

Abstract
BACKGROUND:
Neuropsychiatric systemic lupus erythematosus (NPSLE) is a poorly understood and heterogeneous manifestation of SLE. Common major NPSLE syndromes include strokes, seizures, myelitis, and aseptic meningitis. Easily obtainable biomarkers are needed to assist in early diagnosis and improve outcomes for NPSLE. A frequent end-result of major syndromes is neuronal or glial injury. Blood-based neurofilament light (NfL) and glial fibrillary acidic protein (GFAP) have been utilized as markers for monitoring disease activity and/or severity in other neurodegenerative and neuroinflammatory diseases; however, they have not been evaluated in active major NPSLE.
METHODS:
This was a case-control study. We enrolled patients aged 12–60 years with active major NPSLE, SLE without active major NPSLE, and healthy controls. Active NPSLE was defined as being <6 months from last new or worsening neuropsychiatric symptom. Demographics, clinical data, and serum or plasma biosamples were collected.
RESULTS:
Thirteen patients with active major NPSLE, 13 age/sex/kidney function matched SLE controls without active major NPSLE, and 13 age/sex matched healthy controls (mean ages 26.8, 27.3, 26.6 years) were included. 92% of each group were female. Major syndromes included stroke (5), autonomic disorder (3), demyelinating disease (2), aseptic meningitis (2), sensorimotor polyneuropathy (2), cranial neuropathy (1), seizures (1), and myelopathy (2). Mean (standard deviation) blood NfL and GFAP were 3.6 pg/ml (2.0) and 50.4 pg/ml (15.0), respectively, for the healthy controls. Compared to healthy controls, SLE without active major NPSLE had mean blood NfL and GFAP levels 1.3 pg/ml (p = 0.42) and 1.2 pg/ml higher (p = 0.53), respectively. Blood NfL was on average 17.9 pg/ml higher (95% CI: 9.2, 34.5; p <0.001) and blood GFAP was on average 3.2 pg/ml higher (95% CI: 1.9, 5.5; p <0.001) for cases of active major NPSLE compared to SLE without active major NPSLE. In a subset of 6 patients sampled at multiple time points, blood NfL and GFAP decreased after immunotherapy.
CONCLUSIONS:
Blood NfL and GFAP levels are elevated in persons with SLE with active major NPSLE compared to disease matched controls and may lower after immunotherapy initiation. Larger and longitudinal studies are needed to ascertain their utility in a clinical setting.
Introduction
Neuropsychiatric systemic lupus erythematosus (NPSLE) is estimated to occur in 18 to 56% of persons with systemic lupus erythematosus (SLE).1 NPSLE is associated with decreased quality of life and a significant increase in morbidity, mortality, hospitalization, and disease related costs compared to SLE without neuropsychiatric (NP) symptoms.2–5 Major NPSLE syndromes in the central nervous system (CNS) include aseptic meningitis, strokes, seizures, demyelinating disease, chorea, myelopathy, acute confusional state, severe depression, psychosis, or moderate/severe cognitive dysfunction. Major NPSLE syndromes in the peripheral nervous system (PNS) include acute inflammatory demyelinating polyneuropathy, polyneuropathy with nerve conduction study confirmation, mononeuropathy, autonomic neuropathy, cranial neuropathy, plexopathy, or myasthenia gravis. These major NPSLE syndromes are thought to be directly attributable to systemic inflammatory disease activity.6, 7 Appropriate management of NPSLE can successfully modify the potentially catastrophic sequelae of this disease.5, 8
Despite an expanding collection of new diagnostic testing, imaging, and attribution models, challenges in NPSLE diagnosis remain.1, 2, 6, 7, 9–12 Up to 15% of expert-diagnosed NPSLE is eventually revealed as an NP symptom unrelated to lupus, showing the tendency to over-diagnose in the face of uncertainty.1, 13 Hence, the common practice for diagnosis remains “physician judgement” based on clinical presentation, course and paraclinical testing.14 Clinically, an inaccurate diagnosis leads to increased morbidity from either an undertreated disease process or overtreatment with immunosuppression (such as steroids, cyclophosphamide, or rituximab).15–17 The difficulties in defining or diagnosing NPSLE have impaired epidemiologic research into this disease as well.18, 19 As such, identification of new diagnostic biomarkers has been highlighted as one of the core unmet needs in NPSLE research.1
Serum or plasma neurofilament light (NfL) is a neuron-specific cytoskeletal protein released from neurons after neuro-axonal injury,20 and serum or plasma glial fibrillary acidic protein (GFAP) is an astrocytic structural protein released with glial cell damage or microglial activation.21 These biomarkers are elevated in multiple central and peripheral inflammatory, degenerative and ischemic neurological disorders. These biomarkers have shown utility in these disorders for assessment of treatment response, detection of pre-clinical inflammation, and prediction of future risk of relapse.20, 22–28 Age and kidney function are two important known contributors to variability in these measurements.25, 29, 30
Previous studies have shown elevated NfL and GFAP levels in the cerebrospinal fluid (CSF) of patients with NPSLE that decrease after cyclophosphamide therapy.31, 32 Blood NfL in the general adult SLE population has been evaluated in several retrospective or cross-sectional cohort studies. However, these studies included very few patients with active NPSLE and the timing of samples relative to symptom onset was not reported.33–35 Both are necessary to assess its potential utility in evaluation of NPSLE. Neither blood GFAP nor longitudinal change of either of these blood biomarkers after immunotherapy have been evaluated to date in NPSLE.
The primary objective of this study was to assess blood-based NfL and GFAP in active major NPSLE. We hypothesized that blood NfL and GFAP would be elevated in active major NPSLE compared to SLE controls without active major NPSLE and to healthy controls. We also explored the longitudinal change of these biomarkers in a subset of cases after immunotherapy treatment.
Methods
Study Overview
Patients were enrolled prospectively into our neuro-rheumatologic disease registry between September 2022 and November 2023. These patients were evaluated in Neurology or Rheumatology outpatient clinics or inpatient services at Children’s Hospital Colorado or University of Colorado Hospital. Additional patients were retrospectively identified from Neurology, Rheumatology, or Immunology studies performed at the University of Colorado between 2016 and 2023. These studies enrolled patients with SLE from clinics or inpatient or healthy persons from a community population and included biosample collection. Written consent for enrollment in the prospective study or consent for secondary use were obtained from all participants or their legal representatives under approval from the Colorado Multiple Institutional Review Board.
Patient Identification
Patients with SLE were included if 1) their age at time of enrollment was between 12 and 60 years-old and 2) they had a rheumatologist confirmed clinical diagnosis of SLE and met the 2012 Systemic Lupus International Collaborating Clinics Classification Criteria (SLICC)36 or the 2019 European League Against Rheumatism / American College of Rheumatology (EULAR/ACR) criteria.37 The ages of 12 and 60 years old were chosen as age cutoffs as greater variability and elevation of blood NfL is seen in pre-adolescents and in persons > 60 years-old.38, 39 Patients with SLE were excluded if they had renal failure requiring dialysis at the time of enrollment, given the potential for increased biomarker variability from regular dialysis.
