

Abstract
Previously, we demonstrated the importance of low-level-resistant variants to the evolution of resistance in Staphylococcus aureus exposed to ciprofloxacin in an in vitro system and developed a pharmacodynamic model which predicted the emergence of resistance. Here, we examine and model the evolution of resistance to levofloxacin in S. aureus exposed to simulated levofloxacin pharmacokinetic profiles. Enrichment of subpopulations with mutations in grlA and low-level resistance varied with levofloxacin exposure. A regimen producing average steady-state concentrations (Cavg ss) just above the MIC selected grlA mutants with up to 16-fold increases in the MIC and often additional mutations in grlA/grlB and gyrA. A regimen providing Cavg ss between the MIC and the mutant prevention concentration (MPC) suppressed bacterial numbers to the limit of detection and prevented the appearance of bacteria with additional mutations or high-level resistance. Regimens producing Cavg ss above the MPC appeared to eradicate low-level-resistant variants in the cultures and prevent the emergence of resistance. There was no relationship between the time concentrations remained between the MIC and the MPC and the degree of resistance or the presence or type of mutations that appeared in grlA/B or gyrA. Our pharmacodynamic model described the growth and levofloxacin killing of the parent strains and the most resistant grlA mutants in the starting cultures and correctly predicted conditions that enrich subpopulations with low-level resistance. These findings suggest that the pharmacodynamic model has general applicability for describing fluoroquinolone resistance in S. aureus and further demonstrate the importance of low-level-resistant variants to the evolution of resistance.
In previous work, we examined the evolution of resistance when ciprofloxacin-susceptible (S) Staphylococcus aureus strains were exposed in an in vitro hollow-fiber system to simulated clinical and experimental ciprofloxacin pharmacokinetic profiles (4). We found that with increasing average steady-state concentrations (Cavg ss), the rate of initial killing approached a maximum, and the rate of regrowth decreased. Enrichment of subpopulations with mutations in grlA and low-level resistance also varied depending on the pharmacokinetic environment. A regimen producing Cavg ss slightly above the MIC selected resistant (R) variants with grlA mutations that did not evolve to higher levels of resistance. Clinical regimens which provided Cavg ss intermediate between the MIC and mutant prevention concentration (MPC) resulted in the emergence of subpopulations with gyrA mutations and higher levels of resistance and a regimen producing Cavg ss greater than or equal to the MPC selected grlA mutants, but the appearance of subpopulations with higher levels of resistance was delayed. A regimen designed to maintain ciprofloxacin concentrations entirely above the MPC appeared to eradicate low-level-resistant variants in the inoculum and prevent the emergence of high-level-resistant variants. There was no relationship between the time ciprofloxacin concentrations remained in the mutant selection window (TMSW; the interval between the MIC and the MPC) and the degree of resistance or the presence or type of mutations that appeared in grlA or gyrA.
We fit three pharmacodynamic models to the data generated from our experiments and found that a two-population model with unique growth (g) and killing (k) rate constants for the ciprofloxacin-susceptible (gS and kS, respectively) and -resistant (gR and kR, respectively) subpopulations best described the initial killing and subsequent regrowth patterns observed in the in vitro system (5). The model correctly described the enrichment of subpopulations with low-level resistance in the parent cultures. It confirmed the experimental observations that there was no clear relationship between the TMSW and the enrichment of resistant subpopulations. The model indicated that resistance depended upon the selection of low-level-resistant minor subpopulations in bacterial cultures. It predicted that resistant subpopulations would not emerge when a low-density culture with a low probability of mutants was exposed to a simulated clinical dosing regimen (400 mg every 12 h) or when a high-density culture with a higher probability of mutants was exposed to the high concentrations of an experimental regimen designed to rapidly eradicate grlA mutants, followed by the lower concentrations of the clinical regimen. The validity of these predictions was confirmed with in vitro system experiments.
In this study we extend our observations to levofloxacin, which has been shown to select resistant S. aureus variants less frequently than ciprofloxacin (10, 14, 24, 34). This was done by modeling the effect of simulated clinical and experimental levofloxacin regimens on two S. aureus strains and their grlA mutants in the in vitro system. The experiments provided additional information about the succession of mutations that occur in resistance loci as bacteria evolve and allowed us to test the robustness of our previously developed pharmacodynamic model.
MATERIALS AND METHODS
Bacterial strains.
Two methicillin-resistant S. aureus clinical isolates (MRSA 8043 and MRSA 8282) were used as parent strains for the in vitro system experiments and have been described previously (4). S. aureus SA1199 (levofloxacin broth microdilution MIC of 0.125 μg/ml) and SA1199B (a strain that constitutively expresses high levels of the NorA efflux protein and has a levofloxacin MIC of 1 μg/ml) served as controls in efflux screening experiments (19).
Antimicrobial agent.
Analytical-grade levofloxacin (R. W. Johnson Pharmaceutical Research Institute, Raritan, N.J.) was used to prepare stock solutions according to established guidelines (30). The stock solutions were frozen (−80°C) in aliquots and used within 30 days.
Susceptibility tests.
Levofloxacin MICs and minimal bactericidal concentrations (MBCs) were determined by the broth microdilution method (29, 30) in triplicate for each organism before exposure to levofloxacin and for organisms recovered at 0, 24, 48, and 96 h during in vitro system experiments. MICs were also measured in the presence of 20 μg/ml reserpine (Sigma, St. Louis, Mo.), a competitive inhibitor of NorA, to screen for potential efflux of levofloxacin (20).
Mutant prevention concentration determinations.
Levofloxacin MPCs for MRSA 8043 and MRSA 8282 were determined using a previously described agar dilution method (4). Concentrated bacterial suspensions (∼1010 CFU/ml) were prepared, and 200 μl samples were applied to Mueller-Hinton agar (Difco Laboratories, Detroit, Mich.) containing levofloxacin concentrations of 0, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 2.25, and 2.5 μg/ml. The MPC for each strain was defined as the lowest concentration at which no bacteria were detected following 48 h of incubation at 37°C. Nucleotide sequence analysis of the quinolone resistance-determining regions of grlA/B and gyrA/B (see below) was performed using DNA extracted from the most-resistant variants that appeared.
In vitro system experiments.
A two-compartment hollow-fiber in vitro system that allowed the study strains to be exposed to fluctuating concentrations of levofloxacin was used. The main features and general operation of the system and the method for preparing standardized inocula for the experiments have been described previously (4). Log-phase cultures (∼107 CFU/ml) were exposed to a series of monoexponential levofloxacin pharmacokinetic profiles for 96 h. Three simulations were designed to reproduce the concentration-time profiles of clinical levofloxacin doses of 750 mg, 500 mg, and 250 mg administered as intermittent 1-hour infusions every 24 h. The simulated central and peripheral compartment concentrations were intended to mimic the total levofloxacin concentrations observed in plasma and skin blister fluid, respectively, of adult patients with normal renal function (6-9). An additional experimental regimen (125 mg every 24 h) was simulated to produce levofloxacin concentrations below the MPCs of the study strains for the entire duration of the experiment. Growth control experiments for each strain were conducted over 36 h. Every in vitro system experiment was performed in duplicate.
Pharmacokinetic analysis.
