

Abstract
Acute kidney injury (AKI) is a major risk factor for long-term adverse outcomes, including chronic kidney disease. In mouse models of AKI, maladaptive repair of the injured proximal tubule (PT) prevents complete tissue recovery. However, evidence for PT maladaptation and its etiological relationship with complications of AKI is lacking in humans. We performed single-nucleus RNA sequencing of 120,985 nuclei in kidneys from 17 participants with AKI and seven healthy controls from the Kidney Precision Medicine Project. Maladaptive PT cells, which exhibited transcriptomic features of dedifferentiation and enrichment in pro-inflammatory and profibrotic pathways, were present in participants with AKI of diverse etiologies. To develop plasma markers of PT maladaptation, we analyzed the plasma proteome in two independent cohorts of patients undergoing cardiac surgery and a cohort of marathon runners, linked it to the transcriptomic signatures associated with maladaptive PT, and identified nine proteins whose genes were specifically up- or down-regulated by maladaptive PT. After cardiac surgery, both cohorts of patients had increased transforming growth factor–β2 (TGFB2), collagen type XXIII-α1 (COL23A1), and X-linked neuroligin 4 (NLGN4X) and had decreased plasminogen (PLG), ectonucleotide pyrophosphatase/phosphodiesterase 6 (ENPP6), and protein C (PROC). Similar changes were observed in marathon runners with exercise-associated kidney injury. Postoperative changes in these markers were associated with AKI progression in adults after cardiac surgery and post-AKI kidney atrophy in mouse models of ischemia-reperfusion injury and toxic injury. Our results demonstrate the feasibility of a multiomics approach to discovering noninvasive markers and associating PT maladaptation with adverse clinical outcomes.
INTRODUCTION
Acute kidney injury (AKI) can affect 15 to 20% of hospitalized patients (1). Patients with AKI have a twofold increase in the risk of in-hospital death and a fourfold increase in the risk of developing chronic kidney disease (CKD) or experiencing CKD progression (2). Emerging evidence from mouse ischemia-reperfusion injury (IRI) models of AKI suggests that kidney ischemia results in substantial transcriptional changes in the proximal tubule (PT) (3). Although most PT cells under AKI stress can be fully repaired, a distinct subpopulation of PT cells enters a maladaptive, senescent phenotype that may fail to repair, leading to inflammation and fibrosis (3). This pathophysiological process suggests that PT maladaptation may mediate AKI progression, incomplete recovery, and subsequent development of CKD.
Despite the increasingly detailed knowledge derived from mouse AKI models, there may be important distinctions between humans and mice in renal tubular responses to injury (4). Histologically, frank necrosis is commonly seen in mouse kidneys after severe ischemia; however, this is uncommon in patients with AKI from acute tubular injury (5). The distinctions between mouse AKI models and AKI in hospitalized patients may be one of the reasons why promising therapies derived from preclinical animal models have failed to translate into therapeutic success in human trials (6–9). Unfortunately, until the launch of the Kidney Precision Medicine Project (KPMP) (10), large-scale tissue interrogation studies of human AKI have been lacking. In addition, the invasive nature of kidney biopsy and the lack of noninvasive assessment of PT maladaptation have created an opportunity to identify noninvasive markers of PT maladaptation and to associate such markers with adverse clinical endpoints of AKI. Therefore, in this study, we aimed to investigate transcriptional changes in PT cells in response to injury in hospitalized patients with AKI. In addition, we developed a multiomics approach that integrates the kidney transcriptome and plasma proteome to identify markers of PT maladaptation and determine their associations with severe AKI in patients undergoing cardiac surgery. We hypothesized that maladaptive PT cells that are enriched in pro-inflammatory and profibrotic pathways would be observed in hospitalized patients with AKI caused by diverse etiologies. We also postulated that in patients undergoing cardiac surgery, plasma proteins linked to the transcriptomic signatures of maladaptive PT cells would be associated with the development of severe AKI, as well as post-AKI kidney atrophy in mouse models of AKI.
RESULTS
Single-nucleus RNA sequencing reveals diverse PT cell phenotypes in human AKI
We used single-nucleus RNA sequencing (snRNA-seq) to profile 120,985 nuclei from kidney biopsy samples from 17 participants with AKI and seven healthy participants, including data from 13 participants (six with AKI and seven healthy references) that were published previously and 11 participants with AKI whose data were unpublished (data file S1) (10). The median number of unique molecular identifiers per nucleus was 2941 [interquartile range (IQR): 2069 to 3620], and the median number of genes detected per nucleus was 1720 (IQR: 891 to 2843) (data file S2). Among participants with AKI, 11 (64.7%) were male and 6 (35.3%) self-identified as Black. The baseline serum creatinine was 1.23 mg/dl, 13 (76.5%) participants had stage 3 AKI, and 16 (94.1%) participants received a kidney biopsy after their serum creatinine concentration had peaked, with a median time from AKI diagnosis to biopsy at 7 days (IQR: 4 to 10). Fourteen (82.4%) participants had an acute tubular injury from various causes, such as nonsteroidal anti-inflammatory drugs, antibiotics, sepsis, coronavirus disease 2019 (COVID-19), rhabdomyolysis, and oxalate nephropathy. The demographic and clinical characteristics of participants with AKI and healthy references are presented in detail in data file S1.
Using unsupervised clustering, we identified clusters of all major kidney, stromal, and immune cell types in participants with and without AKI (Fig. 1, A and B). We focused our analysis on PT cells, which included six subclusters, two of which (PT.S1S2 and PT.S3) were enriched in mature PT markers (SLC5A12, SLC22A6, SLC22A7, and SLC7A13) (Fig. 1, C to E, and data file S3). Gene set enrichment analysis (GSEA) of these two subclusters demonstrated enrichment in Gene Ontology terms involved in the physiological function of PT cells, such as organic anion transport and fatty acid metabolism (Fig. 1F and data file S4), consistent with their relatively healthy states. The proportion of cells belonging to the healthy PT subclusters was greater in samples from healthy references but diminished in samples from participants with AKI, indicating a shift in the phenotypes of PT cells during AKI (Fig. 1E).
Fig. 1. SnRNA-seq analysis of 17 participants with AKI and seven healthy references from the KPMP cohort identifies PT cells at different states of health.
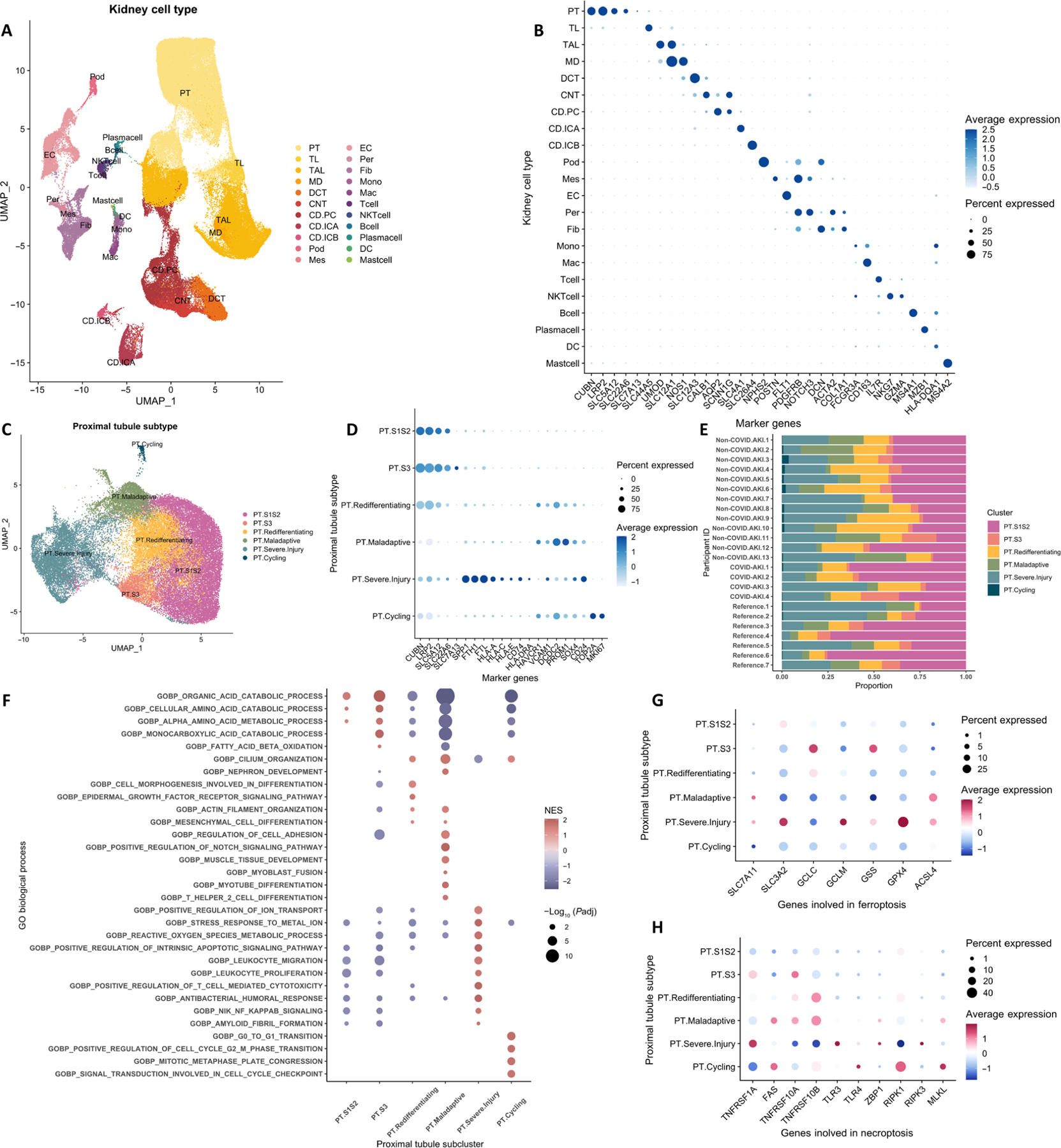
(A) Uniform manifold approximation and projection (UMAP) of 120,985 kidney epithelium, stroma, and immune cell nuclei. (B) Dot plot of canonical marker gene expression of major kidney cell types. (C) UMAP of PT subclusters. (D) Dot plot of marker gene expression of PT subclusters in the overall biopsy cohort. (E) Bar plot of PT subcluster composition in 17 participants with AKI and seven healthy references. (F to H) Dot plot displaying enriched Gene Ontology pathways (F), genes involved in ferroptosis pathways (G), and genes involved in necroptosis pathways (H) among PT subclusters. CD, collecting duct; CNT, connecting tubule; DC, dendritic cell; DCT, distal convoluted tubule; EC, endothelial cell; Fib, fibroblast; Glom, glomerulus; ICA, intercalated cell of collecting duct type A; ICB, intercalated cell of collecting duct type B; Mac, macrophage; MD, macula densa; Mes, mesangial cell; Mono, monocyte; PC, principal cell of collecting duct; Per, pericyte; Pod, podocyte; TAL, thick ascending limb of loop of Henle; TL: thin limb of loop of Henle.
Among the four subclusters of PT cells with decreased expression of mature PT markers (Fig. 1D and data file S3), we observed a subcluster of PT cells enriched in proliferation markers (TOP2A and MKI-67). The other three subclusters exhibited two distinct phenotypes based on gene expression patterns and pathway analysis. In severely injured PT cells, there was substantial up-regulation of markers of cellular stress (SPP1), iron hemostasis (FTH1 and FTL), injury [SRY-Box transcription factor 4 (SOX4) and CD24], MHC class I (HLA-A, HLA-C, and HLA-E), and MHC class II (CD74 and HLA-DRA). GSEA of this subcluster indicated prominent immune system activation and apoptosis associated with severe injury. The other two PT subclusters had lost their differentiated states and expressed the injury markers hepatitis A virus cellular receptor 1 (HAVCR1) and vascular cell adhesion molecule 1 (VCAM1). The expression of canonical PT markers (CUBN and LRP2) and solute transporters was markedly diminished in one cluster (PT.maladaptive), indicating its advanced dedifferentiated state. GSEA of these two subclusters of de-differentiated PT cells demonstrated enrichment in nephron regeneration and Notch signaling pathways (Fig. 1F and data file S4). The terminally dedifferentiated subcluster was additionally enriched in genes associated with immune activation and migration, extracellular matrix adhesion, and fibroblast activation. We refer to the terminally dedifferentiated subcluster hereafter as maladaptive PT because it exhibited similar marker gene expression (VCAM1, HAVCR1, and DCDC2) and pathway enrichment as the mouse “failed to repair” PT described in a previous study in mice (3). In addition to the pro-inflammatory and profibrotic signature, we observed the up-regulation of ACSL4 and down-regulation of GCLC, GSS, and GPX4 (Fig. 1G), indicating potential activation of the ferroptosis pathway and loss of the capacity to remove toxic polyunsaturated fatty acid–phospholipid hydroperoxides (11). There was also a concurrent increase in the expression of necroptosis markers, such as death receptors (FAS, TNFRSF10A, and TNFRSF10B) and the necroptotic executioner MLKL (Fig. 1H).
