

Abstract
HER2 amplification occurs in approximately 5% of colorectal cancer (CRC) cases and is associated only partially with clinical response to combined human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR)-targeted treatment. An alternative approach based on adoptive cell therapy using T cells engineered with anti-HER2 chimeric antigen receptor (CAR) proved to be toxic due to on-target/off-tumor activity. Here we describe a combinatorial strategy to safely target HER2 amplification and carcinoembryonic antigen (CEA) expression in CRC using a synNotch-CAR-based artificial regulatory network. The natural killer (NK) cell line NK-92 was engineered with an anti-HER2 synNotch receptor driving the expression of a CAR against CEA only when engaged. After being transduced and sorted for HER2-driven CAR expression, cells were cloned. The clone with optimal performances in terms of specificity and amplitude of CAR induction demonstrated significant activity in vitro and in vivo specifically against HER2-amplified (HER2amp)/CEA+ CRC models, with no effects on cells with physiological HER2 levels. The HER2-synNotch/CEA-CAR-NK system provides an innovative, scalable, and safe off-the-shelf cell therapy approach with potential against HER2amp CRC resistant or partially responsive to HER2/EGFR blockade.
Keywords: colorectal cancer, immunotherapy, HER2, CEA, CAR, synNotch, NK-92, PDX
Graphical abstract
Medico and colleagues demonstrate that NK cells engineered with a HER2 synNotch/CEA-CAR combinatorial system exhibit selective cytotoxicity and growth inhibition of HER2amp CRC, reducing on-target/off-tumor toxicity. This study highlights a scalable, safe, and accessible off-the-shelf cell therapy approach that holds potential for HER2amp CRC treatment.
Introduction
Colorectal cancer (CRC) is the third most common type of cancer, with a worldwide incidence of 1.9 million new cases per year.1,2 Despite therapy improvements, the 5-year survival rate for patients with metastatic CRC (mCRC) is only 10%–15%.3,4 The mainstay of first-line therapy for advanced CRC is chemotherapy, which can be combined with an anti-vascular endothelial growth factor or anti-epidermal growth factor receptor (EGFR) antibody, based on specific genetic tumor features; however, most tumors progress within 1 year.5,6
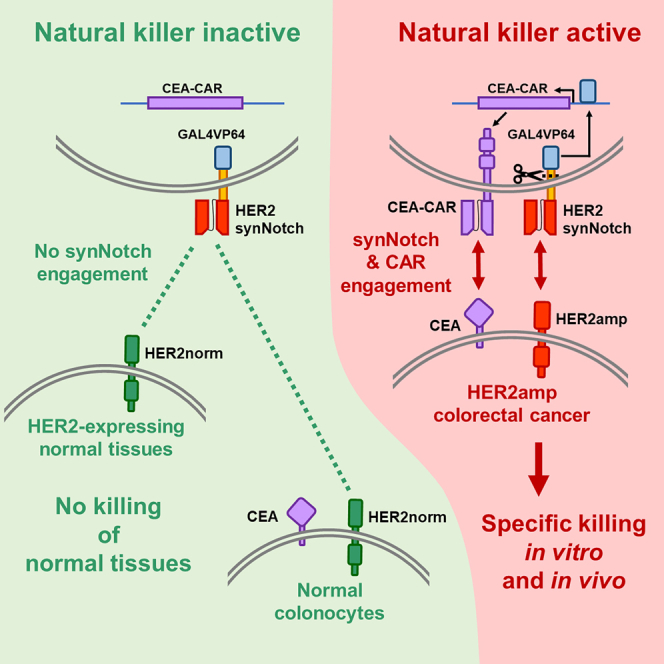
Human ERBB2 is a proto-oncogene that encodes HER2, a member of the EGFR family. The most common mechanism of HER2 oncogenic activation is gene amplification, which leads to overexpression and is a known oncogenic driver of a variety of solid tumors, including CRC.7,8 In particular, HER2 is amplified and overexpressed in approximately 5% of patients with CRC: 100,000 new cases/year globally.9,10 HER2 amplification in CRC is known to promote resistance to EGFR-targeted therapy.11,12 However, multiple studies have shown that combined targeting of HER2 and EGFR is an effective therapeutic strategy in mCRC.13,14,15,16,17,18,19 Despite these encouraging results, two major unmet clinical needs remain unsolved: (1) primary resistance to HER2/EGFR therapy, occurring in almost one-half of the cases16,17,18 and (2) limited duration of the response, due to acquisition of resistance-promoting mutations.14 Consequently, a sizable fraction of HER2-amplified (HER2amp) CRCs needs alternative therapies. In recent years, immune checkpoint inhibitors (ICIs) have produced encouraging results in CRC with microsatellite instability.20,21 However, HER2 amplification occurs more frequently in microsatellite-stable cases,22 where ICIs are ineffective. Another promising alternative is chimeric antigen receptor (CAR)-based adoptive cell therapy (ACT). CAR-based ACT is a highly personalized treatment that involves the infusion of anticancer immune cells engineered with CARs that redirect them against specific antigens expressed by cancer cells.23,24 CAR-based ACT is remarkably effective in the treatment of some hematological malignancies,25 but still requires optimization for solid tumors,26 in particular to address the issue of target antigen specificity.27,28 Indeed, HER2amp CRC exemplifies this drawback: CAR-T cells against HER2 were found to kill not only cancer cells overexpressing HER2, but also cells in normal tissues expressing physiological levels of the protein, leading to severe side effects and even death.29 To overcome this problem, a strategy is needed to ensure selective killing of HER2-overexpressing CRC cells. We, therefore, considered a combinatorial targeting of HER2 and a second antigen with a pan-colon-specific expression, because only HER2amp CRC cells would co-express both at high levels. For combinatorial targeting, we chose the synthetic Notch (synNotch)/CAR artificial network system.30,31,32 In this system, an artificial Notch receptor is engineered to recognize a specific antigen on the surface of the target cell. Upon engagement with the antigen, mechanical tension leads to cleavage of the synNotch intracellular portion, which then reaches the nucleus and promotes specific target gene expression.33 In our case, as previously described, the synNotch intracellular portion is composed of an artificial GAL4VP64 transcription factor, specifically driving expression of a CAR against a second antigen.31 In this way, CAR expression and killer cell activation occur exclusively in the presence of both target antigens. The on-target/off-tumor effect is, therefore, avoided if co-expression of both antigens is restricted to the tumor lesion. The synNotch/CAR combinatorial approach has already been shown to be particularly promising in terms of efficacy and safety.34,35,36,37 To target HER2amp CRC, we chose to adopt an anti-HER2 synNotch, rather than an anti-HER2 CAR, for two main reasons: (1) in HER2amp CRC, expression of the HER2 protein could be heterogeneous,38 as frequently seen in breast cancer,39 while a pan-colon antigen would be expressed by all target cells, leading to better CAR efficacy; and (2) synNotch activation can be better tuned to occur only in the presence of target cells expressing high HER2 levels, by choosing the intermediate-low affinity single chain fragment variable (scFv) of the 4D5-5 monoclonal antibody (mAb),32,40 without compromising CAR efficacy. To choose the CAR target antigen we performed extensive bioinformatic analyses, leading to selection of carcinoembryonic antigen (CEA), encoded by the CEACAM5 gene.
