at other positions. This observation is in keeping with the notion that cell culture adaptation of HCV replicons can be achieved by mutations in different proteins or RNA sequences.
The mechanism of cell culture adaptation is not known. It is possible that some of the HCV proteins are cytotoxic, e.g., via induction of apoptosis, as suggested for BVDV (26, 63), and this effect may be blocked by particular amino acid substitutions. Alternatively, adaptive mutations may counteract the antiviral pathway that is induced or activated by double-stranded RNA generated during replication and activating, e.g., RNase L and the protein kinase PKR. However, this possibility seems less likely because replication of the adapted HCV RNA can readily be blocked by treatment of cells with alpha interferon (R. Bartenschlager and V. Lohmann, unpublished results). Another alternative is that the adaptive mutations directly enhance enzymatic activities of NS3 NTPase/helicase or NS5B RdRp. The fact that the affected amino acid residues are highly conserved between different genotypes would be in agreement with an essential role for protein functions. An inspection of the X-ray crystal structure of the NS3 helicase reveals that the adaptive mutations 1283Gly and 1609Glu are located close together on the surface of the molecule and far away from the active site (Fig. 8A). In NS5B, the highly adaptive mutation 2884Gly is also located on the surface of the molecule (Fig. 8B). The position of this residue close to the end of helix R of the thumb domain and far away from the active site suggests that it is not directly involved in catalysis, although a contribution of this residue to RNA template binding is possible (9). The exposure of these adaptive mutations on the surface of the molecules may suggest that these alterations instead affect interaction sites either between viral proteins within the replicase complex or between viral proteins and activating or inhibiting host cell factors. In the case of activating host cell factors, the mutations might increase the binding, whereas in the case of inhibitors, binding might be lost. Alternatively, adaptive mutations may make RNA replication independent of a particular host cell factor present in limiting amounts in transfected cells. Such a mechanism of adaptation has been described for the bacteriophage Qβ (46). In wild-type phages, replication of viral RNA is dependent on a host cell factor that probably aids in melting the 3′-terminal stem-loop and makes the 3′ end available to Qβ replicase. When grown in Escherichia coli strains that lack this host cell factor, adapted phages develop that carry nucleotide substitutions in the 3′-terminal region. These mutations destabilize the stem and in this way make the 3′ end of the genome accesible to the replicase in the absence of the host cell factor (46).
FIG. 8.

Location of adaptive mutations in the three-dimensional structures of the NS3 helicase (A) and the NS5B RdRp (B). The three domains of the NS3 helicase are color coded according to Kim et al. (32). The two residues altered in the adapted replicons are located on the surface of the molecule, and they are given in ball-and-stick representation. The three-dimensional structure of the NS5B RdRp as viewed from the front is shown in panel B, and individual domains (thumb, palm, and fingers) are marked with different colors. The palm subdomain located at the base is closed on either side by the fingers and thumb subdomains and at the back by the loops. The arginine residue replaced by glycine with the adapted replicon is given in ball-and-stick representation.
Propagation of both the parental and the adapted replicons has so far only been possible in Huh-7 cells but not in other hepatoma cell lines of human origin. This restricted tropism may reflect the dependence of HCV RNA replication on particular host cell factors found only in Huh-7 cells or expressed only in this cell line in sufficient amounts. Therefore, the mutations described in this report may reflect an even closer adaptation to this particular host cell environment, and consequently, HCV RNAs replicating in host cells of other origin might develop adaptive mutations different from the ones described here. In agreement with a host cell-specific adaptation, selection for HCV variants replicating preferentially in certain cell types or organs has been described in several reports (for review, see reference 4). For instance, Sugiyama and coworkers (48) analyzed the HCV population replicating in the human T-cell line MT-2C after inoculation with a patient serum. By comparing several nearly full-length genome sequences found in the serum with those isolated from cultured cells, they found that only a limited HCV population could replicate in these cells. In another analysis, Shimizu and coworkers (47) inoculated a chimpanzee with ∼103 genome equivalents present in the culture supernatants of the human B-cell line Daudi that had been infected with HCV for 58 days. The major HCV variant found in the serum of the animal corresponded to the predominant variant in the patient serum used for infection of Daudi cells. However, in peripheral blood mononuclear cells of the chimpanzee, the major variant corresponded to the dominant variant found in Daudi cells, and this variant was not found in the patient serum. These results suggest the selection of a lymphotropic HCV variant during cell culture passage. However, whether this is due to selection at the level of infection or replication, as is the case with the replicons described here, remains to be determined.