Despite various etiologies and severities of AKI and heterogeneity in the timing of kidney biopsy, we observed similar enrichment of severely injured and maladaptive PT cells as well as diminished proportions of healthy PT cells in participants with AKI, indicating potentially common responses to insult at the transcriptional level in PT cells (Fig. 1E). One participant (non-COVID AKI #8) had completely recovered kidney function at the time of kidney biopsy; however, there was still a considerable proportion of injured and maladaptive PT cells. Furthermore, injured and maladaptive PT cells were seen in healthy controls. Preclinical studies have shown that failed-to-repair, maladaptive PT cells constitute approximately 5% of PT cells in healthy mice (3, 12). The proportion becomes higher with increased age and comorbidities, such as diabetes. The high prevalence of severely injured and maladaptive PT in our healthy controls could be due to advanced aging, subclinical vascular disease, and hemodynamic fluctuation during anesthesia (data file S1).
Gene regulatory network analysis identifies distinct regulators activated in PT cells in healthy and diseased states
We next used pySCENIC to explore whether the gene regulatory structure (regulons) in PT cells in diseased states was deranged compared with the healthy state in participants with AKI. Similar to several mouse AKI studies, we observed enrichment of regulons [hepatic nuclear factor 1α (HNF1A), hepatic nuclear factor 4α (HNF4A), nuclear receptor subfamily 1 group H member 3 (NR1H3), retinoid X receptor α (RXRA), MAF, and MLXIPL] involved in promoting and maintaining PT cell differentiation, stabilizing mitochondrial structure, and maintaining mitochondrial lipid metabolism in PT cells in healthy states (Fig. 2A and data files S5 and S6) (13–17). RXRA is also known to protect tubules from oxidative stress and prevent the AKI-to-CKD transition (18). Regulon enrichment altered substantially as PT cells progressed toward the maladaptive state. Specifically, maladaptive PT cells were enriched in SOX4, a key regulator promoting nephrogenesis, and signal transducer and activator of transcription 5α (STAT5A), which drives abnormal tubular cell growth (19–21). Consistent with the profibrotic and pro-inflammatory signatures from GSEA, maladaptive PT up-regulated TEA domain transcription factor 2 (TEAD2), a regulator governing the epithelial-to-mesenchymal transition, and interferon regulatory factor 8 (IRF8), a transcription factor promoting inflammatory responses (22, 23). As a comparison, severely injured PT cells activated regulators of the response to an inflammatory milieu [CEBPB and high mobility group box 2 (HMGB2)] as well as regulators driving endoplasmic reticulum stress (HES1 and XBP1) and apoptosis in diseased states (HMGB2 and IRF6) (24–27). The expression of regulons across PT cells in healthy and diseased states was consistent with the cellular enrichment estimated from pySCENIC (Fig. 2B). Examining the 10 most-enriched regulons across each PT subcluster demonstrated distinctions in the regulatory networks across PT cells at different states of health (Fig. 2C).
Fig. 2. Gene regulatory network analysis of PT subclusters in 17 participants with AKI demonstrates distinct regulatory networks in PT cells at different states of health.
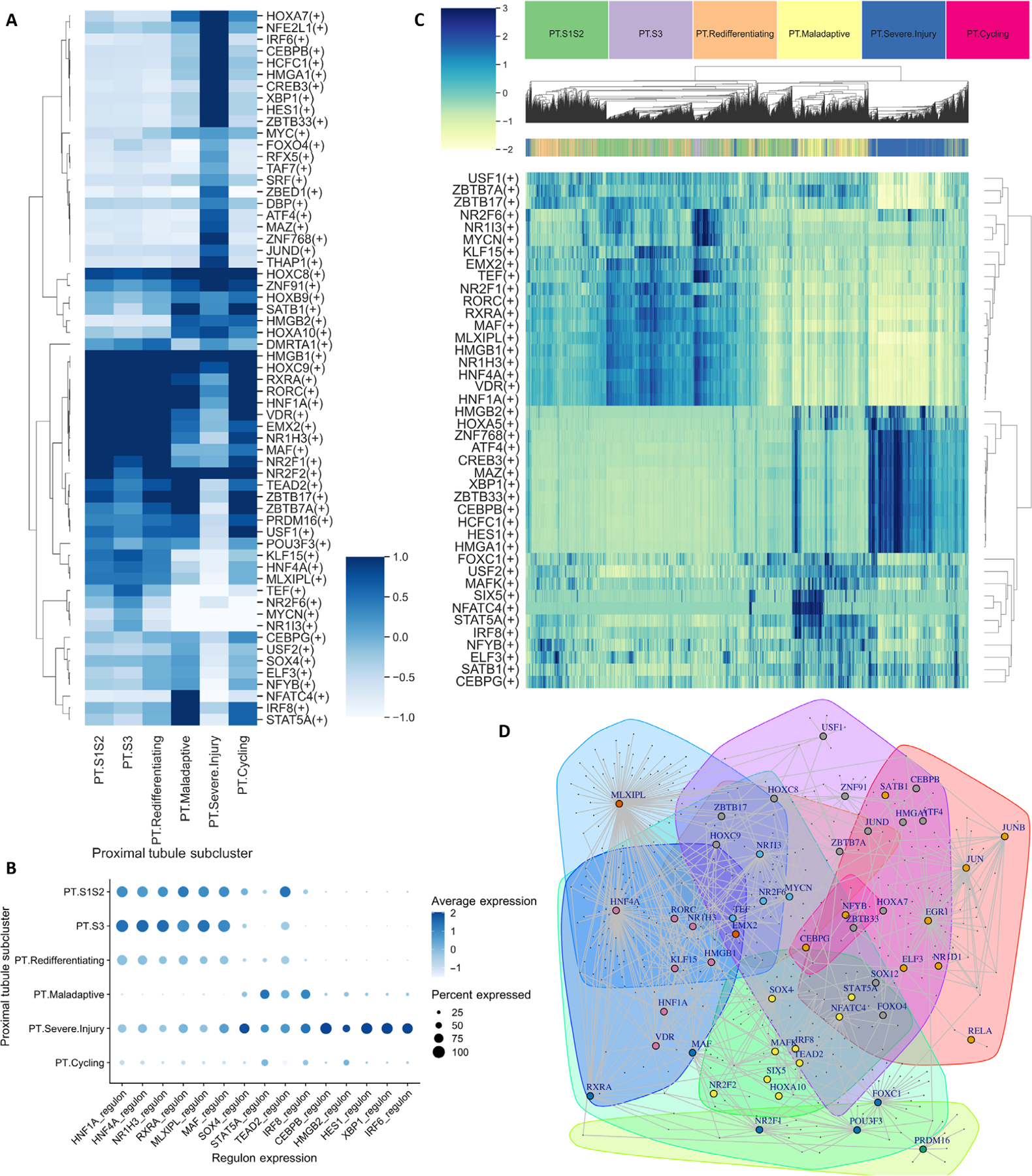
(A) Heatmap depicting average regulon enrichment in each PT subcluster. (B) Average expression of selected regulons enriched in each PT subcluster. (C) Heatmap demonstrating unsupervised clustering of PT subclusters by the top 10 regulons from each subcluster. (D) Louvain clustering of the top 10% of transcription factor–target gene pairs demonstrates clusters of transcription factors (nodes depicted by colors) forming coregulatory networks.
We next examined the top 10% of transcription factor–target gene pairs reconstructed by Epoch and applied Louvain unsupervised clustering for community detection of key regulatory modules (28). Each community represented a group of transcription factors coregulating a set of target genes (Fig. 2D). Consistent with previous findings, we observed coregulation of target genes by HNF4A, HNF1A, and NR1H3, all of which were enriched in healthy PT cells. In maladaptive PT cells, SOX4 and STAT5A formed a coregulation network with TEAD2 and IRF8 and governed the expression of a group of target genes, suggesting that inflammation and fibrosis are associated with the activation of aberrant nephron regeneration during the repair process.
A multiomic investigation identifies markers of PT maladaptation and PT cells in healthy states
To determine the associations between PT maladaptation and severe AKI, we next aimed to determine whether we could identify plasma proteins that are specific to maladaptive PT cells. Using differentially expressed genes from maladaptive PT cells in KPMP participants with AKI and proteins measured in 322 adults undergoing cardiac surgery in the translational investigation of biomarker end-point of AKI (TRIBE-AKI) cohort, we developed a workflow to identify candidate markers of PT maladaptation (Fig. 3, A and B). For genes up-regulated by PT maladaptation, we identified 293 genes that had plasma proteins measured using SomaScan aptamer assays. Among these 293 proteins, 122 increased after cardiac surgery, and 39 were higher in patients who developed stages 2 and 3 AKI (hereafter referred to as severe AKI). We further examined the gene expression of these 39 proteins in KPMP participants and identified four proteins that were relatively specific to maladaptive PT cells as candidate markers of PT maladaptation (Fig. 4A and fig. S1). We also identified 320 genes that were down-regulated as PT cells progressed from healthy to maladaptive states and measured the encoded proteins using SomaScan. Among these, 192 proteins were lower postoperatively, and 38 were lower in patients who developed severe AKI. We further examined the expression of the genes encoding these 38 proteins in KPMP participants and identified 5 that were specifically enriched in healthy PT cells and down-regulated in maladaptive PT cells (Fig. 4A and fig. S2). These proteins may be viewed as markers of PT cells in healthy states and inverse markers of PT maladaptation (a higher level indicates a lower degree of PT maladaptation). The expression patterns of these proteins were consistently seen in kidney biopsy tissues in three recently enrolled KPMP participants with AKI who were not included in the tissue interrogation and marker discovery phase, as well as in postmortem kidney tissues in an independent cohort of eight critically ill patients with AKI (figs. S3 and S4) (29).
Fig. 3. Integration of the kidney tissue transcriptome in participants with AKI and plasma proteome in patients undergoing cardiac surgery.