Chart review was performed to identify potential active major NPSLE cases or SLE controls. Cases of “active major” NPSLE were defined as persons with a defined major NPSLE syndrome9 for whom secondary or non-SLE-related causes of their NP symptoms had been excluded and who had experienced new onset or worsening of these symptoms within the 6 months prior to sampling. This duration was chosen to balance the practicality of sample obtainment within a given time frame (due to variability in presentation to care, or to evaluation at quaternary regional hospitals) with an approximation of expected biomarker kinetics after monophasic injury.40–43 Major NPSLE syndromes included all CNS and PNS NPSLE syndromes previously described by the ACR,9 with the exception of the minor/non-specific syndromes of headaches, anxiety, mild depression, mild cognitive impairment, or polyneuropathy without electrophysiological confirmation.6, 7 These major NPSLE syndromes were also classified as either ischemic or inflammatory NPSLE.44 Potential SLE controls had not experienced any major NPSLE syndromes within the 6 months prior to sampling, although they could have minor NPSLE syndromes. These SLE controls were matched to the cases by age, sex, and kidney function.
The clinical symptoms, diagnostic testing, and clinical course of these identified potential active major NPSLE and SLE controls were retrospectively reviewed by a multi-disciplinary team consisting of one neuroimmunologist and two rheumatologists. Patients were adjudicated as active major NPSLE (probable or definite), SLE controls (without active major NPSLE), or unclear status by this team, prior to biosample testing. At least two of the three reviewers were required to agree on a designation of “probable” or “definite” active major NPSLE for cases, and for SLE controls were required to agree that there was definitely no evidence of active major NPSLE. Those designated as probable or definite active major NPSLE or SLE controls were included in further analysis.
Healthy controls were recruited from the general population through academic internal medicine clinics or community sites in Aurora, Colorado. They underwent self-reported medical screening questionnaires, vital signs, and blood collection. They were excluded if review of self-reported medical history disclosed any serious prior or current medical, autoimmune, or neurologic conditions (including SLE, epilepsy, traumatic brain injury, stroke, or hospitalization for SARS-CoV-2 infection).45, 46 These healthy controls were matched to the active major NPSLE cases by age and sex.
Data Collection
All patients with SLE included in the study underwent chart review for demographics, medical history, neuropsychiatric history and symptoms, and results of relevant systemic, CSF, and other neurodiagnostic testing at the time of biosample collection. All healthy controls included in the study had relevant demographic information obtained. Blood samples (serum and/or plasma) via venipuncture were obtained from each patient and transferred to the lab for centrifugation and aliquoting; CSF samples were obtained from a subset of patients. Samples were frozen at −80° Celsius (°C) until biomarker analysis; several patients had clinical pre-treatment samples stored initially at −20°C, but were transferred to a −80°C research freezer within 6 months. All biosamples underwent measurement of NfL and GFAP concentrations using the Neurology 4-PLEX B assay kit and its associated protocols (Quanterix SR-X™ by Simoa® platform, Billerica, MA, USA). Serum samples were only utilized if no pre-immunotherapy plasma sample was available. To control for variations between levels of analytes in serum versus plasma, serum measurements were converted to plasma equivalents using a linear regression equation derived from 144 internal matched plasma and serum samples from healthy controls (unpublished). Plasma and converted plasma-equivalent NfL and GFAP are referred to as blood NfL and GFAP hereafter.
Statistical Analysis
Demographics, SLE disease history, neuropsychiatric history, and relevant additional potential confounders (including Body Mass Index [BMI] and serum creatinine) were compared between persons with SLE with and without active major NPSLE and healthy controls using ANOVA, Kruskal-Wallis, or Student’s t-test for continuous variables, and Pearson’s χ2 or Fisher’s exact tests for categorical variables.
Blood NfL and GFAP levels between the NPSLE and SLE groups were compared using generalized linear models with a gamma distribution to account for non-normal data distributions. Pearson’s correlation was used to assess the relationship between log-transformed NfL and GFAP for the subset of patients with NPSLE who had temporally paired blood and CSF samples. For the subset of patients with active major NPSLE, we evaluated the differences between those with ischemic and inflammatory NPSLE and the proportion above or below the median for each biomarker using Fisher’s exact test. Receiver operating characteristic (ROC) analysis was done for all SLE patients to assess the area under the curve (AUC) for distinguishing between active major NPSLE and SLE without active major NPSLE for each blood biomarker. For the subset of patients with active major NPSLE who had longitudinal samples, we performed an exploratory descriptive analysis of the blood-based biomarker trends over time.
All statistical analyses were performed using SAS v9.4 (SAS Institute, Cary, NC, USA).
Results
A total of 13 patients with definite (9) or probable (4) active major NPSLE, 13 age, sex, and kidney function matched SLE controls, and 13 age and sex matched healthy controls were identified and included in further analysis. Four of the 13 patients with active major NPSLE had serum biosamples only, while the remainder of the active major NPSLE patients and all the SLE controls and healthy controls had plasma biosamples. Conversion of serum values to plasma equivalents resulted in lower NfL and GFAP values for each of the four patients. Seven patients with active major NPSLE had temporally paired blood and CSF samples.
The demographics, clinical characteristics, and blood-based diagnostic testing for these groups are shown in Table 1. There were no statistically significant differences between these groups. Biosamples for patients with active major NPSLE were collected a median of 64 days from the onset of the first NP symptom (range 9–1872 days), a median of 3 days after the most recent new or worsening NP symptom (range 0–188 days), and a median of 2 days (range 0–188 days) after initiation or escalation of immunotherapy (pre-immunotherapy collection was designated as 0 days). All patients had improvement of their NP symptoms following immunotherapy. The distribution of blood NfL and GFAP for individuals within each group are shown in Figure 1a and 1b. The clinical factors, diagnostic testing, and biomarker levels for each NPSLE case are shown in Table 2.
Table 1.
| Active Major NPSLE, N = 13 | SLE without Active Major NPSLE, N = 13 | Healthy Non-SLE Controls, N = 13 | |
|---|---|---|---|
| Demographics | |||
| Age, mean (SD) | 26.8 (13.9) | 27.3 (12.7) | 26.6 (13.3) |
| Female sex, n (%) | 12 (92) | 12 (92) | 12 (92) |
| Race and Ethnicity | |||
| Asian | 1 (8) | 2 (15) | 0 (0) |
| Black | 1 (8) | 1 (8) | 1 (8) |
| Hispanic or Latino | 2 (15) | 5 (38) | 3 (23) |
| White | 7 (54) | 5 (38) | 8 (62) |
| Multi-racial | 1 (8) | 0 (0) | 1 (9) |
| Prefer not to answer | 1 (8) | 0 (0) | 0 (0) |
| Medical and Disease Characteristics | |||
| Body Mass Index (BMI), mean (SD) | 22.1 (45.1) | 26.6 (8.6) | 24.5 (6.7) |
| Time since SLE diagnosis, years, median (range) | 2.75 (0–31) | 4.5 (0–33) | |
| Prior history of NPSLE, n (%) | 2 (15) | 0 (0) | |
| SLEDAI-2K without neuro scores, mean (SD) | 7.2 (4.3) | 4.5 (3.8) | |
| Concurrent Sjogren’s disease, n (%) | 5 (38) | 0 (0) | |
| Prior or concurrent APLS, n (%) | 4 (31) | 2 (15) | |
| Serum/Urine Diagnostic Testing | |||
| Anti-dsDNA positivity, n (%) | 9 (69) | 9 (69) | |
| Low C3, n/N (%) | 7/12 (58) | 6/13 (46) | |
| Low C4, n/N (%) | 8/12 (67) | 7/13 (54) | |
| Elevated ESR, n/N (%) | 8/12 (67) | 4/11 (36) | |
| Anti-ribosomal P positivity, n/N (%) | 1/10 (10) | 0/2 (0) | |
| APLA positivity, n/N (%) | 5/11 (45) | 6/9 (67) | |
| Serum creatinine, mg/dL, mean (SD) | 0.61 (0.20) | 0.66 (0.10) | |
| Impaired eGFR, n (%) | 0 (0) | 0 (0) | |
| Presence of proteinuria, n (%) | 2 (15) | 1 (8) | |
There were no statistically significant differences between the groups for any of the clinical or diagnostic factors. Diagnostic testing was at time of biosampling. APLA = antiphospholipid antibodies; APLS = antiphospholipid antibody syndrome; BMI = body mass index; eGFR = estimated glomerular filtration rate; ESR = erythrocyte sedimentation rate; NPSLE = neuropsychiatric systemic lupus erythematosus; SD = standard deviation; SLE = systemic lupus erythematosus; SLEDAI = SLE disease activity index
Figure 1.
