

Abstract
Dysregulated T cell activation underpins the immunopathology of rheumatoid arthritis (RA), yet the machineries that orchestrate T cell effector program remain incompletely understood. Herein, we leveraged bulk and single-cell RNA sequencing data from RA patients and validated protein disulfide isomerase family A member 3 (PDIA3) as a potential therapeutic target. PDIA3 is remarkably upregulated in pathogenic CD4 T cells derived from RA patients and positively correlates with C-reactive protein level and disease activity score 28. Pharmacological inhibition or genetic ablation of PDIA3 alleviates RA-associated articular pathology and autoimmune responses. Mechanistically, T cell receptor signaling triggers intracellular calcium flux to activate NFAT1, a process that is further potentiated by Wnt5a under RA settings. Activated NFAT1 then directly binds to the Pdia3 promoter to enhance the expression of PDIA3, which complexes with STAT1 or PKM2 to facilitate their nuclear import for transcribing T helper 1 (Th1) and Th17 lineage-related genes, respectively. This non-canonical regulatory mechanism likely occurs under pathological conditions, as PDIA3 could only be highly induced following aberrant external stimuli. Together, our data support that targeting PDIA3 is a vital strategy to mitigate autoimmune diseases, such as RA, in clinical settings.
Keywords: rheumatoid arthritis, effector T cell program, nuclear transport, PDIA3, non-canonical function
Graphical abstract
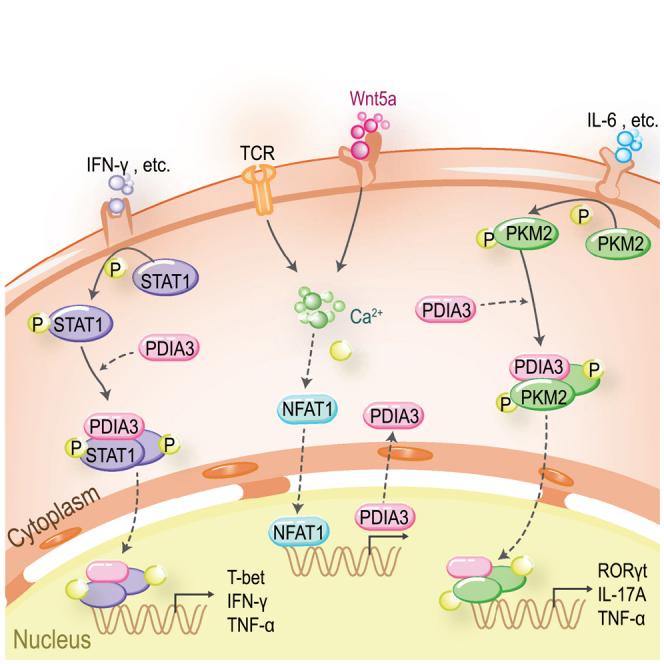
Wang and colleagues reveal that TCR and Wnt5a stimulation collectively trigger intracellular calcium flux to activate NFAT1 in CD4 T cells. NFAT1 promotes the transcription and expression of PDIA3, which potentiates Th1 and Th17 programs by facilitating the nuclear import of STAT1 and PKM2 under distinct polarizing conditions, respectively.
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disorder characterized by the synovium hyperplasia and joint destruction, which affects approximately 1% of the population worldwide.1,2 Despite the remarkable advancement in RA treatment, including the application of biologic agents (e.g., anti-tumor necrosis factor [TNF]-α) and disease-modifying drugs (DMARDs) to mitigate disease progression; approximately 20% of RA patients, however, fail to achieve satisfactory remission.3,4,5 Furthermore, the current therapies are commonly coupled with a heightened risk of infection, which limits their long-term use.6 These facts warrant an urgent need for the clarification of pathoetiologies underlying RA, which would enable the characterization of effective targets and the development of novel therapeutic strategies in clinical settings.
Effector CD4 T cells (Teffs) are the major culprits responsible for RA-related immune dysregulation; they secrete pro-inflammatory cytokines and coordinate the function of cytotoxic CD8 T cells, B cells, and myeloid cells.7,8 Naive CD4 T cells (Tn cells) skew into functionally distinct effector T cells, such as T helper 1 (Th1), Th2, and Th17, and regulatory T (Treg) cells, once they are exposed to a microenvironment enriched in certain particular cytokines.9,10,11 Among the diverse Teff subsets, Th1 and Th17 are found to be the two salient components in RA pathogenesis.12,13 Th1 cells are a major source of type 1 cytokines (interferon [IFN]-γ, TNF-α, and IL-1β), while Th17 cells produce large amounts of inflammatory mediators including IL-17, IL-23, granulocyte macrophage colony stimulating factor, and receptor activators of nuclear factor κ-B ligand.14 Those pro-arthritic factors collectively contribute to the hyperproliferation of fibroblast-like synoviocytes (FLSs) and the excessive activation of osteoclasts, leading to synovium swelling and bone erosion.15,16 Those findings rendered us to assume that some key modulators of the Teff program could play a critical role in predisposing to RA initiation and progression. To address this notion, we analyzed the bulk and single-cell RNA sequencing (scRNA-seq) data derived from peripheral blood mononuclear cells (PBMCs) of RA patients and interrogated the differentially expressed genes between naive and memory T cells, by which the protein disulfide isomerase family A member 3 (PDIA3) was noted to be highly enriched in pathogenic T cells relevant to RA severity.
PDIA3 was initially discovered as a stress-responsive molecule upregulated upon glucose deprivation, and later re-recognized as a PDI family member with redox regulation and chaperone activity.17 Endoplasmic reticulum (ER)-localized PDIA3 participates in the unfolded protein response by regulating the correct folding and quality control of newly synthesized glycoproteins through the interaction with calnexin and calreticulin.18,19 Previously, we demonstrated that SUMOylation of PDIA3 exacerbates proinsulin misfolding and ER stress in islet β cells.20 However, a considerable proportion of PDIA3 may escape ER retention and get access to other subcellular compartments, where it exerts non-canonical biological functions.21,22 PDIA3 could also be released into the circulation and function as a disease biomarker.23 Membrane-associated PDIA3 is an alternative receptor for the active form of 1, 25-dihydroxyvitamin D3 [1,25(OH)2D3], which mediates a rapid (seconds to minutes) ignition of calcium peak.24 Additionally, cytoplasmic PDIA3 assists in the assembly of the mammalian target of rapamycin C1 complex.25 Although the pathophysiological functions of PDIA3 have been investigated in several cell types, including neuronal cells, chondrocytes, and tumor cells,18,26,27 its immune regulatory roles in disease settings are less appreciated.27,28 Previous studies suggested feasible evidence that PDIA3 acts as an autoantigen involved in peptide-loading onto major histocompatibility complex-I molecules in antigen-presenting cells,28,29 while its impact on other immune components remains largely unexplored.
In this study, we characterized that arthritogenic CD4 T cells were featured by the marked upregulation of PDIA3, which was positively correlated with RA severity. Depletion of Pdia3 in CD4 T cells or administration of nitazoxanide (NTZ), an PDIA3 inhibitor approved by the U.S. Food and Drug Administration, mitigated joint destruction and autoimmune responses in collagen-induced RA mice. Moreover, studies in co-cultured T cells and adoptively transferred T cells revealed an intrinsic role for PDIA3 in boosting effector T cell program. Mechanistically, T cell receptor (TCR) signaling and Wnt5a stimulation converge on the calcium (Ca2+)-NFAT1 pathway, thereby enhancing PDIA3 expression. PDIA3 then acts as a chaperone to facilitate the nuclear import of STAT1 and PKM2, by which it confers heightened Th1 and Th17 program. Together, our data support that suppression of PDIA3 is likely to be a promising therapeutic strategy against RA and other autoimmune diseases.
Results
PDIA3 displays enhanced expression in arthritogenic CD4 T cells and positively correlates with RA severity
We first analyzed the bulk RNA-seq (GSE118829) and scRNA-seq (GSE159117) datasets of PBMCs derived from RA patients.30,31 Cells were classified into 13 distinct clusters based on the differential expression pattern of specific markers (Figures 1A and 1B) in dataset GSE159117. Indeed, a marked discrepancy in terms of PDIA3 expression between Tn cells and effector memory CD4 T cells was unraveled through analysis of bulk RNA-seq data (Figures S1A and S1B). Moreover, scRNA-seq also revealed a biologically notable difference regarding the expression levels of PDIA3 between Tn (cluster 2) and memory T cells (cluster 4), although the difference did not reach a statistical significance due to the enormous heterogeneity brought by the sequencing approach (Figure S1C). CD4 T cells were next stratified into PDIA3high and PDIA3low subgroups based on PDIA3 expressions. As compared with the PDIA3low subgroup, the PDIA3high CD4 T cells were featured by the increased expression of Th1 signature genes, such as TBX21, IFNG, TNF, CXCR3, CXCR6, GBP2, and GBP3, along with higher levels of Th17 markers BCL6 and RORC (Figure 1C). To validate those findings, we determined PDIA3 expression in murine Tn cells following TCR stimulation and found a time-dependent upregulation of PDIA3 in TCR-stimulated T cells (Figure 1D). Furthermore, Th1 and Th17 cells manifested a preferential overexpression of Pdia3 as compared with that of Th2 cells, while Treg cells were characterized by the repressed Pdia3 expression as compared with that of Th0 cells (Figure 1E).
Figure 1.

PDIA3 is upregulated in arthritogenic CD4 T cells and positively correlates with RA severity
(A) UMAP on PBMCs from an RA patient. Cell type annotations were labeled for each cluster. DC, dendritic cell; MAIT, mucosal-associated invariant T cell; NK, natural killer cell; pDC, plasmacytoid dendritic cell. (B) Dot plot of key markers used to define the identified cell populations. The color of dot represents the average expression, while the size of the dot represents the percent expression. (C) Dot plot of hallmark genes in PDIA3low and PDIA3high CD4 T cells. (D) Western blots showing PDIA3 expression in murine CD4 T cells stimulated with anti-CD3/CD28 antibodies at the indicated time points. (E) Quantification of Pdia3 mRNA in different T cell subsets by qRT-PCR. (F) Western blot analysis of PDIA3 expression in CD4 T cells from 8- to 10-week-old control and CIA mice. (G) Representative western blot analysis of PDIA3 expression in CD4 T cells from RA patients and healthy controls. (H) Quantification of PDIA3 mRNA levels in CD4 T cells from healthy controls (HCs) (n = 21) versus those from RA patients (n = 43). (I and J) The correlation between PDIA3 mRNA expression levels in CD4 T cells from RA patients (n = 43) with CRP (I) and disease activity score 28 (DAS-28) (J). (K and L) Tn cells from WT mice were cultured in the presence or absence of 5 μM TIZ in Th1- or Th17-skewed conditions. Flow cytometry data for IFN-γ (K, left) and IL-17A (L, left) staining, and the percentages of IFN-γ+ Th1 (K, right) and IL-17A+ Th17 (L, right) cells. (M and N) PBMCs from RA patients were cultured in vitro for 48 h in the presence or absence of TIZ. CD4+IFN-γ+ (M) and CD4+IL-17A+ (N) T cells were then evaluated by flow cytometry. (O) The concentration of Wnt5a in the serum from healthy individuals (n = 8), RA patients (n = 15), as well as in the RA synovial fluid (n = 9) measured by ELISA. HC-S, sera from healthy controls; RA-S, sera from RA patients; RA-JF, joint fluid from RA patients. (P) The correlation between Wnt5a expression levels in the serum and PDIA3 expression levels in CD4 T cells from RA patients (n = 22). Data are expressed as mean ± SEM and images are representative of at least three independent experiments. Statistical significance was calculated by unpaired Student’s t test. The correlation determined by Pearson’s correlation analysis was for (I), (J), and (P). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. ns, not significant.