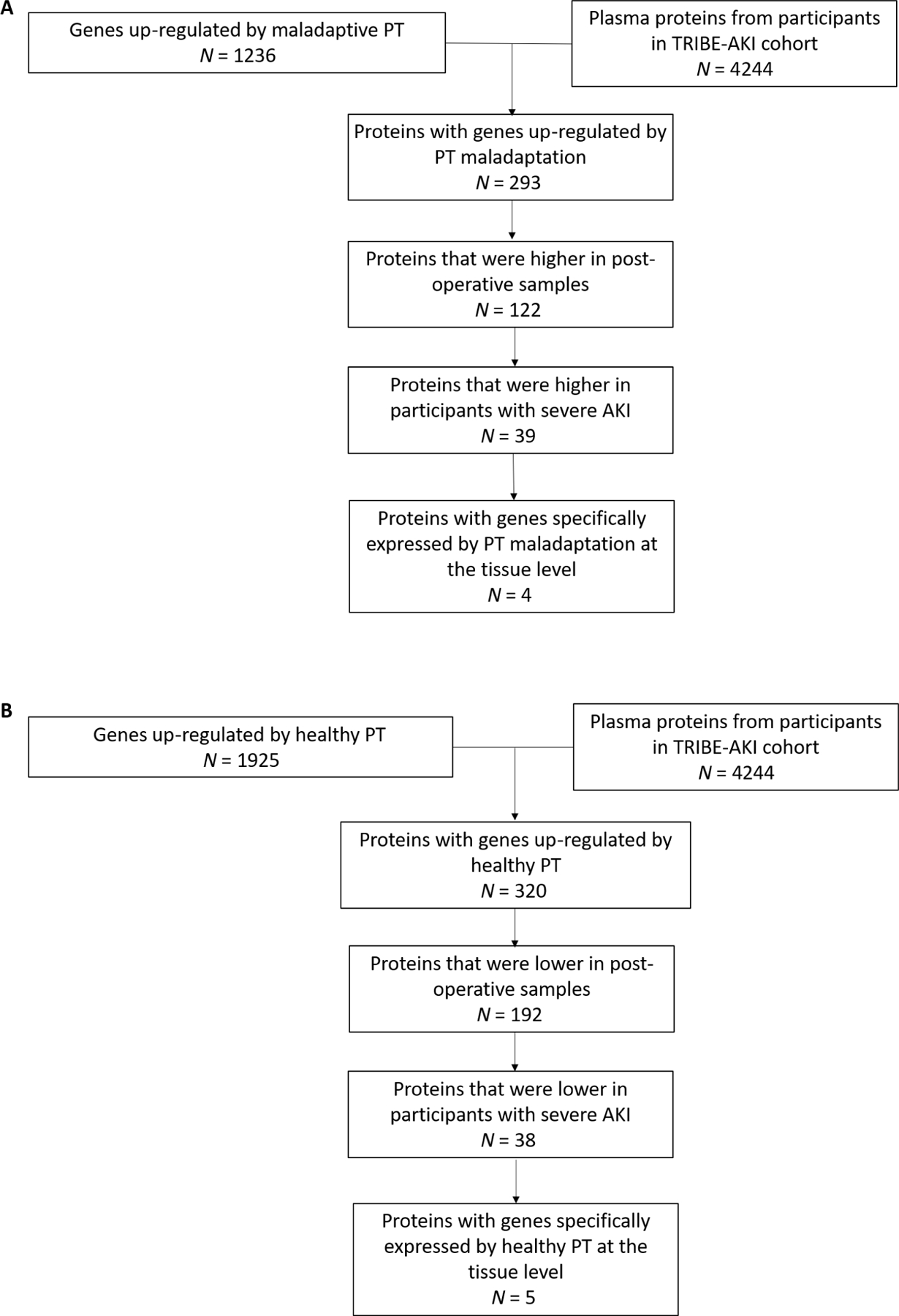
(A) Workflow for identifying markers of PT maladaptation. (B) Workflow for identifying markers of PT cells in healthy states.
Fig. 4. Kidney gene expression of markers of PT maladaptation and PT cells in healthy states in 17 participants with AKI and in mouse models of AKI.
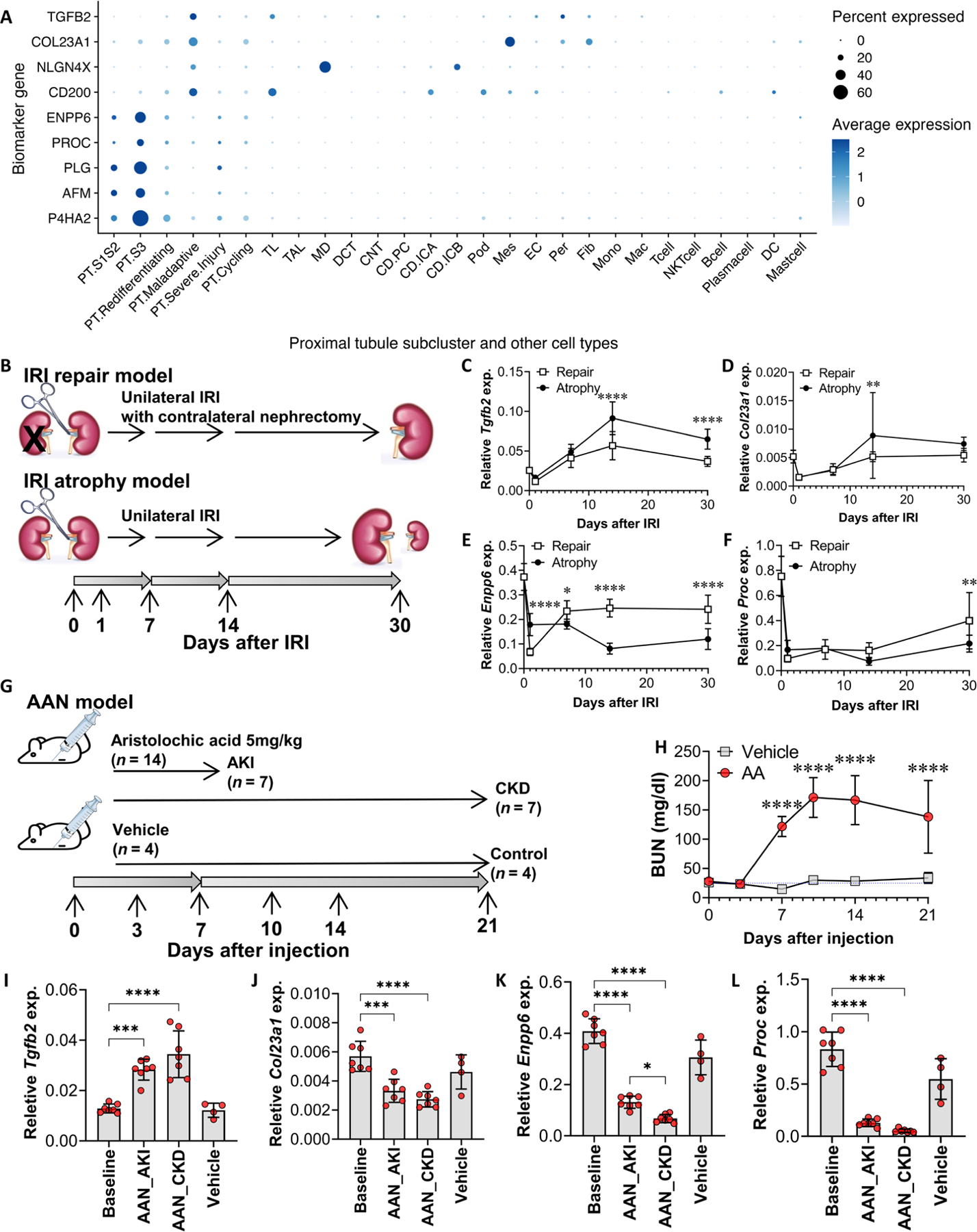
(A) Tissue gene expression of identified markers in 17 participants with AKI from the KPMP cohort at single-nucleus resolution. (B) Wild-type mice were subjected to unilateral IRI (atrophy model) or IRI with contralateral nephrectomy (repair model) (31). Quantitative RT-PCR analysis was performed on whole-kidney RNA harvested 0 (healthy control), 1, 7, 14, and 30 days after injury; n = 9 or 10 kidneys per time point per model. (C to F) Gene expression of Tgfb2 (C), Col23a1 (D), Enpp6 (E), and Proc (F) was compared across time course and between the atrophy versus repair model, using two-way ANOVA followed by Šidák’s posttests (*P < 0.05; **P < 0.01; ****P < 0.0001 at the indicated time points). (G and H) Wild-type mice were subjected to intraperitoneal injection of aristolochic acid (5 mg/kg ) (n = 14) or vehicle (n = 4). Blood samples were collected on days 0, 3, 7, 10, 14, and 21 for blood urea nitrogen (BUN) measurements. Quantitative RT-PCR analysis was performed on whole-kidney RNA harvested on days 0 (baseline), 7 (AKI phase, n = 7), and 21 (CKD phase, n = 7) after aristolochic acid injection, and day 21 (controls, n = 4) after vehicle injection. (I to L) Gene expression of Tgfb2 (I), Col23a1 (J), Enpp6 (K), and Proc (L) was compared using ANOVA followed by the Tukey’s test for subgroup comparison (*P < 0.05, ***P < 0.001, and ****P < 0.0001 comparing gene expression at AKI and CKD time points to baseline by Student’s t tests).
Table 1 displays the baseline characteristics of TRIBE-AKI adult participants with and without severe AKI. Among 322 participants, 47 developed severe AKI. The median age of all participants was 73 years, and 71.7% were male and 94.1% were white. The median baseline estimated glomerular filtration rate (eGFR) was 68.2 ml/min per 1.73 m2, and the median baseline urine albumin-creatinine ratio (ACR) was 15 mg/g. Pre- and postoperative protein concentrations and their fold changes are shown in table S1 and Table 2, respectively.
Table 1. Clinical characteristics in patients from the TRIBE-AKI adult cohort with and without AKI after cardiac surgery.
Continuous variables are presented as median (IQR), and categorical variables are presented as N (%).
| Characteristics | No AKI (N = 275) | AKI* (N = 47) | P value |
|---|---|---|---|
| Age (year) | 74 (66–78) | 72 (61.5–76) | 0.05 |
| Male sex | 195 (70.9%) | 36 (76.6%) | 0.42 |
| Race | 0.95 | ||
| White | 258 (93.8%) | 45 (95.7%) | |
| Black | 7 (2.5%) | 1 (2.1%) | |
| Others and unknown | 10 (3.6%) | 1 (2.1%) | |
| Baseline eGFR (ml/min/1.73 m2) | 68.6 (54.8–83.1) | 66 (47.6–79.7) | 0.13 |
| Baseline ACR (mg/g) | 14 (7–58) | 21 (9–70) | 0.17 |
| Hypertension | 214 (77.8%) | 39 (83.0%) | 0.54 |
| Diabetes mellitus | 105 (38.2%) | 23 (48.9%) | 0.33 |
| Congestive heart failure | 58 (21.1%) | 17 (36.2%) | 0.06 |
| Myocardial infarction | 67 (25.2%) | 14 (29.8%) | 0.51 |
AKI is defined as severe (KDIGO stages 2 and 3) AKI.
Table 2. Protein fold change in the TRIBE-AKI adult cohort and two independent validation cohorts.
All proteins are presented by names of aptamers used in the SomaScan assay. Fold change is based on the comparison of postoperative versus preoperative concentrations for the TRIBE-A KI adult cohort and pediatric cardiac surgery cohort and postrace versus prerace concentrations in the marathon cohort using paired Wilcoxon tests. P4HA2, prolyl 4-hydroxylase.
| TRIBE-AKI adult (N = 322) | Pediatric cardiac surgery (N = 68) | Marathon (N = 39) | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| Protein name | Fold change | P value | Fold change | P value | Fold change | P value |
| NLGN4X.5357.60 | 1.55 | 1.68 × 10−49 | 1.51 | 4.58 × 10−11 | 1.13 | 1.01 × 10−07 |
| COL23A1.4543.65 | 1.51 | 1.16 × 10−32 | 1.45 | 5.35 × 10−08 | 1.08 | 6.00 × 10−01 |
| TGFB2.4156.74 | 1.73 | 1.66 × 10−47 | 1.44 | 8.68 × 10−09 | 1.34 | 5.90 × 10−08 |
| CD200.5112.73 | 1.2 | 3.54 × 10−37 | 1.09 | 6.67 × 10−06 | 1.22 | 1.09 × 10−11 |
| ENPP6.15579.26 | 0.81 | 9.15 × 10−53 | 0.78 | 6.56 × 10−11 | 1.09 | 3.01 × 10−05 |
| PLG.3710.49 | 0.77 | 3.14 × 10−49 | 0.88 | 1.83 × 10−06 | 0.93 | 3.02 × 10−04 |
| PROC.2961.1 | 0.97 | 1.3 × 10−04 | 0.96 | 1.59 × 10−02 | 0.87 | 1.35 × 10−09 |
| P4HA2.11348.132 | 0.95 | 2.19 × 10−12 | 0.95 | 2.09 × 10−04 | 0.93 | 1.12 × 10−03 |
| AFM.4763.31 | 0.97 | 1.31 × 10−03 | 1.08 | 1.11 × 10−07 | 0.98 | 1.20 × 10−04 |
Using multivariable logistic regression with sequential adjustment of covariates, we observed strong and positive associations between postoperative maladaptation markers and the development of severe AKI. X-linked neuroligin 4 (NLGN4X), collagen type XXIII-α1 (COL23A1), and transforming growth factor–β2 (TGFB2) were significantly associated with increased odds of severe AKI in all models (P < 0.05 for all models; Table 3), including model 5, which adjusted for preoperative marker concentrations. In general, the increase of plasma markers in kidney disease may be caused by the decrease in GFR and thus may lead to spurious associations (reverse causation). However, the preoperative concentrations of these proteins were not correlated with preoperative eGFR, suggesting a lack of reverse causation (table S2). In a subset of 54 TRIBE participants who had urine samples collected before and after surgery for proteomic profiling, there were significant increases in urine NLGN4X and COL23A1 postoperatively (P < 0.001 for both markers) but no change in TGFB2 excretion (table S3).