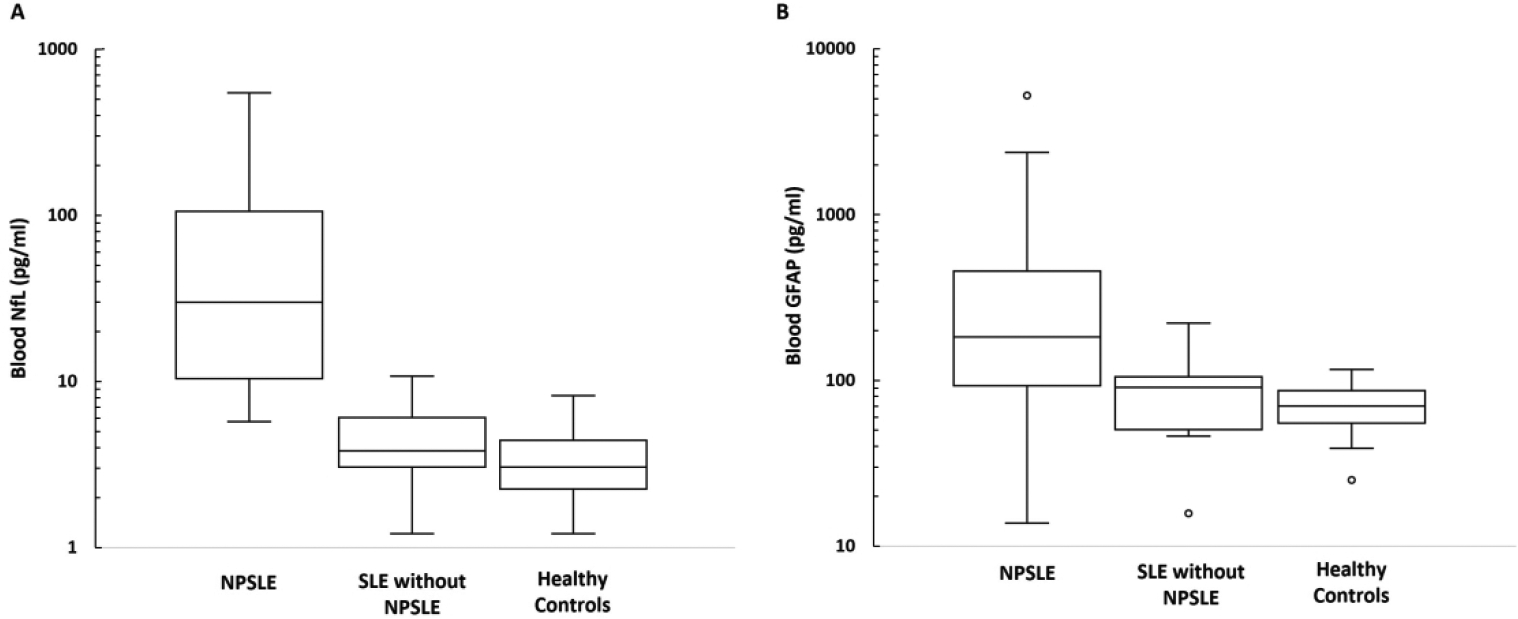
Box and whisker plots showing the distribution of blood neurofilament light (NfL) (A) and glial fibrillary acidic protein (GFAP) (B) between patients with active major neuropsychiatric lupus (NPSLE), systemic lupus erythematosus (SLE) without active major NPSLE, and healthy controls. Y-axis is shown on a logarithmic scale.
Table 2.
| Patient | Age (years) | Concurrent Autoimmune Disease | Level of Suspicion for NPSLE | NPSLE Syndrome(s) | Time Interval from Initial NP Symptom Onseta | Time Interval from Last New or Worsening NP Symptom* | SLEDAI-2K Score (Without Neurologic Sub-Scores) | Pertinent Neurodiagnostic Testing | Blood, CSF NfL (pg/ml) | Blood, CSF GFAP (pg/ml) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 15 | Sjogren’s | Definite | Autonomic disorder, cranial neuropathy | <1 month | <1 month | 11 |
MRI: Mild cerebral/cerebellar atrophy, pachymeningitis CSF: Elevated CSF protein and unique CSF OCBs |
7.4 257.0 |
110.3 2,212.4 |
| 2 | 12 | None | Definite | Demyelinating disorder, myelopathy | <1 month | <1 month | 13 |
MRI: Longitudinally extensive transverse myelitis, grey-matter predominant CSF: Low glucose, elevated protein, unique CSF OCBs, and IgG index Blood: Anti-AQP4 antibody neg |
65.1 8,483.0 |
472.8 22,218.3 |
| 3 | 15 | Sjogren’s | Definite | Seizure disorder, moderate/severe cognitive dysfunction, mild mood disorder | 1–2 years | <1 month | 10 |
MRI: Normal CSF: Mild pleocytosis EEG: Diffuse and focal temporal slowing, bitemporal spikes |
13.3 | 75.2 |
| 4 | 15 | None | Definite | Aseptic meningitis, moderate/severe cognitive dysfunction, mild mood disorder | <1 month | <1 month | 10 |
MRI: Normal CSF: Moderate pleocytosis, elevated protein |
449.3 8,543.0 |
784.5 88,023.8 |
| 5 | 51 | APLSb | Probable | Stroke (ischemic, hemorrhagic), mild mood disorder | 6–12 months | <1 month | 12 |
MRI: Multifocal cerebral white matter and cortical damage, with microhemorrhages. Small focus of parenchymal enhancement. MRA: Luminal irregularities of the intracranial arteries, multifocal. |
34.0 1,908.1 |
178.7 9,443.5 |
| 6 | 23 | APLSb | Definite | Stroke (ischemic) | 6 months | 6 months | 2 |
MRI: Multifocal areas of acute infarction and microhemorrhages in the brain MRA: Luminal irregularities of the intracranial arteries, multifocal. |
144.1 2,495.9 |
211.0 6,060.4 |
| 7 | 30 | None | Definite | Demyelinating disorder | <1 month | <1 month | 4 |
MRI: T2 hyperintensity in dorsal medulla CSF: Low glucose, +antineuronal antibodies Blood: Anti-AQP4 antibody neg. |
29.9 1,534.6 |
112.4 9,800.6 |
| 9 | 55 | Sjogren’s | Probable | Autonomic disorder, polyneuropathy, moderate/severe cognitive dysfunction | 5–6 years | <1 month | 2 |
MRI: Normal CSF: Elevated unique CSF OCBs EMG/NCS: Sensorimotor polyneuropathy |
11.9 612.8 |
80.0 8,588.6 |
| 10 | 24 | APLS, Sjogren’s | Probable | Aseptic meningitis, sensory neuropathy, optic neuritis | <1 month | <1 month | 5 |
MRI: Normal (orbits not done) CSF: Elevated protein and IgG Index Blood: Anti-AQP4 neg EMG/NCS: Normal |
9.0 | 13.1 |
| 11 | 37 | Sjogren’s | Definite | Headache, stroke (ischemic) | <1 month | <1 month | 4 |
MRI: Multifocal acute and subacute infarcts CSF: Mild pleocytosis, borderline IgG index |
29.5 | 16.9 |
| 13 | 20 | None | Probable | Headache, stroke (hemorrhagic), mild cognitive dysfunction | 2–3 months | 1–2 months | 12 |
MRI: Small subarachnoid hemorrhage CSF: Elevated unique CSF OCBs |
5.8 | 53.9 |
| 14 | 18 | APLS | Definite | Stroke (ischemic), mononeuritis multiplex | 2–3 months | <1 month | 6 |
MRI: Multifocal acute and chronic infarcts EMG/NCS: Sensory greater than motor axonal changes, asymmetric |
245.6 | 276.7 |
| 15 | 33 | None | Definite | Headache, transverse myelitis | 3–4 months | 3–4 months | 2 |
MRI: Longitudinally extensive, patchy T2 cord signal abnormality CSF: Elevated opening pressure, low CSF glucose, elevated CSF protein and neutrophilic pleocytosis Blood: Anti-AQP4 antibody neg |
51.7 | 112.4 |
Until the time of sampling.