Samples collected from the central and peripheral compartments of the in vitro system during each dosing regimen experiment were analyzed for levofloxacin concentrations using a previously validated ion pair high-performance liquid chromatography method (4). Standard curves were linear (R2 > 0.999) over the concentration range tested (0.1 to 50 μg/ml). The within-run relative standard deviations (SD) (n = 5) for levofloxacin quality control concentrations of 25, 2.5, and 0.25 μg/ml were 1.51%, 1.38%, and 0.97%. Between-run relative SD (n = 16) for the same quality control concentrations were 1.88%, 1.82%, and 1.23%. The reliable lower limit of quantification (defined as percent relative SD and percent deviation from nominal concentration that was <15%) was 0.06 μg/ml.
Central and peripheral compartment concentration-time data for levofloxacin were analyzed by compartmental and noncompartmental methods (5, 13). A one-compartment pharmacokinetic model, with first-order elimination from the central compartment, was fitted to levofloxacin concentrations in the central compartment using a nonlinear regression program (WinNonlin version 4.0; Pharsight Corporation, Mountain View, Calif.). All pharmacokinetic data were weighted by the inverse of squared predicted concentrations based on the precision of the high-performance liquid chromatography assay.
Pharmacodynamic analysis.
Viable counts were determined for samples collected from the peripheral compartment of the in vitro system using previously described drop and membrane filter count methods (4). Experiments performed to assess the effect of levofloxacin carryover indicated that dilutions of at least 100-fold were necessary to prevent a significant (P < 0.05, paired t test) lowering of colony counts in samples containing levofloxacin concentrations of 0.5 to 16 μg/ml compared to those of drug-free controls. When bacterial counts were unreliable because of levofloxacin carryover, the membrane filter count method was used. Previous experiments established reliable lower limits of detection (coefficients of variation < 20%) of 3.0 × 102, 1.0 × 102, and 7.0 × 101 CFU/ml for filtered samples of 100, 250, and 500 μl, respectively (4).
Resistant subpopulation analysis.
Viable cells were counted to quantify resistant bacterial subpopulations in samples removed from the peripheral compartment of the in vitro system at 0, 24, and 36 h (growth control experiments) and at 0, 24, 48, and 96 h (levofloxacin experiments). The frequency of resistant bacteria in the starting cultures at each levofloxacin concentration was calculated by dividing the number of colonies that appeared by the inoculum applied to each plate. For the purposes of this paper, we defined levofloxacin-susceptible bacteria as those recovered only on drug-free agar. Bacteria with low-level levofloxacin resistance were defined as those recovered only on agar containing a levofloxacin concentration of 0.5 μg/ml. Bacteria with high-level resistance were defined as those recovered on agar containing levofloxacin concentrations of ≥1 μg/ml.
Nucleotide sequence analysis of grlA/B and gyrA/B.
Colonies appearing on levofloxacin-containing Mueller-Hinton agar in the MPC studies and the starting cultures during in vitro system experiments were analyzed for nucleotide changes within the grlA/B and gyrA/B genes as described previously (4). Colonies recovered on drug-free agar at the end of the in vitro system experiments were replica plated onto agar containing increasing levofloxacin concentrations, and colonies that appeared on plates with the highest concentrations were selected for sequence analysis. All colonies were examined when three or fewer colonies were present on a plate. Three colonies were randomly chosen for sequence analysis when more than three colonies were present. Genomic DNA was isolated using lysostaphin (Sigma) and the DNeasy tissue kit (QIAGEN, Valencia, Calif.). Primers, PCR conditions, the sequencing procedure, and interpretation of results were identical to those reported previously (4).
Pharmacodynamic modeling.
The candidate two-population pharmacodynamic models and their equations were presented in an earlier paper (5). The model equations were simultaneously fit to viable-cell count data obtained from the in vitro system experiments with the two levofloxacin-susceptible parent strains (MRSA 8043 and MRSA 8282) and from additional in vitro system studies that characterized the growth and killing of the most prevalent grlA mutants detected in the parent cultures (MRSA 8043L0-1 and MRSA 8282L0-1). Two-population pharmacodynamic models with equivalent (model 1) and different (model 2) net growth (g) and levofloxacin killing (k) rate constants for levofloxacin-susceptible (S) and -resistant (R) subpopulations were evaluated. The initial equation conditions and the approach for deriving initial parameter estimates were the same as previously described (5).
Pharmacodynamic model equations were fitted to equally weighted log-transformed viable counts as a function of time using a nonlinear regression program (WinNonlin). The goodness of fit was evaluated by determining correlation coefficients (r), performing residual analysis, and assessing the precision of parameter estimates (12). The runs test was used to determine whether model-fitted curves deviated systematically from observed viable-cell count data. The pharmacodynamic model of best fit was determined by the second-order Akaike information criterion (AICc) (3). Mathematical simulations were performed, using mean parameter estimates from the model of best fit, to explore the relationship between the levofloxacin TMSW and the emergence of resistant subpopulations.
RESULTS
Susceptibilities and MPCs.
The levofloxacin MICs of MRSA 8043 and MRSA 8282 were 0.5 μg/ml, and the MBCs were 1 μg/ml. Reserpine decreased the levofloxacin MICs of MRSA 8043 (n = 10) by twofold or less (Fig. 1) and had no effect on MRSA 8282 MICs (n = 10). Reserpine lowered the levofloxacin MIC of SA1199B (n = 3) by twofold but did not decrease the MIC of SA1199 (n = 3). The levofloxacin MPCs of MRSA 8043 ranged from 1.50 to 1.75 μg/ml (geometric mean, 1.66 μg/ml; n = 3) and of MRSA 8282 from 1.75 to 2.00 μg/ml (geometric mean, 1.83 μg/ml; n = 3). The most-resistant variants had the S80F, S80Y, or E84K mutation in grlA.
FIG. 1.

Total MRSA 8043 (left) and MRSA 8282 (right) bacterial counts (levofloxacin, 0 μg/ml) and subpopulations resistant to levofloxacin at 0.5 to 16 μg/ml in samples collected from the in vitro system at the indicated times during growth control and simulated levofloxacin dosage regimen experiments. Asterisks above the subpopulation bars indicate that bacteria were recovered in numbers below the reliable limit of detection (100 CFU/ml); no symbol above the bars denotes that no bacteria were recovered at the indicated concentration. Broth microdilution MICs of the total bacterial population in the absence (presence) of reserpine are indicated within the population profiles. Results are expressed as means of results of two separate experiments. The key for all panels appears in the top left panel.
Resistant variant subpopulations in the starting cultures during in vitro system experiments.
The majority (>99.9%) of cells in the starting populations were susceptible to levofloxacin concentrations of <0.5 μg/ml (Fig. 1). Resistant subpopulations were detected in the starting cultures of all 10 experiments with both strains at frequencies ranging from 5.7 × 10−8 to 1.5 × 10−6 when the bacteria were subcultured on agar containing levofloxacin concentrations of 0.5 μg/ml. No bacteria were recovered on agar containing more than 0.5 μg/ml of levofloxacin.
Sequencing of DNA from 30 MRSA 8043 and 30 MRSA 8282 colonies recovered on drug-free agar showed silent mutations or wild-type grlA/B and gyrA/B in all instances. Sequencing of 25 MRSA 8043 and 24 MRSA 8282 colonies recovered on agar containing 0.5 μg/ml of levofloxacin revealed no mutations, silent mutations, or point mutations in grlA and grlB (Table 1). Point mutations in grlA were found in 48% of MRSA 8043 and 83% of MRSA 8282 variants with low-level resistance. The grlB mutations were detected in all three low-level-resistant MRSA 8282 colonies sequenced from the starting population during experiments with the regimen of 125 mg every 24 h. No bacteria with point mutations in gyrA/B were found in the starting cultures of either strain during any experiment.
TABLE 1.