As an effector cell model, we chose the human natural killer (NK) cell line NK-92,41,42,43 for two reasons: (1) they can be easily expanded and repeatedly sorted and cloned, to select engineered effectors with optimal activity and specificity, and (2) engineered effectors can be further expanded for off-the-shelf therapy, with lower production costs.44,45 Indeed, NK-92 cells are already approved by the U.S. Food and Drug Administration for phase 1 and 2 trials.46,47,48
Here we describe an ACT strategy based on the activity of a CEA-CAR conditionally expressed by a synNotch receptor targeting HER2 suitable for the treatment of HER2-amplified CRC.
Results
Identification of CEA as an optimal target for combinatorial CAR approaches against HER2amp CRC
To identify an optimal second antigen to be combined with HER2 in the synNotch/CAR system, after an extensive bioinformatic analysis, we finally selected as main candidates a set of antigens already validated as CAR targets in CRC: CEA, encoded by the CEACAM5 gene49; epithelial cell adhesion molecule (EPCAM), encoded by the EPCAM gene,50 CD133, encoded by the PROM1 gene,51 mucin 1 (MUC1), encoded by the MUC1 gene52; and EGFR, encoded by the EGFR gene.53 Initially, mRNA expression of the candidates was evaluated in normal tissues on the Entrez Gene database.54 The analysis showed that only CEA mRNA was specifically expressed at high levels in the colon, with very low levels in the esophagus and stomach. All other antigens were also expressed at high levels in many other normal tissues, including some that also express HER2 at significant levels, like the esophagus, duodenum, stomach, small intestine, kidney, lung, and skin (Figure 1A). Expression of these CAR targets in normal tissues increases the risk of on-target/off-tumor toxicity. To evaluate in detail HER2 and CEA expression in normal tissue, their mRNA and protein expressions were also assessed on the Protein Atlas database.55,56 The analysis confirmed that HER2 expression was physiologically high in almost all the tissues and that CEA expression is physiologically high only in the colon, with low levels in the proximal digestive tract, bone marrow, and lymphoid tissues (Figure 1B). As a further confirmation, previous immunohistochemical analyses showed that CEA cannot be detected in most normal adult tissues, except in the gastrointestinal tract at low levels, restricted to the apical surface of epithelial cell.57 It should also be noted that, although CEA is already expressed in the normal colon, it is increasingly overexpressed during colorectal carcinogenesis.58,59 To identify suitable patient-derived CRC models for preclinical testing of a possible HER2 synNotch/CEA-CAR-based therapy, mRNA expression of HER2 and CEA was evaluated in 450 CRC samples from The Cancer Genome Atlas (TCGA),60 119 CRC cell lines, and 602 CRC patient-derived xenografts (PDXs).61 As shown in Figure 1, all patient tumors and models overexpressing HER2 also express very high levels of CEA (Figure 1C). HER2 and CEA surface protein expression was further confirmed in selected cell lines by flow cytometry. The HER2amp breast cancer cell line SKBR3 was selected as positive control for HER2 overexpression62 and negative control for CEA expression (CEA−). Three CRC cell lines were also selected, all positive for CEA expression (CEA+): LS180 with physiological/normal HER2 levels (HER2norm), and two cell lines, CRC0080 and CRC0186, carrying HER2 amplification and overexpression15 (Figure 1D). Two additional, tightly controlled models were also used: DIFI and NCI-H508 CRC cells, either expressing physiological HER2 levels or transduced with a HER2 lentiviral vector to constitutively overexpress HER263 (Figure S1).
Figure 1.

SynNotch/CAR target expression analysis
(A) Bar graphs representing normal tissue mRNA expression data from the Entrez Gene database (Gene: ERBB2, CEACAM5, EPCAM, PROM1, and MUC1). RPKM, reads per kilobase of transcript per million. In the charts, red writing highlight organs in which HER2 mRNA is physiologically expressed at high levels. Notably, only CEA presents selective co-expression with HER2 restricted to the colon. (B) mRNA and protein expression of HER2 (top) and CEA (bottom) in normal tissues from the Human Protein Atlas database (ProteinAtlas: ERBB2 and CEACAM5); the red rectangles highlight mRNA and protein expression of HER2 and CEA in the gastrointestinal tract. (C) Dot plots showing HER2 (x axis) and CEA (y axis) mRNA expression in (left to right): human CRC samples, CRC cell lines, and CRC PDXs. Green boxes highlight HER2amp samples, all expressing high CEA levels. (D) Flow cytometry histograms reporting surface expression of HER2(PE) (left) and CEA(APC) (right) in different cell lines used as models (x axis: log10 expression) (three independent experiments).
Construction and preliminary validation of a GAL4-inducible CEA-CAR
The CEA-CAR was cloned in a lentiviral vector downstream of the inducible promoter 5xGAL4-UAS-minCMV, recognized by the transcription factor GAL4VP64 (Figure S2A). A series of preliminary tests, to verify specific CEA-CAR expression in response to GAL4, were conducted in Jurkat cells (Figure S2 and Methods).
To build and preliminarily validate the HER2 synNotch/CEA-CAR system (Figure 2A), Jurkat cells were transduced with 4D5-5 HER2 synNotch,32,40 sorted for synNotch expression, and then transduced with GAL4 CEA-CAR, using low CAR vector amounts to decrease basal expression. Transduced cells were subsequently co-cultured with HER2amp/CEA+ CRC0080 target cells, and two controls: SKBR3 (breast cancer, HER2amp/CEA−) and LS180 (CRC, HER2norm/CEA+). In this case, CEA-CAR expression was basally lower (5%–10% positive cells), and still significantly increased (20%–35% positive cells) in the presence of HER2-overexpressing cells (Figure 2B). These results indicate the greater specificity of the system, with CAR-positive cells increased by more than 4-fold. To check functional activation of Jurkat cells in response to CEA-CAR expression and engagement, CD69 expression64 was evaluated by flow cytometry. Indeed, CD69-positive cells raised from 5% in basal conditions to 35% upon co-culture with CRC0080 cells, indicating functional activation of the vast majority of CEA-CAR-expressing Jurkat cells (Figure 2C).
Figure 2.

CEA-CAR induction in Jurkat cells
(A) The HER2 synNotch/CEA-CAR system scheme. (B) Bar graph showing the percentage of CEA-CAR positive cells after co-culture with target cells expressing different levels of HER2, as indicated (basal = without target cells; two independent experiments). (C) Bar graph showing the percentage of CD69 positive cells after co-culture with CRC0080 HER2amp cells (basal = without target cells). Statistical significance was calculated by one-way ANOVA (B) or t test (D). ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001). Error bars report standard deviation (two independent experiments).
Generation, cloning, and characterization of HER2 synNotch/CEA-CAR-expressing NK-92 cells
After preliminary validation in Jurkat cells, the HER2 synNotch/CEA-CAR system was introduced in NK-92 NK cells.42,44 To this aim, NK-92 cells were subjected to multiple rounds of transduction, cell sorting, and cloning, as described in Figures 3A and 3B. First, cells were transduced with HER2 synNotch and subjected to two rounds of cell sorting, achieving 90% HER2 synNotch-positive cells. The sorted population was then transduced with GAL4 CEA-CAR and subjected to one round of negative sorting for CEA-CAR expression after co-culture with HER2norm cells, decreasing basally expressing cells from 4% to 0.1%, followed by one round of positive sorting for CEA-CAR expression after co-culture with HER2amp cells. In concomitance with this last sorting, individual cells were also seeded for cloning.