To further confirm the above data, we assessed PDIA3 expression in CD4 T cells derived from collagen-induced arthritic (CIA) or control mice. Indeed, CD4 T cells originating from CIA mice displayed significantly higher levels of PDIA3 expression (Figure 1F). We next checked PDIA3 expression in human samples and found that CD4 T cells from RA patients were characterized by increased PDIA3 expression at both protein and mRNA levels as compared with that of healthy controls (Figures 1G and 1H). Moreover, PDIA3 expression in CD4 T cells was noted to be positively correlated with both C-reactive protein (CRP) and disease activity score 28 (Figures 1I and 1J), the two indicative parameters for RA severity.
The above results prompted us to check whether PDIA3 inhibition modulates CD4 T cell polarization. To this end, tizoxanide (TIZ), the active metabolite of NTZ, was employed for the in vitro study.32 Addition of TIZ significantly attenuated the polarization of Tn cells toward Th1 and Th17 cells (Figures 1K and 1L). Moreover, treatment of arthritogenic CD4 T cells from RA patients with TIZ markedly decreased the proportion of IFN-γ+ Th1 cells and IL-17A+ Th17 cells (Figures 1M and 1N). These findings support that PDIA3 plays a critical role in the polarization of effector T cells and the maintenance of their functionality.
Given that Wnt5a was found to be a pivotal inflammatory mediator causing synovial destruction in RA patients,33,34 we further conducted an ELISA to assess circulating and synovial Wnt5a levels and their correlation with PDIA3 expression. As expected, serum Wnt5a levels were markedly higher than those of healthy controls, and the highest Wnt5a levels were detected in the synovial fluid from RA patients (Figure 1O). Intriguingly, a strong positive correlation was characterized between PDIA3 expression in CD4 T cells and circulating Wnt5a levels in RA patients (Figure 1P). These results suggest that PDIA3 can be combined with Wnt5a to serve as biomarkers for RA progression.
Ablation of PDIA3 hampers RA progression by restraining Teff program
Based on the above observations, CD4 T cell-specific Pdia3 knockout (KO) mice, in which the CD4Cre mice were backcrossed with the Pdia3flox mice to produce the CD4CrePdia3flox/flox mice (defined as KO thereafter), were generated.35 The KO mice and their control littermates were next subjected to collagen immunization for RA induction. The development of arthritis, as assessed by hind paw swelling, was monitored for 3 weeks. We observed decreased clinical scores and decreased edema of the hind paws in KO mice (Figures 2A and 2B). Histological staining confirmed lower inflammatory damage in the joints of KO mice (Figure 2C). Consistently, the circulating IFN-γ, IL-17A, and TNF-α levels were markedly decreased in KO mice as compared with control mice (Figures 2D–2F). The KO mice were characterized by the reduced IFN-γ-producing Th1 cells, lower IL-17A-producing Th17 cells, and increased CD4+CD25+Foxp3+ Treg cells as compared with the control littermates (Figures 2G–2I).
Figure 2.

Pharmacological inhibition or genetic ablation of PDIA3 protects mice against collagen-induced arthritis
(A) Clinical scores in WT and KO mice after collagen-induced arthritis. (B) Representative picture of the ankle of WT and KO mice at day 42 after CIA induction. (C) Hematoxylin and eosin (H&E) staining of the ankle section from the arthritic mice. (D–F) Plasma IFN-γ (D), IL-17A (E), and TNF-α (F) levels in WT and KO arthritic mice 42 days after CIA induction. (G–I) Axillary lymph nodes from WT and KO arthritic mice were harvested at the same time and subjected to flow cytometry analysis. The frequencies of CD4+IFN-γ+ (Th1) (G), CD4+IL-17A+ (Th17) (H), and CD4+Foxp3+ (Treg) (I) subsets are shown as representative dot plot graphs. (J) The scheme for the RA mouse model with NTZ administration. (K) Clinical scores in control and NTZ-treated mice after collagen-induced arthritis. (L) Representative picture of the ankle of the arthritic mice day 42 after CIA induction. (M) H&E staining of ankle sections from the arthritic mice. (N–P) Plasma IFN-γ (N), IL-17A (O), and TNF-α (P) levels in WT and KO arthritic mice on 42 days after CIA induction. (Q–S) Axillary lymph nodes from these arthritic mice were harvested and subjected to flow cytometry analysis. The frequencies of CD4+IFN-γ+ (Th1) (Q), CD4+IL-17A+ (Th17) (R), and CD4+Foxp3+ (Treg) (S) subsets are shown as representative dot plot graphs. Data are expressed as mean ± SEM (n = 5 per group) and images are representative of at least three independent experiments. Statistical significance was calculated by unpaired Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. ns, not significant.
To further test whether inhibition of PDIA3 attenuates RA progression in CIA mice, the mice were treated with NTZ by intragastrical administration every 3 days for four times beginning from day 22 right after the second collagen challenge (Figure 2J). Lower disease severity scores along with relieved hind paw swelling were found in the treatment group (Figures 2K and 2L). Histological examination showed mitigated damage of joint synovium in NTZ-treated mice (Figure 2M). Furthermore, we examined pro-inflammatory cytokines in the serum of RA mice and found that the treatment group had significantly lower levels of IFN-γ, IL-17A, and TNF-α (Figures 2N–2P). Given the central role of CD4 T cells in RA pathogenesis, we checked T cell profiles in the axillary lymph nodes and detected reduced IFN-γ-producing Th1 and IL-17A-producing Th17 cells, coupled with increased Foxp3+ Treg cells in NTZ-treated mice (Figures 2Q–2S). Collectively, these findings support that pharmacological inhibition or genetic ablation of PDIA3 defers RA progression by impairing the Th1 and Th17 program.
Pdia3 deficiency intrinsically impairs Th1 and Th17 program
Next, we investigated how PDIA3 affects the functionality of CD4 T cells. Of note, no discernible phenotypic alterations in terms of common immunological parameters were observed in KO mice under physiological conditions. Specifically, the sizes of the thymus, spleen, and lymph nodes were comparable between the KO and control mice (Figure 3A). We then examined CD4 T cell proliferation and apoptosis, the two biological processes essential for T cell survival. Tn cells were traced by carboxy-fluorescein succinimidyl amino ester (CFSE) labeling or stained with Ki67 to check their proliferative capability. There was no difference in terms of proliferation between the KO and control CD4 T cells (Figures 3B and 3C). PDIA3 also failed to show a perceptible impact on the apoptosis level of CD4 T cells, as evidenced by a comparable percentage of live cells between the KO and control cells (Figure 3D). Similarly, no discernable difference was observed for the number and subsets of splenic CD4 T cells (Figures S2A, S2B, and S2D–S2F), as well as their activation status (Figure S2C). Those data suggest that PDIA3 might specifically regulate the Teff program upon polarizing stimulation or pathological insults.
Figure 3.

Pdia3 deficiency intrinsically impairs Th1 and Th17 program
(A) Representative pictures of the thymus, mesenteric lymph nodes (mLN), and spleen from 8- to 12-week-old WT and KO mice. (B–F) Splenic Tn cells isolated from WT and KO mice aged 10 weeks were activated with plate-coated anti-CD3/CD28 (10 μg/mL) antibodies for 72 h. (B and C) The percentage of proliferated CD4 T cells was defined by Ki67 staining (B) and CFSE assay (C). (D) Apoptosis of T cells was determined by Annexin V/PI staining. (E and F) Expression of IFN-γ (E) and IL-17A (F) was examined in activated CD4 T cells. (G and H) Tn cells purified from WT and KO mice were cultured under Th1- or Th17-polarized conditions in vitro. Flow cytometry analysis of Th1 (G) and Th17 (H) cell frequencies. (I) The scheme for the co-culture experiments. (J) The percentage of proliferated CD4 T cells was defined by CFSE or cell-trace violet (CTV) staining. (K and L) The percentage of IFN-γ-producing Th1 cells (K) and IL-17A-producing Th17 cells (L) after co-culturing. The experiments were repeated at least three times.
To address the above issue, CD4 T cells were subjected to anti-CD3/CD28 stimulation in the presence of IL-2 (Th0 condition) for 48 h followed by flow cytometry and immunoblotting analysis. Pdia3 deficiency significantly attenuated the production of IFN-γ and IL-17A (Figures 3E and 3F). Interestingly, we noted a marked decrease in the uptake of the glucose analog 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino)-2-deoxyglucose (2-NBDG), along with reduced expression of GLUT1 and key glycolytic enzymes in PDIA3-deficient CD4 T cells (Figures S3A and S3C). In contrast, the absence of PDIA3 did not affect the uptake of fluorescently labelled palmitate (C16 BODIPY), as well as the expression levels of CD36 and CPT1α, suggesting a relatively limited impact on lipid metabolism (Figures S3B and S3C). Next, Tn cells were cultured under Th1 and Th17 conditions, and we noticed that Pdia3 deficiency substantially impaired Th1 and Th17 differentiation (Figures 3G and 3H). To dissect whether PDIA3 intrinsically impairs the CD4 T cell polarization, cell-trace violet-labeled KO (CD45.2) and CFSE-labeled wild-type (WT, CD45.1) Tn cells were mixed at a 1:1 ratio and then co-cultured under either Th1 or Th17 condition for 2–3 days (Figure 3I). Similar to the above findings, no perceptible difference was noted in cell proliferation (Figure 3J), yet the Pdia3-deficient CD4 T cells had an impaired capability to skew into Th1 and Th17 program, respectively (Figures 3K and 3L).
Next, Rag1−/− mice were employed to further explore the impact of PDIA3 on Th1 and Th17 program, as Rag1−/− mice lack functional T and B cells but are susceptible to colitis once they are adoptively transferred functional CD4 T cells.36,37 CD4 T cells from WT or KO mice were transferred into Rag1−/− recipients via tail vein injection (Figure 4A). The mice receiving KO cells displayed an improved body weight loss as compared with those mice transferred with WT cells (Figure 4B). Consistently, the KO cell recipients exhibited a reduced colon shortening and less pronounced destruction of intestinal villi than WT cell recipients (Figures 4C and 4D). Although both groups of mice displayed a comparable total number of CD4 T cells (Figure 4E), decreased proportions of Th1 and Th17 cells along with elevated Treg cells, however, were noted in the mesenteric lymph nodes of mice transferred with KO cells (Figures 4F–4H). In consideration of the indispensability of antigen-specific CD4 T cells in adaptive immune responses, we then inoculated KO mice and their littermate controls with ovalbumin (OVA)-conjugated Candida albicans (Figure 4I). In this infectious model, a plunge in I-Ab-restricted OVA-specific (tetramer+) CD4 T cells was found in the KO mice (Figure 4J). In particular, the KO mice exhibited a steep decrease in the productivity of OVA-reactive IFN-γ+ Th1 and IL-17A+ Th17 cells (Figures 4K and 4L). Together, these findings provide convincing evidence that PDIA3 intrinsically coordinates Th1 and Th17 effector programs.