Point mutations detected in the grlA, grlB, gyrA, and gyrB genes for colonies recovered on agar containing the indicated levofloxacin concentration at the indicated times during in vitro system experiments with MRSA 8043 and MRSA 8282a
| Strain | Experimentc | LVX concn (μg/ml) | Point mutation detected at 0 h in:
|
LVX concn (μg/ml) | Point mutation detected at 24 or 96 h inb:
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| grlA | grlB | gyrA | gyrB | grlA | grlB | gyrA | gyrB | |||||||||||||
| MRSA 8043 | Growth control #1 | 0.5 | None (3) | None (3) | None (3) | None (3) | 0.5 | S80F (3) | None (3) | None (3) | None (3) | |||||||||
| Growth control #2 | 0.5 | S80Y (2) | None (3) | None (3) | None (3) | 1 | S80Y (3) | None (3) | None (3) | None (3) | ||||||||||
| None (1) | ||||||||||||||||||||
| LVX, 125 mg every 24 h, #1 | 0.5 | E84K (1) | None (3) | None (3) | None (3) | 1 | E84K (3) | None (3) | None (3) | None (3) | ||||||||||
| None (2) | ||||||||||||||||||||
| LVX, 125 mg every 24 h, #2 | 0.5 | None (3) | None (3) | None (3) | None (3) | 2 | None (3) | N470I (3) | S84A (1) | None (3) | ||||||||||
| None (2) | ||||||||||||||||||||
| LVX, 250 mg every 24 h, #1 | 0.5 | None (3) | None (3) | None (3) | None (3) | 0 | None (3) | None (3) | None (3) | None (3) | ||||||||||
| LVX, 250 mg every 24 h, #2 | 0.5 | S80F (2) | None (2) | None (2) | None (2) | 0.5 | S80F (1) | None (3) | None (3) | None (3) | ||||||||||
| None (2) | ||||||||||||||||||||
| LVX, 500 mg every 24 h, #1 | 0.5 | E84K (2) | None (2) | None (2) | None (2) | 0 | None (3) | None (3) | None (3) | None (3) | ||||||||||
| LVX, 500 mg every 24 h, #2 | 0.5 | S80F (3) | None (3) | None (3) | None (3) | 0 | None (3) | None (3) | None (3) | None (3) | ||||||||||
| LVX, 750 mg every 24 h, #1 | 0.5 | None (1) | None (1) | None (1) | None (1) | 0 | None (3) | None (3) | None (3) | None (3) | ||||||||||
| LVX, 750 mg every 24 h, #2 | 0.5 | S80Y (2) | None (2) | None (2) | None (2) | 0 | None (3) | None (3) | None (3) | None (3) | ||||||||||
| MRSA 8282 | Growth control #1 | 0.5 | S80Y (2) | None (2) | None (2) | None (2) | 1 | S80Y (3) | None (3) | None (3) | None (3) | |||||||||
| Growth control #2 | 0.5 | S80F (3) | None (3) | None (3) | None (3) | 0.5 | S80F (3) | None (3) | None (3) | None (3) | ||||||||||
| LVX, 125 mg every 24 h, #1 | 0.5 | P144S (3) | E422D (3) | None (3) | None (3) | 4 | S80F (3) | E422D (3) | S84L (2) | None (3) | ||||||||||
| P144S (3) | None (1) | |||||||||||||||||||
| LVX, 125 mg every 24 h, #2 | 0.5 | S80F (1) | E422D (3) | None (3) | None (3) | 8 | S80F (3) | E422D (3) | S84L (3) | None (3) | ||||||||||
| A116P (1) | P144S (3) | |||||||||||||||||||
| P144S (3) | ||||||||||||||||||||
| LVX, 250 mg every 24 h, #1 | 0.5 | None (1) | None (1) | None (1) | None (1) | 0 | None (3) | None (3) | None (3) | None (3) | ||||||||||
| LVX, 250 mg every 24 h, #2 | 0.5 | S80Y (2) | None (2) | None (2) | None (2) | 0.5 | S80Y (2) | None (3) | None (3) | None (3) | ||||||||||
| None (1) | ||||||||||||||||||||
| LVX, 500 mg every 24 h, #1 | 0.5 | S80F (3) | None (3) | None (3) | None (3) | 0 | None (3) | None (3) | None (3) | None (3) | ||||||||||
| LVX, 500 mg every 24 h, #2 | 0.5 | S80Y (1) | None (1) | None (1) | None (1) | 0 | None (3) | None (3) | None (3) | None (3) | ||||||||||
| LVX, 750 mg every 24 h, #1 | 0.5 | None (3) | None (3) | None (3) | None (3) | 0 | None (3) | None (3) | None (3) | None (3) | ||||||||||
| LVX, 750 mg every 24 h, #2 | 0.5 | S80F (3) | None (3) | None (3) | None (3) | 0 | None (3) | None (3) | None (3) | None (3) | ||||||||||
DNA sequence analysis was performed (i) on all colonies when three or fewer colonies were recovered or (ii) on three colonies when more than three colonies were recovered at a given levofloxacin concentration. Numbers in parentheses represent the number of colonies with the corresponding mutation.
Colonies were recovered at 24 h during growth control experiments and at 96 h during simulated levofloxacin dosage regimen experiments.
Results are presented for the initial (#1) and repeat (#2) experiments.
Levofloxacin pharmacokinetics.
Desired levofloxacin pharmacokinetic profiles for all regimens were accurately and reproducibly simulated in the in vitro system (Fig. 2). Peak levofloxacin concentrations in the peripheral compartment, attained 15 to 30 min after the end of the 1-hour intermittent infusions, were 86 to 97% of the corresponding central compartment concentrations. The extent of levofloxacin penetration into the peripheral compartment, estimated by the ratio of the area under the concentration-time curve (AUC) in the peripheral and central compartments, was between 97 and 104% for all experiments.
FIG. 2.

Measured central (filled symbols) and peripheral (open symbols) compartment concentrations for simulated levofloxacin dosage regimens. The fitted curves from the one-compartment pharmacokinetic model are superimposed on the symbols. The levofloxacin MICs for MRSA 8043 and MRSA 8282 are depicted by the lower dot-and-dash horizontal line. The levofloxacin MPCs for MRSA 8043 (solid line) and MRSA 8282 (dashed line) are also shown.
A one-compartment pharmacokinetic model accurately described observed central compartment levofloxacin concentration-time profiles. The correlation (r) between measured and model-predicted central compartment concentrations ranged from 0.981 to 0.998 for all dosage regimen simulations. No systematic deviation in the residuals between observed and model-predicted concentrations was observed. The mean (± standard error) central compartment estimates for elimination rate constant (kel), half-life, and volume of distribution, obtained by compartmental analysis using pooled pharmacokinetic data from all dosage regimen simulations, were 0.099 ± 0.002 h−1, 7.03 ± 0.03 h, and 162.4 ± 0.8 ml, respectively. The model-predicted central compartment pharmacokinetic profiles mimicked pharmacokinetics in the peripheral compartment (Fig. 2). The one-compartment pharmacokinetic model slightly over- and underpredicted the measured maximum concentrations of the drug in serum (Cmax) and minimum concentrations of the drug in serum in the peripheral compartment by a mean of 5.6% (range, 1.9 to 8.7%) and 4.9% (range, 0.8 to 8.9%), respectively, but the extents of drug exposure were similar, with a mean difference in AUCs at 24 h (AUC24) of only −0.2% (range, −4.6 to 3.3%).
Bacterial population dynamics.