Figure 3.

Workflow for NK-92 transduction, selection, and cloning
NK-92 were first transduced with the HER2 synNotch lentiviral vector and sorted twice for constitutive HER2 synNotch expression. Subsequently, cells were transduced with the GAL4-driven CEA-CAR lentiviral vector and sorted to exclude those with basal CEA-CAR expression after co-culture with HER2-normal CRC cells. Finally, cells were co-cultured with HER2amp cells and sorted for positive CEA-CAR expression, to concomitantly generate a sorted population and individual clones. (A) Transduction and sorting workflow. (B) Flow cytometry plots before and after each sorting step, as indicated. Red boxes represent the selected populations. y axis, log10 expression; x axis, forward scatter; %, percent of cell above the depicted positivity threshold; MFI, mean fluorescence intensity.
CEA-CAR induction was assessed in detail after expansion of the sorted population (Figure 4C). The cells displayed a very low/negative basal CEA-CAR expression (ranging from 0% to 10% in different experiments) after co-culture with LS180 HER2norm control cells and a variable CEA-CAR induction after co-culture with the HER2amp SKBR3, CRC0080, and CRC0186 cells. The variability in the extent of CEA-CAR expression was probably due to the different HER2 surface expression in the tested cell models.
Figure 4.

CEA-CAR expression in the NK-92 sorted population and in the clones
(A) Flow cytometry analysis of CEA-CAR basal expression in each clone; the red box indicates clones with the lowest CEA-CAR basal expression, selected for subsequent induction experiments against HER2amp cells (two independent experiments). (B) Flow cytometry analysis of CEA-CAR induction in selected clones after co-culture with HER2amp SKBR3; the red box indicates the clones with the highest induction, selected for subsequent induction experiments against all target cells. Bars are SDs (three independent experiments). (C) Flow cytometry plots displaying CEA-CAR induction in NK-92 HER2 synNotch/CEA-CAR after co-culture with HER2 normal and amplified cells (numbers in the square indicate the fraction of CEA-CAR positive cells).
Eleven clones were successfully grown from the last sorting and initially tested for basal CEA-CAR expression, keeping seven clones with less than 5% positive cells (Figure 4A). These clones were then tested for CEA-CAR induction by co-culture with SKBR3 cells, leading to further selection of three clones (Figure 4B). The three clones (NK.92.5F, NK-92.4C, and NK-92.4G) were subsequently expanded and further characterized, confirming low basal CAR expression (ranging from 0% to 5% in different experiments) and high CAR induction after co-culture with different target cell lines (up to 80%–90% with SKBR3) (Figures 4C and S3A). Finally, the NK-92.5F clone was selected for further experiments, in view of its superior induction performances (Figure 4C). The NK-92.5F clone showed better induction also when co-cultured with DIFI or NCI-NCI-H508 cells artificially overexpressing HER2, confirming higher specificity and sensitivity respect to the sorted population (Figure S2B). To be used in human patients, NK-92 cells have to be irradiated.43 Therefore, CEA-CAR induction by the NK-92.5F clone upon co-culture with HER2amp cells was assessed also after irradiation and found to be only marginally decreased (Figure S2C).
To verify whether the NK-92.5F clone maintained the NK-92 typical features, immunophenotyping was performed by flow cytometry (Figure 5A), highlighting no major differences in the levels of the NK markers CD56, NKG2D, NKp30, NKp46, and NKp44, with the DNAM-1 receptor expressed by a lower fraction of cells. Also, the NK-92.5F clone and parental cells were subjected to global RNA sequencing to assess gene expression. A 134-gene signature related to NK cells downloaded from IMMPORT65 was evaluated and displayed no major differences in gene expression between the NK-92.5F clone and wild-type (WT) cells (Figure 5B). These results indicate that the NK-92.5F clone does not show a significant genetic and functional drift and should behave similarly to parental NK-92 cells in basal conditions.
Figure 5.

NK-92.5F clone characterization
(A) Flow cytometry analysis of the major NK/T cell antigen markers on the NK-92 WT and on the NK-92.5F clone. (two independent experiments). (B) Dot plot showing the correlation between NK-92 WT and 5F clone mRNA expression. Black dots = all genes; red dots = NK-specific gene signature. (C) Flow cytometry histograms of CEA-CAR induction after co-culture for different times with HER2amp cells, of either the sorted population (left) or the NK-92.5F clone (right) (three independent experiments). (D) Flow cytometry histograms of CEA-CAR induction and time course suppression after removal of HER2amp target cells, of either the sorted population (left) or the NK-92.5F clone (right). %, percent of cells above the depicted positivity threshold; MFI, mean fluorescence intensity of all cells (three independent experiments).
To evaluate the kinetics and extent of CEA-CAR induction and repression by the sorted population and the NK-92.5F clone, time course co-culture experiments were performed using SKBR3 as target cells. Indeed, the NK-92.5F clone showed a consistently better CEA-CAR on/off profile. In particular, the induction was higher and faster with respect to the sorted population, indeed was already detectable at 12 h and reaching 80%–90% of positive cells at 24–48 h (Figure 5C). When SKBR3 cells were removed after maximal induction, CEA-CAR expression was almost completely abrogated by the NK-92.5F clone within 48 h, while 10%–15% of the sorted population cells remained positive (Figure 5D). As expected, the two additional clones 4C and 4G also displayed slightly worse on/off kinetics (Figures S2D and S2E).
NK-92.5F clone therapeutic efficacy in vitro and in vivo
The killing assays showed consistent and significant cytotoxicity of the irradiated NK-92.5F clone compared with controls (NK-92 WT and NK-92 HER2 synNotch only) against HER2amp/CEA+ CRC models, at all effector:target ratios (Figure 6A). In the case of HER2amp/CEA− SKBR3 control target cells, low killing with no significant difference between the NK-92.5F clone and control effectors was observed. Also, HER2norm/CEA+ CRC cells (LS180) were not preferentially killed by the NK-92.5F clone. Indeed, LS180 displayed higher basal killing by NK-92 controls, indicating the greater sensitivity of these cells to NK-92 killing. As an additional readout of NK-92 cell activation and killing, degranulation upon interaction with target cells was evaluated by measuring the surface expression of CD107a66 and the release in the supernatant of interferon-γ (IFN-γ) and granzyme B. Massive degranulation and CD107a increase by NK-92.5F clone were observed only upon co-culture with HER2amp/CEA+ CRC cells (Figure 6B). Notably, the observed results were similar to those obtained with non-irradiated effectors (Figure S4), indicating that irradiation does not compromise NK-92 activation and killing. Moreover, higher basal killing by WT NK-92 cells was observed at 1:1 effector:target ratio without irradiation, most likely due to expression by the CRC cell lines of the activating ligands for the NKP30, DNAM1, and NKG2D receptors, as observed by RNA sequencing (RNA-seq) (data not shown), that in the absence of irradiation may favor cell proliferation and basal activity.
Figure 6.