Table 3. Associations between postoperative markers of PT maladaptation, PT cells in healthy states, and severe AKI after cardiac surgery in patients from the TRIBE-AKI adult cohort.
All proteins are presented by names of aptamers used in the SomaScan assay, and measurements were log2-transformed so that the odds ratio represents an increase in the odds of AKI per doubling of protein concentrations.
| Odds ratio (OR, 95% CI) | Model 1* | Model 2 | Model 3 | Model 4 | Model 5 |
|---|---|---|---|---|---|
| NLGN4X.5357.60 | 1.87 (1.09–3.48) | 2.09 (1.18–4.07) | 2.48 (1.35–5.07) | 2.65 (1.37–6.1) | 6.29 (2.04–20.95) |
| COL 23A1.4543.65 | 2.04 (1.01–4.24) | 2.23 (1.08–4.74) | 2.85 (1.32–6.45) | 2.35 (1.08–5.38) | 2.54 (1.13–6.01) |
| TGFB2.4156.74 | 2.27 (1.14–4.66) | 2.53 (1.24–5.34) | 3.25 (1.5–7.4) | 2.4 (1.07–5.61) | 2.35 (1.03–5.63) |
| CD200.5112.73 | 1.94 (0.78–5) | 2.18 (0.85–5.62) | 2.42 (0.92–6.56) | 1.63 (0.54–4.46) | 3.2 (0.77–13.28) |
| ENPP6.15579.26 | 0.29 (0.05–1.09) | 0.18 (0.03–0.89) | 0.21 (0.03–0.91) | 0.29 (0.04–1.09) | 0.04 (0.003–0.39) |
| PLG.3710.49 | 0.13 (0.04–0.44) | 0.09 (0.02–0.31) | 0.12 (0.03–0.44) | 0.14 (0.03–0.54) | 0.07 (0.01–0.44) |
| PROC 2961.1 | 0.23 (0.07–0.69) | 0.15 (0.04–0.49) | 0.24 (0.06–0.88) | 0.23 (0.06–0.89) | 0.17 (0.03–0.83) |
| P4HA2.11348.132 | 0.14 (0.03–0.67) | 0.11 (0.02–0.57) | 0.18 (0.03–0.87) | 0.17 (0.03–0.84) | 0.25 (0.02–2.35) |
| AFM.4763.31 | 0.43 (0.19–1.01) | 0.42 (0.18–0.97) | 0.61 (0.25–1.52) | 0.6 (0.23–1.64) | 0.47 (0.06–3.59) |
Covariates were sequentially added to the regression models: Model 1 comprised postoperative markers alone; model 2 additionally adjusted for age, sex, and race; model 3 additionally adjusted for hypertension, diabetes mellitus, congestive heart failure, myocardial infarction, and baseline eGFR; model 4 additionally adjusted for baseline ACR ; model 5 additionally adjusted for preoperative marker concentrations.
Regarding the five markers of PT cells in healthy states, we observed that postoperative ectonucleotide pyrophosphatase/phosphodiesterase 6 (ENPP6), protein C (PROC), and plasminogen (PLG) were inversely associated with the development of severe AKI (a lower plasma concentration indicated a higher risk of severe AKI; Table 3). Three plasma markers of PT cells in healthy states could also be measured in the urine by SomaScan aptamer assays. Excretion of both PLG and afamin (AFM) significantly increased after cardiac surgery (P < 0.01 for both markers), and excretion of PROC was significantly higher (P < 0.01) after being indexed to urine creatinine to account for urine dilution (table S3), which may be due to shedding from healthy PT cells after injury. When added to known markers of kidney diseases, markers of PT maladaptation and PT cells in healthy states could further enhance the predictive performance for AKI (table S4).
We validated these plasma proteins as indicators of PT maladaptation and PT cells in healthy states in two independent cohorts. The pediatric cardiac surgery cohort comprised 68 participants undergoing surgery for the repair of congenital heart disease, with a median age of 41 months (IQR: 6 to 84), including 37 girls (54.4%). We observed significant increases in plasma TGFB2, COL23A1, and NLGN4X (proteins of PT maladaptation) and significant decreases in plasma ENPP6, PLG, and PROC (proteins of PT cells in healthy states) after cardiac surgery, similar to the findings in their adult counterparts (P < 0.05 for all markers; Table 2 and table S1). Next, we compared these proteins in 39 participants at high risk of exercise-associated tubular injury before and immediately after running a marathon (30). The median age of the marathon runners was 42 (IQR: 33 to 51) years, 21 (53.8%) were women, 2 (5.1%) participants had hypertension, and 1 (2.6%) had diabetes. We observed significant increases in plasma TGFB2 and NLGN4X and significant decreases in plasma PLG and PROC after the marathon, consistent with our findings from the two cardiac surgery cohorts (P < 0.0001 for these four markers; Table 2 and table S1).
Reverse translational investigation validates markers of PT maladaptation and PT cells in healthy states in mouse models of ischemic and toxic AKI
To further validate the associations between these proteins and the maladaptive response to AKI of different causes, we conducted a reverse translational investigation and compared the mRNA expression of these markers (Col23a1, Tgfb2, Enpp6, and Proc) in two different mouse models of IRI followed by either repair or atrophy, as well as in a mouse model of toxic aristolochic acid nephropathy (AAN) presenting as AKI-to-CKD transition (31). Plg and Nlgn4 were excluded from this analysis because their expression was very low and undetectable, respectively, based on publicly available single-cell and snRNA-seq datasets from injured mouse kidneys (fig. S5) (3, 32).
In the setting of mouse kidney IRI, reverse transcription polymerase chain reaction (RT-PCR) of whole-kidney mRNA revealed that Tgfb2 had higher expression in the setting of progressive kidney fibrosis and atrophy (P < 0.0001 for both time and model factors using two-way ANOVA; Fig. 4, B to F), whereas the expression of Enpp6 and Proc (markers expressed by PT cells in healthy states) increased in the setting of kidney repair [P < 0.0001 (time factor) and P = 0.0916 (model factor) for Proc, P < 0.0001 for both time and model factors for Enpp6; Fig. 4, B to F]. The continuous increase in gene expression of Tgfb2 and the sustained decreases in Enpp6 and Proc during progressive kidney fibrosis indicated an unresolved, maladaptive response in the kidney, beyond the immediate repair phase of IRI. Moreover, Tgfb2 had strong, positive correlations with known fibrosis markers (Col1a1, Col3a1, Fn1, Pdgfb, and Acta2), and Enpp6 and Proc had moderate to strong negative correlations with fibrosis (fig. S6 and table S5). In the AAN model, we also observed a progressive increase in Tgfb2 expression, as well as continuing decreases in Enpp6 and Proc expression as mice progressed from AKI to CKD (Fig. 4, G to L). These RT-PCR results were consistent with results from a different model of repeated AKI leading to CKD from aristolochic acid (33) and results from our IRI models (fig. S7). These results in mouse models of AKI were largely similar to findings from human AKI kidney biopsies and further supported the association of these markers with maladaptive repair after diverse causes of injury to the kidney. Col23a1 expression in both the IRI and AAN models was low. It did not differ between the repair and atrophy models at 30 days after IRI and decreased from baseline in the AAN model.
In summary, our transcriptomic investigation demonstrated that maladaptive repair in PT cells is a shared response to injury in hospitalized patients with AKI of diverse causes. Furthermore, we identified plasma NLGN4X, COL23A1, and TGFB2 as markers that increased in the setting of PT maladaptation after cardiac surgery, whereas plasma ENPP6, PLG, and PROC serve as markers of PT cells in healthy states that diminish in the setting of maladaptive repair after injury.
DISCUSSION
During the process of AKI progression, incomplete recovery, and AKI-to-CKD transition in mice, some injured PT cells from AKI undergo dedifferentiation but then fail to redifferentiate and recover normal function. These PT cells exhibit a maladaptive profile, lose their physiological function, enter a senescent cell-cycle arrest phase, activate programmed cell death pathways, and eventually form atrophic tubules (32, 34). In addition, maladaptive PT cells persistently produce and secrete profibrotic factors and recruit and activate the transition of pericytes and fibroblasts into myofibroblasts, leading to the production of matrix material and fibrosis (35). Our study demonstrated that PT maladaptation is similarly present in human AKI. In these maladaptive PT cells, we further identified that the activation of regulators involved in maladaptive tubular cell growth, such as SOX4 and STAT5A, was accompanied by a close coregulation network with pro-inflammatory and profibrotic mediators (36). Whether therapeutic intervention halting this maladaptive repair process can attenuate the risk of severe AKI and the AKI-to-CKD transition requires further investigation.
PT maladaptation was widely present in participants with AKI of diverse etiologies, suggesting a shared response to injury in tubular cells. This is consistent with observations from mouse models of ischemic and toxic AKI, in which injured PT cells enter the senescent and maladaptive phenotype and mediate interstitial fibrosis (33, 37). In addition, a large proportion of maladaptive PT cells were observed in the participants who had completely recovered kidney function at the time of kidney biopsy. In mice, serum creatinine may return to near baseline within 7 days after kidney injury; however, the maladaptive process may persist for weeks (37, 38). Taking these results together, therapies may need to be initiated and maintained for an extended period, perhaps months after AKI, to prevent the AKI-to-CKD transition.
Kidney tissue interrogation allows the identification of PT maladaptation for in-depth mechanistic investigation. However, the invasive nature of the kidney biopsy procedure makes it challenging to apply this tissue interrogation to large cohorts of patients. This highlights the importance of developing sensitive and noninvasive markers to measure this maladaptive repair process occurring in the kidney tissue. These markers may characterize the prevalence of PT maladaptation and establish its etiological associations with clinical complications of AKI in large cohorts of patients. They may also serve as potential pharmacodynamic endpoints for early investigation of targeted therapeutics in preventing complications of AKI.
With this motivation, we developed a workflow to integrate findings from snRNA-seq with the plasma proteome in patients at high risk of AKI from cardiac surgery. The discovery of markers by integrating these two different cohorts, instead of using only the kidney biopsy cohort, is primarily due to the small sample size of patients with available transcriptomic data. Our snRNA-seq analysis demonstrates that the PT maladaptation phenotype may be shared across different patient populations with diverse causes of AKI, further suggesting the feasibility of this integrative approach. We identified multiple markers of PT maladaptation associated with AKI progression. Tubular epithelial expression of TGF-β plays a critical role in developing interstitial fibrosis after AKI (39). Although the TGFB2 isoform is not well described in AKI, it is implicated in the epithelial-to-mesenchymal transition in cancer cells, potentially by interacting with the urokinase PLG activator (40). NLGN4X and COL23A1, although not yet described in kidney disease, may be involved in cellular junction and extracellular matrix formation. These proteins may potentially arise from post-injury fibrosis (41, 42). The associations between AKI and these markers of cell adhesion, migration, and extracellular matrix material are consistent with the profibrotic profile of PT maladaptation. In addition, we identified multiple markers of the successful repair of injured PT cells. PLG and PROC have been shown to alleviate fibrosis and inflammation after renal IRI (43, 44). ENPP6 is involved in the extracellular degradation of glycerophosphocholine to provide choline intracellularly but has not yet been reported in kidney disease (45). These results suggest that the markers found in our study may not only have prognostic value but may also provide potential mechanistic insights into the maladaptive repair of PT cells in the progression of AKI.