Figure 4.

Pdia3-deficient CD4 T cells are comprised in Th1 and Th17 program in vivo
(A) The scheme for the adoptive transfer experiments. (B) The percentage of body weight change of WT and KO CD4 T cell recipients. (C) Representative colon images at day 9 after adoptive transfer and quantitative analysis of colon length. (D) Hematoxylin and eosin (H&E) staining of representative distal colon sections. (E–H) Mesenteric lymph nodes from WT and KO colitic mice were harvested and subjected to flow cytometry analysis. Frequencies of CD4+ T(E), CD4+IFN-γ+ (Th1) (F), CD4+IL-17A+ (Th17) (G), and CD4+Foxp3+ (Treg) (H) subsets are shown as representative dot plot graphs. (I) The scheme for C. albicans infection mouse model. (J–L) Splenocytes from infected WT and KO mice were analyzed by flow cytometry. Frequencies of CD4+ I-Ab OVA323-339 Tetramer+ CD4 T cells (J), CD4+ IFN-γ+ Th1 (K), and CD4+ IL-17A+ Th17 (L) subsets were shown as representative dot plot graphs. Data are expressed as mean ± SEM. Statistical significance was calculated by unpaired Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. ns, not significant.
The TCR/Wnt5a-Ca2+-NFAT1 axis promotes PDIA3 expression in CD4 T cells
We next sought to figure out the upstream pathways that regulate PDIA3 expression in CD4 T cells, especially under the setting of RA. Bioinformatic analysis of the Pdia3 promoter sequence identified a putative NFAT1 binding motif (−617 to −611, start codon as +1) (Figure 5A). Next, chromatin immunoprecipitation (ChIP) was conducted to enrich NFAT1-bound DNA fragments, and primers flanking the potential NFAT1-binding site on Pdia3 promoter were utilized for PCR analysis. The ChIP-PCR results unequivocally verified the binding of NFAT1 onto the Pdia3 promoter (Figure 5B). To further corroborate the above finding, a WT luciferase reporter plasmid containing the Pdia3 promoter and a mutant plasmid (MUT) harboring the NFAT1 binding motif-depleted Pdia3 promoter were constructed. Luciferase reporter assays revealed that cells co-transfected with NFAT1 and MUT Pdia3 exhibited much lower luciferase signal as compared with those co-transfected with NFAT1 and WT counterparts (Figure 5C), supporting the role of NFAT1 as an upstream transcription factor of Pdia3. Moreover, RA-derived CD4 T cells were featured by the increased NFAT1 expression, which manifested a robust positive correlation with PDIA3 expression (Figures S4A and S4B). There is evidence that NFAT1 also transduces signaling elicited by environmental cues and Wnt ligands,38 other than acting as a mediator of TCR signaling. Given that Wnt5a was found to be enriched in the joint fluid of RA patients (Figure 1O), and the circulating Wnt5a was positively correlated with PDIA3 levels (Figure 1P), we thus reasoned that Wnt5a synergizes with TCR signaling to promote PDIA3 expression in CD4 T cells. Indeed, Tn cells exhibited a heightened propensity to differentiate into Th1 and Th17 cells upon treatment with exogenously supplemented Wnt5a (Figures 5D and 5E). However, such augmenting effects were substantially abrogated following the KO of Pdia3 (Figures 5D and 5E). In addition, the expression level of PDIA3 and nuclear accumulation of NFAT1 in CD4 T cells were both enhanced by Wnt5a treatment in a dose-dependent manner (Figure 5F).
Figure 5.

PDIA3 is upregulated in CD4 T cells via the TCR/Wnt5a-Ca2+-NFAT1 axis
(A) The putative binding site in the Pdia3 promoter region for NFAT1 was predicted with a relative profile score threshold of 80%. (B) ChIP-PCR analysis of the NFAT1 binding activity to the Pdia3 promoter. (C) Luciferase reporter assays for the Pdia3 promoter conducted in HEK 293T cells. (D and E) Tn cells from WT and KO mice polarized to Th1(D) and Th17(E) cells in the presence of recombinant Wnt5a. (F) The expression of NFAT1 and PDIA3 was measured by immunoblotting in the presence of recombinant Wnt5a at the indicated concentrations. (G) The scheme to validate the upstream pathway of PDIA3. (H) The expression of NFAT1 and PDIA3 detected by Western blot analysis in the presence of recombinant Wnt5a and/or iNFAT1. (I–L) Tn cells from WT mice were skewed to Th1 (I and J) and Th17 (K and L) in the presence of recombinant Wnt5a and/or iNFAT1. (M and N) Flow cytometry analysis of calcium ions by detecting Fluo-4 (M) and Rhod-2 (N). (O) Western blot results of NFAT1 and PDIA3 expression in the presence of recombinant Wnt5a and/or EDTA. (P) The protein levels of NFAT1 and PDIA3 in the presence of recombinant Wnt5a and/or MSAB.
As Ca2+ flux is an important intermediate of both TCR and Wnt5a signaling, while β-catenin also mediates the classical pathway downstream of Wnt5a, inhibitors for each of the pathways, were, therefore, employed to address how TCR signaling synergizes with Wnt5a to drive PDIA3 expression (Figure 5G).39,40,41 In line with our expectation, administration of NFAT1 inhibitor (iNFAT1) attenuated Wnt5a-induced NFAT1 expression and its nuclear accumulation along with decreased PDIA3 expression (Figure 5H). Consistently, iNFAT1 potently repressed Wnt5a-potentiated IFN-γ and IL-17A production (Figures 5I–5L). Moreover, flow cytometry analysis uncovered elevated Ca2+ flux in the presence of TCR stimulation alone or in combination with recombinant Wnt5a as determined by Fluo-4 and RhoD-2 staining (Figures 5M and 5N). Once Ca2+ was chelated by EDTA, CD4 T cells expressed much lower levels of NFAT1 and PDIA3 with or without Wnt5a treatment, and NFAT1 nuclear translocation was almost completely abolished (Figure 5O). Importantly, the addition of MSAB, a β-catenin inhibitor, did not result in a perceptible impact on NFAT1 and PDIA3 expression following TCR and Wnt5a stimulation (Figure 5P). Altogether, the enhanced PDIA3 expression under RA settings can be ascribed to the synergistic effect of TCR and Wnt5a stimulation, which collectively activates the Ca2+-NFAT1 signaling pathway.
PDIA3 interacts with STAT1 in Th1 cells and PKM2 in Th17 cells
To dissect the mechanisms by which PDIA3 regulates Th1 response, WT and KO Tn cells were skewed to Th1 condition and then subjected to RNA deep sequencing. Comparative analysis identified a total of 7,353 differentially expressed genes, among which 3,374 were upregulated and 3,979 were downregulated in KO cells (Figure 6A). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses revealed that Th1 cell differentiation ranked among the top pathways of the differentially expressed genes (Figure 6B), and some of the signature genes were shown in the heatmap (Figure 6C), confirming the essential role of PDIA3 in Th1 polarization. Intriguingly, the TNF signaling pathway, a key pathway involved in the pathogenesis of RA, was also highly enriched (Figure 6B), and flow cytometry analysis confirmed that the Pdia3-deficient Th1 cells produced less TNF-α (Figure 6D). To identify the Th1 program related protein(s) acting downstream of PDIA3, we constructed a His-PDIA3 fusion protein to pulldown the interacting protein(s) in Th1 cells. A total of 274 targets were detected by mass spectrometry analysis. Among these captured prey proteins, STAT1, a critical transcription factor in Th1 differentiation, ranked among the top hits (Figure 6E).42 We performed molecular docking with Autodock to detect the potential interaction between PDIA3 and STAT1. There were multiple groups of residues adopted to form stable hydrogen bonds between them (Figure 6F), indicating the possible binding between the two proteins. Indeed, western blot analysis of co-immunoprecipitated products verified the interaction between PDIA3 and STAT1 (Figure 6G).
Figure 6.

PDIA3 interacts with STAT1 or PKM2 in effector T cells
(A–C) RNA-seq revealed differentially expressed genes (DEGs) in WT vs. KO Th1 cells. (A) Volcano plot showing DEGs between WT and KO Th1 cells. (B) KEGG pathway analysis on DEGs between WT and KO Th1 cells. (C) Representative heatmap results for selected DEGs between WT and KO Th1 cells. (D) Representative flow cytometry staining of INF-γ and TNF-α in differentiated Th1 cells from WT and KO mice. (E) Top 10 candidate binding partners of PDIA3 in Th1 cells. (F) Representative image of autodocking between PDIA3 and STAT1 protein. (G) Immunoprecipitation assay of the lysates of WT Th1 cells using anti-PDIA3 antibody. (H–J) RNA-seq revealed differentially expressed genes (DEGs) in WT vs. KO Th17 cells. (H) Volcano plot showing DEGs between WT vs. KO Th17 cells. (I) KEGG pathway analysis on DEGs between WT vs. KO Th17 cells. (J) Representative heatmap results for selected DEGs between WT vs. KO Th17 cells. (K) Representative flow cytometry results of IL-17A and TNF-α in differentiated Th17 cells from WT and KO mice. (L) Top 10 candidate binding partners of PDIA3 in Th17 cells. (M) Representative image of autodocking between PDIA3 and PKM2 protein. (N) Immunoprecipitation assay of the lysates of WT Th17 cells using anti-PDIA3 antibody. All in vitro studies were repeated at least three times. Data are expressed as mean ± SEM. Statistical significance was analyzed by unpaired Student’s t test. ∗∗p < 0.01.
Similarly, to dissect the mechanisms underlying PDIA3-mediated regulation of Th17 response, Th17-polarized WT and KO cells were harvested for RNA deep sequencing. A total of 980 differentially expressed genes were identified, with 180 and 800 mRNAs upregulated and downregulated in KO cells, respectively (Figure 6H). KEGG pathway enrichment analysis revealed that Th17 differentiation was the top-ranked pathway (Figure 6I). This was corroborated by the downregulation of hallmark genes related to the Th17 program in Pdia3-deficient cells (Figure 6J). Strikingly, the TNF signaling pathway was also identified in Th17 cells (Figure 6I), and flow cytometry analysis confirmed that the KO-derived Th17 cells produced less TNF-α (Figure 6K). These results provide transcriptomic evidence supporting the role of PDIA3 in Th17 program. Pulldown assays were then conducted in Th17 cells, and the resulting products were analyzed by mass spectrometry. PKM2, an essential regulator for Th17 differentiation, was identified as the most significant one (Figure 6L). Molecular docking results indicated a potential interaction between PKM2 and PDIA3 (Figure 6M), which was further corroborated by co-immunoprecipitation (Co-IP) assays (Figure 6N). Taken together, these results indicate that PDIA3 regulates Th1 and Th17 effector program, likely through interacting with STAT1 and PKM2, respectively.