Bacteria were in log-phase growth at the start of each experiment. In the absence of levofloxacin, the bacteria continued to grow at an exponential rate until the carrying capacity of the in vitro system was reached (∼109 CFU/ml for MRSA 8043 and ∼1010 CFU/ml for MRSA 8282), and counts remained near this plateau for the remainder of each experiment (Fig. 3).
FIG. 3.

Observed viable counts of MRSA 8043 (filled symbols, top panel), MRSA 8043L0-1 (open symbols, top panel), MRSA 8282 (filled symbols, bottom panel), and MRSA 8282L0-1 (open symbols, bottom panel) during growth control experiments and following exposure to simulated levofloxacin pharmacokinetic profiles. Viable counts are plotted as the means and ranges from two separate experiments. The reliable limit of detection for these experiments was 70 CFU/ml. The pharmacodynamic model-predicted viable count-versus-time profiles are shown by the solid (MRSA 8043 and MRSA 8282) and dashed (MRSA 8043L0-1 and MRSA 8282L0-1) curves. The key for both panels appears in the top panel. The slopes of the killing and regrowth portions of the viable-cell count curves were similar for replicate dosage regimen simulations with a given strain (error bars).
During levofloxacin dosage regimen simulations, densities of the starting cultures ranged from 7.6 × 106 to 2.6 × 107 CFU/ml for MRSA 8043 and from 9.7 × 106 to 2.0 × 107 CFU/ml for MRSA 8282. This corresponded to a total population of ∼1.5 × 108 to 5.2 × 108 CFU in the 20-ml peripheral compartment for both bacteria. Exposure of cultures to the simulated regimens of 750 and 500 mg every 24 h resulted in a decline in viable counts to below the reliable limit of detection (70 CFU/ml). Small numbers of viable bacteria persisted below this limit for the remainder of the experiments. When the bacteria were exposed to the regimen of 250 mg every 24 h, viable counts decreased to the limit of detection and then increased to slightly above this limit toward the end of the experiments. When the cultures were exposed to a regimen of 125 mg every 24 h, viable counts declined initially but then increased and approached the carrying capacity of the system. The slopes of the killing and regrowth portions of the viable-cell count curves were similar for replicate dosage regimen simulations with a given strain (Fig. 3).
Changes in susceptibilities of bacterial populations.
No change in levofloxacin MICs (Fig. 1) was observed during growth control experiments with MRSA 8043 and MRSA 8282 or when the cultures were exposed to simulated levofloxacin regimens of 750 mg, 500 mg, and 250 mg every 24 h. In contrast, the MICs increased 2- to 16-fold when the bacteria were exposed to the levofloxacin regimen of 125 mg every 24 h. Reserpine did not decrease the MIC for resistant MRSA 8282 bacteria recovered from the latter experiments but lowered the MIC for resistant MRSA 8043 in one of the replicate experiments by twofold.
Changes in resistant subpopulations and grlA/B and gyrA/B genotypes.
During growth control experiments, the number of low-level-resistant variants with or without mutations in grlA increased (Fig. 1 and Table 1). Resistant subpopulations did not emerge when the cultures were exposed to the 750 and 500 mg every 24 h levofloxacin regimens. Bacteria that persisted at the ends of these experiments did not have mutations in grlA/B or gyrA/B. The grlA mutants that were present in some of the starting cultures were still detected at 96 h after exposure to the regimen of 250 mg every 24 h, but no additional mutations were found. In contrast, when the cultures were exposed to the regimen of 125 mg every 24 h, subpopulations with high-level resistance and mutations in grlA, grlB, and gyrA emerged. Different patterns of nucleotide sequence changes in grlA/B and gyrA/B were observed during the latter experiments. In some cases, no mutants were detected in the starting cultures, and resistant bacteria were recovered at 96 h with mutations in grlB and gyrA. In other cases, grlA mutants were present in the starting populations, and resistant bacteria were recovered at 96 h with no additional mutations. In yet other experiments, grlA and grlB mutants were present in the starting populations, and resistant bacteria were recovered at 96 h with additional gyrA mutations. The pattern of nucleotide sequence changes varied with different dosage regimen simulations and between replicate experiments of the same simulation (Table 1).
Pharmacokinetic-pharmacodynamic values and the emergence of resistance.
The simulated levofloxacin dosage regimens of 500 mg and 750 mg every 24 h, which provided Cmax/MIC ratios between 10.6 and 18.9 and AUC24/MIC ratios between 113 h and 188 h (Table 2), did not result in the emergence of resistance to levofloxacin. The TMSW for a 24-h period ranged from 19.6 to 45.9% during these experiments. The regimen of 250 mg every 24 h, which produced Cmax/MIC ratios between 5.33 and 6.17 and AUC24/MIC ratios between 56.6 h and 64.1 h, failed to eradicate grlA mutants that were detected in the starting cultures (Table 1) but did not result in a change in levofloxacin MICs at 96 h (Fig. 1). The TMSW was between 55.7 and 62.2% in these experiments. The regimen of 125 mg every 24 h, which produced the lowest Cmax/MIC (2.64 to 3.22) and AUC24/MIC (28.1 h to 32.9 h) ratios resulted in the emergence of subpopulations with mutations in gyrA and high-level resistance (Fig. 1 and Table 1). The TMSW ranged between 46.1 and 57.5% in these experiments.
TABLE 2.
Levofloxacin pharmacokinetic parameters relative to MRSA 8043 and MRSA 8282 MICs and MPCs during in vitro system experimentsa
| Simulation (Cavg ss)b | MRSA strain | Parameter during first 24 h
|
Steady-state parameter
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
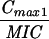 |
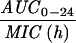 |
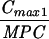 |
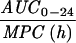 |
T0-24 MSW | 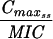 |
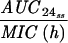 |
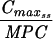 |
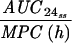 |
T24ss MSW | ||
| 125 mg every 24 h (0.68 μg/ml) | 8043 | 2.66 | 28.1 | 0.80 | 8.46 | 47.5 | 3.22 | 32.9 | 0.97 | 9.90 | 53.1 |
| 8282 | 2.64 | 28.2 | 0.72 | 7.70 | 46.1 | 3.05 | 32.4 | 0.83 | 8.84 | 57.5 | |
| 250 mg every 24 h (1.33 μg/ml) | 8043 | 5.48 | 56.8 | 1.65 | 17.1 | 55.7 | 6.17 | 63.3 | 1.86 | 19.1 | 56.0 |
| 8282 | 5.33 | 56.6 | 1.46 | 15.5 | 58.9 | 5.99 | 64.1 | 1.64 | 17.5 | 62.2 | |
| 500 mg every 24 h (2.62 μg/ml) | 8043 | 10.7 | 113 | 3.22 | 34.0 | 41.9 | 12.1 | 127 | 3.64 | 38.2 | 37.0 |
| 8282 | 10.6 | 114 | 2.89 | 31.1 | 45.9 | 11.8 | 125 | 3.22 | 34.2 | 41.7 | |
| 750 mg every 24 h (3.93 μg/ml) | 8043 | 16.1 | 164 | 4.85 | 49.5 | 22.2 | 18.9 | 188 | 5.69 | 56.8 | 19.6 |
| 8282 | 16.2 | 167 | 4.43 | 45.7 | 28.3 | 18.5 | 188 | 5.05 | 51.5 | 24.4 | |
Expressed as the means of results of two separate experiments. Cmax1, Cmax attained with the first dose; Cmax ss, Cmax at steady state; T0-24 MSW, the percentage of time levofloxacin was in the MSW during the first 24-h period; T24ssMSW, the percentage of time levofloxacin was in the MSW during a 24-h period at steady state.