Although we could not validate the correlation between these plasma proteins and gene expression in our kidney biopsy cohort because of its limited sample size, post–cardiac surgery changes in these markers were highly consistent between the two cohorts of adult and pediatric patients undergoing cardiac surgery. The discrepancies in two markers, COL23A1 and ENPP6, between marathon runners and patients undergoing cardiac surgery may be due to differences in the kidney’s response to these two distinct insults. Although marathons may be associated with volume depletion, IRI, and heat stress, cardiac surgery may additionally involve an enhanced inflammatory milieu from cardiopulmonary bypass (46, 47). In addition, upon validating markers of PT maladaptation and PT cells in healthy states in mouse IRI models with different repair capacities as well as a model of toxic kidney injury, we found that three of the four markers were associated with maladaptive changes, interstitial fibrosis, and kidney atrophy. The consistency of the gene expression patterns in ischemic and toxic AKI models further supports the stereotypical maladaptive response observed in human AKI of diverse etiologies. The low expression of Col23a1 and its lack of correlation with fibrosis, and the lack of Nlgn4 and Plg expression in mouse kidneys, could be due to transcriptomic differences between humans and mice and highlight the importance and benefit of direct interrogation of human kidney tissues.
Our snRNA-seq analysis demonstrated that maladaptive repair of PT cells may be a stereotypical response to injury in participants with diverse etiologies and severities of AKI. In addition, a link between maladaptive tissue regeneration and inflammation may be explored further in mechanistic studies for potential therapeutic agent development. By integrating the kidney tissue transcriptome and plasma proteome, we found multiple proteins that reflect PT cells’ maladaptive and healthy states, which can be measured noninvasively to establish the etiological association between PT maladaptation and adverse outcomes of AKI in large cohorts of patients. Future studies can determine whether these plasma proteins can serve as pharmacodynamic endpoints in early-stage clinical trials investigating drugs targeting PT maladaptation. Our multiomics marker development pipeline may also be adopted in research aiming to establish associations between diseased cell states and clinical outcomes in other kidney diseases.
We recognize several limitations of this study. We could not assess gene expression changes along the temporal trajectory of PT maladaptation. This is due to the cross-sectional nature of kidney biopsy procedures after AKI diagnosis and interindividual variations of PT cells with diverse disease pathophysiology and clinical courses, which makes it challenging to use trajectory analysis tools. Proteomic and snRNA-seq analysis was performed in two different cohorts, preventing any direct correlation between plasma proteins with tissue gene expression. Proteomic profiling in the marker discovery cohort was performed using the first postoperative plasma samples. Proteins that would be released in the plasma later in the course of injury and maladaptation and proteins that are more likely to be excreted in the urine, such as kidney injury molecule-1, may not be adequately captured and may lead to false-negative results (48). Future studies may investigate whether additional markers can be identified using plasma and urine samples obtained later in the course of AKI. Although we validated the plasma proteomic findings in two independent cohorts of patients at risk of kidney injury, the small sample size of the validation cohorts and the relatively lower risk for clinical AKI events limit our ability to determine associations between these proteins and clinical AKI in the validation cohorts. Plasma proteins of PT maladaptation can also originate from tissues other than the kidney. However, the correlation between kidney expression of these markers and fibrosis in mouse models of ischemic and toxic AKI was reassuring. Last, protein concentration quantified by SomaScan may not align perfectly with those measured using targeted immunoassays, necessitating further validation for implementation in research and clinical practice.
Our transcriptomic investigation in human and preclinical experiments demonstrated that maladaptive repair in PT cells is a shared response to injury from various causes. Furthermore, we identified three plasma proteins—NLGN4X, COL23A1, and TGFB2—that can serve as markers of PT maladaptation, whereas plasma proteins ENPP6, PLG, and PROC signify healthy states of PT cells. These markers may improve the prognostication of complications after AKI and potentially serve as pharmacodynamic endpoints to facilitate drug development in AKI. Our discovery approach combining multiomics methods can also assist with etiological associations between healthy or altered cell states and clinical outcomes in other kidney diseases.
MATERIALS AND METHODS
Study design
This study aimed to elucidate the transcriptomic landscape of PT cells in human AKI, identify plasma proteomic signatures linked to the maladaptive process, and determine their associations with progression to severe AKI after cardiac surgery. We performed snRNA-seq analysis of hospitalized participants with AKI in the KPMP study. KPMP is an ongoing, prospective, observational cohort of participants with AKI and CKD receiving kidney biopsies sponsored by the National Institute of Diabetes, Digestive and Kidney Diseases (10). Participants with AKI were recruited if they developed AKI during hospitalization and had baseline eGFR > 45 ml/min per 1.73 m2. AKI was defined as an increase in serum creatinine by 50% from their baseline, defined as the nearest outpatient serum creatinine concentration 7 to 365 days before hospitalization. Kidney biopsies were obtained from 13 hospitalized participants with AKI who consented to research biopsies at four recruitment sites across the United States: Johns Hopkins Hospital, Yale–New Haven Hospital, University of Pittsburgh Medical Center, and Columbia University Medical Center. Additional kidney biopsies were obtained from four hospitalized participants with COVID-19–associated AKI at Johns Hopkins Hospital. Healthy reference tissues were obtained from nontumor regions of kidney tissue after tumor nephrectomy in three participants and from intraoperative kidney biopsy in four participants undergoing urological procedures for nephrolithiasis removal in the Human BioMolecular Atlas Program (HuBMAP) consortium at Washington University at St. Louis. Tissue processing and single-nucleus isolation were performed at Washington University in St. Louis according to the published protocol of the KPMP consortium (10). SnRNA-seq data from 6 of 17 participants with AKI were published in a kidney atlas study by the KPMP consortium (10), whereas data from the other 11 participants with AKI are new.
Our next goals were to link the maladaptive response in kidney tissue to the plasma proteome to identify PT maladaptation markers and to determine their associations with AKI. Although it may be more biologically plausible to discover markers in the urine, which the tubular cells directly face, the coverage of proteomic profiling may be low when the overall urine protein concentration is low. In addition, a recent study demonstrated that many markers of kidney injury, initially found in the urine, can be measured in the blood and may have better prognostic performance (49). We used samples from the TRIBE-AKI adult study cohort, a multicenter prospective cohort study of adults who underwent cardiac surgery in North America from July 2007 to December 2010 (48). Patients were recruited before cardiac surgery if they were at high risk of developing postoperative AKI and were followed from enrollment until death or development of end-stage kidney disease. Although the TRIBE-AKI study was among the first few pioneering studies exploring the prognostic values of kidney disease markers in AKI, the proteomic data are newly generated, and this is the first investigation of proteomic data with AKI in this cohort.
We validated the proteomic findings in two independent cohorts. The first validation cohort was the pediatric cardiac surgery study cohort, a multicenter prospective cohort of children who underwent cardiac surgery for the repair of congenital heart disease in North America from 2007 to 2010. Children were excluded if they had a history of kidney transplantation or dialysis (50). The second validation cohort comprised marathon runners participating in the 2015 Hartford Marathon (CT, US) (30). Adult runners with normal body mass indexes (18.5 to 24.9 kg/m2) and at least 3 years of running experience and regular training were included. Runners were excluded if they had any history of kidney disease or participated in another marathon within 4 weeks before the 2015 Hartford Marathon. Given the observational nature of our cohort studies, randomization, blinding, and sample size calculation were not performed.
We validated the gene expression of markers of maladaptive PT and PT cells in healthy states in three KPMP participants with AKI who were recently enrolled and were not included in the discovery phase, as well as in kidney autopsy tissues from a cohort of critically ill patients with AKI published by Hinze et al. (29). We further validated the gene expression of identified markers in two mouse models with different repair capacities after IRI (repair and atrophy models) (31) and in a mouse model of AAN presenting as AKI-to-CKD transition. Samples from all human participant research studies were collected after informed consent and with the approval of the local IRBs, and the Yale University Animal Care and Use Committee approved all animal protocols.
Human snRNA-seq data library preparation, preprocessing, and analysis
We used the Cell Ranger 7.0 pipeline to align snRNA-seq FASTQ files to the human hg38 reference genome. We used CellBender to remove ambient RNA contamination and DoubletDetection to remove doublets (51, 52), then excluded low-quality nuclei with fewer than 200 or more than 7500 genes detected (10). Details of the snRNA-seq processing procedure are described in Supplementary Materials and Methods (53).
We focused our analysis on PT subclusters. We obtained differentially expressed genes for each PT subcluster by comparing gene expression in that subcluster with that of other subclusters using the Wilcoxon test and accounting for multiple comparisons using the false discovery rate. We included all differentially expressed genes and performed GSEA using the FGSEA package (54) (preprint). We defined PT maladaptation as PT cells with near-complete dedifferentiation and enrichment of pro-inflammatory and profibrotic genes and pathways.
Gene regulatory network analysis
We used pySCENIC for gene regulatory network analysis for PT cells after retaining 4247 highly variable genes (minimal dispersion 0.4 using Scanpy) (55, 56). We generated coexpression networks via Epoch and cis-regulatory networks of PT nuclei with the human hg38 reference genome (28). We obtained the gene-motif ranking of 10 kb around the transcription start site (57). We plotted the top 10% of transcription factor–target gene pairs using igraph and performed community detection using Louvain clustering. All snRNA-seq analyses were performed in R 4.1.2 and Python 3.7.
Discovery and validation of plasma markers of PT maladaptation and PT cells in healthy states
Sample collection, processing, and proteomic measurements in the three patient cohorts are described in detail in Supplementary Materials and Methods (30, 48, 50). The primary outcome of our analysis is severe AKI [Kidney Disease Improving Global Outcome (KDIGO) stages 2 and 3] in adult patients undergoing cardiac surgery in the TRIBE-AKI cohort. We define stage 2 AKI as a twofold increase in serum creatinine from baseline to the peak postoperative value. We define stage 3 AKI as a threefold increase in serum creatinine from baseline to the peak post-operative value or requiring kidney replacement therapy.
Validation of markers of PT maladaptation and PT cells in healthy states in kidney tissues from individuals with AKI
The prospective KPMP cohort allowed us to internally validate the expression of identified markers in three recently enrolled participants whose snRNA-seq data were not included in the discovery phase. In these three participants, snRNA-seq libraries were prepared using the approach described earlier. We used the metadata from 17 participants with AKI from the discovery phase as a reference and predicted PT cell subtypes using pySingleCellNet, a random forest–based classifier (58). In addition, we downloaded an snRNA-seq library of postmortem kidney tissues from an independent cohort of eight critically ill patients with AKI published by Hinze et al. (GSE210622) (29) and performed clustering of PT cells using the approach discussed by these investigators. Gene expression patterns of these markers were explored in the internal and external validation cohorts.
Animal surgery and experimental protocol
The Yale University Animal Care and Use Committee approved all animal protocols. Male C57BL/6 (Envigo, Indianapolis, IN) wild-type mice (aged 9 to 11 weeks) were used in this work.
To establish the unilateral IRI (atrophy) model, warm renal ischemia was induced using a nontraumatic microaneurysm clip (FST Micro Clamps, Foster City, CA) on the left renal pedicle for 27 min, leaving the right kidney intact. To establish the unilateral IRI with the contralateral nephrectomy (repair) model, the right kidney was surgically removed at the time of left kidney ischemia, as we have previously described (31). Mice were euthanized on days 1, 7, 14, and 30 after surgery (n = 9 or 10 per time point for each model). Control mice were euthanized and represented as day 0 (n = 9).
To establish a mouse model of AAN, 14 mice were treated with a single 5 mg/kg body weight dose of aristolochic acid (Sigma-Aldrich) intraperitoneally, and four mice were treated with a vehicle. After injection, blood was collected on days 3, 7, 10, 14, and 21. Seven AAN-treated mice were euthanized, and kidneys were harvested after injection on days 7 and 21. Mice treated with the vehicle were used as experimental controls and were offered on day 21 after injection. Blood urea nitrogen was measured using Stanbio Diagnostic Set (Thermo Fisher Scientific). In addition, we downloaded a publicly available snRNA-seq dataset of mouse AAN kidneys (33) and compared gene expression of markers between the kidney fibrosis phase (day 28) and baseline.