PDIA3 serves as a chaperone to facilitate STAT1 and PKM2 nuclear import
PDIA3 is considered to primarily localize in the ER in various cell types, however, confocal microscopy of CD4 T cells demonstrated that PDIA3 predominantly resides in the nucleus and its presence increased over time following TCR stimulation (Figure 7A), suggesting a non-canonical role of PDIA3 in T cell function. Indeed, a nuclear localization signal (NLS) was predicted through the NLS_Mapper (https://nls-mapper.iab.keio.ac.jp/cgibin/NLS_Mapper_form.cgi), which is highly conserved across different species (Figure 7B). These observations prompted us to assume that PDIA3 carries its interacting partners for nuclear shuttling during the course of effector T cell polarization. To address this assumption, a Pdia3 mutant (PDIA3-MUT) was first employed for the study, in which lysine residues within the NLS region at position 493–500 were deleted (Figure 7C), as lysine residues are crucial for the nuclear import signals and their alterations would trap proteins within the cytoplasmic compartment.43 Indeed, the transduction of naive Pdia3-deficient CD4 T cells with retroviruses carrying PDIA3-WT constructs markedly promoted Th1 and Th17 program (Figures 7D–7G). In sharp contrast, the PDIA3-MUT-transduced naive Pdia3-deficient CD4 T cells were characterized by the significantly attenuated Th1 and Th17 program (Figures 7D–7G). These results support that PDIA3 regulates effector T cell program dependent on its nuclear localization.
Figure 7.

PDIA3 facilitates STAT1 and PKM2 nuclear import to favor the effector T cell program
(A) Confocal microscopy of PDIA3 (green) in Tn cells stimulated with anti-CD3/CD28 antibodies at the indicated time points. Scale bar, 10 μm. (B) Alignment of NLS sequences of PDIA3 among different species. (C) PDIA3 mutation with deletion of its NLS was established based on the NLS_mapper prediction. (D–G) Tn cells were transduced with vector, PDIA3-WT, or PDIA3-MUT retroviruses under Th1- and Th17-polarizing conditions. Flow cytometry data for IFN-γ (D) and IL-17A (F) staining, and the percentages of IFN-γ+ Th1 (E) and IL-17A+ Th17 (G) cells. (H and I) WT and KO CD4 T cells were exposed to IL-12 (H) or IL-6 (I) for 6 h and then analyzed by native PAGE. (J) Western blot analysis of p-STAT1 in nuclear fraction from WT and KO CD4 T cells following IL-12 stimulation for 30 min. (K) Western blot analysis of p-PKM2 and p-STAT3 in the nuclear fraction from WT and KO CD4 T cells following IL-6 stimulation for 30 min. (L) HEK 293T cells were transfected with a Flag tag-labeled STAT1-Y701D plasmid combined with a His tag-labeled WT Pdia3 plasmid (PDIA3 WT-His) or a His tag-labeled mutant Pdia3 plasmid (PDIA3 MUT-His). Western blot analysis of PDIA3 and STAT1 in whole cell lysate and nuclear fraction, respectively. (M) HEK 293T cells were transfected with Flag tag-labeled PKM2-Y105D plasmid combined with PDIA3 WT-His or PDIA3 MUT-His, respectively. Western blot analysis of PDIA3 and PKM2 in whole cell lysate and nuclear fraction. (N) KO Tn cells were transduced with Vector, Retro-STAT1-Y701D or Retro-STAT1-Y701F and cultured under Th1 condition for 3 days. The frequencies of IFN-γ+ Th1 cells were shown in representative dot plots. (O) KO Tn cells were transduced with vector, Retro-PKM2-Y105D, or Retro-PKM2-Y105F and cultured under Th17 condition for 3 days. The frequencies of IL-17A+ Th17 cells are shown in representative dot plots. All in vitro studies were repeated at least three times.
To further address that PDIA3 mainly forms a cargo for STAT1 and PKM2 nuclear import, we first checked whether PDIA3 impacts their polymerization. The patterns of monomeric and dimeric STAT1 or monomeric, dimeric, and tetrameric PKM2 were identical between the PDIA3-null and WT CD4 T cells (Figures 7H and 7I), indicating that PDIA3 does not affect the formation of STAT1 or PKM2 polymers. Immunoblotting was next conducted to check their phosphorylation states. It was noted that Pdia3-deficient Th1 cells were manifested by the reduced phosphorylated STAT1 in the nucleus (Figure 7J). Unlike STAT1, PKM2 is upstream of STAT3 signaling in the Th17 program,44 but similar to STAT1 phosphorylation, Pdia3-deficient Th17 cells were featured by the decreased phosphorylation of both PKM2 and STAT3 in the nucleus (Figure 7K). Those observations rendered us to postulate that PDIA3 selectively binds to the activated (phosphorylated) STAT1 and PKM2 without affinity to the inactive form of STAT1 and PKM2. In contrast with our assumption, PDIA3 not only interacted with the constitutively active form of STAT1 (STAT1-Y701D) and PKM2 (PKM2-Y105D), but also with their inactive counterparts (Figures S5A and S5B).
The next key question is whether PDIA3 facilitates the nuclear import of STAT1 and PKM2. We first confirmed that PDIA3 lost nuclear localization once its NLS was disrupted (Figures 7L and 7M). Phosphorylation-active STAT1 and PKM2 were barely detectable in the nucleus of cells co-transduced with PDIA3-MUT as compared with those co-transduced with PDIA3-WT (Figures 7L and 7M). Of note, the inactive form of STAT1 (STAT1-Y701F) and PKM2 (PKM2-Y105F) were also detected in the nucleus (Figures S5C and S5D), implying that PDIA3 also facilitates the nuclear import of non-phosphorylated STAT1 or PKM2. However, only ectopic expression of the active form of STAT1 (STAT1-Y701D) rescued the Th1 program in Pdia3-deficient CD4 T cells, but not the inactive form of STAT1 (STAT1-Y701F) (Figure 7N); similar results of PKM2 were observed in Th17 program in Pdia3 KO CD4 T cells (Figure 7O).
Finally, we sought to address whether NTZ represses PDIA3 activity by inhibiting its nuclear translocation. For this purpose, TIZ, the active metabolite of NTZ, was used for the study. It was interestingly noted that TIZ not only repressed PDIA3 nuclear translocation (Figures S6A and S6B) but also significantly reduced nuclear accumulation of STAT1 (Figure S6A) and PKM2 (Figure S6B). Co-IP assays further demonstrated that TIZ disrupted the interaction between PDIA3 and STAT1 (Figure S6C) or PKM2 (Figure S6D). Those results support that NTZ could be a viable therapeutic drug for the treatment of RA in clinical settings. Overall, our data pinpointed PDIA3 as a chaperone to facilitate the nuclear import of STAT1 and PKM2, thereby regulating Th1 and Th17 programs. However, the phosphorylation of STAT1 and PKM2, induced by cytokine stimulation, serves as a prerequisite for the optimization of the PDIA3 regulatory effect. Such a two-layered modulatory mechanism might be evolutionarily important to prevent excessive T cell activation and explains why PDIA3 expresses at a low level in naive T cells, but exhibits a time-dependent increase upon sustained TCR stimulation (Figure 1D).
Discussion
RA is a prototypical autoimmune disease, in which multiple hematopoietic and non-hematopoietic cell types are engaged at different disease stages. The etiology and pathogenesis of RA remain largely unknown but are suggested to rely heavily on the interplay between FLS and immune cells. Immune-derived cytokines activate FLS, which in turn produce metalloproteinases and inflammatory mediators to damage bone and cartilage.45,46 At the same time, FLS further activates immune cells, especially macrophages and T cells, to exacerbate autoimmune responses. Such a vicious crosstalk explains at least in part the limited therapeutic effect of strategies that are solely based on immunosuppressants or biological agents. For example, TNF-α has been considered the major culprit in RA pathogenesis, yet anti-TNF-α therapy fails to cure RA, although it can improve the related symptoms in clinical settings. Worse still, long-term use of existing immunotherapies bears the risk of infection and/or tumor development.47 The dysregulation of immune responses, particularly the aberrant activation of Th1 and Th17 cells, plays a critical role in RA initiation and progression. As such, drugs targeting Th1/Th17 cells have shown great translational value in randomized clinical trials.8,48,49,50,51,52,53 It is, therefore, imperative to investigate the primary pathological prompts in the breakdown of immune homeostasis to advance RA therapy. Thus, in the current study, we scrutinized bulk and single-cell RNA-seq data from RA patients and discovered PDIA3 to be a promising target.
NFAT has been recognized as a key driver of inflammatory responses,54 and the Ca2+-NFAT signaling is hyperactivated in RA synovial inflammatory cells.55 The expression level of PDIA3 is tightly controlled by NFAT1, and we proved that NFAT1 directly binds to the Pdia3 promoter, whereby it promotes the transcription of Pdia3. Intriguingly, Wnt5a, an inducer of NFAT1, was found to be correlated with PDIA3 expression in CD4 T cells. Based on these observations, we further confirmed that the surge in calcium ions evoked by Wnt5a and TCR signaling motivates NFAT1 activity, thereby leading to the upregulation of PDIA3. Nevertheless, the source of Wnt5a is worthy of further investigation.
STAT1 and PKM2 were identified as the key downstream effectors of PDIA3. STAT1 is a fundamental orchestrator for Th1 cell differentiation by fostering the expression of IL-12R and the master transcription factor T-bet, both of which are required for Th1 cell development.56 Additionally, STAT1 also promotes the production of IFN-γ and other Th1 cytokines such as TNF-α.57,58 In general, STAT1 is retained in the cytoplasm at the quiescent state, while it becomes activated and phosphorylated upon cytokine stimulation. Through the interaction of its SH2 domains, two phosphorylated STAT1 form a homodimer, which then translocates into the nucleus to transcribe target genes.59 However, the mechanisms responsible for the nuclear import of STAT1 dimers remain poorly understood. We now unveiled that PDIA3 is capable of binding to both active and inactive forms of STAT1 without affecting STAT1 dimerization. Upon binding, PDIA3 functions as a molecular chaperone to facilitate STAT1 nuclear translocation, thereby driving the expression of Th1 lineage-specific genes. Similarly, PKM2 exists in three forms—monomer, dimer, and tetramer—to execute different biological functions. PKM2 primarily presents as an inactive monomer. In response to synthetic compounds (e.g., TEPP-46) and endogenous factors including F1,6BP and certain amino acids, PKM2 is allosterically activated to form stable tetramers,60,61 which are located in the cytoplasm and exhibit high glycolytic activity. Beyond its canonical metabolic function, dimeric PKM2 also coordinates gene expression in the nucleus and shows protein kinase activity.62,63,64 There is feasible evidence that PKM2 dimer directly binds to STAT3 (a Th17 key transcription factor) to enhance its phosphorylation and activation, thereby enhancing the Th17 program.65 Phosphorylation of PKM2 at tyrosine 105 (Y105) restrains tetramer formation, but promotes PKM2 dimerization.66 We now demonstrated that PDIA3 has no impact on PKM2 polymerization and binds to PKM2 regardless of its phosphorylation state. However, enforced expression of constitutively active STAT1 or PKM2 is able to rescue the impaired Th1/Th17 program induced by Pdia3 deficiency, suggesting that PDIA3 only constitutes a part of the nuclear-importing machineries.