Cell culture-adaptive mutations have also been described for several other viruses. For instance, replication of Sindbis virus, a member of the alphavirus group, is cytolytic in many different cell lines. In a search for cell culture-adapted noncytolytic RNAs of the Sindbis virus, Frolov and coworkers (15) constructed selectable replicons that allowed the isolation of cell colonies carrying persistently replicating Sindbis virus RNAs. Adapted replicons were isolated from selected BHK-21 cells, and the adaptive mutations were mapped to two positions in nonstructural protein (nsp) 2 downstream of its papain-like proteinase domain. Both mutations were found to reduce RNA replication to a level that was no longer harmful to the cell. Interestingly, such replication-repressing mutations led to hyperprocessing of the viral nsp123 replicase, resulting in downregulation of negative-strand RNA synthesis (15). For the Hepatitis A virus, a member of the Picornaviridae family, several cell culture-adapted isolates have been described that differ from the nonadapted primary isolate at a number of positions scattered throughout the genome (19, 29). Important growth-enhancing mutations could be mapped to nonstructural proteins 2B and 2C as well as the 5′ NTR (11, 16), but the mechanisms underlying cell culture adaptation remain to be determined.
One of the most surprising observations that we made during this study was that the combination of the highly adaptive NS5B mutation with the adaptive mutations contained in the SfiI fragment of clone 9-13F led to a replicon that no longer gave rise to G418-resistant colonies. In fact, this phenomenon masked the identification of cell culture-adaptive mutations, because our initial analyses were performed with replicon fragments isolated from a selected cell line and containing the conserved NS5B substitution (38). One possibility is that the combination of certain up-mutations generates RNAs replicating to a level that is harmful for the cell. Although high-level replication of a viral RNA may be cytotoxic, as suggested recently for the pestivirus BVDV (40), our results did not support this possibility. Since a cytopathogenic replicon should have a dominant phenotype, we performed a series of cotransfection experiments and found that inclusion of this replicon in the transfection reduced neither the expression of a marker gene nor the number of G418-resistant colonies generated with another replicon. Therefore, our results support a model in which particular mutations within the replicon sequence are not compatible. This was found for the combinations of the highly adaptive NS5B substitution with either the NS4B or the NS5A mutation (1936 P→S or 2163 E→G, respectively). In contrast, the adaptive mutations within the NS3 helicase were compatible both with this particular NS5B substitution and with the other mutations contained in the SfiI fragment. While additive or synergistic effects of adaptive mutations have been described for several virus systems, to our knowledge the incompatibility of adaptive mutations that we describe here is without precedent. The mechanisms underlying this phenomenon are not known. We can assume that the HCV proteins form a highly ordered multiprotein complex that contains additional cellular factors and that mutations within this complex may disturb interactions required for replicase activity. In the simplest model, the mutations in NS5A and NS5B described here are mutually exclusive because they affect a contact site between both proteins. Alternatively, the substitutions could alter the folding of the polyprotein and affect processing. Certainly more biochemical and genetic studies are required to clarify this important point.
In summary, we have developed cell culture-adapted, highly efficient HCV replicons. The possibility of generating RNAs with a CFU/μg of >10,000 colonies permits the use of genetics to study HCV replication in cell culture. This powerful method should allow the identification of cis-acting RNA elements required for RNA synthesis as well as the viral and cellular factors involved.
ACKNOWLEDGMENTS
We are grateful to Neera Borkakoti for helpful discussions and for providing the 3D models presented in Fig. 8 and to Rene Devos, Hilary Overton, and Julian Symons for a critical reading of the manuscript. We also thank U. Herian for excellent technical assistance, Hartmut Kleinert for the gift of the plasmid used to generate β-actin-specific riboprobes, and Nicole Krieger for stimulating discussions.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 490; Teilprojekt A2) and Roche Products Ltd. A.D. was supported by the Deutscher Akademischer Austauschdienst e.V.