Quantitative PCR analysis
Whole-kidney RNA was extracted with an RNeasy Mini kit (Qiagen, Germantown, MD) and reverse-transcribed using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories). Gene expression analysis was determined by quantitative real-time PCR using an iCycler iQ (Bio-Rad Laboratories) and normalized to hypoxanthine-guanine phosphoribosyltransferase (Hprt). Primers used include mouse Col23a1 (forward: GGCATAAGTGATCCTCAGACATAA and reverse: AGTTGGCGCATCCCATAAA), Enpp6 (forward: GGAACACATGACCGTGTATGA and reverse: TCTCTCGACTCT CTGCTATGAA), Tgfb2 (forward: AGAGGGATCTTGGATGGAAATG and reverse: TGAGGACTTTGGTGTGTTGAG), Proc (forward: CCTCAAACGAGACACAGACAGACTTAG and reverse: GATCATACTCACCAAGCCTCAC), and Hprt (forward: CAGTACAGCCCCAAAATGGT and reverse: CAAGGGCATATCCAACAACA). Data were expressed using the comparative threshld cycle (ΔCT) method and mRNA ratios were given by 2-ΔCT.
Statistical analysis
We reported descriptive characteristics of 322 TRIBE-AKI participants with and without the primary outcome using medians (IQR) and proportions. We compared descriptive characteristics using Wilcoxon tests and chi-square tests.
To identify markers of PT maladaptation and PT cells in healthy states, we first selected differentially expressed genes that have an average log2 fold change > 0.25 in the maladaptive PT cell sub-cluster and subclusters of PT cells in healthy states and selected corresponding proteins measured by SomaScan. We used pairwise Wilcoxon tests to compare the median concentration of plasma proteins in postoperative versus preoperative samples and used Wilcoxon tests to compare the median concentration of proteins in patients who did versus did not develop severe AKI in TRIBE-AKI participants. We additionally determined correlations between preoperative maladaptation markers and baseline eGFR using Spearman correlation.
To determine associations between markers of PT maladaptation and markers of PT cells in healthy states and the primary outcome in TRIBE-AKI adult participants, we used logistic regression with sequential adjustment of covariates. Model 1 was the univariable model, including postoperative markers only. Model 2 adjusted for age, sex, and race. Model 3 additionally adjusted for baseline eGFR, hypertension, diabetes mellitus, myocardial infarction, and heart failure. Model 4 additionally adjusted for baseline urine ACR. Model 5 additionally adjusted for preoperative marker concentrations.
We next determined whether markers of PT maladaptation and PT cells in healthy states can further improve the prediction of severe AKI beyond other kidney disease markers (kidney injury molecule-1, neutrophil gelatinase–associated lipocalin, and soluble urokinase plasminogen activator receptor) and other clinical variables (age, sex, Black race, preoperative eGFR, ACR, hypertension, diabetes, myocardial infarction, and heart failure). We compared the area under the curve of the logistic regression models using 1000 bootstrap samples. We compared the protein concentrations in the first postoperative urine samples with the preoperative samples for the subset of 54 TRIBE-AKI adult participants who had urine collected for proteomic profiling. For validation in pediatric cardiac surgery participants, we compared protein concentrations in the first postoperative samples versus preoperative samples. For validation in marathon runners, we compared protein concentrations in the immediate postrace samples versus the prerace samples. We performed these comparisons using pairwise Wilcoxon tests.
For our mouse models of IRI followed by repair and atrophy, we used two-way analysis of variance (ANOVA) (GraphPad Prism 8) for model comparison to test whether there was a difference between the models and in the time course, followed by Šidák posttests for subgroup comparison at each time point. In addition, we determined Pearson’s correlation in gene expression of identified markers with fibrosis markers in the recovery phase of AKI (days 7, 14, and 30). For the mouse model of AAN, we used ANOVA followed by Tukey tests for comparisons across subgroups and Student’s t tests to determine the difference between gene expression in the AKI (7 days) and CKD phases (21 days) compared with baseline. We conducted a complete case analysis and considered a two-sided P < 0.05 as statistically significant. All statistical analyses were performed using R version 4.1.2.
Supplementary Material
Funding:
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases grant UH3DK114866 (to C.R.P.), National Heart, Lung and Blood Institute: R01HL085757 (to C.R.P.), National Institute of Diabetes and Digestive and Kidney Diseases grant UH3DK114861 (to P.M.P.), National Institute of Diabetes and Digestive and Kidney Diseases grant K01DK120783 (to L.X.), National Institute of Diabetes and Digestive and Kidney Diseases grant R01DK093771 (to L.G.C.), and National Institute of Diabetes and Digestive and Kidney Diseases grants U01DK133081, U01DK133091, U01DK133092, U01DK133093, U01DK133095, U01DK133097, U01DK114866, U01DK114908, U01DK133090, U01DK133113, U01DK133766, U01DK133768, U01DK114907, U01DK114920, U01DK114923, U01DK114933, and U24DK114886 (to the KPMP Consortium).
The KPMP Consortium:
In addition to KPMP Consortium members who are authors (Y.W., S.M., D.G.M., W.O., P.M.P., L.G.C., and C.R.P.), the following KPMP Consortium members are collaborators who have contributed to study design, data analysis, and interpretation: Mohamed G. Atta1, Lauren Bernard1, Celia P. Corona-Villalobos1, Jose M Monroy-Trujillo1, Avi Z. Rosenberg1, Sonya Shah1, Jeanine Hernandez1, C. John Sperati1, Noralinda B. Viloria1, Ashley R. Wang1, Alan Xu1, Tanima Arora3, Vijayakumar R Kakade3, Gilbert W. Moeckel3, Melissa M. Shaw3, Ugochukwu Ugwuowo3, Angela M. Victoria-Castro3, F. Perry Wilson3, Adam Burgess4, Michele M Elder4, Matthew Gilliam4, Daniel E. Hall4, John A. Kellum4, Raghavan Murugan4, Matthew R. Rosengart4, Roderick Tan4, Tina Vita4, James Winters4, Blue B. Lake7, Richard Knight8, Stewart H. Lecker9, Alexander Morales9, Mark E. Williams9, Humra Athar10, Stephanie J. Aw10, Laurence H Beck Jr10, Joel M Henderson10, Courtney Huynh10, Shana Maikhor10, Ingrid F Onul10, Insa M Schmidt10, Ashish Upadhyay10, Ashish Verma10, Sushrut S. Waikar10, Pranav Yadati10, Guanghao Yu10, Steve Bogen11, Mia R. Colona12, Gearoid Michael McMahon12, Helmut Rennke12, M. Todd Valerius12, Astrid Weins12, Anna Greka13, Nir Hacohen13, Jamie L. Marshall13, Mark P. Aulisio14, William S. Bush14, Yijiang Chen14, Dana C. Crawford14, Lakeshia Bush15, Leslie Cooperman15, Crystal A Gadegbeku15, Leal Herlitz15, Vivian Jeffers15, Stacey Jolly15, Charles O′Malley15, John F. O′Toole15, Ellen Palmer15, Emilio D Poggio15, John R. Sedor15, Dianna Sendrey15, Kassandra Spates-Harden15, Jonathan J Taliercio15, Paul S. Appelbaum16, Olivia Balderes16, Jonathan Barasch16, Andrew S. Bomback16, Pietro A. Canetta16, Vivette. D′Agati16, Krzysztof Kiryluk16, Karla Mehl16, German Varela16, Joana P. Gonçalves17, Roy Lardenoije17, Lukasz G. Migas17, Raf Van de Plas17, Laura Barisoni18, Anant Madabhushi19, Andrew Janowczyk19, Theodore Alexandrov20, Nils Gehlenborg21, Mark S. Keller21, Seymour Rosen21, Mahla Asghari22, Tarek M. El-Achkar22, Daria Barwinska22, Sharon B Bledsoe22, William S. Bowen22, Ying-Hua Cheng22, Pierre c. Dagher22, Kenneth W. Dunn22, Michael T Eadon22, Michael Ferkowicz22, Debora Gisch22, Katherine J. Kelly22, Ricardo Melo Ferreira22, Angela R. Sabo22, Timothy A. Sutton22, Abraham Verdoes22, Curtis Warfield22, James C. Williams, Jr. 22, Stephanie Wofford22, Devin M. Wright22, Katy Börner23, Bruce W. Herr II23, Ellen M. Quardokus23, Sophia A. Angus24, Sarah W Chen24, Isabel Donohoe24, Camille Johansen24, Jenny Molina-Guzman24, Sylvia E Rosas24, Neil Roy24, Melissa D. Rubinsky24, Paolo S. Silva24, Jennifer K. Sun24, Gabriel Zeinoun24, Joseph Ardayfio25, Jack Bebiak25, Keith Brown25, Taneisha Campbell25, Catherine E. Campbell25, Lynda Hayashi25, Nichole Jefferson25, Glenda V. Roberts25, John Saul25, Anna Shpigel25, Christy Stutzke25, Robert Koewler25, and Roy Pinkeney25, Evren U Azeloglu26, Kirk N Campbell26, Steven G. Coca26, Jens Hansen26, Jonathan Haydak26, John Cijiang He26, Ravi Iyengar26, Gina Koch26, Sean Lefferts26, Kristin Meliambro26, Girish N Nadkarni26, Marissa Patel26, Isaac E Stillman26, Joji Tokita26, Stephen C Ward26, Pottumarthi Prasad27, Samir V Parikh28, Brad H. Rovin28, Christopher R Anderton29, Brittney L. Gorman29, Jessica Lukowski29, Ljiljana Paša-Tolić29, Dusan Velickovic29, George (Holt) Oliver30, Weiguang Mao31, Rachel S. G. Sealfon31, Olga G Troyanskaya31, Aaron Wong31, Ari Pollack32, Brandon Ginley33, Brendon Lutnick33, David H. Beyda34, Erika R Bracamonte34, Frank C. Brosius34, Baltazar Campos34, Nicole Marquez34, Katherine Mendoza34, Raymond Scott34, Bijin Thajudeen34, Rebecca Tsosie34, Gregory Woodhead34, Kavya Anjani35, Zoltan G. Laszik35, Tariq Mukatash35, Minnie M Sarwal35, Tara K Sigdel35, Milda R. Saunders36, Ashley R. Burg37, Petter Bjornstad38, Hsieh EWY38, Jessica Kendrick38, Laura Pyle38, Joshua M. Thurman38, Carissa Vinovskis38, Julia Wrobel38, Fuyong Xing38, Nicholas Lucarelli39, Pinaki Sarder39, James T Bui40, Eunice Carmona-Powell40, Monica L. Fox40, Ron C. Gaba40, Tanika N. Kelly40, James P. Lash40, Natalie Meza40, Devona Redmond40, Amada Renteria40, Ana C. Ricardo40, Suman Setty40, Anand Srivastava40, Fadhl Alakwaa41, Heather K. Ascani41, Ulysses G. J. Balis41, Markus Bitzer41, Victoria M. Blanc41, Nikole Bonevich41, Ninive Conser41, Nathan Creger41, Dawit Demeke41, Rachel Dull41, Sean Eddy41, Renee Frey41, John Hartman41, Yongqun He41, Jeffrey B. Hodgin41, Wenjun Ju41, Matthias Kretzler41, Chrysta C Lienczewski41, Laura H. Mariani41, Phillip J. McCown41, Rajasree Menon41, Viji Nair41, Edgar A. Otto41, Rebecca Reamy41, Michael P. Rose41, Jennifer A. Schaub41, Becky Steck41, Haneen Tout41, Zach Wright41, Oyedele A. Adeyi42, Cathy A Bagne42, Jerica M. Berge42, M. Luiza Caramori42, Alyson Coleman42, Yanli Ding42, Paul Drawz42, Siobhan M. Flanagan42, Tasma Harindhanavudhi42, Dori Henderson42, Christopher J. Jones42, Rachel R. Kaspari42, Susan Klett42, Sisi Ma42, Patrick H. Nachman42, Via Rao42, Nicolas J Rauwolf42, Sami Safadi42, Michelle L. Snyder42, Susan M. Wolf42, Seth Winfree43, Peter R. Bream44, Jr., Tashas Cameron-Wheeler44, Anne Froment44, Jennifer L. Jones44, Dhatri Kakarla44, Sara S. Kelley44, Sora Lee44, Priya Mody44, Amy K. Mottl44, Fernanda Ochoa Toro44, Prabir Roy-Chaudhury44, Evan M. Zeitler44, Annapurna Pamreddy45, Nagarjunachary Ragi45, Kumar Sharma45, Manjeri Venkatachalam45, Hongping Ye45, Guanshi Zhang45, Qi Cai46, Catherine Campbell46, S. Susan Hedayati46, Allen R Hendricks46, Asra Kermani46, Simon C. Lee46, Shihong Ma46, R. Tyler Miller46, Harold Park46, Jiten Patel46, Anil Pillai46, Samuel Rice46, Jose R. Torrealba46, Robert D Toto46, Miguel A. Vazquez46, Nancy Wang46, Natasha Wen46, Charles E. Alpers47, Ashley C Berglund47, Brooke Berry47, Kristina N Blank47, Keith D. Brown47, Jonas M Carson47, Ian H. de Boer47, Matthew Dekker47, Ashveena L Dighe47, Frederick Dowd47, Stephanie M. Grewenow47, Lynda Hayashi47, Jonathan Himmelfarb47, Andrew N Hoofnagle47, Nichole M. Jefferson47, Cienn N. Joyeux47, Richard A. Knight47, Brandon G Larson47, Christine P Limonte47, Robyn L. McClelland47, Sean D. Mooney47, Yunbi Nam47, Christopher Park47, Jimmy Phuong47, Alexa Plisiewicz47, Kasra A Rezaei47, Glenda V. Roberts47, Natalya Sarkisova47, Jaime Snyder47, Katherine R. Tuttle47, Artit Wangperawong47, Adam Wilcox47, Kayleen Williams47, Bessie A. Young47, Jamie L Allen48, Madeline E. Colley48, Yarieli Cuevas-Rios48, Mark P de Caestecker48, Katerina V. Djambazova48, Martin Dufresne48, Melissa A Farrow48, Agnes B. Fogo48, Audra M. Judd48, Angela R.S. Kruse48, Jeffrey M. Spraggins48, Jeannine Basta49, Kristine Conlon49, Joseph P. Gaut49, Reetika Ghag49, Sanjay Jain49, Madhurima Kaushal49, Amanda Knoten49, Amy McMurray49, Brittany C Minor49, Gerald Nwanne49, Michael Rauchman49, Anitha Vijayan49, Bo Zhang49.