Diagnosed at time of presentation by presence of strokes and positive APLAs. AQP4 = aquaporin-4; APLA = antiphospholipid antibody; APLS = antiphospholipid antibody syndrome; CSF = cerebrospinal fluid; EEG = electroencephalogram; EMG/NCS = electromyography/nerve conduction studies; GFAP = glial fibrillary acidic protein; IgG = immunoglobulin G; MRA = magnetic resonance angiography; MRI = magnetic resonance imaging; neg = negative; NfL = neurofilament light; NP = neuropsychiatric; NPSLE = neuropsychiatric systemic lupus erythematosus; OCB = oligoclonal bands; SLEDAI = systemic lupus erythematosus disease activity index.
Correlation of Blood and CSF Biomarker Levels
Log-transformed blood and CSF NfL were significantly correlated for active major NPSLE, as shown in Supplementary Figure 1 (Pearson’s r = 0.88, p = 0.01). Log-transformed blood and CSF GFAP were also significantly correlated for active major NPSLE, as shown in Supplementary Figure 2 (Pearson’s r = 0.81, p = 0.03).
Comparison between Inflammatory and Ischemic NPSLE Phenotypes
Five of the NPSLE patients presented with strokes and a predominantly ischemic NPSLE phenotype, while eight presented with predominantly inflammatory NPSLE phenotypes (Table 2).44 The median blood NfL level for all 13 cases of NPSLE was 29.9 pg/ml. 40% of those with ischemic NPSLE and 50% of those with inflammatory NPSLE were below this median blood NfL level – this was not significantly different (p = 0.59). The median blood GFAP level for all 13 cases of NPSLE was 112.4 pg/ml. 40% of those with ischemic NPSLE and 50% of those with inflammatory NPSLE were below this median blood GFAP level – this was also not significantly different between groups (p = 0.59).
Comparison between NPSLE, SLE without NPSLE and Healthy Controls
Mean blood NfL was 3.6 pg/ml (standard deviation [SD] 2.0) for the non-SLE healthy controls. Mean blood NfL was 1.3 pg/ml higher for SLE without active major NPSLE compared to the non-SLE healthy controls (95% CI: 0.7, 2.5; p=0.42). Mean blood NfL was 17.9 pg/ml higher for cases of active major NPSLE than for SLE controls without active major NPSLE (95% CI: 9.2, 34.5; p <0.001). Every NPSLE case had a higher blood NfL level than its matched SLE disease control.
Mean blood GFAP was 50.4 pg/ml (SD 15.0) for the non-SLE healthy controls. Mean blood GFAP was 1.2 pg/ml higher for SLE without active major NPSLE compared to the non-SLE healthy controls (95% CI: 0.7, 2.0; p=0.53). Blood GFAP was on average 3.2 pg/ml higher for cases of active major NPSLE than for SLE controls without active major NPSLE (95% CI: 1.9, 5.5; p <0.001).
Distinguishing between NPSLE and SLE without NPSLE
The AUC for blood NfL for detection of active major NPSLE was 0.96, indicating an excellent ability to distinguish between active major NPSLE and SLE without active major NPSLE.47 Using the median blood NfL level for all SLE patients (6.8 pg/ml) as the threshold, blood NfL ≥ 6.8 pg/ml had a sensitivity and specificity of 92.3% for identifying active major NPSLE in the SLE patients.
The AUC for blood GFAP for detection of active major NPSLE was 0.77, indicating an acceptable ability to distinguish between active major NPSLE and SLE without active major NPSLE.47 Using the median blood GFAP level for all SLE patients (70.7 pg/ml) as the threshold, blood GFAP ≥ 70.7 pg/ml had a sensitivity and specificity of 76.9% for identifying active major NPSLE in the SLE patients.
Longitudinal Trends after Immunotherapy
An exploratory analysis of longitudinal trends in the blood-based biomarkers after initiation of immunotherapy was performed with 6 patients with active major NPSLE who had biosamples from two timepoints (Figure 2). The interval between the first and second time points was highly variable (median 430.5, range 112 – 1490 days), as was the time of the first sample after initial immunotherapy (median 11.5, range 0 – 188 days). In all cases, both NfL and GFAP trended downward over time. However, for the two patients with ischemic NPSLE presentations they remained elevated compared to SLE without NPSLE patients several years after diagnosis and treatment, whereas for the inflammatory NPSLE presentations the biomarker levels at the second time point were close to or at the range seen in SLE without NPSLE.
Figure 2.