REFERENCES
- 1.Asabe S I, Tanji Y, Satoh S, Kaneko T, Kimura K, Shimotohno K. The N-terminal region of hepatitis C virus-encoded NS5A is important for NS4A-dependent phosphorylation. J Virol. 1997;71:790–796. doi: 10.1128/jvi.71.1.790-796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: John Wiley and Sons; 1987. [Google Scholar]
- 3.Bartenschlager R, Ahlborn-Laake L, Mous J, Jacobsen H. Kinetic and structural analyses of hepatitis C virus polyprotein processing. J Virol. 1994;68:5045–5055. doi: 10.1128/jvi.68.8.5045-5055.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J Gen Virol. 2000;81:1631–1648. doi: 10.1099/0022-1317-81-7-1631. [DOI] [PubMed] [Google Scholar]
- 5.Beard M R, Abell G, Honda M, Carroll A, Gartland M, Clarke B, Suzuki K, Lanford R, Sangar D V, Lemon S M. An infectious molecular clone of a Japanese genotype 1b hepatitis C virus. Hepatology. 1999;30:316–324. doi: 10.1002/hep.510300137. [DOI] [PubMed] [Google Scholar]
- 6.Behrens S E, Tomei L, De Francesco R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996;15:12–22. [PMC free article] [PubMed] [Google Scholar]
- 7.Blight K J, Rice C M. Secondary structure determination of the conserved 98-base sequence at the 3′ terminus of hepatitis C virus genome RNA. J Virol. 1997;71:7345–7352. doi: 10.1128/jvi.71.10.7345-7352.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyer J C, Haenni A L. Infectious transcripts and cDNA clones of RNA viruses. Virology. 1994;198:415–426. doi: 10.1006/viro.1994.1053. [DOI] [PubMed] [Google Scholar]
- 9.Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale R L, Mathieu M, De Francesco R, Rey F A. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc Natl Acad Sci USA. 1999;96:13034–13039. doi: 10.1073/pnas.96.23.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 11.Emerson U S, Huang Y K, McRill C, Lewis M, Purcell R H. Mutations is both the 2B and 2C genes of hepatitis A virus are involved in adaptation to growth in cell culture. J Virol. 1992;66:650–654. doi: 10.1128/jvi.66.2.650-654.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Izumi N, Marumo F, Sato C. Comparison of full-length sequences of interferon-sensitive and resistant hepatitis C virus 1b: sensitivity to interferon is conferred by amino acid substitutions in the NS5A region. J Clin Investig. 1995;96:224–230. doi: 10.1172/JCI118025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Ogura Y, Izumi N, Marumo F, Sato C. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N Engl J Med. 1996;334:77–81. doi: 10.1056/NEJM199601113340203. [DOI] [PubMed] [Google Scholar]
- 14.Failla C, Tomei L, De Francesco R. Both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural proteins. J Virol. 1994;68:3753–3760. doi: 10.1128/jvi.68.6.3753-3760.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frolov I, Agapov E, Hoffman T A, Prágai B M, Lippa M, Schlesinger S, Rice C M. Selection of RNA replicons capable of persistent noncytopathic replication in mammalian cells. J Virol. 1999;73:3854–3865. doi: 10.1128/jvi.73.5.3854-3865.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Funkhouser A W, Purcell R H, D'Hondt E, Emerson S. Attenuated hepatitis A virus: genetic determinants of adaptation to growth in MRC-5 cells. J Virol. 1994;68:148–157. doi: 10.1128/jvi.68.1.148-157.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gale M J, Blakely S M, Kwieciszewski B, Tan S L, Dossett M, Tang N M, Korth M J, Polyak S J, Gretch D R, Katze M G. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanism of kinase regulation. Mol Cell Biol. 1998;18:5208–5218. doi: 10.1128/mcb.18.9.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gale M J, Korth M J, Tang N M, Tan S L, Hopkins D A, Dever T E, Polyak S J, Gretch D R, Katze M G. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology. 1997;230:217–227. doi: 10.1006/viro.1997.8493. [DOI] [PubMed] [Google Scholar]