Affiliations: Affiliations 1 to 4 can be found on the first page of the paper. 7San Diego Institute of Science, Altos Labs, San Diego, CA 92121, USA. 8American Association of Kidney Patients, Tampa, FL 33613, USA. 9Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02215, USA. 10Boston University School of Medicine, Boston Medical Center, Boston, MA 02118, USA. 11Boston Cell Standards, Boston, MA 02111, USA. 12Brigham and Women’s Hospital, Boston, MA 02115, USA. 13Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA. 14Case Western Reserve University School of Medicine, Cleveland, OH 44106, USA. 15Cleveland Clinic, Cleveland, OH 44103, USA. 16Vagelos College of Physicians and Surgeons, Columbia University, New York, NY 10032, USA. 17Delft Center for Systems and Control, Delft University of Technology, Delft, Netherlands. 18Duke University, Durham, NC 27708, USA. 19Emory University and Georgia Institute of Technology, Atlanta, GA 30322, USA. 20European Molecular Biology Laboratory, Heidelberg, Germany. 21Harvard University Medical School, Boston, MA 02115, USA. 22Indiana University School of Medicine, Indianapolis, IN 46202, USA. 23Indiana University, Bloomington, IN 47405, USA. 24Joslin Diabetes Center, Boston, MA 02215, USA. 25KPMP Patient Partner, University of Washington, Seattle, WA 98195, USA. 26Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA. 27Northwestern University, Chicago, IL 60611, USA. 28Ohio State University, Columbus, OH 43210, USA. 29Pacific Northwest National Laboratories, Richland, WA 99354, USA. 30Parkland Health, Dallas, TX 75235, USA. 31Princeton University, Princeton, NJ 08544, USA. 32Seattle Children’s Hospital, Seattle, WA 98105, USA. 33State University of New York Buffalo, Buffalo, NY 14260, USA. 34University of Arizona, Tucson, AZ 85721, USA. 35University of California San Francisco, San Francisco, CA 94143, USA. 36University of Chicago, Chicago, IL 60637, USA. 37University of Cincinnati, Cincinnati, OH 45221, USA. 38University of Colorado, Boulder, CO 80309, USA. 39University of Florida, Gainesville, FL 32611, USA. 40University of Illinois Chicago, Chicago, IL 60607, USA. 41University of Michigan, Ann Arbor, MI 48109, USA. 42University of Minnesota, Minneapolis, MN 55455, USA. 43University of Nebraska Medical Center, Omaha, NE 68198, USA. 44University of North Carolina, Chapel Hill, NC 27599, USA. 45University of Texas Health Science Center at San Antonio, San Antonio, TX, USA. 46University of Texas Southwestern, Dallas, TX 78229, USA. 47University of Washington, Seattle, WA 98195, USA. 48Vanderbilt University, Nashville, TN 37235, USA. 49Washington University St. Louis, St. Louis, MO 63130, USA.
The TRIBE-AKI Consortium:
In addition to TRIBE-AKI Consortium members who are authors (Y.W., S.M., D.G.M., W.O., S.G.M., P.D., and C.R.P.), the following TRIBE-AKI Consortium members are collaborators who have contributed to study design, data analysis, and interpretation: Michael Zappitelli50, Amit X. Garg51,52,53.
Affiliations: 50Division of Paediatric Nephrology, Department of Paediatrics, Hospital for Sick Children, Toronto, ON, Canada. 51ICES, Toronto, Ontario, Canada. 52Department of Epidemiology and Biostatistics, Western University, London, ON, Canada. 53Division of Nephrology, Department of Medicine, Western University, London, ON, Canada.
Footnotes
Supplementary Materials
This PDF file includes:
Reference (60)
Other Supplementary Material for this manuscript includes the following:
Data files S1 to S6
MDAR Reproducibility Checklist
Competing interests: C.R.P. is a member of the advisory board of and owns equity in RenalytixAI. He also serves as a consultant for Genfit and Novartis. Y.W. and C.R.P. are named inventors on provisional patent number 63/371,894 titled “Diagnosis and Treatment of Acute Kidney Injury”. D.G.M. and C.R.P. are named coinventors on a pending patent number US-20200072847-A1 titled “Methods and Systems for Diagnosis of Acute Interstitial Nephritis.” D.G.M. and C.R.P. are founders of the diagnostics company Predict AIN, LLC. P.M.P. serves as a consultant for Janssen Research & Development, LLC. The other authors declare that they have no competing interests. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. The code used for analysis has been deposited at Zenodo (DOI: 10.5281/zenodo.8280678) (59). Raw sequencing data from KPMP participants are under controlled access because they are potentially identifiable. The raw snRNA-seq data are available under a material agreement with University of Washington. These can be requested from KPMP by contacting info@kpmp.org and are available by signing a data user agreement (DUA) promising to abide by KPMP security standards and to not reidentify participants, share data outside those named on the DUA, or sell the data. Data access is granted to anyone signing the KPMP DUA as is. Processed sequencing data by the KPMP consortium are publicly available at http://atlas.kpmp.org/ repository and compiled (https://doi.org/10.48698/3z31-8924) (10). The proteomic data analyzed in this work are not publicly available because the widespread sharing of TRIBE-AKI study data was not stipulated in the ethics approval for the study. The TRIBE-AKI principal investigator (C.R.P., chirag.parikh@jhmi.edu) may be contacted for requests for the deidentified analytical dataset.
REFERENCES AND NOTES
- 1.Ronco C, Bellomo R, Kellum JA, Acute kidney injury. Lancet 394, 1949–1964 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Coca SG, Singanamala S, Parikh CR, Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int 81, 442–448 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirita Y, Wu H, Uchimura K, Wilson PC, Humphreys BD, Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc. Natl. Acad. Sci. U.S.A 117, 15874–15883 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosen S, Samuel NH, Difficulties in understanding human acute tubular necrosis: Limited data and flawed animal models. Kidney Int 60, 1220–1224 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Wen Y, Yang C, Menez SP, Rosenberg AZ, Parikh CR, A systematic review of clinical characteristics and histologic descriptions of acute tubular injury. Kidney Int. Rep 5, 1993–2001 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharp CN, Siskind LJ, Developing better mouse models to study cisplatin-induced kidney injury. Am. J. Physiol. Renal Physiol 313, F835–F841 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allgren RL, Marbury TC, Rahman SN, Weisberg LS, Fenves AZ, Lafayette RA, Sweet RM, Genter FC, Kurnik BR, Conger JD, Sayegh MH, Anaritide in acute tubular necrosis. Auriculin anaritide acute renal failure study group. N. Engl. J. Med 336, 828–834 (1997). [DOI] [PubMed] [Google Scholar]
- 8.Pickkers P, Mehta RL, Murray PT, Joannidis M, Molitoris BA, Kellum JA, Bachler M, Hoste EAJ, Hoiting O, Krell K, Ostermann M, Rozendaal W, Valkonen M, Brealey D, Beishuizen A, Meziani F, Murugan R, de Geus H, Payen D, van den Berg E, Arend J; STOP-AKI Investigators, Effect of human recombinant alkaline phosphatase on 7-day creatinine clearance in patients with sepsis-associated acute kidney injury: A randomized clinical trial. JAMA 320, 1998–2009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tumlin JA, Finkel KW, Murray PT, Samuels J, Cotsonis G, Shaw AD, Fenoldopam mesylate in early acute tubular necrosis: A randomized, double-blind, placebo-controlled clinical trial. Am. J. Kidney Dis 46, 26–34 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Lake BB, Menon R, Winfree S, Hu Q, Ferreira RM, Kalhor K, Barwinska D, Otto EA, Ferkowicz M, Diep D, Plongthongkum N, Knoten A, Urata S, Mariani LH, Naik AS, Eddy S, Zhang B, Wu Y, Salamon D, Williams JC, Wang X, Balderrama KS, Hoover PJ, Murray E, Marshall JL, Noel T, Vijayan A, Hartman A, Chen F, Waikar SS, Rosas SE, Wilson FP, Palevsky PM, Kiryluk K, Sedor JR, Toto RD, Parikh CR, Satija R, Greka A, Macosko EZ, Gaut JP, Hodgin JB; KPMP Consortium MT Eadon, P. C. Dagher, T. M. El-Achkar, K. Zhang, M. Kretzler, S. Jain, An atlas of healthy and injured cell states and niches in the human kidney. Nature 619, 585–594 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stockwell BR, Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401–2421 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muto Y, Wilson PC, Ledru N, Wu H, Dimke H, Waikar SS, Humphreys BD, Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. Nat. Commun 12, 2190 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martovetsky G, Tee JB, Nigam SK, Hepatocyte nuclear factors 4α and 1α regulate kidney developmental expression of drug-metabolizing enzymes and drug transporters. Mol. Pharmacol 84, 808–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR, Downregulation of liver X receptor-α in mouse kidney and HK-2 proximal tubular cells by LPS and cytokines. J. Lipid Res 46, 2377–2387 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Shirota S, Yoshida T, Sakai M, Kim JI, Sugiura H, Oishi T, Nitta K, Tsuchiya K, Correlation between the expression level of c-maf and glutathione peroxidase-3 in c-maf−/− mice kidney and c-maf overexpressed renal tubular cells. Biochem. Biophys. Res. Commun 348, 501–506 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Suzuki S, Yokoyama A, Noro E, Aoki S, Shimizu K, Shimada H, Sugawara A, Expression and pathophysiological significance of carbohydrate response element binding protein (ChREBP) in the renal tubules of diabetic kidney. Endocr. J 67, 335–345 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Stoeckman AK, Ma L, Towle HC, Mlx is the functional heteromeric partner of the carbohydrate response element-binding protein in glucose regulation of lipogenic enzyme genes. J. Biol. Chem 279, 15662–15669 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Cai J, Jiao X, Fang Y, Yu X, Ding X, The orphan nuclear receptor RORα is a potential endogenous protector in renal ischemia/reperfusion injury. FASEB J., Exp. Biol 33, 5704–5715 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Huang J, Arsenault M, Kann M, Lopez-Mendez C, Saleh M, Wadowska D, Taglienti M, Ho J, Miao Y, Sims D, Spears J, Lopez A, Wright G, Hartwig S, The transcription factor Sry-related HMG box-4 (SOX4) is required for normal renal development in vivo. Dev. Dyn 242, 790–799 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Pak S, Kim W, Kim Y, Song C, Ahn H, Dihydrotestosterone promotes kidney cancer cell proliferation by activating the STAT5 pathway via androgen and glucocorticoid receptors. J. Cancer Res. Clin. Oncol 145, 2293–2301 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Fragiadaki M, Lannoy M, Themanns M, Maurer B, Leonhard WN, Peters DJM, Moriggl R, Ong ACM, STAT5 drives abnormal proliferation in autosomal dominant polycystic kidney disease. Kidney Int 91, 575–586 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Diepenbruck M, Waldmeier L, Ivanek R, Berninger P, Arnold P, van Nimwegen E, Christofori G, Tead2 expression levels control the subcellular distribution of Yap and Taz, zyxin expression and epithelial-mesenchymal transition. J. Cell Sci 127, 1523–1536 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Contursi C, Wang IM, Gabriele L, Gadina M, O’Shea J, Morse HC 3rd, Ozato K, IFN consensus sequence binding protein potentiates STAT1-dependent activation of IFNγ-responsive promoters in macrophages. Proc. Natl. Acad. Sci. U.S.A 97, 91–96 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Li X, Yang J, Zhang D, LncRNA ENSMUST_147219 mediates the progression of ischemic acute kidney injury by targeting the miR-221–5p/IRF6 axis. Apoptosis 27, 531–544 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin R, Zhao A, Han S, Zhang D, Sun H, Li M, Su D, Liang X, The interaction of S100A16 and GRP78 actives endoplasmic reticulum stress-mediated through the IRE1α/XBP1 pathway in renal tubulointerstitial fibrosis. Cell Death Dis 12, 942 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu W, Zhang H, Zhang Q, Xu J, β-Amyrin ameliorates diabetic nephropathy in mice and regulates the miR-181b-5p/HMGB2 axis in high glucose-stimulated HK-2 cells. Environ. Toxicol 37, 637–649 (2022). [DOI] [PubMed] [Google Scholar]