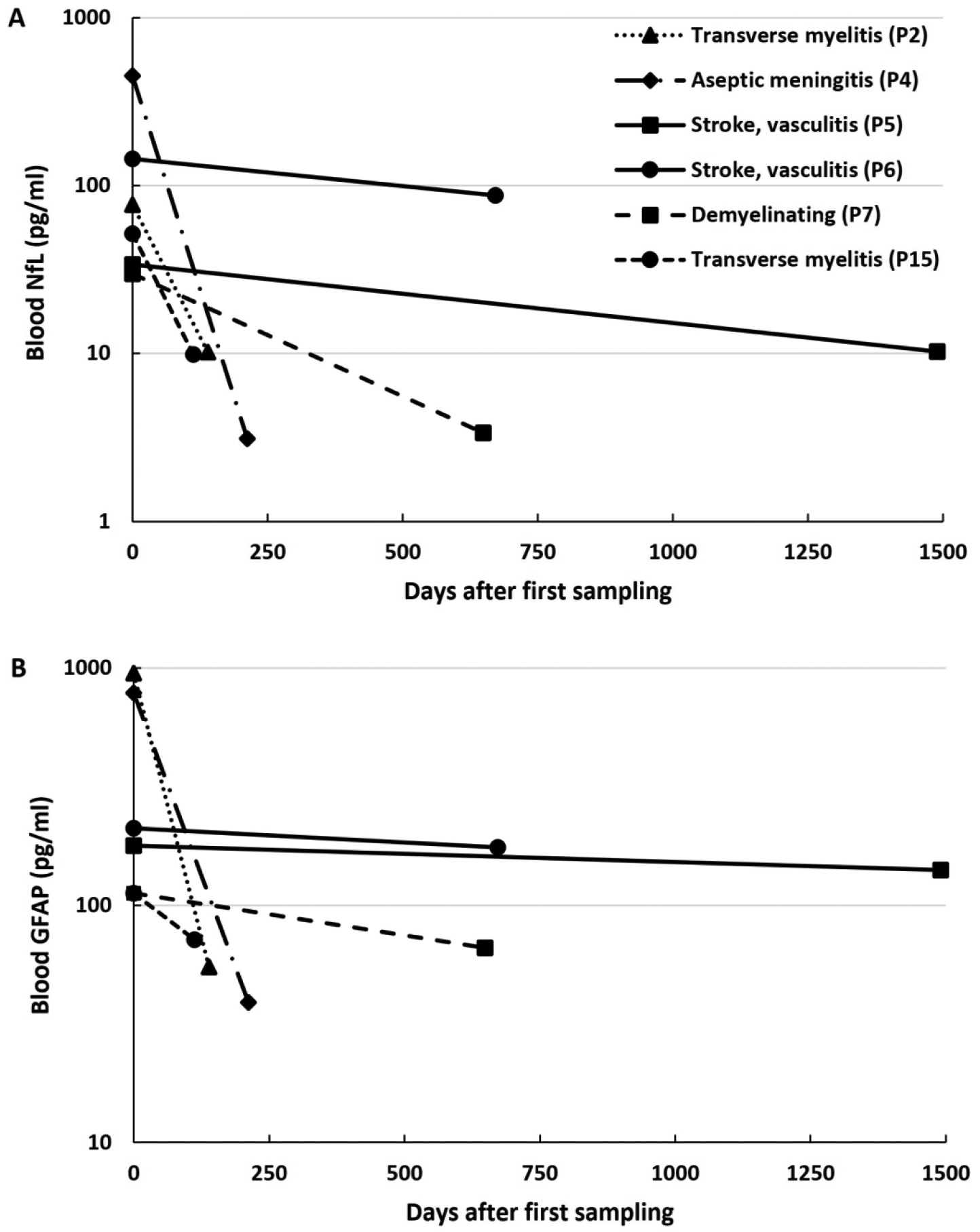
Longitudinal trends of plasma neurofilament light (NfL) (A) and glial fibrillary acidic protein (GFAP) (B) in active major neuropsychiatric lupus after immunotherapy treatment. Y-axis is shown on a logarithmic scale.
The interval immunotherapy treatments between samples for each patient included: Patient 2 - intravenous (IV) steroids followed by oral steroid taper, IV cyclophosphamide (CYC) every 2 weeks for 5 doses, two doses of IV rituximab, IV immunoglobulin, and 5 sessions of plasma exchange; Patient 4 – IV steroids followed by oral steroid taper, IV CYC every 2 weeks for 6 doses followed by mycophenolate mofetil (MMF) maintenance therapy; Patient 5 – IV CYC monthly for 6 months followed by MMF maintenance therapy; Patient 6 – continuation of maintenance rituximab, oral steroid taper; Patient 7 – IV CYC monthly for 6 months, followed by MMF maintenance therapy, oral steroid taper, and rituximab initiation and maintenance; and Patient 15 – completed IV CYC monthly for 3 months, followed by azathioprine maintenance therapy and oral steroid taper. By the second time point, neurologic symptoms had improved for Patients 2, 4, 5, and 15, and had fully resolved for Patients 6 and 7.
Discussion
To the best of our knowledge, this is the first study to demonstrate that blood NfL and GFAP concentrations are elevated in active major NPSLE compared to SLE without active major NPSLE, with blood NfL in particular having an excellent ability to distinguish between the two. This study is also the first to evaluate blood GFAP in the SLE population and to evaluate these biomarkers in a cohort that includes adolescent patients. Blood and CSF biomarker levels were significantly correlated, similar to that found in prior cross-sectional studies of adults with SLE.34, 35 Both blood NfL and GFAP are decreased following immunotherapy treatment, although persistent significant elevations may continue years after treatment even after resolution of NP symptoms. There was not a significant difference between biomarker levels for ischemic and inflammatory NPSLE. Our results suggest that in the right context, these blood-based biomarkers may serve a role as easily obtainable markers of end-organ nervous system damage in SLE to aid in NPSLE evaluation along with traditional neurodiagnostic testing, such as magnetic resonance imaging (MRI), electroencephalogram, and cerebrospinal fluid (CSF) analysis.
Our study complements prior studies and helps clarifies the relationship of these biomarkers with NPSLE. One retrospective cohort study noted higher serum NfL levels in patients with focal CNS NPSLE compared to those with SLE without NPSLE. However, in a multiple linear regression model, age and serum creatinine concentration were strong significant predictors of serum NfL levels, while presence of NP symptoms was not.33 A cross-sectional study noted higher plasma NfL in patients with current cognitive dysfunction, mood disorder, or polyneuropathy compared to patients with past NP manifestations. However, these symptoms were not new or recent in onset and the association did not remain significant after correction for multiple comparisons.34 Another cross-sectional study noted higher plasma NfL levels in persons with SLE compared to age/sex matched healthy controls, but no difference between those with SLE with or without NPSLE. This study had fewer patients with recent or “focal” NPSLE compared to the other studies, although one patient with recent myelitis had the highest plasma NfL level in their study. However, the study was not designed to look at active or acute NPSLE; the authors noted the need for future work to assess NfL in acute NPSLE.35
In our study, we focused on active NPSLE using a case-control design instead of retrospective cohort or cross-sectional designs. The strength of this approach is that we were able to identify cases with well-characterized and active disease, report the specific timing of biosamples relative to symptom onset to assess those earlier in their course of NPSLE, and control through matching for some of the major and most common potential confounders for blood NfL levels noted in prior studies in SLE or populations without neurologic disease (age and kidney function).29, 33–35 By focusing on major NPSLE syndromes, where NPSLE may be most apparent or most likely due to direct systemic SLE activity6, 7 as well as by performing multi-disciplinary adjudication, we also increased our confidence that our cases were truly cases of active major NPSLE.
While both blood NfL and GFAP were significantly elevated in active major NPSLE compared to SLE without NPSLE, this elevation was most pronounced for blood NfL. Autoantibodies have also been evaluated in multiple studies for their role in the pathogenesis of NPSLE, with a recent study demonstrating that brain-reactive autoantibodies in NPSLE are directed against neuronal targets.48 This may be part of the reason for the blood NfL findings in this study – if autoantibodies are being directed towards neuronal targets and causing neuronal injury, the levels would be expected to increase more than blood GFAP levels. However, an impaired blood brain barrier or injury from ischemia in cases of ischemic NPSLE may also contribute to levels, and further work would be needed to evaluate the relationship of the biomarker levels with specific autoantibodies in active major NPSLE and its phenotypes.
Our study did not note a difference in NfL levels between persons with SLE without NPSLE and age/sex matched healthy controls, in contrast to a prior study in which SLE patients had higher levels.35 This may be due to the lower median SLE disease duration in our study compared to theirs (4.6 vs 10 years), though prior studies have conflicting results on whether a correlation exists between NfL and SLE disease duration.33, 35 It may also be that our study was underpowered to detect a difference between these groups.