- 19.Graff J, Normann A, Feinstone S M, Flehmig B. Nucleotide sequence of wild-type hepatitis A virus GBM in comparison with two cell culture-adapted variants. J Virol. 1994;68:548–554. doi: 10.1128/jvi.68.1.548-554.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grakoui A, McCourt D W, Wychowski C, Feinstone S M, Rice C M. A second hepatitis C virus-encoded proteinase. Proc Natl Acad Sci USA. 1993;90:10583–10587. doi: 10.1073/pnas.90.22.10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grakoui A, McCourt D W, Wychowski C, Feinstone S M, Rice C M. Characterization of the hepatitis C virus-encoded serine proteinase: determination of proteinase-dependent polyprotein cleavage sites. J Virol. 1993;67:2832–2843. doi: 10.1128/jvi.67.5.2832-2843.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grakoui A, Wychowski C, Lin C, Feinstone S M, Rice C M. Expression and identification of hepatitis C virus polyprotein cleavage products. J Virol. 1993;67:1385–1395. doi: 10.1128/jvi.67.3.1385-1395.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hijikata M, Kato N, Ootsuyama Y, Nakagawa M, Shimotohno K. Gene mapping of the putative structural region of the hepatitis C virus genome by in vitro processing analysis. Proc Natl Acad Sci USA. 1991;88:5547–5551. doi: 10.1073/pnas.88.13.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hijikata M, Mizushima H, Akagi T, Mori S, Kakiuchi N, Kato N, Tanaka T, Kimura K, Shimotohno K. Two distinct proteinase activities required for the processing of a putative nonstructural precursor protein of hepatitis C virus. J Virol. 1993;67:4665–4675. doi: 10.1128/jvi.67.8.4665-4675.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho S N, Hunt H D, Horton R M, Pullen J K, Pease L R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 26.Hoff H S, Donis R O. Induction of apoptosis and cleavage of poly(ADP-ribose) polymerase by cytopathic bovine viral diarrhea virus infection. Virus Res. 1997;49:101–113. doi: 10.1016/s0168-1702(97)01460-3. [DOI] [PubMed] [Google Scholar]
- 27.Honda M, Ping L H, Rijnbrand R A, Amphlett E, Clarke B, Rowlands D, Lemon S M. Structural requirements for initiation of translation by internal ribosome entry within genome-length hepatitis C virus RNA. Virology. 1996;222:31–42. doi: 10.1006/viro.1996.0395. [DOI] [PubMed] [Google Scholar]
- 28.Ito T, Lai M M C. Determination of the secondary structure of and cellular protein binding to the 3′-untranslated region of the hepatitis C virus RNA genome. J Virol. 1997;71:8698–8706. doi: 10.1128/jvi.71.11.8698-8706.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jansen R W, Newbold J E, Lemon S M. Complete nucleotide sequence of a cell culture-adapted variant of hepatitis A virus: comparison with wild-type virus with restricted capacity for in vitro replication. Virology. 1988;163:299–307. doi: 10.1016/0042-6822(88)90270-x. [DOI] [PubMed] [Google Scholar]
- 30.Kaneko T, Tanji Y, Satoh S, Hijikata M, Asabe S, Kimura K, Shimotohno K. Production of two phosphoproteins from the NS5A region of the hepatitis C viral genome. Biochem Biophys Res Commun. 1994;205:320–326. doi: 10.1006/bbrc.1994.2667. [DOI] [PubMed] [Google Scholar]
- 31.Kim D W, Gwack Y, Han J H, Choe J. C-terminal domain of the hepatitis C virus NS3 protein contains an RNA helicase activity. Biochem Biophys Res Commun. 1995;215:160–166. doi: 10.1006/bbrc.1995.2447. [DOI] [PubMed] [Google Scholar]
- 32.Kim J L, Morgenstern K A, Griffith J P, Dwyer M D, Thomson J A, Murcko M A, Lin C, Caron P R. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure. 1998;6:89–100. doi: 10.1016/s0969-2126(98)00010-0. [DOI] [PubMed] [Google Scholar]