Cavg ss for the simulated dosage regimens are expressed as the means of results of two separate experiments.
In vitro system experiments with grlA mutants
Additional in vitro system studies were performed to characterize the growth and levofloxacin killing rates of the most-prevalent grlA mutants (MRSA 8043L0-1 and MRSA 8282L0-1) detected in the parent cultures. Both variants had the same grlA mutation (S80F) and a levofloxacin MIC of 1 μg/ml. Reserpine did not decrease the MIC for either bacterium. The net growth rate of these bacteria in the absence of levofloxacin was lower than that of the corresponding parent strains (Fig. 3), and the net killing rate of these mutants when exposed to a levofloxacin regimen of 1,000 mg every 24 h (Cmax attained with the first dose [Cmax1]/MIC of 11.0 to 11.3 and AUC0-24/MIC of 111 h to 113 h) was lower than that of the parent strains exposed to a regimen of 500 mg every 24 h, which provided a similar drug exposure (Cmax1/MIC of 10.6 to 10.7 and AUC0-24/MIC of 113 h to 114 h).
Pharmacodynamic modeling.
The AICc and other model selection statistics indicated that model 2 best described the trends in the viable-cell count data (Table 3). Model 1 was associated with significant bias (P < 0.05, runs test) in model-predicted versus observed viable counts, with the model underpredicting the growth rate of MRSA 8043 and MRSA 8282 and overpredicting the killing rate of MRSA 8043L0-1 and MRSA 8282LC0-1 during the levofloxacin simulations with 1,000 mg every 24 h. There was also a systematic deviation (P < 0.05) in observed viable counts above or below the fitted regrowth curves for replicate experiments with regimens of 250 mg and 125 mg every 24 h. Model 2 better characterized the trends in bacterial killing and growth observed during in vitro system experiments (Fig. 3) and had less bias and more-precise parameter estimates than model 1. The coefficient of variation (CV) of all model parameter estimates, except R0 (the initial size of the R subpopulation), for both strains was ≤13.8% (Table 4). Estimates of R0 were less precise than the other estimates (CVs, 66.4% for MRSA 8043/MRSA 8043L0-1 and 46.1% for MRSA 8282/MRSA 8282L0-1). There was a strong positive correlation between gS and kS values for both MRSA 8043 (r = 0.990) and MRSA 8282 (r = 0.984). The high interdependence of these parameter estimates was anticipated since the model was fit to viable count data that reflected the net result of growth and killing. The univariate 95% confidence intervals for g and k estimates for S and R subpopulations did not coincide for MRSA 8282/MRSA 8282L0-1, but the confidence intervals of gS and gR estimates overlapped for MRSA 8043/MRSA 8043L0-1. The univariate and planar 95% confidence intervals of the EC50, the concentration that results in 50% of the maximal k, for susceptible and resistant isolates did not coincide.
TABLE 3.
Selection statistics for the two pharmacodynamic modelsa
| Strain | Model | K | log(L) | AICc | Δi | wi |
|---|---|---|---|---|---|---|
| MRSA 8043 | 1 | 6 | −379.43 | 771.26 | 90.70 | 0.00 |
| 2 | 8 | −331.93 | 680.56 | 0.00 | 1.00 | |
| MRSA 8282 | 1 | 6 | −342.84 | 698.06 | 134.75 | 0.00 |
| 2 | 8 | −273.32 | 563.31 | 0.00 | 1.00 |
K, number of estimated parameters in the model; log(L), maximized log-likelihood function; Δi, simple AICc differences; wi, Akaike weights for models in the set. See also reference 5.
TABLE 4.
Parameter estimates for the two-population pharmacodynamic model (model 2)a
| Strain | gS (h−1) | gR (h−1) | kS (h−1) | kR (h−1) | EC50,S (μg/ml) | EC50,R (μg/ml) | R0 (CFU/ml) | Nmax (CFU/ml) |
|---|---|---|---|---|---|---|---|---|
| MRSA 8043 | 0.87 (0.09) | 0.72 (0.04) | 1.53 (0.09) | 1.12 (0.04) | 0.20 (0.02) | 0.72 (0.09) | 1.14 (0.76) | 1.1 (0.2) × 109 |
| MRSA 8282 | 0.87 (0.05) | 0.66 (0.02) | 1.47 (0.05) | 1.02 (0.02) | 0.20 (0.01) | 0.75 (0.06) | 1.32 (0.61) | 7.5 (0.8) × 109 |
Values are means ± standard errors. EC50,S and EC50,R, EC50 of the S and R subpopulations, respectively; Nmax, carrying capacity of the in vitro system.
Pharmacodynamic model predictions.
Model 2 predicted that, in the absence of levofloxacin, the majority S subpopulation would grow exponentially until the carrying capacity of the in vitro system was reached (Fig. 3). The competing R subpopulation would also increase exponentially but was predicted to plateau at numbers that were ∼7 to 8 log10 lower than those for the S subpopulation. In contrast, the model predicted that R populations grown in the absence of S populations would reach the carrying capacity of the in vitro system. Model predictions for the growth of parent strains and their corresponding grlA mutants were in concordance with total and resistant subpopulation profiles observed during growth control experiments (Fig. 1 and 3).
The model predicted that the S subpopulation would be eradicated (i.e., decline to < 1 CFU) during all levofloxacin dosage simulations, with the time required for extinction decreasing as the intensity of levofloxacin exposure increased. For instance, 82 h (MRSA 8043) to 98 h (MRSA 8282) would be required to extinguish S populations at the lowest levofloxacin exposure (125 mg every 24 h) but only 36 h (MRSA 8043) to 39 h (MRSA 8282) would be necessary to eradicate S populations at the highest exposure (750 mg every 24 h).
The model predicted that the levofloxacin regimens of 125 mg and 250 mg every 24 h would result in enrichment of the R subpopulation, with the R populations constituting 0.62% and 0.55% of the total MRSA 8043 population at 24 h for each of the regimens, respectively. The observed percentage of the total MRSA 8043 population resistant to levofloxacin at ≥0.5 μg/ml for the two regimens was 3.3% and <8.1% (Fig. 1). The model predicted that R would comprise 0.21 and 0.26% of the total MRSA 8282 population by 24 h for the same levofloxacin exposures. The observed percentages were 0.4% and <1.6%. The model predicted that the R population would constitute the entire population by 96 h with both regimens. The observed percentage of bacteria resistant to levofloxacin concentrations of ≥0.5 μg/ml at 96 h with the regimen of 125 mg every 24 h ranged from 96 to 101%, but the percentage for the regimen of 250 mg every 24 h ranged from <1.6 to 39%.
Mathematical simulations with the pharmacodynamic model identified no clear relationship between TMSW and the enrichment of the R subpopulation. Constant and fluctuating pharmacokinetic profiles that failed to eradicate the R subpopulation and that produced concentrations entirely outside the mutant selection window (TMSW, 0%) or within the window for variable periods (0% < TMSW ≤ 100%) were predicted to result in the selection of R populations.
DISCUSSION
In this study, we examined the relationship between pharmacokinetics and the evolution of resistance when S. aureus was exposed to simulated levofloxacin regimens in an in vitro system. We fit several two-population pharmacodynamic models, previously developed for ciprofloxacin (5), to the viable-cell count data to determine if they described the killing and regrowth seen in the in vitro system.