In clinical practice, the outcome of NPSLE is best if evaluated, diagnosed, and treated quickly. However, NPSLE may be difficult to diagnose, especially if initial neurodiagnostic testing is inconclusive or only minor symptoms are present, which have a higher chance of being caused by non-SLE factors. A biomarker that could 1) complement neurodiagnostic testing at initial presentation, 2) assist in distinguishing between less prominent or conclusive features of NPSLE and NP symptoms from non-SLE etiologies, or 3) predict the short-term risk of developing NPSLE would have a great impact in improving early diagnosis and treatment of NPSLE. The simple threshold used here for blood NfL did provide high sensitivity and specificity for detection of active major NPSLE in this SLE cohort, but is limited by lack of standard collection intervals and the low number of patients studied. In other autoimmune neuropsychiatric diseases such as autoimmune encephalitis, elevations in blood NfL may be seen independent of abnormalities on standard CSF or MRI studies,22, 28 and blood NfL is able to distinguish autoimmune encephalitis from primary psychotic disorders.49 This gives promise for similar utility in SLE and NPSLE; however, given the lack of specificity of the biomarkers to SLE, they may not be able to distinguish between primary NPSLE and causes of secondary NPSLE or NP symptoms from non-SLE related causes if these other causes are expected to result in neuroinflammation or neurologic injury. Future studies may need to evaluate individual primary NPSLE syndromes and carefully matched mimicking conditions to determine if accurate differentiation between these is possible with biomarker thresholds alone or in conjunction with other clinical attribution criteria.10 Given the high sensitivity of blood NfL in this study for NPSLE though, a blood NfL level below the threshold may assist in excluding NPSLE or other mimicking conditions that cause injury. While this study represents a first step by showing the elevations of these biomarkers and their potential in definite or probable active major NPSLE, it is not able to assess the utility of these biomarkers for the 3 impactful goals above or assess the correlation of these biomarkers with CSF or MRI abnormalities. These will need to be the focus of future studies in SLE and NPSLE.
Persistent NP symptoms post-treatment for NPSLE may result in additional or repeated neurodiagnostic assessments, clinical monitoring and/or additional treatments. An easily obtainable biomarker that correlates with therapeutic response or resolution of neurodiagnostic abnormalities would be a useful tool for assessing if persistent symptoms were due to partially or undertreated active NPSLE, compared to chronic sequelae of the injury sustained during the episode of NPSLE. Our exploration of biomarker trends after immunotherapy noted all levels decreased over time and is consistent with literature in other neurologic diseases21, 22 suggesting blood NfL and GFAP should be evaluated in larger longitudinal studies as possible therapeutic biomarkers in NPSLE. Our study showed potential differences between biomarker trajectories in ischemic and inflammatory NPSLE, consideration should be given to evaluating these NPSLE subtypes separately.
The uncertainty in true prevalence of NPSLE has hindered research into diagnostic strategies, biomarker identification, and treatment outcomes for NPSLE – if there is variability in what is grouped under NPSLE, then studies on aspects of NPSLE will be expected to also have variable results with a lack of generalizability. Given that blood NfL and GFAP were elevated in active major NPSLE syndromes, future studies should further examine the role and clinical implication of these biomarkers in both major and minor NPSLE syndromes and describe the clinical profile of patients with elevated biomarkers but with less prominent NP symptoms.
Limitations
This study has several limitations. First, the generalizability of this study’s findings may be limited. This study was conducted at a single academic center with a small sample size, and the study population may not be representative of the greater SLE population. For example, kidney function is known to alter blood NfL levels, both in the general population and in adult SLE, but no patients in our study had impaired renal function.29, 33–35 Our study was small, limiting our ability to adjust for potential confounders. Instead, we matched cases and controls using three demographic and clinical factors known to most influence blood NfL levels (age, sex, and kidney function), minimizing these confounding factors. The small sample size may have also limited our ability to detect differences in demographic or clinical variables between groups, as well as the differences between ischemic and inflammatory NPSLE. We also only had CSF samples for a subset of patients with NPSLE, limiting the ability to assess correlation of blood and CSF biomarker levels or correlation with other markers of CSF inflammation.
We did not exclude persons with SLE with positive anti-phospholipid antibodies or secondary Sjogren’s disease, each of which may independently cause NP symptoms. When co-occurring, SLE is a likely driver of the inflammation and antibody generation for these secondary autoimmune diseases in addition to contributing to the NP symptoms independently through multiple other systemic mechanisms.1, 4 When causing neurologic symptoms, each of these diseases are also treated with immunotherapy. Therefore, we did not exclude these patients. Studying a single NPSLE-related syndrome is difficult, as these syndromes frequently overlap or co-exist, and each individual syndrome is significantly rarer than as a group. Therefore, for this initial study we chose to group our patients based on major NPSLE features where there is higher suspicion that SLE is directly driving the underlying pathophysiology.6, 7 We hypothesized that many if not all of these major syndromes would have a common end pathway – neuronal injury or glial injury/activation – that would be detected by our choice of biomarkers. We did not have cases for every major NPSLE syndrome, however, such as psychosis. We also were not able to compare to causes of secondary NPSLE or non-SLE related major NP symptoms that may occur in persons with SLE – such as overlapping multiple sclerosis or neuromyelitis optica, steroid-induced neuropathy, etc. Future studies will need to evaluate if it is possible to distinguish between these causes based solely on the level of neurologic or glial injury present and measured, or in conjunction with other clinical attribution criteria.10
We chose to define active NP syndromes as 6 months or less from the last new or worsening NP symptom. However, depending on the individual NPSLE syndrome, whether ischemic or inflammatory phenotype, or the exact timing of the samples within those 6 months, there may be a wide variability in the biomarker concentrations. Elevations in NfL and GFAP are time-dependent after injury. Even after more homogeneous and monophasic diseases such as stroke or traumatic brain injury, there is variability in reported time to peak NfL levels (between 7 days and 3 months) and the trajectory after a peak (returning to normal within 1 year, or remaining significantly elevated for years after).40–42 For GFAP, a peak is expected within a few days, and a half-life of 24–48 hours is estimated.43 It is not clear if these kinetics would also hold true in NPSLE, where injury may occur episodically along a more prolonged period of time, and where the SLE disease and systemic inflammation may alter the permeability of the blood brain barrier.4 Therefore, variability in the timing of the sample collections in our study due to patient presentation may have also contributed to variability in levels. Kinetic analyses of these biomarkers over standardized intervals in future studies would be ideal, as a biomarker’s clinical utility will be greatest if it is predictive of NPSLE at initial presentation rather than at a delayed peak.
Given the rarity of active major NPSLE, we chose to include serum or plasma biosamples from both prospective and retrospective studies. Studies looking at serum and plasma NfL or GFAP note high correlation in concentrations between blood sample types, and for our three serum samples we converted them to plasma equivalents using 144 internal healthy control samples with a conversion that was more conservative than published equations.50, 51 As no conversion between serum and plasma GFAP has been published, we used a similar internal procedure for GFAP. The converted plasma-equivalent levels for both NfL and GFAP for these samples were lower than the serum levels and did not greatly impact the significance or magnitude of our findings for the active major NPSLE group. Variability from several samples being stored initially at −20° before transfer to −80°C is thought to be minimal given the known pre-analytic stability of these biomarkers.46, 52 Future studies would benefit from minimizing differences in blood sample type or storage, or from external validation of the method of conversion between serum and plasma samples.
Lastly, the gold standard of diagnosis for NPSLE remains multidisciplinary consensus. In our study, we adjudicated each NPSLE case and SLE control carefully with this gold standard to increase confidence in identification of true cases or controls. However, even experts are noted to have difficulties with accurate diagnosis,13 and these may not have been adjudicated appropriately. Despite this, there was still a significant difference seen between biomarker levels for the two SLE groups.