- 33.Kolykhalov A A, Agapov E V, Blight K, Mihalik K, Feinstone S M, Rice C M. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science. 1997;277:570–574. doi: 10.1126/science.277.5325.570. [DOI] [PubMed] [Google Scholar]
- 34.Kolykhalov A A, Feinstone S M, Rice C M. Identification of a highly conserved sequence element at the 3′ terminus of hepatitis C virus genome RNA. J Virol. 1996;70:3363–3371. doi: 10.1128/jvi.70.6.3363-3371.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolykhalov A A, Mihalik K, Feinstone S M, Rice C M. Hepatitis C virus-encoded enzymatic activities and conserved RNA elements in the 3′ nontranslated region are essential for virus replication in vivo. J Virol. 2000;74:2046–2051. doi: 10.1128/jvi.74.4.2046-2051.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin C, Pragai B M, Grakoui A, Xu J, Rice C M. Hepatitis C virus NS3 serine proteinase: trans-cleavage requirements and processing kinetics. J Virol. 1994;68:8147–8157. doi: 10.1128/jvi.68.12.8147-8157.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lohmann V, Körner F, Herian U, Bartenschlager R. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J Virol. 1997;71:8416–8428. doi: 10.1128/jvi.71.11.8416-8428.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lohmann V, Körner F, Koch J O, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 39.Melkonyan H, Sorg C, Klempt M. Electroporation efficiency in mammalian cells is increased by dimethyl sulfoxide (DMSO) Nucleic Acids Res. 1996;24:4356–4357. doi: 10.1093/nar/24.21.4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mendez E, Ruggli N, Collett M S, Rice C M. Infectious bovine viral diarrhea virus (strain NADL) RNA from stable cDNA clones: a cellular insert determines NS3 production and viral cytopathogenicity. J Virol. 1998;72:4737–4745. doi: 10.1128/jvi.72.6.4737-4745.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murphy F A, Fauquet C M, Bishop D H L, Ghabrial S A, Jarvis A W, Martelli G P, Mayo M A, Summers M D, editors. Classification and nomenclature of viruses: sixth report of the International Committee on Taxonomy of Viruses. New York, N.Y: Springer Verlag; 1995. pp. 424–426. [Google Scholar]
- 42.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42:3858–3863. [PubMed] [Google Scholar]
- 43.Reed K E, Rice C M. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr Top Microbiol Immunol. 2000;242:55–84. doi: 10.1007/978-3-642-59605-6_4. [DOI] [PubMed] [Google Scholar]
- 44.Reed K E, Xu J, Rice C M. Phosphorylation of the hepatitis C virus NS5A protein in vitro and in vivo: properties of the NS5A-associated kinase. J Virol. 1997;71:7187–7197. doi: 10.1128/jvi.71.10.7187-7197.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rijnbrand R, Bredenbeek P, van der Straaten T, Whetter L, Inchauspe G, Lemon S, Spaan W. Almost the entire 5′ non-translated region of hepatitis C virus is required for cap-independent translation. FEBS Lett. 1995;365:115–119. doi: 10.1016/0014-5793(95)00458-l. [DOI] [PubMed] [Google Scholar]
- 46.Schuppli D, Georgijevic J, Weber H. Synergism of mutations in bacteriophage Qβ RNA affecting host factor dependence of Qb replicase. J Mol Biol. 2000;295:149–154. doi: 10.1006/jmbi.1999.3373. [DOI] [PubMed] [Google Scholar]
- 47.Shimizu Y K, Igarashi H, Kiyohara T, Shapiro M, Wong D C, Purcell R H, Yoshikura H. Infection of a chimpanzee with hepatitis C virus grown in cell culture. J Gen Virol. 1998;79:1383–1386. doi: 10.1099/0022-1317-79-6-1383. [DOI] [PubMed] [Google Scholar]
- 48.Sugiyama K, Kato N, Mizutani T, Ikeda M, Tanaka T, Shimotohno K. Genetic analysis of the hepatitis C virus (HCV) genome from HCV-infected human T cells. J Gen Virol. 1997;78:329–336. doi: 10.1099/0022-1317-78-2-329. [DOI] [PubMed] [Google Scholar]