The starting cultures of all in vitro system experiments consisted mainly of levofloxacin-susceptible bacteria, although minor subpopulations with low-level resistance (MIC, 1 μg/ml) were reproducibly detected at low frequencies (5.7 × 10−8 to 1.5 × 10−6). Resistance may have been due to efflux, although our assays with reserpine suggested that this played a minor role. Genotyping of the levofloxacin-resistant variants in the starting cultures revealed a variety of mutations in grlA/B. The S80Y, S80F, E84K, and A116P grlA mutations observed have been associated with phenotypic levofloxacin resistance in S. aureus (15, 22, 35), but the P144S grlA and the E422D grlB mutations do not appear to contribute to resistance (16, 26, 33). Other resistant subpopulations did not have mutations in the sequenced regions of grlA/B or gyrA/B. Resistance may have been due to mutations in other regions of these genes. The presence of minor subpopulations of low-level-resistant variants in the starting cultures and the variety of mutations among these bacteria are consistent with the concept that mutations arise randomly from wild-type cells during nonselective growth, and these findings are similar to our experience with ciprofloxacin (4).
Different patterns of killing and regrowth were observed in the in vitro system during the levofloxacin simulations. The initial rate of bacterial killing approached a maximum, and the rate of regrowth decreased with increasing Cavg ss. The emergence of levofloxacin resistance also varied in the different pharmacokinetic environments. The greatest increases in MICs were observed with the simulated experimental dosing regimen of 125 mg every 24 h, providing Cavg ss that were just above the MIC but within the mutant selection windows of both strains. In these experiments, low-level-resistant variants present in the starting cultures became a greater proportion of the population within 24 h. During the remainder of the experiment, their numbers increased and bacteria with a variety of mutations in grlA/B or gyrA and high-level resistance appeared. With the simulated clinical regimen of 250 mg given every 24 h, which also produced a Cavg ss within the MSW but closer to the MPC, bacterial numbers initially fell to the limit of detection but then slowly increased. Bacteria recovered at 96 h did not have an increased MIC measured by broth microdilution, although a slight increase (less than twofold) was detected when the MIC was measured by the Etest (AB Biodisk, Piscataway, N.J.). No additional mutations in grlA/B or gyrA/B were detected. Resistance did not develop when the Cavg ss exceeded the MPC. With these clinical regimens (500 and 750 mg every 24 h), viable counts declined to the limit of detection within 24 h, and the minor subpopulations of grlA mutants that were present in the starting populations appeared to be eliminated. These findings are consistent with our ciprofloxacin experience (4) and the suggestion of others that the key to successful antimicrobial therapy is eradication of minor low-level-resistant subpopulations present in the inoculum (1, 2).
A variety of different grlA/B and gyrA/B genotypes that emerged with exposure to levofloxacin were seen in resistant bacteria, and there was no apparent pattern to the presence or type of mutations that appeared. Genotypic differences occurred in several cases, even though the bacteria had been exposed to the same simulated levofloxacin regimen, and bacteria with the same MIC often had different mutations. As with ciprofloxacin (4), these data suggest that bacteria follow heterogeneous pathways of evolution and that the sequence of resistance determinants is not predetermined.
Our work supports the concept that resistant subpopulations of S. aureus are selectively enriched when levofloxacin concentrations fall within the MSW, although we could not find a relationship between the TMSW and the degree of resistance as reported by others (11). In our in vitro system, high-level-resistant variants did not appear with the regimen of 250 mg every 24 h, producing a TMSW that was slightly higher than that of the regimen of 125 mg every 24 h, with which resistant variants appeared. It is also of note that the TMSW for the regimens of 125 mg every 24 h and 500 mg every 24 h were very similar, and yet resistance did not appear with the latter regimen. Drug concentrations outside the MSW were below the MIC in the former regimen but above the MPC in the latter regimen. These findings are consistent with those of our previous work with ciprofloxacin (4) and again suggest that pharmacokinetic parameters other than TMSW are important for the selection of resistant variants.
Our data showed that resistance did not occur with the Cmax/MIC ratios above 5.33 and AUC24/MIC ratios above 56.6 h afforded by the dosing simulations of 250, 500, and 750 mg every 24 h but occurred with Cmax/MICs up to 3.22 and AUC24/MICs up to 32.9 h provided by the regimen of 125 mg every 24 h. These findings are consistent with there being a relationship between antimicrobial exposure and the selection of resistant variants. Additional experiments using a wider range of concentrations and incremental changes in Cmax/MIC or AUC24/MIC would be useful to fully evaluate these pharmacodynamic concepts and determine optimal values for levofloxacin in the in vitro system. The Cmax/MIC or AUC24/MICs obtained in the in vitro system represent free (unbound) drug exposures. We acknowledge that our experimental design may have overestimated the killing activity for a given levofloxacin regimen because the pharmacokinetic profiles simulated may not reflect the free-drug exposures in normal volunteers or infected patients if protein binding (∼30%) is considered.
Our findings are consistent with an earlier study where no resistance occurred when S. aureus was exposed to levofloxacin daily doses of 400 to 800 mg in an in vitro glass infection model (21). Our results are similar to those of another study where a levofloxacin-susceptible (MIC, 0.5 μg/ml) S. aureus strain was exposed to a simulated levofloxacin regimen of 500 mg every 24 h in a hollow-fiber in vitro system (25). Viable counts for this susceptible strain declined to below the lower limit of detection when cells were exposed to this dosage regimen simulation as in our experiments. In the same study, when strains with reduced susceptibility to levofloxacin (MICs of 2 to 4 μg/ml) were exposed to the identical regimen, viable counts decreased but then increased. These results are similar to ours with the levofloxacin simulation of 125 mg every 12 h, perhaps because the fluoroquinolone exposures relative to the MIC were similar. A different relationship between levofloxacin exposure and increases in MIC was reported by others (11). Increases in MICs of up to 2.5-fold occurred with simulated regimens producing Cmax/MIC and AUC24/MIC ratios closely approximating those of our 125-mg, 250-mg, and 500-mg levofloxacin simulations. The emergence of resistance within 72 h with the 250-mg and the 500-mg simulations in this study may have been due to the larger inocula used (10- to 40-fold higher than ours). This would increase the likelihood that more spontaneously occurring low-level-resistant variants would be present at the outset of the experiments. Our pharmacodynamic model predicted that if R0 for MRSA 8043 and MRSA 8282 was increased by 40-fold, resistant subpopulations would emerge within 72 h with a levofloxacin regimen of 500 mg every 24 h.
The two-population pharmacodynamic models of this study were developed using an approach similar to that used to model the evolution of ciprofloxacin resistance (5). Other investigators have published similar analyses of multipopulation responses to antimicrobial pressure both in animals (17) and in an in vitro system (18). Low-level-resistant grlA mutants of each strain were incorporated into our models as the resistant subpopulation because they represented the most prevalent resistant phenotype and genotype detected in the starting cultures. This approach differed from that of other investigations where model parameter estimates for the resistant subpopulation were derived solely from the regrowth phase of the total viable count-time curves and not from additional independent experiments using resistant bacteria (23, 28, 36). Although both of our candidate pharmacodynamic models described the patterns of bacterial growth and killing observed in the in vitro system, the simpler model (model 1), with equivalent growth and levofloxacin killing rate constants, had more biased parameter estimates. It underpredicted the growth of the susceptible population and overpredicted the killing rate of the resistant subpopulation by levofloxacin. The alternate model, with unique growth and killing rate constants for susceptible and resistant subpopulations (model 2), resulted in parameter estimates with less bias and greater precision. We did not consider more-complex pharmacodynamic models (e.g., ones that allowed for adaptive resistance during levofloxacin exposure or that had more than two subpopulations with different susceptibility phenotypes or genotypes) because our previous experience with ciprofloxacin suggested that it would be difficult to obtain precise estimates for the additional parameters from the viable count data (5).