- 27.Bechara R, Amatya N, Bailey RD, Li Y, Aggor FEY, Li D-D, Jawale CV, Coleman BM, Dai N, Gokhale NS, Taylor TC, Horner SM, Poholek AC, Bansal A, Biswas PS, Gaffen SL, The m6A reader IMP2 directs autoimmune inflammation through an IL-17- and TNFα-dependent C/EBP transcription factor axis. Sci. Immunol 6, eabd1287 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su EY, Spangler A, Bian Q, Kasamoto JY, Cahan P, Reconstruction of dynamic regulatory networks reveals signaling-induced topology changes associated with germ layer specification. Stem Cell Reports 17, 427–442 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinze C, Kocks C, Leiz J, Karaiskos N, Boltengagen A, Cao S, Skopnik CM, Klocke J, Hardenberg J-H, Stockmann H, Gotthardt I, Obermayer B, Haghverdi L, Wyler E, Landthaler M, Bachmann S, Hocke AC, Corman V, Busch J, Schneider W, Himmerkus N, Bleich M, Eckardt K-U, Enghard P, Rajewsky N, Schmidt-Ott KM, Single-cell transcriptomics reveals common epithelial response patterns in human acute kidney injury. Genome Med 14, 103 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mansour SG, Verma G, Pata RW, Martin TG, Perazella MA, Parikh CR, Kidney injury and repair biomarkers in marathon runners. Am. J. Kidney Dis 70, 252–261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Puthumana J, Thiessen-Philbrook H, Xu L, Coca SG, Garg AX, Himmelfarb J, Bhatraju PK, Ikizler TA, Siew E, Ware LB, Liu KD, Go AS, Kaufman JS, Kimmel PL, Chinchilli VM, Cantley L, Parikh CR, Biomarkers of inflammation and repair in kidney disease progression. J. Clin. Invest 131, e139927 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balzer MS, Doke T, Yang Y-W, Aldridge DL, Hu H, Mai H, Mukhi D, Ma Z, Shrestha R, Palmer MB, Hunter CA, Susztak K, Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration. Nat. Commun 13, 4018 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu Y-A, Liao C-T, Raybould R, Talabani B, Grigorieva I, Szomolay B, Bowen T, Andrews R, Taylor PR, Fraser D, Single-nucleus RNA sequencing identifies new classes of proximal tubular epithelial cells in kidney fibrosis. J. Am. Soc. Nephrol 32, 2501–2516 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK, Failed tubule recovery AKI -CKD transition, and kidney disease progression. J. Am. Soc. Nephrol 26, 1765–1776 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lan R, Geng H, Polichnowski AJ, Singha PK, Saikumar P, McEwen DG, Griffin KA, Koesters R, Weinberg JM, Bidani AK, Kriz W, Venkatachalam MA, PTEN loss defines a TGF-β-induced tubule phenotype of failed differentiation and JNK signaling during renal fibrosis. Am. J. Physiol. Renal Physiol 302, F1210–F1223 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, Meyer-Schaller N, Schübeler D, van Nimwegen E, Christofori G, Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell 23, 768–783 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV, Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med 16, 535–543 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu L, Guo J, Moledina DG, Cantley LG, Immune-mediated tubule atrophy promotes acute kidney injury to chronic kidney disease transition. Nat. Commun 13, 4892 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pozdzik AA, Giordano L, Li G, Antoine M-H, Quellard N, Godet J, De Prez E, Husson C, Declèves A-E, Arlt VM, Goujon J-M, Brochériou-Spelle I, Ledbetter SR, Caron N, Nortier JL, Blocking TGF-β signaling pathway preserves mitochondrial proteostasis and reduces early activation of PDGFRβ+ pericytes in aristolochic acid induced acute kidney injury in wistar male rats. PLOS ONE 11, e0157288 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santibanez JF, Krstic J, Transforming growth factor-β and urokinase type plasminogen interplay in cancer. Curr. Protein Pept. Sci 19, 1155–1163 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard M-P, Raynaud M, Ronce N, Lemonnier E, Calvas P, Laudier B, Chelly J, Fryns J-P, Ropers H-H, Hamel BCJ, Andres C, Barthélémy C, Moraine C, Briault S, X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet 74, 552–557 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu F, Chang K, Ma J, Qu Y, Xie H, Dai B, Gan H, Zhang H, Shi G, Zhu Y, Zhu Y, Shen Y, Ye D, The oncogenic role of COL23A1 in clear cell renal cell carcinoma. Sci. Rep 7, 9846 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li X, Kimura H, Hirota K, Kasuno K, Torii K, Okada T, Kurooka H, Yokota Y, Yoshida H, Synergistic effect of hypoxia and TNF-α on production of PAI-1 in human proximal renal tubular cells. Kidney Int 68, 569–583 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Nazir S, Gadi I, Al-Dabet MM, Elwakiel A, Kohli S, Ghosh S, Manoharan J, Ranjan S, Bock F, Braun-Dullaeus RC, Esmon CT, Huber TB, Camerer E, Dockendorff C, Griffin JH, Isermann B, Shahzad K, Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood 130, 2664–2677 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morita J, Kano K, Kato K, Takita H, Sakagami H, Yamamoto Y, Mihara E, Ueda H, Sato T, Tokuyama H, Arai H, Asou H, Takagi J, Ishitani R, Nishimasu H, Nureki O, Aoki J, Structure and biological function of ENPP6, a choline-specific glycerophosphodiester-phosphodiesterase. Sci. Rep 6, 20995 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mansour SG, Martin TG, Obeid W, Pata RW, Myrick KM, Kukova L, Jia Y, Bjornstad P, El-Khoury JM, Parikh CR, The role of volume regulation and thermoregulation in AKI during marathon running. Clin. J. Am. Soc. Nephrol 14, 1297–1305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Bellomo R, Cardiac surgery-associated acute kidney injury: Risk factors, pathophysiology and treatment. Nat. Rev. Nephrol 13, 697–711 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Parikh CR, Thiessen-Philbrook H, Garg AX, Kadiyala D, Shlipak MG, Koyner JL, Edelstein CL, Devarajan P, Patel UD, Zappitelli M, Krawczeski CD, Passik CS, Coca SG; TRIBE-AKI Consortium, Performance of kidney injury molecule-1 and liver fatty acid-binding protein and combined biomarkers of AKI after cardiac surgery. Clin. J. Am. Soc. Nephrol 8, 1079–1088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu C, Debnath N, Mosoyan G, Chauhan K, Vasquez-Rios G, Soudant C, Menez S, Parikh CR, Coca SG, Systematic review and meta-analysis of plasma and urine biomarkers for CKD outcomes. J. Am. Soc. Nephrol 33, 1657–1672 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenberg JH, Devarajan P, Thiessen-Philbrook HR, Krawczeski C, Parikh CR, Zappitelli M, Kidney injury biomarkers 5 years after AKI due to pediatric cardiac surgery. Pediatr. Nephrol 33, 1069–1077 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gayoso A, Shor J, JonathanShor/DoubletDetection: Doubletdetection v4.2. (2022); 10.5281/zenodo.6349517. [DOI] [Google Scholar]
- 52.Fleming SJ, Chaffin MD, Arduini A, Akkad A-D, Banks E, Marioni JC, Philippakis AA, Ellinor PT, Babadi M, Unsupervised removal of systematic background noise from droplet-based single-cell experiments using CellBender. Nat. Methods 20, 1323–1335 (2022). [DOI] [PubMed] [Google Scholar]
- 53.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R, Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A, Fast gene set enrichment analysis. bioRxiv 10.1101/060012 (2021). 10.1101/060012. [DOI] [Google Scholar]
- 55.Wolf FA, Angerer P, Theis FJ, SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar N, Mishra B, Athar M, Mukhtar S, Inference of gene regulatory network from single-cell transcriptomic data using pySCENIC. Methods Mol. Biol 2328, 171–182 (2021). [DOI] [PubMed] [Google Scholar]
- 57.Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, van den Oord J, Atak ZK, Wouters J, Aerts S, SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tan Y, Cahan P, SingleCellNet: A computational tool to classify single cell RNA-seq data across platforms and across species. Cell Syst 9, 207–213.e2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wen Y, KPMP snRNAseq and TRIBE-AKI proteomics analysis (2023); doi: 10.5281/zenodo.8280678. [DOI] [Google Scholar]
- 60.Williams SA, Kivimaki M, Langenberg C, Hingorani AD, Casas JP, Bouchard C, Jonasson C, Sarzynski MA, Shipley MJ, Alexander L, Ash J, Bauer T, Chadwick J, Datta G, DeLisle RK, Hagar Y, Hinterberg M, Ostroff R, Weiss S, Ganz P, Wareham NJ, Plasma protein patterns as comprehensive indicators of health. Nat. Med 25, 1851–1857 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. The code used for analysis has been deposited at Zenodo (DOI: 10.5281/zenodo.8280678) (59). Raw sequencing data from KPMP participants are under controlled access because they are potentially identifiable. The raw snRNA-seq data are available under a material agreement with University of Washington. These can be requested from KPMP by contacting info@kpmp.org and are available by signing a data user agreement (DUA) promising to abide by KPMP security standards and to not reidentify participants, share data outside those named on the DUA, or sell the data. Data access is granted to anyone signing the KPMP DUA as is. Processed sequencing data by the KPMP consortium are publicly available at http://atlas.kpmp.org/ repository and compiled (https://doi.org/10.48698/3z31-8924) (10). The proteomic data analyzed in this work are not publicly available because the widespread sharing of TRIBE-AKI study data was not stipulated in the ethics approval for the study. The TRIBE-AKI principal investigator (C.R.P., chirag.parikh@jhmi.edu) may be contacted for requests for the deidentified analytical dataset.