- 49.Suzich J A, Tamura J K, Palmer H F, Warrener P, Grakoui A, Rice C M, Feinstone S M, Collett M S. Hepatitis C virus NS3 protein polynucleotide-stimulated nucleoside triphosphatase and comparison with the related pestivirus and flavivirus enzymes. J Virol. 1993;67:6152–6158. doi: 10.1128/jvi.67.10.6152-6158.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanaka T, Kato N, Cho M J, Shimotohno K. A novel sequence found at the 3′ terminus of hepatitis C virus genome. Biochem Biophys Res Commun. 1995;215:744–749. doi: 10.1006/bbrc.1995.2526. [DOI] [PubMed] [Google Scholar]
- 51.Tanaka T, Kato N, Cho M J, Sugiyama K, Shimotohno K. Structure of the 3′ terminus of the hepatitis C virus genome. J Virol. 1996;70:3307–3312. doi: 10.1128/jvi.70.5.3307-3312.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanji Y, Hijikata M, Satoh S, Kaneko T, Shimotohno K. Hepatitis C virus-encoded nonstructural protein NS4A has versatile functions in viral protein processing. J Virol. 1995;69:1575–1581. doi: 10.1128/jvi.69.3.1575-1581.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tanji Y, Kaneko T, Satoh S, Shimotohno K. Phosphorylation of hepatitis C virus-encoded nonstructural protein NS5A. J Virol. 1995;69:3980–3986. doi: 10.1128/jvi.69.7.3980-3986.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tomei L, Failla C, Santolini E, De Francesco R, La Monica N. NS3 is a serine protease required for processing of hepatitis C virus polyprotein. J Virol. 1993;67:4017–4026. doi: 10.1128/jvi.67.7.4017-4026.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tsukiyama K K, Iizuka N, Kohara M, Nomoto A. Internal ribosome entry site within hepatitis C virus RNA. J Virol. 1992;66:1476–1483. doi: 10.1128/jvi.66.3.1476-1483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van den Hoff M J, Moorman A F, Lamers W H. Electroporation in ‘intracellular’ buffer increases cell survival. Nucleic Acids Res. 1992;20:2902. doi: 10.1093/nar/20.11.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang C, Sarnow P, Siddiqui A. Translation of human hepatitis C virus RNA in cultured cells is mediated by an internal ribosome-binding mechanism. J Virol. 1993;67:3338–3344. doi: 10.1128/jvi.67.6.3338-3344.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yamada N, Tanihara K, Takada A, Yorihuzi T, Tsutsumi M, Shimomura H, Tsuji T, Date T. Genetic organization and diversity of the 3′ noncoding region of the hepatitis C virus genome. Virology. 1996;223:255–261. doi: 10.1006/viro.1996.0476. [DOI] [PubMed] [Google Scholar]
- 59.Yamashita T, Kaneko S, Shirota Y, Qin W, Nomura T, Kobayashi K, Murakami S. RNA-dependent RNA polymerase activity of the soluble recombinant hepatitis C virus NS5B protein truncated at the C-terminal region. J Biol Chem. 1998;273:15479–15486. doi: 10.1074/jbc.273.25.15479. [DOI] [PubMed] [Google Scholar]
- 60.Yanagi M, Purcell R H, Emerson S U, Bukh J. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc Natl Acad Sci USA. 1997;94:8738–8743. doi: 10.1073/pnas.94.16.8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yanagi M, St. Claire C M, Emerson S U, Purcell R H, Bukh J. In vivo analysis of the 3′ untranslated region of the hepatitis C virus after in vitro mutagenesis of an infectious cDNA clone. Proc Natl Acad Sci USA. 1999;96:2291–2295. doi: 10.1073/pnas.96.5.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yao N H, Reichert P, Taremi S S, Prosise W W, Weber P C. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure Folding Design. 1999;7:1353–1363. doi: 10.1016/s0969-2126(00)80025-8. [DOI] [PubMed] [Google Scholar]
- 63.Zhang G, Aldridge S, Clarke M C, McCauley J W. Cell death induced by cytopathic bovine viral diarrhoea virus is mediated by apoptosis. J Gen Virol. 1996;77:1677–1681. doi: 10.1099/0022-1317-77-8-1677. [DOI] [PubMed] [Google Scholar]