The net growth and levofloxacin killing rate constants of the resistant subpopulation were lower, and the EC50 estimates were higher than the respective parameters of the susceptible subpopulation. This was similar to our findings with ciprofloxacin (5). The planar confidence intervals for EC50s of the susceptible and resistant subpopulations did not overlap, unlike those for g and k, suggesting that the lower rate of killing of the resistant bacteria at comparable levofloxacin exposures was mainly due to differences in EC50s. Estimates of R0s were less precise than other parameter estimates, as was the case when we applied the model to ciprofloxacin pharmacodynamics. The R0 estimates were consistent with the observed resistance frequencies in our starting cultures and with measured resistance frequencies reported by others (10, 14, 24, 34).
The parameter estimates developed with the model using grlA mutants may not be applicable to bacteria with different resistance mechanisms (e.g., efflux or small-colony variants). As with ciprofloxacin (5), however, the model correctly characterized the emergence of subpopulations with low-level resistance even though grlA mutants were not detected in all of the starting cultures. This suggests that knowing the susceptibility phenotypes of the resistant subpopulations may be sufficient for designing regimens to prevent the emergence of resistance.
Predictions of the two-population model were often correct, but limitations were evident. For instance, the model correctly predicted that resistance would not occur with the regimens of 750 mg and 500 mg every 24 h, but it failed to predict the persistence of low numbers of susceptible bacteria. The model correctly predicted the elimination of susceptible bacteria and the enrichment of low-level-resistant variants during exposure to 125 mg every 24 h exposures but did not explain the origin or appearance of additional subpopulations with high-level resistance and mutations in gyrA. The model also predicted that low-level-resistant variants would be enriched with the regimen of 250 mg every 24 h. Bacterial numbers did increase toward the end of the 96-h experiment (Fig. 3), but the number of grlA mutants and other resistant variants present in the starting cultures did not increase above the reliable limit of detection (Fig. 1). It is possible that the bacteria with slightly increased MICs (detected by the Etest but not broth microdilution) detected at the end of the experiment may have become the predominant subpopulation if the experiments had been continued beyond 96 h.
The ability of the model to describe the population dynamics of bacteria exposed to both levofloxacin and ciprofloxacin suggests that the model is robust and may have general applicability to describing and predicting the relationship between fluoroquinolone exposure and the evolution of resistance in S. aureus. The model may even be applicable to other antimicrobials, such as rifampin or linezolid, where resistance occurs by point mutations (27, 31, 32).
Resistance did not emerge with simulated levofloxacin pharmacokinetic profiles that provided lower Cmax/MIC and AUC24/MIC ratios than those of the ciprofloxacin profiles that resulted in the selection of resistant variants (4). This finding suggests that the pharmacokinetic-pharmacodynamic ratios required to prevent the emergence of resistance in S. aureus vary for different fluoroquinolones. Our robust pharmacodynamic model provides a theoretical framework for understanding the bacterial strain and antimicrobial characteristics that influence the emergence of resistance in S. aureus populations. Mathematical simulations with the model are warranted to examine strain and fluoroquinolone factors that influence the pharmacokinetic-pharmacodynamic indices that are required to prevent the evolution of resistance in S. aureus.
Acknowledgments
This work was supported by grant GM066072 (to M.E.E.) from the National Institutes of Health and by funding from Ortho-McNeil Pharmaceuticals, Inc., Raritan, N.J. Jeffrey Campion and Philip Chung were supported by the American Foundation for Pharmaceutical Education and the University of Kentucky Research Challenge Trust.
We thank Glenn A. Kaatz (Wayne State University, School of Medicine) for kindly providing the S. aureus control strains, SA1199 and SA1199B, used in the fluoroquinolone efflux screening studies.
REFERENCES
- 1.Baquero, F., M. C. Negri, M. I. Morosini, and J. Blazquez. 1998. Antibiotic-selective environments. Clin. Infect. Dis. 27(Suppl. 1):S5-S11. [DOI] [PubMed] [Google Scholar]
- 2.Baquero, F., M. C. Negri, M. I. L. Morosini, and J. Blazquez. 1997. The antibiotic selective process: concentration-specific amplification of low-level resistant populations, p. 93-111. In S. B. Levy (ed.), Antibiotic resistance: origins, evolution, selection and spread. John Wiley & Sons, New York, N.Y. [DOI] [PubMed]
- 3.Burnham, K. P., and D. R. Anderson. 2002. Model selection and multimodel inference: a practical information-theoretic approach, 2nd ed. Springer, New York, N.Y.
- 4.Campion, J. J., P. J. McNamara, and M. E. Evans. 2004. Evolution of ciprofloxacin-resistant Staphylococcus aureus in in vitro pharmacokinetic environments. Antimicrob. Agents Chemother. 48:4733-4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campion, J. J., P. J. McNamara, and M. E. Evans. 2005. Pharmacodynamic modeling of ciprofloxacin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 49:209-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chien, S. C., M. C. Rogge, L. G. Gisclon, C. Curtin, F. Wong, J. Natarajana, R. R. Williams, C. L. Fowler, W. K. Cheung, and A. T. Chow. 1997. Pharmacokinetic profile of levofloxacin following once-daily 500-milligram oral or intravenous doses. Antimicrob. Agents Chemother. 41:2256-2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chien, S. C., F. A. Wong, C. L. Fowler, S. V. Callery-D'Amico, R. R. Williams, R. Nayak, and A. T. Chow. 1998. Double-blind evaluation of the safety and pharmacokinetics of multiple oral once-daily 750-milligram and 1-gram doses of levofloxacin in healthy volunteers. Antimicrob. Agents Chemother. 42:885-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Child, J., D. Mortiboy, J. M. Andrews, A. T. Chow, and R. Wise. 1995. Open-label crossover study to determine pharmacokinetics and penetration of two dose regimens of levofloxacin into inflammatory fluid. Antimicrob. Agents Chemother. 39:2749-2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chow, A. T., C. L. Fowler, R. R. Williams, N. Morgan, S. Kaminski, and J. Natarajana. 2001. Safety and pharmacokinetics of multiple 750-milligram doses of intravenous levofloxacin in healthy volunteers. Antimicrob. Agents Chemother. 45:2122-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans, M. E., and W. B. Titlow. 1998. Levofloxacin selects fluoroquinolone-resistant methicillin-resistant Staphylococcus aureus less frequently than ciprofloxacin. J. Antimicrob. Chemother. 40:285-288. [DOI] [PubMed] [Google Scholar]
- 11.Firsov, A. A., S. N. Vostrov, I. Y. Lubenko, K. Drlica, Y. A. Portnoy, and S. H. Zinner. 2003. In vitro pharmacodynamic evaluation of the mutant selection window hypothesis using four fluoroquinolones against Staphylococcus aureus. Antimicrob. Agents Chemother. 47:1604-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gabrielsson, J., and D. Weiner. 2000. Pharmacokinetic/pharmacodynamic data analysis: concepts and applications, 3rd ed. Apotekarsocieteten, Stockholm, Sweden.
- 13.Gibaldi, M., and D. Perrier. 1982. Pharmacokinetics, 2nd ed. Marcel Dekker, Inc., New York, N.Y.