PDIA3 has been documented as a member of the PDI family and exhibits redox regulation and PDI activity in the ER.22 Interestingly, PDIA3 is also expressed in various subcellular compartments, where it exerts diverse biological functions.18,28,67,68,69,70 The role of PDIA3 in CD4 T cells, however, is almost undetermined. We unveiled an altered PDIA3 expression in pathogenic CD4 T cells both in mice and humans. Pdia3 ablation does not affect the proliferation and apoptosis of CD4 T cells, but intrinsically impairs Th1 and Th17 polarization, demonstrating that elevated PDIA3 in CD4 T cells might be a key pathological factor in chronic autoimmune settings. Furthermore, we provided experimental evidence in support of PDIA3-targeted therapies. Genetic ablation and pharmacological inhibition of PDIA3 halted RA progression by repressing Th1 and Th17 differentiation both in the CIA mouse model and in immune cells obtained from RA patients. Notably, the FDA-approved PDIA3 inhibitor, NTZ, manifested a good therapeutic effect in CIA model, which paved the avenue for clinical trials aiming at repurposing NTZ for RA treatment.
Recent studies revealed that the ER chaperone GRP78/BiP could translocate to the nucleus, acting as a transcriptional regulator in response to the stress signals.71 Similarly, we unveiled a nuclear enrichment of PDIA3 in CD4 T cells, particularly following sustained TCR stimulation. PDIA3 binds to STAT1 or PKM2, by which it facilitates their nuclear import to transcribe Th1/Th17 lineage-related genes. As such, overexpression of PDIA3 boosted Th1 and Th17 program, while deletion of the NLS sequence blunted its nuclear presence and prevented PDIA3 from amplifying Th1 and Th17 program. In summary, our results support that targeting PDIA3 represents a promising strategy to attenuate RA progression in clinical settings. It is worthy of note that the deletion of PDIA3 in CD4 T cells or pharmacological inhibition of PDIA3 by NTZ only protected mice from RA to a certain extent. Therefore, it is plausible that strategies combining PDIA3 inhibitors and other therapies could be more effective in RA treatment.
Materials and methods
Antibodies and reagents
Recombinant murine PDIA3 and Wnt5a were generated by the Design Gene Biotechnology (Wuhan, China). BD HorizonTM Fixable Viability Stain 780 (#565388, RRID: AB_2869673) was obtained from BD Biosciences (San Diego, CA, USA). Purified NA/LE Hamster Anti-mouse CD3e (#553057, RRID: AB_394590) and hamster anti-mouse CD28 (#553295, RRID: AB_394764), recombinant mouse IL-2 (#575404), IL-12 (#577004), IL-6 (#575706) and TGF-β (#763102) , and FITC-conjugated anti-mouse CD4 (#100406, RRID: AB_312690), Brilliant Violet 421-conjugated anti-mouse CD4 (#100437, RRID: AB_10900241), APC-conjugated anti-mouse CD4 (#100412, RRID: AB_312696), APC-conjugated anti-mouse CD45.1 (#110714, RRID: AB_313502), phycoerythrin (PE)-conjugated anti-mouse CD44 (#103008, RRID: AB_312958), APC-conjugated anti-mouse CD62L (#104412. RRID: AB_313099), PE/Cy7-conjugated anti-mouse CD25 (#101916, RRID: AB_261676), PE-conjugated anti-human/mouse Ki67 (#151210, RRID: AB_2716014), PE/Cy7-conjugated anti-mouse IFN-γ (#505826, RRID: AB_2295770), Brilliant Violet 421-conjugated anti-mouse IL-17A (#506926, RRID: AB_10900442), APC-conjugated anti-mouse IL-17A (#506916, RRID: AB_536017), PE-conjugated anti-mouse IL-17A (#506904, RRID: AB_315463), and Alexa Fluor 647-conjugated anti-mouse Foxp3 (#126408, RRID: AB_1089115) antibodies were purchased from BD Biosciences and BioLegend (San Diego, CA, USA). Anti-PDIA3 (#2887, RRID: AB_2160837), anti-STAT1 (#14994, RRID: AB_2737027), anti-Phospho-STAT1 (Tyr701, #9167, RRID: AB_561284), anti-Phospho-STAT3 (#9145, RRID: AB_2491009), anti-PKM2 (#4053, RRID:AB_1904096), anti-Phospho-PKM2 (#3827, RRID: AB_1950369), and anti-NFAT1 (#4389, RRID: AB_1950418) antibodies were ordered from Cell Signaling Technology (Danvers, MA, USA). The anti-Lamin B1 (#12987-1-AP, RRID: AB_2136290), anti-His tag (#10001, RRID: AB_11232228), anti-Flag tag (#20543, RRID: AB_11232216), and anti-ACTB (#20536-1-AP, RRID: AB_10700003) antibodies were obtained from Proteintech (Wuhan, China). The anti-PDIA3 (#ab13506, RRID: AB_1140700) antibody was obtained from Abcam (Cambridge, UK). I-Ab OVA323-339 Tetramer-ISQAVHAAHAEINEAGR were purchased from BML (Tokyo, Japan). The HEK 293T cell line was purchased from the American Type Culture Collection (Manassas, VA, USA) (CRL-3216). The PDIA3 inhibitor NTZ (S1627) was obtained from Selleck (Houston, TX, USA) and used for the in vivo experiments, while TIZ (S5394), the active metabolite of NTZ, was ordered from Selleck for the in vitro studies.
Human samples
Patients with RA were classified according to the 2010 RA classification criteria, and they were all in a naive state regarding the use of the DMARDs. The disease activity of RA was determined by the DAS-28 and measurement of CRP using the clinical and laboratory data. Fresh anticoagulant peripheral blood samples from RA patients and healthy volunteers were collected, and serum and blood cells were separated immediately. The studies were approved by the Institutional Review Board of Tongji Hospital (TJ-IRB20220973).
Mice
C57BL/6, CD4-Cre (CD45.2), WT (CD45.1), and Rag1 KO mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). The Pdia3flox/flox mice were generated by BRL Medicine Inc. (Shanghai, China), followed by crossing with the CD4-Cre mice to generate the CD4-Cre+Pdia3flox/flox (CD4CrePdia3f/f) KO mice. The littermate Pdia3flox/flox mice were used as controls. All mice were housed in a specific pathogen-free facility under climate-controlled conditions with a 12-h light/12-h dark cycle and were provided with water and a standard diet at Tongji Medical College. All animal experiments were conducted under the approval of Tongji Hospital Animal Care and Use Committee according to the National Institutes of Health guidelines (TJH-202204016).
Collagen-induced arthritis mouse model
For collagen-induced arthritis, the KO mice and control littermates were immunized with 100 μg chicken type II collagen (#20011, Chondrex, Washington, DC, USA) emulsified in complete Freund’s adjuvant containing 4 mg/mL Mycobacterium tuberculosis (#7001, Chondrex) by intradermal injection at the base of the tail, followed by a booster injection of collagen in complete Freund’s adjuvant 21 days after the first injection. Three blinded investigators monitored the development of swelling by measuring clinical scores.
NTZ administration in collagen-induced arthritis mouse model
For NTZ administration, NTZ was suspended in 1% carboxymethyl cellulose (2.5 mL/kg) and administered freshly via oral gavage (200 mg/kg body weight) every 3 days following the second immunization until day 35. The mice in the control group received an equivalent volume of solvent (1% sodium carboxymethyl cellulose). Three investigators masked to the groups monitored the development of swelling by measuring clinical scores.72
Adoptive transfer mouse model
Tn cells were purified from the spleen of WT and KO mice using the naive CD4 T cell Isolation Kit (130-104-453; Miltenyi Biotec, Auburn, CA, USA) according to the manufacturer’s instructions. CD4 T cells (1 × 106/mouse) were injected through the tail vein into the 8-week-old recombination-activating gene 1-deficient (Rag1−/−) mice. All recipients were monitored for body weight and sacrificed on day 9 after following transfer for histological and flow cytometry analyses.
Candida albicans infection model
OVA-conjugated C. albicans strain SC5314 was a kind gift from Dr. Haifeng Zhou (Department of Integrated Traditional Chinese and Western Medicine, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China). The yeasts were grown in lipid yeast extract-peptone-dextrose medium that was shaken at 150 rpm overnight. After centrifugation and washes, blastospores were suspended in PBS at an appropriate concentration. Mice were kept in a separate room for 7 days before the experiments. For experimental infections, the mice were intravenously inoculated with 3,000 spores per gram via the tail vein. Spleen samples were collected 1 week after infection for flow cytometry analysis.
Cell isolation and in vitro polarization
Naive T cells were enriched with a Naive CD4 T cell Isolation Kit for mice (130-104-453; Miltenyi Biotec), and CD4 T cells were enriched with either a mouse CD4 T cell Isolation Kit (130-049-201; Miltenyi Biotec) or a human CD4 T cell Isolation Kit (130-045-101; Miltenyi Biotec) according to the manufacturer’s instructions. Th1 and Th17 cell polarization were conducted as previously described.9 Briefly, Tn cells were activated with plate-coated 10 μg/mL anti-CD3 (145-2C11) and anti-CD28 (37.51) for 3 days in the presence of Th1 skewing condition (10 ng/mL IL-2 and 10 ng/mL IL-12) and Th17 polarizing condition (10 ng/mL IL-2, 20 ng/mL IL-6 and 5 ng/mL TGF-β). For Wnt5a and TIZ administration, 10 ng/mL Wnt5a or 5 μM TIZ was added into the medium at the beginning of the cell culture.
T cell proliferation assay
Tn cells purified from the spleens of WT and KO mice were stained with Cell Tracer CFSE (Life Technologies, Carlsbad, CA) or Cell Trace Violet (Thermo Fisher Scientific, Waltham, MA, USA). The cells (1 × 106/mL) were plated in anti-CD3- and anti-CD28-coated 96-well plates in triplicates. Cell proliferation was determined after 72 h by detecting CSFE dye dilution or Cell Trace Violet using a Miltenyi MACSQuant Analyzer 10 flow cytometer.
RNA-seq analysis
Differentiated Th1 and Th17 cells from WT and KO mice were collected. High-quality total RNA was obtained from these cells using RNeasy spin column kits and verified using an Agilent 2100 Bioanalyzer. RNA-seq libraries were prepared using an Illumina TruSeq Stranded mRNA LT Sample Prep kit according to the manufacturer’s instructions. The libraries were subsequently validated for an average size of approximately 311–328 bp using a 2100 Bioanalyzer and an Agilent DNA1000 kit. Sequencing of paired-end reads (75 bp) was performed with a MiSeq Reagent kit V3 150 cycle on a MiSeq system. Sequence data were mapped on the mouse genome (mm10) using TopHat and analyzed using Cufflinks.