NK-92.5F in vitro biologic activity
(A) Specific killing activity of irradiated NK-92.5F against SKBR3, LS180, CRC0080, and CRC0186 cells after 48 h of co-culture at different effector:target ratio. Bars are SD (three independent experiments). Two-way ANOVA p-values: ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001. (B) Degranulation and cytokine release upon co-culture with SKBR3, LS180, CRC0080, and CRC0186 with irradiated effectors. Bars are SDs. Two-way ANOVA p-values: ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001 (three independent experiments for CD107a; four independent experiments for IFN-γ; three independent experiments for granzyme B). (C) Representative confocal microscopy images of CRC0080 organoids grown for 3 days in the presence of different NK-92 effectors, as indicated. Cancer cells were stained with NucBlue (blue signal), and NK-92 cells were stained with PHK26 (red signal). Original magnification ×20; scale bars, 100 μm.
Moreover, the irradiated NK-92.5F clone showed significant killing activity also when co-cultured with DIFI or NCI-H508 cells artificially overexpressing HER2 but not against the parental DIFI and NCI-H508 with normal levels of HER2, confirming high specificity and sensitivity of the system (Figure S5A). To further extend exploration of HER2-induced CEA-CAR expression and target killing, we selected five additional cell lines expressing detectable HER2 but below the levels reached upon HER2 amplification. In all cases CEA-CAR induction was negligible and no increased killing by the NK-92.5F clone with respect to NK-92 controls was observed (Figures S5B and S5C). These data showed that the NK-92.5F clone is selectively activated only by HER2amp/CEA+ CRC cells.
We then considered whether the 5F clone could be effective also in other tumor types expressing HER2 and CEA at high levels. We started exploring the co-expression of HER2 and CEA at the RNA level using cBioportal67 in other gastrointestinal tumors. Indeed, we found that a fraction of gastric cancers in the TCGA panCancer Atlas displays high HER2 and CEA co-expression (Figure S6A). We, thus, obtained a PDX-derived, HER2amp cell line, GTR0233,68 expressing very high HER2 and medium-high CEA levels (Figure S6B). This line was efficiently killed by NK-92.5F cells in vitro (Figure S6C). To further extend the analysis on tumors with frequent HER2 amplification,69 we investigated also HER2 and CEA expression in breast cancer. Again, we found that a fraction of TCGA breast cancer samples express both at high levels (Figure S6D) and obtained a breast cancer cell line with HER2 amplification, HCC2218, that displayed medium CEA expression, verified by flow cytometry (Figure S6E). Interestingly, NK-92.5F cells displayed specific, although suboptimal, enhancement of killing (Figure S6F).
To be fully active in vivo, effector cells need to efficiently perform tumor homing and penetration. Indeed, higher infiltration of the NK-92.5F clone with respect to controls was observed in a transwell assay when HER2amp/CEA+ CRC0080 CRC cells were used as attractors (Figure S7A). Moreover, specific homing and penetration by the NK-92.5F clone with respect to control NK-92 cells were observed by immunofluorescence upon co-culture with HER2amp/CEA+ CRC organoids (Figures 6C and S7B–S7D).
After confirming the specific activity of the NK-92.5F clone in vitro, the in vivo therapeutic efficacy was assessed on the HER2amp/CEA+ models: cell line xenograft for CRC0186 and the PDX of origin for CRC0080.13 First, in vivo induction of CEA-CAR expression was assessed by injection of irradiated NK-92.5F cells directly into the tumor mass generated by HER2amp CRC0080 subcutaneous implants. The CEA-CAR signal was clearly associated with intratumorally detected NK-92.5F cells (Pearson’s correlation between cells and CAR signals = 0.5) (Figure 7A). These data demonstrate that, when NK-92.5F cells get in contact with HER2amp target cells in vivo, CEA-CAR expression is induced. Subsequently, the irradiated NK-92.5F clone was tested for efficacy against the two HER2amp/CEA+ CRC model xenografts by tail vein injection. In both models, the clone induced a significant and sustained reduction of tumor growth (Figure 7B), leading to increased survival (Figure 7C). Control effectors (NK-92 WT and HER2 synNotch only) displayed only marginal, not significant, activity. Furthermore, no activity of NK-92.5F was observed against LS180 (HER2norm/CEA+) xenografts (Figures 7B and 7C).
Figure 7.

NK-92.5F in vivo therapeutic efficacy
(A) Representative immunofluorescence images of CEA-CAR expression by the NK-92.5F clone after injection into CRC0080 xenograft tumors; the tumor was explanted 3 days after intratumoral inoculation of NK-92.5F (red: pkh26-labeled NK-92.5F cells; green: CEA-CAR antibody; blue: NucBlue-labeled cancer cells). Original magnification ×10; scale bars, 100 μm (two independent experiments). (B) In vivo growth of CRC0080, CRC0186, and LS180 xenografts treated with different NK-92 effectors, as indicated. We intravenously injected 5 × 106 irradiated effectors twice a week for a total of six injections. CTRL, PBS. Bars are SEM. Growth curves are stopped when the first mouse of the control cohort has reached the humane endpoint. Two-way ANOVA p-values: ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, between NK-92.5F clone vs. CTRL or WT NK-92 in CRC0080, and between NK-92.5F vs. all control cohorts in CRC0186. (C) Survival curves of mice implanted with CRC0080, CRC0186 or LS180 xenografts and intravenously injected with 5 × 106 NK-92 irradiated effector cells, as indicated. Statistical significance between CTRL and 5F clone was calculated by log rank test for trend. ∗p ≤ 0.05, ∗∗p ≤ 0.01.
These results show that the NK-92.5F clone is specifically effective only against HER2amp/CEA+ CRC models both in vitro and in vivo.
Additional safety and efficacy studies
When maximally induced by HER2 synNotch, the CEA-CAR is active on any cell expressing sufficient levels of CEA, including normal colonocytes adjacent to the tumor. As shown in Figure 5D, CEA-CAR expression is substantially decreased 24 h after detachment of cancer cells. To further evaluate the possibility of bystander effects at even shorter times, NK-92.5F cells were preactivated by incubation for 24 h on a monolayer of fixed CRC0080 HER2amp cells and tested for killing either CRC0080 or LS180 cells, at different time intervals after removal from the CRC0080 monolayer. While both cells express high levels of CEA, LS180 express levels of HER2 recapitulating those of normal colorectal cells. As shown in Figure 8A, the fixed CRC0080 monolayer was even better at inducing CEA-CAR expression, probably because fixed target cells cannot be killed, leading to more sustained synNotch engagement, like with HER2amp/CEA− cells such as SKBR3 (Figure 5C). As shown in Figure 8B, killing of CRC0080 cells was not significantly modified by pre-activation at any time. Only minor, barely significant enhancements were observed on LS180 at early time points (0 or 6 h) and only at a 1:5 effector:target ratio, confirming that very early after detachment from HER2amp cancer cells most of the CEA-CAR activity is lost within few hours.
Figure 8.