Future Directions
Larger, prospective, and multi-center studies are needed to further assess the clinical utility of these biomarkers in the SLE population. These studies should provide increased assessment of potential confounders to these biomarker levels that are common in the SLE population, such as kidney function or systemic SLE disease activity. They should also evaluate these biomarkers within NPSLE subgroups (such as between major or minor syndromes, CNS and PNS syndromes, or inflammatory vs ischemic pathophysiologies), individual NPSLE syndromes, or in cases with less prominent clinical NP symptoms or inconclusive diagnostic testing. Longitudinal studies may provide informative data including trends and peak elevation after onset of NP symptoms and after initiation of treatment, therefore allowing us to assess whether these biomarkers can be a reliable early marker of disease activity and of treatment response, and provide an optimal threshold to maximize sensitivity and specificity of these biomarkers for detecting active major NPSLE.
Conclusions
Blood NfL and GFAP are elevated in active major NPSLE compared to SLE controls without active major NPSLE and to healthy controls. In a small subset with longitudinal samples, these biomarkers were observed to trend downward after immunotherapy treatment was initiated. Larger, longitudinal and multi-center studies evaluating the clinical utility of these biomarkers in assisting in the diagnosis, prediction, and monitoring of NPSLE are needed.
Supplementary Material
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported prospectively by the University of Colorado Neurology Intradepartmental Grant; and REDCap use in clinical data collection supported by NIH/NCATS Colorado CTSA Grant Number UM1 TR004399. Secondary use was made of data collected through funding by the National Institutes of Health (K23AR070897); the CARRA/LFA award; the Rocky Mountain Multiple Sclerosis Center; and the University of Colorado Neurology departmental funds. Contents are the author’s sole responsibility and do not necessarily represent official NIH views.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of the article.
Contributor Information
Ryan Kammeyer, Departments of Pediatrics and Neurology, University of Colorado School of Medicine.
Kimberly Chapman, Department of Neurology, University of Colorado School of Medicine.
Anna Furniss, Adult and Child Center for Outcomes Research and Delivery Science (ACCORDS), University of Colorado School of Medicine.
Elena Hsieh, Department of Pediatrics, Section of Allergy and Immunology; Department of Immunology and Microbiology, University of Colorado School of Medicine.
Robert Fuhlbrigge, Department of Pediatrics-Rheumatology, University of Colorado School of Medicine.
Ekemini A. Ogbu, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH, USA; University of Cincinnati College of Medicine, Cincinnati, OH, USA; Johns Hopkins University School of Medicine, Baltimore, MD, USA
Susan Boackle, Physician-Scientist, Rheumatology.
JoAnn Zell, Department of Medicine-Rheumatology, University of Colorado School of Medicine.
Kavita V Nair, Department of Neurology, University of Colorado School of Medicine.
Tyler L Borko, Department of Neurology, University of Colorado School of Medicine.
Jennifer C Cooper, Department of Pediatrics-Rheumatology, University of Colorado School of Medicine.
Jeffrey L Bennett, Departments of Neurology and Ophthalmology, Programs in Neuroscience and Immunology, University of Colorado School of Medicine.
Amanda L Piquet, Department of Neurology, University of Colorado School of Medicine.
References
- 1.Govoni M and Hanly JG. The management of neuropsychiatric lupus in the 21st century: still so many unmet needs? Rheumatology (Oxford) 2020; 59: v52–v62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Govoni M, Bombardieri S, Bortoluzzi A, et al. Factors and comorbidities associated with first neuropsychiatric event in systemic lupus erythematosus: does a risk profile exist? A large multicentre retrospective cross-sectional study on 959 Italian patients. Rheumatology (Oxford) 2012; 51: 157–168. 20111110. [DOI] [PubMed] [Google Scholar]
- 3.Zhu TY, Tam LS, Lee VW, et al. Systemic lupus erythematosus with neuropsychiatric manifestation incurs high disease costs: a cost-of-illness study in Hong Kong. Rheumatology (Oxford) 2009; 48: 564–568. 20090305. [DOI] [PubMed] [Google Scholar]
- 4.Fanouriakis A, Boumpas DT and Bertsias GK. Pathogenesis and treatment of CNS lupus. Curr Opin Rheumatol 2013; 25: 577–583. [DOI] [PubMed] [Google Scholar]
- 5.Zirkzee EJ, Huizinga TW, Bollen EL, et al. Mortality in neuropsychiatric systemic lupus erythematosus (NPSLE). Lupus 2014; 23: 31–38. 20131115. [DOI] [PubMed] [Google Scholar]
- 6.Bortoluzzi A, Scirè CA, Bombardieri S, et al. Development and validation of a new algorithm for attribution of neuropsychiatric events in systemic lupus erythematosus. Rheumatology (Oxford) 2015; 54: 891–898. 20141021. [DOI] [PubMed] [Google Scholar]
- 7.Ainiala H, Hietaharju A, Loukkola J, et al. Validity of the new American College of Rheumatology criteria for neuropsychiatric lupus syndromes: a population-based evaluation. Arthritis Rheum 2001; 45: 419–423. [DOI] [PubMed] [Google Scholar]
- 8.Ceccarelli F, Perricone C, Pirone C, et al. Cognitive dysfunction improves in systemic lupus erythematosus: Results of a 10 years prospective study. PLoS One 2018; 13: e0196103. 20180503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.ACR. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. In: Arthritis Rheum Apr 1999, pp. 599–608. [DOI] [PubMed] [Google Scholar]
- 10.Bortoluzzi A, Fanouriakis A, Appenzeller S, et al. Validity of the Italian algorithm for the attribution of neuropsychiatric events in systemic lupus erythematosus: a retrospective multicentre international diagnostic cohort study. BMJ Open 2017; 7: e015546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monov S and Monova D. Classification criteria for neuropsychiatric systemic lupus erythematosus: do they need a discussion? Hippokratia 2008; 12: 103–107. [PMC free article] [PubMed] [Google Scholar]
- 12.Zardi EM, Giorgi C and Zardi DM. Diagnostic approach to neuropsychiatric lupus erythematosus: what should we do? Postgrad Med 2018; 130: 536–547. 20180709. [DOI] [PubMed] [Google Scholar]
- 13.Magro-Checa C, Zirkzee EJ, Beaart-van de Voorde LJJ, et al. Value of multidisciplinary reassessment in attribution of neuropsychiatric events to systemic lupus erythematosus: prospective data from the Leiden NPSLE cohort. Rheumatology (Oxford, England) 2017; 56: 1676–1683. [DOI] [PubMed] [Google Scholar]
- 14.Bortoluzzi A, Scirè CA and Govoni M. Attribution of Neuropsychiatric Manifestations to Systemic Lupus Erythematosus. Frontiers in Medicine 2018; 5. Mini Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tokunaga M, Saito K, Kawabata D, et al. Efficacy of rituximab (anti-CD20) for refractory systemic lupus erythematosus involving the central nervous system. Ann Rheum Dis 2007; 66: 470–475. 20061115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barile-Fabris L, Ariza-Andraca R, Olguín-Ortega L, et al. Controlled clinical trial of IV cyclophosphamide versus IV methylprednisolone in severe neurological manifestations in systemic lupus erythematosus. Ann Rheum Dis 2005; 64: 620–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stojanovich L, Stojanovich R, Kostich V, et al. Neuropsychiatric lupus favourable response to low dose i.v. cyclophosphamide and prednisolone (pilot study). Lupus 2003; 12: 3–7. [DOI] [PubMed] [Google Scholar]