Pulldown mass spectrometry
Tn cells were isolated from the spleen of WT mice and polarized into Th1 and Th17 as described above. After washes in ice-cold PBS, the cells were lysed on ice for 30 min. The cell lysates were then incubated with His-tagged PDIA3 overnight at 4°C and PDIA3-interacting proteins were enriched with anti-His antibody. The resulting protein complexes were passed through SPA columns and eluted for mass spectrometry analysis.
ChIP assay
ChIP assays were performed as previously reported.73 Briefly, 1 × 106 WT CD4 T cells were cross-linked with 1% formaldehyde followed by sonication on ice. The NFAT1-DNA complexes were next precipitated using an anti-NFTA1 antibody. The eluted DNA was subjected to ChIP-PCR with indicated primers. The primers used for PDIA3 in the ChIP assay were: forward 5′-TTC CAC ATT TGC CTG GTC TAT-3' and reverse 5′-TGC TGG GCA AGT GAT CCA AC-3'.
Plasmid constructs and luciferase reporter assay
The Pdia3 promoter (−2,000 to −1, the transcriptional start site as +1, WT), and the mutated promoter (MUT), in which the NFAT1 binding motif ATGGAAA was deleted, were synthesized by the Tsingke Biological Technology (Wuhan, China). Both promoters were cloned into the pGL3 vector as reported.73 HEK 293T cells were transfected with either WT or MUT PDIA3 plasmids along with the NFAT1 plasmid using a Lipofectamine 3000 kit (Invitrogen, Waltham, MA, USA). After 48 h of transfection, cell lysates were prepared and subjected to analysis of reporter activity using a Dual-Luciferase Reporter Assay System Kit (Promega, Madison, WI, USA).
Glucose and lipid uptake assays
To evaluate the uptake of glucose and lipids, CD4 T cells activated by plate-coated anti-CD3 and anti-CD28 were washed and incubated with 2-NBDG and BODIPY (Thermo Fisher Scientific) at 37°C for 20 min prior to flow cytometry analysis, and their uptake was analyzed by flow cytometry, respectively.
ELISA
IFN-γ, IL-17A, and TNF-α concentration levels in mouse serum were measured using mouse IFN-γ, IL-17A, and TNF-α ELISA kits (Biolegend). Absorbance at 450 nm was measured and protein concentration was calculated with a standard curve according to the manufacturer’s instructions.
RNA extraction and quantitative PCR
Total RNA was isolated from murine and human CD4 T cells using the Trizol reagent (Takara, Osaka, Japan). For mRNA analysis, an aliquot containing 1 μg of total RNA was reversely transcribed using a cDNA synthesis kit (Takara). Real-time PCR was performed using the SYBR Green PCR master mix (Applied Biosystems, South San Francisco, CA, USA) in the ABI Prism 7500 Sequence Detection System (Applied Biosystems). The relative expression level of each gene was calculated with the 2−ΔΔCt method as previously reported and normalized to the β-actin expression level.42
Flow cytometry
Single-cell suspension was prepared from the spleen or lymph nodes as reported previously.8 The cells were stained for surface markers on ice for 20 min and washed with FACS buffer (2% BSA in PBS). For intracellular staining, the cells were cultured for 6 h in a complete medium containing 50 ng/mL PMA and 500 ng/mL ionomycin and GolgiPlug (BD Bioscience). The lymphocytes were then stained with mAb to cell surface markers, followed by intracellular staining of IFN-γ, IL-17A, TNF-α, and Foxp3 with the Fixation/Permeabilization Solution (BD Bioscience). The stained samples were analyzed with MACSQuant Analyzer 10 flow cytometer (Miltenyi Biotec) and FlowJo software, version 8.1.
Immunofluorescence staining
Immunofluorescence staining of CD4 T cells was carried out as described previously.73 Briefly, CD4 T cells were stained with anti-CD4 antibody for 20 min on ice, washed and fixed with 4% paraformaldehyde (PFA) for 5–10 min, and then permeabilized with 0.5% Triton X-100 PBS for 5 min before blocking with 1% BSA and 0.02% Triton X-100 PBS. Primary antibodies were added at 1:100 (anti-PDIA3) and incubated for 16 h at 4°C. After washing, secondary antibodies were incubated with 5 μg/mL DAPI for 30 min at room temperature. The stained CD4 T cells were imaged and analyzed by confocal microscopy (FV1000, Olympus, Tokyo, Japan).
SDS-PAGE and native PAGE for immunoblotting
Cell lysates were prepared using the radio-immunoprecipitation assay (RIPA) buffer (Servicebio, Wuhan, China) containing a protease inhibitor cocktail (Roche, Indianapolis, IN, USA). Immunoblotting analysis of target proteins was conducted as described using the appropriate primary antibodies, followed by probing with the corresponding horseradish peroxidase-conjugated secondary antibodies, respectively.74 For the native PAGE, total proteins were extracted from cells with mild lysis buffer and not heat-denatured. Non-denatured gel sample loading buffer (Beyotime Institute of Biotechnology, Nantong, China) was used, and electrophoresis was performed using native PAGE running buffer (Beyotime Institute of Biotechnology) and non-denatured gels without SDS and DTT. The remaining experimental steps were the same as those of conventional immunoblotting experiments. The reactive bands were visualized using ECL plus reagents (Servicebio).
Histology
Colonic tissue was harvested and fixed in 4% PFA and then embedded in paraffin. Ankle tissues were obtained and fixed overnight with 4% PFA, followed by decalcification with 0.5 M EDTA for 2 weeks. Sections were prepared and stained with hematoxylin and eosin as previously reported.9
Retroviral transduction
Tn cells from KO mice were transduced with retroviruses containing the PDIA3-WT, PDIA3-MUT, STAT1-Y701D, STAT1-Y701F, PKM2-Y105D, and PKM2-Y105F as described previously.42 Briefly, Tn cells were first cultured under Th1 or Th17 conditions as described for 24 h. The concentrated retrovirus was added (multiplicity of infection = 50) in the presence of 5 μg/mL polybrene. The cells were immediately centrifuged with virus in the plate at 1,800 rpm for 2 h and then cultured at 37°C and 5% CO2 for 6 h. After washing with culture medium, the cells were continually cultured under Th1 or Th17 condition as described earlier for 2 days, followed by intracellular staining for flow cytometry analysis.
Statistical analysis
All in vitro studies were conducted at least three times. Comparisons between multiple groups were conducted by one-way ANOVA. Correlations were determined by Pearson’s correlation analysis. All other results were expressed as mean ± SEM, and their comparisons were accomplished by the unpaired Student’s t test to determine the significant differences between two groups. A paired Student’s t test was used to determine the significant differences between human T cell response upon NTZ treatment. Statistical analysis of the data was conducted using the GraphPad Prism 8 software (GraphPad Software Inc., San Diego, CA, USA), and the results were expressed as mean ± SEM. In all cases, a p value of less than 0.05 was considered statistically significant.
Data and code availability
All data needed to evaluate the conclusions in this article are included in the paper and/or its supplemental information. Additional data within the manuscript are available from the corresponding author upon request.
Acknowledgments
We thank Yang Li and Wen-Ye Mo for animal breeding. We also thank the affected patients and healthy volunteers in this study. This study was supported by funding from the National Key R&D Program of China 2022YFA0806101 (C.Y.W.); National Natural Science Foundation of China [82130023, 81920108009, and 91749207 (C.Y.W.); 82100823 (F.X.W.); 82070808 (S.Z.); 81873656 (F.X.); 81770823 (P.Y.); 82200923 (F.S.); and 82270885 (Q.L.Y.); Postdoctoral Science Foundation of China 2023M731200 (J.H.L.); Key Laboratory of Endocrine and Metabolic Diseases of Shanxi Province 202104010910009 (S.W.L.); Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College, Huazhong University of Science and Technology; and the Innovative Funding for Translational Research from Tongji Hospital.
Author contributions
Conceptualization: F.X.W., F.S., and C.-Y.W.; methodology: F.X.W., C.L.Y., J.H.L., L.L.D., P.Y., F.S., and C.-Y.W.; investigation: F.X.W., C.L.Y., J.H.L., S.J.R., Q.J.C., W.Y.L., T.W., D.N.S., Q.M., Y.C., L.L.D., F.S., and C.-Y.W.; data curation: F.X.W., C.L.Y., J.H.L., L.L.D., F.S., and C.-Y.W.; formal analysis: F.X.W., C.L.Y., J.H.L., S.J.R., Q.J.C., W.Y.L., L.L.D., J.X., Y.N.W., F.S., and C.-Y.W.; visualization: F.X.W., C.L.Y., T.Y., W.G.K., S.Z., F.X., Q.L.Y., Z.Z.Y., L.L.D., F.S., and C.-Y.W.; validation: J.H.L., L.L.D., P.Y., F.S., and C.-Y.W.; funding acquisition: F.X.W, J.H.L., S.W.L., S.Z., F.X., Q.L.Y., P.Y., F.S , and C.-Y.W.; project administration: F.X.W., L.L.D., F.S., and C.-Y.W.; resources: S.W.L., C.L.Y., J.H.L., L.L.D., F.S., and C.-Y.W.; supervision: F.X.W., L.L.D., F.S., and C.-Y.W.; writing – original draft: F.X.W. and C.L.Y.; writing – review and editing: F.X.W., C.L.Y., J.H.L., D.E., L.L.D., F.S., and C.-Y.W.
Declaration of interests
The authors declare that they have no conflict of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2024.05.038.
Contributor Information
Ling-Li Dong, Email: tjhdongll@163.com.
Fei Sun, Email: phil_sunfei@163.com.
Cong-Yi Wang, Email: wangcy@tjh.tjmu.edu.cn.