Additional NK-92.5F safety and efficacy studies
(A) Flow cytometry plots displaying CEA-CAR induction in NK-92.5F after co-culture with HER2amp cells, either alive or fixed, as indicated. Numbers in the squares indicate the fraction of CEA-CAR positive-cells (two independent experiments). (B) Killing activity of irradiated NK-92.5F preactivated by incubation for 24 h on top of a monolayer of fixed HER2amp CRC0080, against CRC0080, or LS180 cells, at different time intervals after pre-activation and at different effector:target ratio, as indicated. Bars are SD (three independent experiments). (C) Killing activity of irradiated NK-92.5F and NK-92.F3 clones, against SKBR3, LS180, CRC0080, and CRC0186 cells after 48 h of co-culture at different effector:target ratios. Bars are SD (two independent experiments). (D) In vivo growth of CRC0186 xenografts treated with NK-92.5F (HER2 synNotch/CEA-CAR) or with NK-92.F3 (constitutive CEA-CAR), as indicated. We intravenously injected 5 × 106 irradiated effectors twice a week for a total of six injections, CTRL, PBS. Bars are SEM. Growth curves are stopped when the first mouse of the control cohort has reached the humane endpoint. Two-way ANOVA p values (B, C, and D): ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, between NK-92.5F or NK-92.F3 vs. CTRL. (E) Survival curves of CRC0186 xenografts intravenously injected with 5 × 106 NK-92 irradiated effector cells, as indicated. Statistical significance between CTRL and 5F/F3 clones was calculated by log rank test for trend. ∗p ≤ 0.05.
Despite inducing marked and significant growth inhibition, NK-92.5F cells did not induce in vivo tumor regression. One possible explanation could be the loss of HER2 expression by target cells during in vivo treatment. This possibility was ruled out by the absence of any difference in HER2 immunohistochemistry (IHC) signal at the end of the treatment with NK-92.5F cells (Figure S8). We then considered the possibility of testing a different effector type, i.e., primary T cells. In these cells, multiple sorting and cloning to achieve optimal and specific CAR induction is not feasible. A simplified approach was therefore chosen, involving sorting the double-transduced cells only for synNotch expression (see Methods). As shown in Figure S9A, basal CAR expression in the absence of exposure to HER2amp cells is non negligible, and CAR induction is considerably less pronounced than in NK-92.5F. However, in vitro killing activity was mostly directed against HER2amp cells (Figure S9B), which renders this approach promising if better induction is achieved. A third possibility is suboptimal activity due to transient CEA-CAR expression in the synNotch system. We, therefore, generated an additional NK-92 clone (F3 clone) expressing high levels of the CEA-CAR under the EF1A promoter (Figure S9C), to verify if constitutive CAR expression could lead to more pronounced killing activity. As shown in Figures 8C–8E, no significant differences in killing activity, both in vitro and in vivo, were observed between the 5F and F3 clones, with the exception of the expected killing of LS180 by the F3 clone, confirming its independence from HER2 amplification.
Discussion
The main aim of this study was to set up and validate a combinatorial antigen-targeting approach based on the synNotch/CAR system, to engineer killer cells against HER2amp CRC without directing them also against normal tissues known to express these targets at potentially critical levels.29 CEA, the second target antigen of the synNotch/CAR system, is highly expressed in carcinomas as well as in normal cells of the gastrointestinal tract, but it is virtually absent in all other tissues of the human body.58,70 CEA is a negative prognostic biomarker in CRC,71,72 and its measurement is the most sensitive detector of liver metastasis.73 Based on its expression profile, several trials targeting CEA for ACT were used. Indeed, in a phase 1 trial, CAR T cells targeting CEA were tested in three patients with mCRC, and all experienced severe colitis, with one achieving tumor regression.74 In a second study, patients in the treated cohort had transient, acute respiratory toxicity that, in combination with a lack of prolonged CAR-T cell persistence, resulted in premature trial closure.75 In only one trial (NCT02349724) on patients with refractory mCRC, no significant side effects were observed.49 Despite several clinical trials being in phase 1, some of them also with promising clinical outcomes, some reservations remain given the on-target/off-tumor side effects associated with strategies based on constitutive CARs.76 These clinical findings corroborate our choice of hitting CEA-positive CRC with combinatorial strategies.
The bioinformatic analysis pointed out that, even if individual expression of HER2 and CEA is not completely restricted to tumor cells, only CRCs carrying HER2 amplification could overexpress both HER2 and CEA at the optimal levels required for combinatorial targeting. In this way, the on-target/off-tumor toxicity is kept under control, because the synNotch system allows CEA-CAR expression only in the presence of HER2 amplification, which is absent in a physiological state.
Two recent works also explored the synNotch/CAR system in solid tumors. Hernandez-Lopez and colleagues77 built an HER2 synNotch with low affinity for the antigen and a GAL4-inducible HER2 CAR with high affinity for the antigen. Indeed, such a circuit relies on invariably high HER2 expression by all target cells for optimal efficacy. It is known that HER2 expression levels can vary within the tumor, also in the context of HER2 amplification.38,39 We used a HER2 synNotch with slightly higher affinity, which could prompt stronger yet selective CAR induction. Hyrenius-Wittesten and colleagues34 also chose HER2 as CAR target, driven by an anti-ALPPL2 synNotch, again requiring homogeneously high HER2 levels within the tumor. The choice of the CEA-CAR presented here, targeting a lineage antigen expressed by the vast majority of CRC cancer cells, should lead to increased efficacy and reduced probability of escape.
The preliminary series of experiments in Jurkat cells confirmed that the HER2 synNotch receptor, when engaged with its cognate antigen, correctly drives expression of the CEA-CAR under the 5xGAL4-UAS-mCMV promoter, although with some basal activity and suboptimal induction levels. This could be due either to intrinsic limits of the vector system, or to random integration in unfavorable genome regions. The second hypothesis turned out to be the correct one: sorting and cloning of transduced effectors could lead to substantial improvements of the expression dynamic range: undetectable CEA-CAR basal expression and robust induction upon engagement of the HER2 synNotch. Such ON-OFF behavior is similar to that obtained with the SNIP-CAR system,78 but without the need for drug administration to induce CAR activity. However, when applied to primary T cells, the synNotch-CAR system displayed worse performances, confirming the need for improvements in vectors and procedures to avoid multiple sortings of primary T or NK cells. The synNotch/CAR NK-92 system, being based on an immortalized cell line, allowed complex handling procedures, such as multiple transductions, sorting, and cloning, leading to very high and specific CAR induction in the NK-92.5F clone. Most likely, the key factor in determining the optimal behavior of the 5F clone is the lentiviral integration site, which can affect both basal expression levels and the extent of induction.79 In addition, the system’s ability to completely abolish CAR expression within just 24–48 h after removal of HER2amp target cells indicates excellent safety in a future clinical setting: CEA-CAR-expressing cells moving away from the tumor do not maintain CEA-CAR expression, thereby lowering the risk of on-target/off-tumor toxicities. Residual CAR expression after detachment from target cells could be very dangerous in the clinical context because the effectors could also target normal tissues; all effectors should become CEA-CAR negative when detached from target cells as fast as possible, to reduce the possibility of on-target/off-tumor toxicity. Previous findings with the synNotch/CAR system in T cells indeed highlighted some bystander effect, with toxicity against nearby normal cells expressing the CAR antigen but not the synNotch engager.80 Our in vitro data based on effector cell pre-activation show that the NK-92.5F clone turns down CAR expression quite rapidly after detachment from HER2amp cells and loses, within the first few hours, almost all efficacy against cells expressing high levels of CEA but normal HER2 levels. These results show that the risk of killing normal colonocytes by activated, CEA-CAR positive 5F cells, once migrated away from cancer cells, is extremely low. Yet, formal disproval of on-target/off-tumor side effects was not possible with these models, because there is no mouse CEACAM5 orthologue and the 4D5 scFv is not cross-reactive against mouse ERBB2.