- 14.Gilbert, D. N., S. J. Kohlhepp, K. A. Slama, G. Gurnkemeier, G. Lewis, R. J. Dworkin, S. E. Slaughter, and J. E. Legett. 2001. Phenotypic resistance of Staphylococcus aureus, selected Enterobacteriaceae, and Pseudomonas aeruginosa after single and multiple in vitro exposures to ciprofloxacin, levofloxacin, and trovafloxacin. Antimicrob. Agents Chemother. 45:883-892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gootz, T. D., R. Zaniewski, S. Haskell, F. S. Kaczmarek, and A. E. Maurice. 1999. Activities of trovafloxacin compared with those of other fluoroquinolones against purified topoisomerase and gyrA and grlA mutants of Staphylococcus aureus. Antimicrob. Agents Chemother. 43:1845-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guirao, G. Y., M. C. M. Toldos, B. M. Peris, M. A. A. Manzanares, M. N. G. Zifiaurre, J. A. M. Andres, J. L. Munoz Bellido, J. A. Garcia-Rodriguez, and M. S. Hernandez. 2001. Molecular diversity of quinolone resistance in genetically related clinical isolates of Staphylococcus aureus and susceptibility to newer fluoroquinolones. J. Antimicrob. Chemother. 47:157-161. [DOI] [PubMed] [Google Scholar]
- 17.Gumbo, T., A. Louie, M. R. Deziel, L. M. Parsons, M. Salfinger, and G. L. Drusano. 2004. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J. Infect. Dis. 190:1642-1651. [DOI] [PubMed] [Google Scholar]
- 18.Jumbe, N., A. Louie, R. Leary, W. Liu, M. R. Deziel, V. H. Tam, R. Bachhawat, C. Freeman, J. Kahn, K. Bush, M. N. Dudley, M. H. Miller, and G. L. Drusano. 2003. Application of a mathematical model to prevent in vivo amplification of antibiotic-resistant bacterial populations during therapy. J. Clin. Investig. 112:275-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaatz, G. W., and S. M. Seo. 1997. Mechanisms of fluoroquinolone resistance in genetically related strains of Staphylococcus aureus. Antimicrob. Agents Chemother. 41:2733-2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaatz, G. W., S. M. Seo, and C. A. Ruble. 1993. Efflux-mediated fluoroquinolone resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 37:1086-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang, S. L., M. J. Rybak, B. J. McGrath, G. W. Kaatz, and S. M. Seo. 1994. Pharmacodynamics of levofloxacin, ofloxacin, and ciprofloxacin, alone and in combination with rifampin against methicillin-susceptible and -resistant Staphylococcus aureus in an in vitro infection model. Antimicrob. Agents Chemother. 38:2702-2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawrence, L. E., M. Frosco, B. Ryan, S. Chaniewski, H. Yuang, D. C. Hooper, and J. F. Barrett. 2002. Bactericidal activities of BMS-284756, a novel des-F(6)-quinolone, against Staphylococcus aureus strains with topoisomerase mutations. Antimicrob. Agents Chemother. 46:191-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li, R. C., D. E. Nix, and J. J. Schentag. 1994. Pharmacodynamic modeling of bacterial kinetics: β-lactam antibiotics against Escherichia coli. J. Pharm. Sci. 83:970-975. [DOI] [PubMed] [Google Scholar]
- 24.Limoncu, M. H., S. Ermertcan, C. B. Cetin, G. Cosar, and G. Dinc. 2003. Emergence of phenotypic resistance to ciprofloxacin and levofloxacin in methicillin-resistant and methicillin-sensitive Staphylococcus aureus strains. Int. J. Antimicrob. Agents 21:420-424. [DOI] [PubMed] [Google Scholar]
- 25.Lister, P. D. 2001. Pharmacodynamics of moxifloxacin and levofloxacin against Staphylococcus aureus and Staphylococcus epidermidis in an in vitro pharmacodynamic model. Clin. Infect. Dis. 32(Suppl. 1):S33-S38. [DOI] [PubMed] [Google Scholar]
- 26.Low, D. E., M. Muller, C. L. Duncan, B. M. Willey, J. C. de Azavedo, A. McGeer, B. N. Kreiswirth, S. Pong-Porter, and D. J. Bast. 2002. Activity of BMS-284756, a novel des-fluoro(6) quinolone, against Staphylococcus aureus, including contributions of mutations to quinolone resistance. Antimicrob. Agents Chemother. 46:1119-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meka, V. G., S. K. Pillai, G. Sakoulas, C. Wennersten, L. Venkataraman, P. C. DeGirolami, G. M. Eliopoulos, R. C. Moellering, Jr., and H. S. Gold. 2004. Linezolid resistance in sequential Staphylococcus aureus isolates associated with the T2500A mutation in the 23S rRNA gene and loss of a single copy of rRNA. J. Infect. Dis. 190:311-317. [DOI] [PubMed] [Google Scholar]
- 28.Mouton, J. W., A. A. T. M. M. Vinks, and N. C. Punt. 1997. Pharmacokinetic-pharmacodynamic modeling of activity of ceftazidime during continuous and intermittent infusion. Antimicrob. Agents Chemother. 41:733-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.National Committee for Clinical Laboratory Standards. 1999. Methods for determining bactericidal activity of antimicrobial agents; approved guideline, M26-A, vol. 19. National Committee for Clinical Laboratory Standards, Wayne, Pa.
- 30.National Committee for Clinical Laboratory Standards. 2003. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard M7-A6, vol. 23. National Committee for Clinical Laboratory Standards, Wayne, Pa.
- 31.Pillai, S. K., G. Sakoulas, C. Wennersten, G. T. Eliopoulos, R. C. Mollering, Jr., M. J. Ferraro, and H. S. Gold. 2002. Linezolid resistance in Staphylococcus aureus: characterization and stability of resistant phenotype. J. Infect. Dis. 186:1603-1607. [DOI] [PubMed] [Google Scholar]
- 32.Schmitz, F. J., A. C. Fluit, D. Hafner, A. Beeck, M. Perdikouli, M. Boos, S. Scheuring, J. Verhoef, K. Kohrer, and C. von Eiff. 1999. Development of resistance to ciprofloxacin, rifampin, and mupirocin in methicillin-susceptible and -resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 44:3229-3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmitz, F. J., M. E. Jones, B. Hofmann, B. Hansen, S. Scheuring, M. Luckefahr, A. Fluit, J. Verhoef, U. Hadding, H. P. Heinz, and K. Kohrer. 1998. Characterization of grlA, grlB, and gyrB mutations in 116 unrelated isolates of Staphylococcus aureus and effects of mutations on ciprofloxacin MIC. Antimicrob. Agents Chemother. 42:1249-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmitz, F. J., C. von Eiff, M. Gondolf, A. C. Fluit, J. Verhoef, G. Peters, U. Hadding, H.-P. Heinz, and M. E. Jones. 1999. Staphylococcus aureus small colony variants: rate of selection and MIC values compared to wild-type strains, using ciprofloxacin, ofloxacin, levofloxacin, sparfloxacin, and moxifloxacin. Clin. Microbiol. Infect. 5:376-378. [DOI] [PubMed] [Google Scholar]
- 35.Wang, T., M. Tanaka, and K. Sato. 1998. Detection of grlA and gyrA mutations in 344 Staphylococcus aureus strains. Antimicrob. Agents Chemother. 42:236-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yano, Y., T. Oguma, H. Nagata, and S. Sasaki. 1998. Application of logistic growth model to pharmacodynamic analysis of in vitro bactericidal kinetics. J. Pharm. Sci. 87:1177-1183. [DOI] [PubMed] [Google Scholar]