Supplemental information
References
- 1.Scherer H.U., Häupl T., Burmester G.R. The etiology of rheumatoid arthritis. J. Autoimmun. 2020;110 doi: 10.1016/j.jaut.2019.102400. [DOI] [PubMed] [Google Scholar]
- 2.Cross M., Smith E., Hoy D., Carmona L., Wolfe F., Vos T., Williams B., Gabriel S., Lassere M., Johns N., et al. The global burden of rheumatoid arthritis: estimates from the global burden of disease 2010 study. Ann. Rheum. Dis. 2014;73:1316–1322. doi: 10.1136/annrheumdis-2013-204627. [DOI] [PubMed] [Google Scholar]
- 3.Becede M., Alasti F., Gessl I., Haupt L., Kerschbaumer A., Landesmann U., Loiskandl M., Supp G.M., Smolen J.S., Aletaha D. Risk profiling for a refractory course of rheumatoid arthritis. Semin. Arthritis Rheum. 2019;49:211–217. doi: 10.1016/j.semarthrit.2019.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Ehrenstein M.R., Evans J.G., Singh A., Moore S., Warnes G., Isenberg D.A., Mauri C. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J. Exp. Med. 2004;200:277–285. doi: 10.1084/jem.20040165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamanaka H., Tanaka E., Nakajima A., Furuya T., Ikari K., Taniguchi A., Inoue E., Harigai M. A large observational cohort study of rheumatoid arthritis, IORRA: Providing context for today's treatment options. Mod. Rheumatol. 2020;30:1–6. doi: 10.1080/14397595.2019.1660028. [DOI] [PubMed] [Google Scholar]
- 6.Richter A., Listing J., Schneider M., Klopsch T., Kapelle A., Kaufmann J., Zink A., Strangfeld A. Impact of treatment with biologic DMARDs on the risk of sepsis or mortality after serious infection in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2016;75:1667–1673. doi: 10.1136/annrheumdis-2015-207838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y., Shen Y., Hohensinner P., Ju J., Wen Z., Goodman S.B., Zhang H., Goronzy J.J., Weyand C.M. Deficient Activity of the Nuclease MRE11A Induces T Cell Aging and Promotes Arthritogenic Effector Functions in Patients with Rheumatoid Arthritis. Immunity. 2016;45:903–916. doi: 10.1016/j.immuni.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou H.F., Wang F.X., Sun F., Liu X., Rong S.J., Luo J.H., Yue T.T., Xiao J., Yang C.L., Lu W.Y., et al. Aloperine Ameliorates IMQ-Induced Psoriasis by Attenuating Th17 Differentiation and Facilitating Their Conversion to Treg. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.778755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun F., Wang F.X., Zhu H., Yue T.T., Yang C.L., Luo J.H., Luo X., Zhou H.F., Rong S.J., Lu W.Y., et al. SUMOylation of PDPK1 Is required to maintain glycolysis-dependent CD4 T-cell homeostasis. Cell Death Dis. 2022;13:181. doi: 10.1038/s41419-022-04622-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boettler T., Choi Y.S., Salek-Ardakani S., Cheng Y., Moeckel F., Croft M., Crotty S., von Herrath M. Exogenous OX40 stimulation during lymphocytic choriomeningitis virus infection impairs follicular Th cell differentiation and diverts CD4 T cells into the effector lineage by upregulating Blimp-1. J. Immunol. 2013;191:5026–5035. doi: 10.4049/jimmunol.1300013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Belle T.L., Nierkens S., Arens R., von Herrath M.G. Interleukin-21 receptor-mediated signals control autoreactive T cell infiltration in pancreatic islets. Immunity. 2012;36:1060–1072. doi: 10.1016/j.immuni.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiang E.Y., Kolumam G.A., Yu X., Francesco M., Ivelja S., Peng I., Gribling P., Shu J., Lee W.P., Refino C.J., et al. Targeted depletion of lymphotoxin-alpha-expressing TH1 and TH17 cells inhibits autoimmune disease. Nat. Med. 2009;15:766–773. doi: 10.1038/nm.1984. [DOI] [PubMed] [Google Scholar]
- 13.Yao C., Sakata D., Esaki Y., Li Y., Matsuoka T., Kuroiwa K., Sugimoto Y., Narumiya S. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat. Med. 2009;15:633–640. doi: 10.1038/nm.1968. [DOI] [PubMed] [Google Scholar]
- 14.Molinero L.L., Cubre A., Mora-Solano C., Wang Y., Alegre M.L. T cell receptor/CARMA1/NF-κB signaling controls T-helper (Th) 17 differentiation. Proc. Natl. Acad. Sci. USA. 2012;109:18529–18534. doi: 10.1073/pnas.1204557109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuller K., Wong B., Fox S., Choi Y., Chambers T.J. TRANCE is necessary and sufficient for osteoblast-mediated activation of bone resorption in osteoclasts. J. Exp. Med. 1998;188:997–1001. doi: 10.1084/jem.188.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yasuda H., Shima N., Nakagawa N., Yamaguchi K., Kinosaki M., Mochizuki S., Tomoyasu A., Yano K., Goto M., Murakami A., et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chichiarelli S., Altieri F., Paglia G., Rubini E., Minacori M., Eufemi M. ERp57/PDIA3: new insight. Cell. Mol. Biol. Lett. 2022;27:12. doi: 10.1186/s11658-022-00315-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bilches Medinas D., Malik S., Yıldız-Bölükbaşı E., Borgonovo J., Saaranen M.J., Urra H., Pulgar E., Afzal M., Contreras D., Wright M.T., et al. Mutation in protein disulfide isomerase A3 causes neurodevelopmental defects by disturbing endoplasmic reticulum proteostasis. EMBO J. 2022;41 doi: 10.15252/embj.2020105531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Risola D., Ricci D., Marrocco I., Giamogante F., Grieco M., Francioso A., Vasco-Vidal A., Mancini P., Colotti G., Mosca L., Altieri F. ERp57 chaperon protein protects neuronal cells from Aβ-induced toxicity. J. Neurochem. 2022;162:322–336. doi: 10.1111/jnc.15655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li N., Luo X., Yu Q., Yang P., Chen Z., Wang X., Jiang J., Xu J., Gong Q., Eizirik D.L., et al. SUMOylation of Pdia3 exacerbates proinsulin misfolding and ER stress in pancreatic beta cells. J. Mol. Med. 2020;98:1795–1807. doi: 10.1007/s00109-020-02006-6. [DOI] [PubMed] [Google Scholar]
- 21.Perez-Vargas J., Teppa E., Amirache F., Boson B., Pereira de Oliveira R., Combet C., Bockmann A., Fusil F., Freitas N., Carbone A., Cosset F.L. A fusion peptide in preS1 and the human protein disulfide isomerase ERp57 are involved in hepatitis B virus membrane fusion process. Elife. 2021;10 doi: 10.7554/eLife.64507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ros M., Nguyen A.T., Chia J., Le Tran S., Le Guezennec X., McDowall R., Vakhrushev S., Clausen H., Humphries M.J., Saltel F., Bard F.A. ER-resident oxidoreductases are glycosylated and trafficked to the cell surface to promote matrix degradation by tumour cells. Nat. Cell Biol. 2020;22:1371–1381. doi: 10.1038/s41556-020-00590-w. [DOI] [PubMed] [Google Scholar]
- 23.Niu Y., Xue J., Wu X., Qu M., Wang L., Liang W., Li T. Clinical Significance of Serum Haptoglobin and Protein Disulfide-Isomerase A3 in the Screening, Diagnosis, and Staging of Colorectal Cancer. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.935500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Staquicini F.I., Hajitou A., Driessen W.H., Proneth B., Cardó-Vila M., Staquicini D.I., Markosian C., Hoh M., Cortez M., Hooda-Nehra A., et al. Targeting a cell surface vitamin D receptor on tumor-associated macrophages in triple-negative breast cancer. Elife. 2021;10 doi: 10.7554/eLife.65145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramirez-Rangel I., Bracho-Valdes I., Vazquez-Macias A., Carretero-Ortega J., Reyes-Cruz G., Vazquez-Prado J. Regulation of mTORC1 complex assembly and signaling by GRp58/ERp57. Mol. Cell Biol. 2011;31:1657–1671. doi: 10.1128/MCB.00824-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rellmann Y., Eidhof E., Hansen U., Fleischhauer L., Vogel J., Clausen-Schaumann H., Aszodi A., Dreier R. ER Stress in ERp57 Knockout Knee Joint Chondrocytes Induces Osteoarthritic Cartilage Degradation and Osteophyte Formation. Int. J. Mol. Sci. 2021;23 doi: 10.3390/ijms23010182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song D., Liu H., Wu J., Gao X., Hao J., Fan D. Insights into the role of ERp57 in cancer. J. Cancer. 2021;12:2456–2464. doi: 10.7150/jca.48707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clement C.C., Osan J., Buque A., Nanaware P.P., Chang Y.C., Perino G., Shetty M., Yamazaki T., Tsai W.L., Urbanska A.M., et al. PDIA3 epitope-driven immune autoreactivity contributes to hepatic damage in type 2 diabetes. Sci. Immunol. 2022;7 doi: 10.1126/sciimmunol.abl3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anczurowski M., Sugata K., Matsunaga Y., Yamashita Y., Wang C.H., Guo T., Murata K., Saijo H., Kagoya Y., Saso K., et al. Chaperones of the class I peptide-loading complex facilitate the constitutive presentation of endogenous antigens on HLA-DP(84GGPM87) J. Autoimmun. 2019;102:114–125. doi: 10.1016/j.jaut.2019.04.023. [DOI] [PubMed] [Google Scholar]
- 30.Zhang B., Zhang Y., Xiong L., Li Y., Zhang Y., Zhao J., Jiang H., Li C., Liu Y., Liu X., et al. CD127 imprints functional heterogeneity to diversify monocyte responses in inflammatory diseases. J. Exp. Med. 2022;219 doi: 10.1084/jem.20211191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takeshita M., Suzuki K., Kondo Y., Morita R., Okuzono Y., Koga K., Kassai Y., Gamo K., Takiguchi M., Kurisu R., et al. Multi-dimensional analysis identified rheumatoid arthritis-driving pathway in human T cell. Ann. Rheum. Dis. 2019;78:1346–1356. doi: 10.1136/annrheumdis-2018-214885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piacentini S., La Frazia S., Riccio A., Pedersen J.Z., Topai A., Nicolotti O., Rossignol J.F., Santoro M.G. Nitazoxanide inhibits paramyxovirus replication by targeting the Fusion protein folding: role of glycoprotein-specific thiol oxidoreductase ERp57. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-28172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacLauchlan S., Zuriaga M.A., Fuster J.J., Cuda C.M., Jonason J., Behzadi F., Duffen J.P., Haines G.K., 3rd, Aprahamian T., Perlman H., Walsh K. Genetic deficiency of Wnt5a diminishes disease severity in a murine model of rheumatoid arthritis. Arthritis Res. Ther. 2017;19:166. doi: 10.1186/s13075-017-1375-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahmoud D.E., Kaabachi W., Sassi N., Mokhtar A., Moalla M., Ammar L.B., Jemmali S., Rekik S., Tarhouni L., Kallel-Sellami M., et al. SFRP5 Enhances Wnt5a Induced-Inflammation in Rheumatoid Arthritis Fibroblast-Like Synoviocytes. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.663683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu H., Liu S., He W., Sun F., Li Y., Yang P., Yu Q., Zhang S. One-Step Genotyping Method in loxP-Based Conditional Knockout Mice Generated by CRISPR-Cas9 Technology. Mol. Biotechnol. 2022;64:1227–1233. doi: 10.1007/s12033-022-00500-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He Q., Gao H., Chang Y.L., Wu X., Lin R., Li G., Lin J., Lu H., Chen H., Li Z., et al. ETS-1 facilitates Th1 cell-mediated mucosal inflammation in inflammatory bowel diseases through upregulating CIRBP. J. Autoimmun. 2022;132 doi: 10.1016/j.jaut.2022.102872. [DOI] [PubMed] [Google Scholar]