The NK-92.5F clone showed specific functional activation and target killing only when co-cultured with HER2amp/CEA+ CRC cells, while sparing CEA+ CRC cells expressing physiological HER2 levels. This behavior is most likely a consequence of the cell sorting criteria: NK-92 were selected to not express the CAR when exposed to cells that express HER2 at high but not extremely high levels and to exhibit extreme CAR induction when exposed to HER2amp cells. Of note, HER2 amplification and CEA overexpression are not limited to CRC: a small fraction of other tumor types displays these features. The observed effectiveness of NK-92.5F cells on gastric and breast cancer cell lines confirmed the possibility of extending its application to other HER2amp tumors expressing sufficient levels of CEA.
The transwell and 3D organoid experiments showed increased homing and penetration of the NK-92.5F clone with respect to NK-92 WT, when exposed to HER2amp/CEA+ CRC cells. Active migration of the NK-92.5F clone toward HER2-overexpressing cells is unlikely to be mediated by the HER2-synNotch alone because it requires cell-cell engagement and mechanical stretching for activation. More likely, when the first NK-92.5F cells encounter their target cells, they get fully activated, degranulate, release chemokines, and kill target cells, which leads also to enhanced attraction of additional NK-92.5F cells to the tumor site.
The NK-92 population transduced with the HER2 synNotch but not with the CEA-CAR provides a very interesting control to investigate if the physical engagement of the synNotch, stabilizing NK-target cell interaction, could lead to an increase in NK-92 killing activity, even in the absence of the CAR. Indeed, the results show that synNotch engagement alone is not sufficient to significantly increase NK-92 activity. A second important control was the challenge of the NK-92.5F clone with HER2amp/CEA− target cells. In fact, it has been shown that the sole CAR expression without engagement with the target antigen could promote a certain degree of effector activation, leading to basal activation and exhaustion in the case of T cells.81 This phenomenon was not observed in the functional activation experiments against HER2amp/CEA− target cells.
As already mentioned, being NK-92 a human lymphoma-derived cell line, irradiation before in vivo application is mandatory and compromises persistence. Indeed, we also observed that irradiation slightly decreases in vitro degranulation and release of IFN-γ. However, it did not significantly impair NK-92 short-term viability, CEA-CAR induction, or target killing both in vitro and in vivo, suggesting potential clinical applicability under Good Manufacturing Process procedures.
In vivo, the NK-92.5F clone successfully controlled the growth of HER2amp/CEA+ CRC xenografts, although it did not cause overt tumor regression, which is in line with what was seen in previous works with CAR-NK-92.82,83 Analysis of in vivo HER2 expression after treatment ruled out the possibility of an escape through partial HER2 loss. Alternative effectors that do not need irradiation, such as primary T or NK cells, could provide better efficacy at the cost of suboptimal specificity and amplitude of CAR induction by the synNotch system. Our results with T cells corroborate this scenario, similar to previous works.32,34,35,36,37 However, the observed in vitro killing activity supports further explorations with T or NK cells, in particular considering improvements in the vector/induction system. In the synNotch/CAR system, CAR expression is strictly limited to the period of contact with synNotch-engaging target cells, which could lead to somewhat lower CAR activity. In the case of HER2/CEA, the most straightforward comparison is between the NK-92.5F clone and the NK-92 clone (F3) constitutively expressing the CEA-CAR. Such a comparison is also more clinically relevant because CEA expression in normal tissues is more restricted than that of HER2. The in vitro and in vivo results presented here demonstrate no differences between constitutive and synNotch-induced CEA-CAR expression, except for the lost selectivity for HER2 amplification, with possible on-target/off-tumor effects on normal CEA-expressing cells. It will be also critical to optimize the frequency of infusions and the number of infused effectors to achieve the best therapeutic outcome with limited toxicity. Indeed, mice treated with the NK-92.5F clone showed no signs of pain, and no organ toxicity was observed during gross autopsies. Surely, irradiated NK-92 effectors cannot be considered as a living drug, able to expand and persist in patients. This limit, however, is counterbalanced by the greatly increased availability as an off-the-shelf product, easy to further improve with additional engineering steps, and to scale up for potential use also in allogeneic settings, as already demonstrated in several clinical studies.47,48
In conclusion, this work showed the design, characterization, and preclinical validation of a combinatorial CEA-CAR-based immunotherapy strategy against CRC with HER2 amplification. The chosen approach for combinatorial targeting, based on the synNotch/CAR system, demonstrated excellent performance, flexibility, and safety, making it ideal for targeting different antigen combinations. This study potentially offers an accessible, off-the-shelf cell therapy approach in the contest of HER2amp CRC, using the HER2 synNotch/CEA-CAR strategy in CRC resistant to targeted therapy and in addition to HER2/EGFR-targeted treatment in partially responsive cases.
Materials and methods
Gene expression data analysis
For normal tissues, expression data were obtained from the Entrez Gene (https://www.ncbi.nlm.nih.gov/gene) and Human Protein Atlas (https://www.proteinatlas.org) online repositories. Entrez Gene provides RNA-seq profiles of tissue samples from 95 human individuals representing 27 different tissues.54 Human Protein Atlas55 provides both mRNA and protein expression profiles, respectively: (1) HPA RNA-seq of 1,206 cell lines, 40 human tissues, and 18 blood cell types, (2) GTEx RNA-seq of 36 human tissues, and (3) protein expression scores from tissue microarrays, including 44 normal tissues from 144 individuals.
For CRC, the following profiles were analyzed: (1) RNA-seq profiles of 450 human tumor samples obtained from TCGA as previously described,84,85 (2) RNA-seq profiles of 602 CRC PDX samples from our laboratory,61 and (3) RNA-seq profiles of 119 CRC cell lines. Additional gene expression data were obtained from the cBioPortal (www.cbioportal.org), selecting gastric and breast cancer datasets from the TCGA panCancer Atlas.
Generation of RNA-seq profiles
NK-92 RNA was extracted, quantified, sequenced, and analyzed as previously described.61 We downloaded 134 NK cell-related genes from Immport (https://www.immport.org/resource).65 Expression values, calculated in counts per million (CPM), were plotted after addition of 1 CPM (to avoid zero values) and log2 transformation.