- 18.Kampylafka EI, Alexopoulos H, Kosmidis ML, et al. Incidence and prevalence of major central nervous system involvement in systemic lupus erythematosus: a 3-year prospective study of 370 patients. PLoS One 2013; 8: e55843. 20130212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarwar S, Mohamed AS, Rogers S, et al. Neuropsychiatric Systemic Lupus Erythematosus: A 2021 Update on Diagnosis, Management, and Current Challenges. Cureus 2021; 13: e17969. 20210914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varhaug KN, Barro C, Bjørnevik K, et al. Neurofilament light chain predicts disease activity in relapsing-remitting MS. Neurol Neuroimmunol Neuroinflamm 2018; 5: e422. 20171128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abdelhak A, Foschi M, Abu-Rumeileh S, et al. Blood GFAP as an emerging biomarker in brain and spinal cord disorders. Nature Reviews Neurology 2022; 18: 158–172. [DOI] [PubMed] [Google Scholar]
- 22.Kammeyer R, Mizenko C, Sillau S, et al. Evaluation of Plasma Neurofilament Light Chain Levels as a Biomarker of Neuronal Injury in the Active and Chronic Phases of Autoimmune Neurologic Disorders. Front Neurol 2022; 13: 689975. 20220302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novakova L, Zetterberg H, Sundström P, et al. Monitoring disease activity in multiple sclerosis using serum neurofilament light protein. Neurology 2017; 89: 2230–2237. 20171027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piehl F, Kockum I, Khademi M, et al. Plasma neurofilament light chain levels in patients with MS switching from injectable therapies to fingolimod. Mult Scler 2018; 24: 1046–1054. 20170619. [DOI] [PubMed] [Google Scholar]
- 25.Chang X, Huang W, Wang L, et al. Serum Neurofilament Light and GFAP Are Associated With Disease Severity in Inflammatory Disorders With Aquaporin-4 or Myelin Oligodendrocyte Glycoprotein Antibodies. Front Immunol 2021; 12: 647618. 20210316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Godelaine J, De Schaepdryver M, Bossuyt X, et al. Prognostic value of neurofilament light chain in chronic inflammatory demyelinating polyneuropathy. Brain Commun 2021; 3: fcab018. 20210309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bischof A, Manigold T, Barro C, et al. Serum neurofilament light chain: a biomarker of neuronal injury in vasculitic neuropathy. Ann Rheum Dis 2018; 77: 1093–1094. 20170725. [DOI] [PubMed] [Google Scholar]
- 28.Mariotto S, Gajofatto A, Zuliani L, et al. Serum and CSF neurofilament light chain levels in antibody-mediated encephalitis. J Neurol 2019; 266: 1643–1648. 20190403. [DOI] [PubMed] [Google Scholar]
- 29.Fitzgerald KC, Sotirchos ES, Smith MD, et al. Contributors to Serum NfL Levels in People without Neurologic Disease. Annals of Neurology 2022; 92: 688–698. 10.1002/ana.26446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stocker H, Beyer L, Trares K, et al. Association of Kidney Function With Development of Alzheimer Disease and Other Dementias and Dementia-Related Blood Biomarkers. JAMA Network Open 2023; 6: e2252387–e2252387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tjensvoll AB, Lauvsnes MB, Zetterberg H, et al. Neurofilament light is a biomarker of brain involvement in lupus and primary Sjögren’s syndrome. J Neurol 2021; 268: 1385–1394. 20201030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trysberg E, Nylen K, Rosengren LE, et al. Neuronal and astrocytic damage in systemic lupus erythematosus patients with central nervous system involvement. Arthritis Rheum 2003; 48: 2881–2887. [DOI] [PubMed] [Google Scholar]
- 33.Engel S, Boedecker S, Marczynski P, et al. Association of serum neurofilament light chain levels and neuropsychiatric manifestations in systemic lupus erythematosus. Ther Adv Neurol Disord 2021; 14: 17562864211051497. 20211022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lauvsnes MB, Zetterberg H, Blennow K, et al. Neurofilament light in plasma is a potential biomarker of central nervous system involvement in systemic lupus erythematosus. J Neurol 2022; 269: 3064–3074. 20211120. [DOI] [PubMed] [Google Scholar]
- 35.Zervides KA, Janelidze S, Nystedt J, et al. Plasma and cerebrospinal fluid neurofilament light concentrations reflect neuronal damage in systemic lupus Erythematosus. BMC Neurology 2022; 22: 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petri M, Orbai AM, Alarcón GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012; 64: 2677–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol 2019; 71: 1400–1412. 20190806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khalil M, Pirpamer L, Hofer E, et al. Serum neurofilament light levels in normal aging and their association with morphologic brain changes. Nat Commun 2020; 11: 812. 20200210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abdelhak A, Petermeier F, Benkert P, et al. Serum neurofilament light chain reference database for individual application in paediatric care: a retrospective modelling and validation study. Lancet Neurol 2023. 20230728. [DOI] [PubMed] [Google Scholar]
- 40.Tiedt S, Duering M, Barro C, et al. Serum neurofilament light: A biomarker of neuroaxonal injury after ischemic stroke. Neurology 2018; 91: e1338–e1347. 20180914. [DOI] [PubMed] [Google Scholar]
- 41.Shahim P, Politis A, van der Merwe A, et al. Neurofilament light as a biomarker in traumatic brain injury. Neurology 2020; 95: e610–e622. 20200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez JD, Martirosian RA, Mun KT, et al. Temporal Patterning of Neurofilament Light as a Blood-Based Biomarker for Stroke: A Systematic Review and Meta-Analysis. Front Neurol 2022; 13: 841898. 20220516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thelin EP, Zeiler FA, Ercole A, et al. Serial Sampling of Serum Protein Biomarkers for Monitoring Human Traumatic Brain Injury Dynamics: A Systematic Review. Front Neurol 2017; 8: 300. 20170703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zirkzee EJ, Steup-Beekman GM, van der Mast RC, et al. Prospective study of clinical phenotypes in neuropsychiatric systemic lupus erythematosus; multidisciplinary approach to diagnosis and therapy. J Rheumatol 2012; 39: 2118–2126. 20120915. [DOI] [PubMed] [Google Scholar]
- 45.Prudencio M, Erben Y, Marquez CP, et al. Serum neurofilament light protein correlates with unfavorable clinical outcomes in hospitalized patients with COVID-19. Science Translational Medicine 2021; 13: eabi7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barro C, Chitnis T and Weiner HL. Blood neurofilament light: a critical review of its application to neurologic disease. Annals of Clinical and Translational Neurology 2020; 7: 2508–2523. 10.1002/acn3.51234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mandrekar JN. Receiver Operating Characteristic Curve in Diagnostic Test Assessment. Journal of Thoracic Oncology 2010; 5: 1315–1316. [DOI] [PubMed] [Google Scholar]
- 48.Cocco C, Manca E, Corda G, et al. Brain-reactive autoantibodies in neuropsychiatric systemic lupus erythematosus. Front Immunol 2023; 14: 1157149. 20230613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guasp M, Martín-Aguilar L, Sabater L, et al. Neurofilament Light Chain Levels in Anti-NMDAR Encephalitis and Primary Psychiatric Psychosis. Neurology 2022; 98: e1489–e1498. 20220210. [DOI] [PubMed] [Google Scholar]
- 50.Rübsamen N, Willemse EAJ, Leppert D, et al. A Method to Combine Neurofilament Light Measurements From Blood Serum and Plasma in Clinical and Population-Based Studies. Front Neurol 2022; 13: 894119. 20220614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huebschmann NA, Luoto TM, Karr JE, et al. Comparing Glial Fibrillary Acidic Protein (GFAP) in Serum and Plasma Following Mild Traumatic Brain Injury in Older Adults. Front Neurol 2020; 11: 1054. 20200918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Lierop ZYGJ, Verberk IMW, van Uffelen KWJ, et al. Pre-analytical stability of serum biomarkers for neurological disease: neurofilament-light, glial fibrillary acidic protein and contactin-1. 2022; 60: 842–850. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.