- 37.Zhang W., Cao X., Zhong X., Wu H., Shi Y., Feng M., Wang Y.C., Ann D., Gwack Y., Yuan Y.C., et al. SRC2 controls CD4(+) T cell activation via stimulating c-Myc-mediated upregulation of amino acid transporter Slc7a5. Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2221352120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fric J., Lim C.X.F., Koh E.G.L., Hofmann B., Chen J., Tay H.S., Mohammad Isa S.A.B., Mortellaro A., Ruedl C., Ricciardi-Castagnoli P. Calcineurin/NFAT signalling inhibits myeloid haematopoiesis. EMBO Mol. Med. 2012;4:269–282. doi: 10.1002/emmm.201100207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shah R., Spektor T.M., Weisenberger D.J., Ding H., Patil R., Amador C., Song X.Y., Chun S.T., Inzalaco J., Turjman S., et al. Reversal of dual epigenetic repression of non-canonical Wnt-5a normalises diabetic corneal epithelial wound healing and stem cells. Diabetologia. 2023;66:1943–1958. doi: 10.1007/s00125-023-05960-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weiß M., Hernandez L.C., Gil Montoya D.C., Löhndorf A., Krüger A., Kopdag M., Uebler L., Landwehr M., Nawrocki M., Huber S., et al. Adhesion to laminin-1 and collagen IV induces the formation of Ca(2+) microdomains that sensitize mouse T cells for activation. Sci. Signal. 2023;16 doi: 10.1126/scisignal.abn9405. [DOI] [PubMed] [Google Scholar]
- 41.Li Y., Kong Y., An M., Luo Y., Zheng H., Lin Y., Chen J., Yang J., Liu L., Luo B., et al. ZEB1-mediated biogenesis of circNIPBL sustains the metastasis of bladder cancer via Wnt/β-catenin pathway. J. Exp. Clin. Cancer Res. 2023;42:191. doi: 10.1186/s13046-023-02757-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yue T., Sun F., Wang F., Yang C., Luo J., Rong S., Zhou H., Xiao J., Wang X., Zhou Q., et al. MBD2 acts as a repressor to maintain the homeostasis of the Th1 program in type 1 diabetes by regulating the STAT1-IFN-gamma axis. Cell Death Differ. 2022;29:218–229. doi: 10.1038/s41418-021-00852-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu J., Wu T., Zhang B., Liu S., Song W., Qiao J., Ruan H. Types of nuclear localization signals and mechanisms of protein import into the nucleus. Cell Commun. Signal. 2021;19:60. doi: 10.1186/s12964-021-00741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Damasceno L.E.A., Prado D.S., Veras F.P., Fonseca M.M., Toller-Kawahisa J.E., Rosa M.H., Públio G.A., Martins T.V., Ramalho F.S., Waisman A., et al. PKM2 promotes Th17 cell differentiation and autoimmune inflammation by fine-tuning STAT3 activation. J. Exp. Med. 2020;217 doi: 10.1084/jem.20190613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Floudas A., Smith C.M., Tynan O., Neto N., Krishna V., Wade S.M., Hanlon M., Cunningham C., Marzaioli V., Canavan M., et al. Distinct stromal and immune cell interactions shape the pathogenesis of rheumatoid and psoriatic arthritis. Ann. Rheum. Dis. 2022;81:1224–1242. doi: 10.1136/annrheumdis-2021-221761. [DOI] [PubMed] [Google Scholar]
- 46.Komatsu N., Takayanagi H. Mechanisms of joint destruction in rheumatoid arthritis - immune cell-fibroblast-bone interactions. Nat. Rev. Rheumatol. 2022;18:415–429. doi: 10.1038/s41584-022-00793-5. [DOI] [PubMed] [Google Scholar]
- 47.Choi B., Park H.J., Song Y.K., Oh Y.J., Kim I.W., Oh J.M. The risk of newly diagnosed cancer in patients with rheumatoid arthritis by TNF inhibitor use: a nationwide cohort study. Arthritis Res. Ther. 2022;24:191. doi: 10.1186/s13075-022-02868-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ewer K.J., Barrett J.R., Belij-Rammerstorfer S., Sharpe H., Makinson R., Morter R., Flaxman A., Wright D., Bellamy D., Bittaye M., et al. T cell and antibody responses induced by a single dose of ChAdOx1 nCoV-19 (AZD1222) vaccine in a phase 1/2 clinical trial. Nat. Med. 2021;27:270–278. doi: 10.1038/s41591-020-01194-5. [DOI] [PubMed] [Google Scholar]
- 49.Joshua R.A. Massive increase in splenic germinal centres of chickens experimentally-infected with Trypanosoma brucei brucei. Vet. Parasitol. 1983;13:101–108. doi: 10.1016/0304-4017(83)90068-7. [DOI] [PubMed] [Google Scholar]
- 50.Miossec P., Kolls J.K. Targeting IL-17 and TH17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012;11:763–776. doi: 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- 51.Burke J.R., Cheng L., Gillooly K.M., Strnad J., Zupa-Fernandez A., Catlett I.M., Zhang Y., Heimrich E.M., McIntyre K.W., Cunningham M.D., et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aaw1736. [DOI] [PubMed] [Google Scholar]
- 52.Grant J.L., Ghosn E.E.B., Axtell R.C., Herges K., Kuipers H.F., Woodling N.S., Andreasson K., Herzenberg L.A., Herzenberg L.A., Steinman L. Reversal of paralysis and reduced inflammation from peripheral administration of beta-amyloid in TH1 and TH17 versions of experimental autoimmune encephalomyelitis. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3004145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gonzalez-Rey E., Chorny A., Del Moral R.G., Varela N., Delgado M. Therapeutic effect of cortistatin on experimental arthritis by downregulating inflammatory and Th1 responses. Ann. Rheum. Dis. 2007;66:582–588. doi: 10.1136/ard.2006.062703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giblin M.J., Smith T.E., Winkler G., Pendergrass H.A., Kim M.J., Capozzi M.E., Yang R., McCollum G.W., Penn J.S. Nuclear factor of activated T-cells (NFAT) regulation of IL-1β-induced retinal vascular inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2021;1867 doi: 10.1016/j.bbadis.2021.166238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park Y.J., Yoo S.A., Kim M., Kim W.U. The Role of Calcium-Calcineurin-NFAT Signaling Pathway in Health and Autoimmune Diseases. Front. Immunol. 2020;11:195. doi: 10.3389/fimmu.2020.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Afkarian M., Sedy J.R., Yang J., Jacobson N.G., Cereb N., Yang S.Y., Murphy T.L., Murphy K.M. T-bet is a STAT1-induced regulator of IL-12R expression in naïve CD4+ T cells. Nat. Immunol. 2002;3:549–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 57.Ivashkiv L.B. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018;18:545–558. doi: 10.1038/s41577-018-0029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang Y., Yu M., Hu X., Han L., Yang K., Ba H., Zhang Z., Yin B., Yang X.P., Li Z., Wang J. STAT1 mediates transmembrane TNF-alpha-induced formation of death-inducing signaling complex and apoptotic signaling via TNFR1. Cell Death Differ. 2017;24:660–671. doi: 10.1038/cdd.2016.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramana C.V., Gil M.P., Schreiber R.D., Stark G.R. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23:96–101. doi: 10.1016/s1471-4906(01)02118-4. [DOI] [PubMed] [Google Scholar]
- 60.Anastasiou D., Yu Y., Israelsen W.J., Jiang J.K., Boxer M.B., Hong B.S., Tempel W., Dimov S., Shen M., Jha A., et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 2012;8:839–847. doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Masaki S., Hashimoto K., Kihara D., Tsuzuki C., Kataoka N., Suzuki K. The cysteine residue at 424th of pyruvate kinase M2 is crucial for tetramerization and responsiveness to oxidative stress. Biochem. Biophys. Res. Commun. 2020;526:973–977. doi: 10.1016/j.bbrc.2020.03.182. [DOI] [PubMed] [Google Scholar]
- 62.Dayton T.L., Jacks T., Vander Heiden M.G. PKM2, cancer metabolism, and the road ahead. EMBO Rep. 2016;17:1721–1730. doi: 10.15252/embr.201643300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.He C.L., Bian Y.Y., Xue Y., Liu Z.X., Zhou K.Q., Yao C.F., Lin Y., Zou H.F., Luo F.X., Qu Y.Y., et al. Pyruvate Kinase M2 Activates mTORC1 by Phosphorylating AKT1S1. Sci. Rep. 2016;6 doi: 10.1038/srep21524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Israelsen W.J., Vander Heiden M.G. Pyruvate kinase: Function, regulation and role in cancer. Semin. Cell Dev. Biol. 2015;43:43–51. doi: 10.1016/j.semcdb.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hitosugi T., Kang S., Vander Heiden M.G., Chung T.W., Elf S., Lythgoe K., Dong S., Lonial S., Wang X., Chen G.Z., et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2009;2 doi: 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Angiari S., Runtsch M.C., Sutton C.E., Palsson-McDermott E.M., Kelly B., Rana N., Kane H., Papadopoulou G., Pearce E.L., Mills K.H.G., O'Neill L.A.J. Pharmacological Activation of Pyruvate Kinase M2 Inhibits CD4(+) T Cell Pathogenicity and Suppresses Autoimmunity. Cell Metab. 2020;31:391–405.e8. doi: 10.1016/j.cmet.2019.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aureli C., Gaucci E., Arcangeli V., Grillo C., Eufemi M., Chichiarelli S. ERp57/PDIA3 binds specific DNA fragments in a melanoma cell line. Gene. 2013;524:390–395. doi: 10.1016/j.gene.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 68.Turano C., Coppari S., Altieri F., Ferraro A. Proteins of the PDI family: unpredicted non-ER locations and functions. J. Cell. Physiol. 2002;193:154–163. doi: 10.1002/jcp.10172. [DOI] [PubMed] [Google Scholar]
- 69.Turano C., Gaucci E., Grillo C., Chichiarelli S. ERp57/GRP58: a protein with multiple functions. Cell. Mol. Biol. Lett. 2011;16:539–563. doi: 10.2478/s11658-011-0022-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coppari S., Altieri F., Ferraro A., Chichiarelli S., Eufemi M., Turano C. Nuclear localization and DNA interaction of protein disulfide isomerase ERp57 in mammalian cells. J. Cell. Biochem. 2002;85:325–333. doi: 10.1002/jcb.10137. [DOI] [PubMed] [Google Scholar]
- 71.Liu Z., Liu G., Ha D.P., Wang J., Xiong M., Lee A.S. ER chaperone GRP78/BiP translocates to the nucleus under stress and acts as a transcriptional regulator. Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2303448120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tu J., Chen W., Fang Y., Han D., Chen Y., Jiang H., Tan X., Xu Z., Wu X., Wang H., et al. PU.1 promotes development of rheumatoid arthritis via repressing FLT3 in macrophages and fibroblast-like synoviocytes. Ann. Rheum. Dis. 2023;82:198–211. doi: 10.1136/ard-2022-222708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang F., Sun F., Luo J., Yue T., Chen L., Zhou H., Zhang J., Yang C., Luo X., Zhou Q., et al. Loss of ubiquitin-conjugating enzyme E2 (Ubc9) in macrophages exacerbates multiple low-dose streptozotocin-induced diabetes by attenuating M2 macrophage polarization. Cell Death Dis. 2019;10:892. doi: 10.1038/s41419-019-2130-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xiao J., Sun F., Wang Y.N., Liu B., Zhou P., Wang F.X., Zhou H.F., Ge Y., Yue T.T., Luo J.H., et al. UBC9 deficiency enhances immunostimulatory macrophage activation and subsequent antitumor T cell response in prostate cancer. J. Clin. Invest. 2023;133 doi: 10.1172/JCI158352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in this article are included in the paper and/or its supplemental information. Additional data within the manuscript are available from the corresponding author upon request.