Cell cultures
SKBR3 human breast adenocarcinoma cells and LS180 human colorectal adenocarcinoma cells, were purchased from Interlab Cell Line Collection (ICLC, IRCCS San Martino Policlinico Hospital). HCT116 human colorectal carcinoma, NCI-H508 colorectal adenocarcinoma were purchased from the National Cancer Institute (NCI). We purchased 293T human kidney cells, Jurkat acute T cell leukemia cells, SKCO1 colorectal adenocarcinoma cells, LS174T Dukes' type B colorectal adenocarcinoma and SW1116 colorectal adenocarcinoma cells, HCC2218 breast ductal carcinoma from the American Type Culture Collection (LGC Standards Srl). DIFI colorectal carcinoma cells, HDC9 colon adenocarcinoma, and RW7213 colon carcinoma cells were kindly provided by Prof. A. Bardelli (Candiolo Cancer Institute, Candiolo, Torino/IFOM ETS, The AIRC Institute of Molecular Oncology). NCI-H508 and DIFI CRC cells, overexpressing HER2, were obtained in Trusolino lab as described.63 CRC0080 and CRC0186 CRC cells were obtained in the Bardelli lab as described.15 CRC0186 cells were further separated from mouse fibroblasts with the Cell Celector Flex (ASL Automated laboratory System), Sartorius. GTR0233 gastric carcinoma cells were a kind gift of Silvia Giordano and Simona Corso.68 NK-92 human malignant non-Hodgkins’s lymphoma cells were purchased from the Leibniz Institute DSMZ. Cells were maintained at 37°C in 5% CO2 in recommended media (Sigma Life Science, GIBCO Thermo Fisher Scientific): RPMI-1640 for HCT116, NCI-H508, Jurkat, LS174T, HCC2218, GTR0233; DMEM for SKBR3; MEM for LS180, SKCO1; IMDM for 293T; Ham’s F12 for DIFI; DMEM/F12 for CRC0080, CRC0186, SW1116, HDC9, RW7213; all supplemented with 10% of fetal bovine serum (FBS, Euroclone SpA), 1% of (Sigma-Aldrich), 1% penicillin-streptomycin (Sigma-Aldrich), and only for CRC0186 also Y-27632 Rock Inhibitor 10 μM (Selleckem). NK-92 cells were grown in alpha MEM supplemented with 12.5% FBS (Euroclone SpA), 12.5% horse serum (Euroclone SpA), 1% L-glutamine, 1% penicillin-streptomycin, and 100 U/mL interleukin (IL)-2 (Miltenyi Biotec). Adherent cells were cultured in Petri dishes (coated with collagen I for CRC0186), suspension cells were cultured in flasks (Corning). CRC0080 and CRC0186 organoids were cultured in 12-well costar in a 200-μL Matrigel droplet (Product Number: 356234, Corning) covered with 1 mL modified DMEM/F12 supplemented with B-27, N-2, and 1 mM N-acetylcysteine (Thermo Fisher Scientific), plus 1% penicillin-streptomycin, 1% L-glutamine (Sigma-Aldrich), and 20 ng/mL EGF (Sigma-Aldrich). All cell cultures were tested for mycoplasma contamination.
Lentiviral vectors
The following lentiviral vectors were purchased from Addgene: sGFP (#79129), LAG16 SynNotch30 (#85421), 4D5-5 HER2 synNotch32 (#85423), 5xGAL4 UAS BFP PGK Cherry30,32 (#79130), 5xGAL4 UAS30 (#79119), and EF1alpha CEA-CAR.86 The GAL4 CEA-CAR was constructed as follows: the 5xGAL4-UAS-mCMV promoter was placed upstream from a second-generation CEA-CAR, composed of the Anti-CEA scFv from the mAb BW431/26, followed by the IgG4 hinge, the CD28 transmembrane and intracellular domains and the CD3ζ domain.87 Promoter and CAR were cloned into a standard lentiviral backbone by Vector Builder. The CMV-luciferase lentiviral vector was designed in house and cloned by Vector Builder. For preliminary testing of CEA-CAR regulated induction, a lentiviral vector constitutively expressing GAL4VP64 under the EFS promoter (EFS-GAL4VP64-PGK-PURO) was designed and cloned in house (Figures S2A and S2B).
Lentiviral vectors preparation
Third-generation lentivirus stock production was obtained through 293T cells calcium phosphate transient transfection as described previously.88 Briefly, 5 × 106 293T cells were seeded in 15-cm Petri dishes. The day after, 37.5 μg of the transfer vector, 16.26 μg of the packaging plasmids pMDLg/pRRE, 6.25 μg of the plasmid pRSV.REV, and 9 μg of the vesicular stomatitis virus (VSV) envelope plasmid pMD2.VSV-G were added in a solution of 125 μL CaCl2, 2.5 M, and 1,250 μL of HBS 2× (for the formation of Ca3(PO4)4 particle). After 16 h from transfection medium was changed and then 1 mM/L Na butyric acid (Sigma-Aldrich) was added. Thirty hours later, the supernatants with the viral particles were purified and concentrated by ultracentrifugation. Determination of the viral p24 antigen concentration was done by HIV-1 p24 Core profile ELISA (PerkinElmer Life Science, Inc.).
Lentiviral transduction
CRC cell lines were plated at 50,000 cells/well in six-well plates in complete medium. After 24 h, the medium was replaced and the cells were transduced with 1 μL of concentrate lentivirus, with polybrene (8 μg/mL).
Jurkat cells were plated at 100,000 cell/well in 24-well plates in RPMI medium plus the lentiviral vector of choice. After overnight incubation, cells were washed and resuspended in culture medium. When applicable, puromycin selection (2 μg/mL) was used to select transduced cells.
NK-92 cells were transduced at 300,000 cells/well in 24-well plates, in NK-92 medium (see above) further supplemented with 1,000 U/mL human IL-2 (Miltenyi Biotec), plus the lentivirus of choice, and [10 μg/mL] protamine sulfate (Sigma-Aldrich). Plates were centrifuged at 1,000×g for 1 h at 32°C and incubated 24 h at 37°C, after which the medium was changed to supplement fresh virus. Plates were again centrifuged at 1,000×g for 1 h, incubated 24 h at 37°C, and finally washed and resuspended in culture medium.
Isolation, activation, transduction, and expansion of HER2 synNotch/CEA-CAR T cells
T cells were purified from peripheral blood mononuclear cells (PBMCs) of healthy donors. PBMCs were isolated by density gradient centrifugation (Lymphosep, Aurogene) and then activated by anti-Biotin MACSi Bead Particles loaded with anti-CD2, anti-CD3, and anti-CD28 antibodies (Miltenyi Biotec) for 24 h. PBMCs were maintained in culture medium + human recombinant IL-2 (100U/mL, Miltenyi Biotech) for a further 24 h. T cells were then transduced at 1,000,000 cells/well in 24-well plates, in RPMI medium further supplemented with 100 U/mL human IL-2 (Miltenyi Biotec), plus polybrene (8 μg/mL) for 12 h with the HER2 synNotch vector. Plates were then centrifuged at 1,000×g for 5 min washed and resuspended in culture medium with the second vector, GAL4 CEA-CAR, again for 12 h. Finally, plates were centrifuge washed and resuspend in culture medium. Non-transduced T cells (WT) were used as a paired control. HER2 synNotch/CEA-CAR or WT T cells were expanded for a maximum of 4 weeks in the presence of IL-2 (100 U/mL).
Flow cytometry and cell sorting
Adherent cells were washed twice with cold PBS (Sigma-Aldrich), detached with 1 mM PBS-EDTA and resuspended (100,000 cells in 200 μL) in PBS-1% BSA plus 1 μL of anti-HER2 (PE Mouse Anti-Human HER-2/neu Clone NEU24.7) or anti-CEA (APC, CD66abce Antibody, anti-human REAaffinity Clone REA876) antibody for 30 min on ice. Cells were then washed with cold PBS, co-stained with DAPI (1 μL of 1 μg/mL working solution, Sigma-Aldrich) for 5 min at 4°C and analyzed by flow cytometry (Cyan ADP, Beckman Coulter s.r.l.) using Summit 4.3 software (Dako). The same procedure was applied to cells in suspension (Jurkat, NK-92), with the exception of detachment. SynNotch expression was evaluated using 2 μL of anti-myc tag mouse Ab (9B11 clone, Cell Signaling). CEA-CAR expression was assessed using 1 μL of mAb specific for the IgG1/CH2CH3 spacer, Alexa Fluor 647 AffiniPure F(ab')2 Fragment Goat Anti-Human IgG, H + L antibody (Jackson ImmunoResearch). For surface marker analysis, the following antibodies were used: 1 μL of anti-CD56 (PE/APCvio770 Mouse Anti-Human, Clone MY31) anti-CD3 (VioBlue mouse anti-human Clone BW264/56), anti-NKG2D (PE/APC Mouse Anti-Human Clone 1D11), anti-DNAM1 (APC mouse anti-human clone DX11), anti-NKp46 (Viobright515 mouse anti-human clone 9E2), anti-NKp30 (PEvio615 anti-human clone REA823), and anti-NKp44 (PEvio770 anti-human clone REA1163). All antibodies were from Miltenyi Biotec. The fluorescence signal derived from the Isotype control or without Ab was set as threshold (0 < mean fluorescent intensity > 101). For cell sorting, Jurkat or NK-92 cells stained as above were sorted using a MoFlo ASTRIOS EQ Cell Sorter (Beckman Coulter). At the final sorting step of NK-92 HER2 synNotch/CEA-CAR cells or constitutive CEA-CAR cells were directed both in a single tube to obtain a population, and on a 96-well plate, to obtain single clones. HER2 synNotch/CEA-CAR T cells were sorted only once for HER2 synNotch expression and expanded for 2 weeks.
Irradiation of NK-92 cells
NK-92 irradiation was optimized to achieve a complete proliferative block without compromising short-term viability.89 Cells were collected by centrifugation, counted, washed, resuspended in fresh growth medium, and irradiated with 5 Gy (RAD GIL, Gilardoni S.p.a.).
CEA-CAR induction experiments
To assess CEA-CAR induction, Jurkat, NK-92 and T cells were co-cultured in vitro with target cells (30,000/well in a 24-well plate or 90,000/well in a 12-well plate) at 1:3 effectors/target ratio, for 24 h for CRC0080, CRC0186, DIFI, NCI-H508 cells, or 48 h for LS180 and SKBR3 cells, in target cell culture medium at 37°C, 5% CO2. CAR induction was assessed by flow cytometry.
For time course CEA-CAR induction analysis, NK-92 cells were co-cultured (as described above) for 0, 3, 6, 12, 18, 24, and 48 h with the SKBR3 HER2amp target cells. For time course CEA-CAR repression analysis, NK-92 cells were cultured for 48 h with SKBR3 HER2amp target cells, then removed from the target cells evaluated at 0, 24, 48, and 72 h for CAR repression. In all experiments, CAR expression was evaluated by flow cytometry as described above.
Pre-activation assays
NK-92.5F clone cells were preactivated by incubation for 24h on top of a monolayer of HER2amp CRC0080 cells, previously fixed with PAF 4% for 15 min, and then tested for killing against target cells (see Cytotoxicity assays), at different intervals after removal from the CRC0080 monolayer.
Cytotoxicity assays
Cytotoxicity of irradiated/non-irradiated NK-92 and T cells was assessed by co-culture with target cells (10,000/well in a 96-well plate) for 48 h at different effectors:target ratios (1:1, 1:5, 1:10), in target cell culture medium at 37°C, 5% CO2. Target cell viability was evaluated either with Cell Titer-Glo Luminescent Cell Viability Assay (Promega) or with Luciferase Assays (D-Luciferin Firefly potassium salt; PerkinElmer Part Number #122799) in the case of LUC-transduced target cells. Luciferase chemiluminescence was detected with a Spark 10M instrument (Tecan).
Cytokine secretion assays
Irradiated/non-irradiated NK-92 cells were incubated with target cell (30,000/well in a 48-well plate, at an effector:target ratio of 1:3) for 48 h in target cell culture medium at 37°C, 5% CO2. Thereafter, supernatants were collected and tested for IFN-γ and granzyme B, respectively using the Human IFN-gamma Quantikine ELISA Kit (Catalog #: DIF50, R&D Systems) and the Human Granzyme B ELISA Kit (Catalog # BMS2027-2, Thermo Fisher Scientific).
Degranulation assays
Degranulation of irradiated/non-irradiated NK-92 cells was assessed after co-culture with target cells (30,000/well in a 24-well plate, E/T 1:1) for 24 h in target cell culture medium at 37°C, 5% CO2, by flow cytometry measurement of surface expression of CD107a with an anti-CD107a detection antibody (PE anti-human clone REA792, Miltenyi Biotec).
Invasion assays
For confocal microscopy invasion assays, CRC0080 or CRC0186 organoids were plated in Matrigel (Product Number: 356234, Corning) domes in eight-well glass-bottom chamber slides (Falcon), covered with modified DMEM/F12. After 48 h, organoids were overnight labeled with NucBlue (NucBlue Live Ready Probes Reagent) directly in the culture chamber slide wells. Then CRC0080 or CRC0186-derived organoids were co-cultured with irradiated NK-92 cells previously stained with PKH26 dye, at an E:T ratio of 2:1 in culture medium in the presence of IL-2 (100 U/mL). After 48 h of co-culture, the medium was removed, and organoids were fixed with a 4% PAF solution for 15 min and covered with mounting medium, to be observed using a TCS SPE Leica microscope. Image acquisition was performed by maintaining the same laser power, gain, offset, and magnification (20×). We generated maximum intensity projections for each analyzed organoid with LAS X Software (Leica) to quantify NK-92 cell recruitment and infiltration. Images of the total PKH26 red fluorescence area present either at the boundary or inside the organoids were analyzed using ImageJ software (in at least five images per condition).
For transwell invasion assays, target cells were plated in the bottom chamber of 24-well Transwell plates (Corning), at 70% confluency in 700 μL. The 8-μm pore membranes of the migration chambers were coated with 50 μL Matrigel (Corning), after which irradiated NK-92 cells previously stained with Pkh26 dye (Sigma-Aldrich) were plated in 500 μL. After 48 h, stained cells migrated to the bottom well were detected by flow cytometry. The fold-increase of migrated cells was calculated against wells in which only NK-92 cells in the upper chamber were plated, with no target cells in the bottom chamber.
In vivo experiments
The antitumor activity of the various irradiated NK-92 effectors was evaluated using CRC xenograft models in immunodeficient mice. In vivo experiments received approval by the competent committee and internal review board (auth. N° 225/2021-PR). CRC xenografts were established in 6- to 7-week-old NOD/SCID (Charles River Laboratories, SRL) male mice by subcutaneous injection with 1 × 106 cells (CRC0186, LS180) or PDX implantation (CRC0080). When tumors were approximately 20 mm3 in volume, mice were randomized and then infused in the tail vein twice a week with 5 × 106 irradiated NK-92 effectors resuspended in 150 μL PBS, for a total of six infusions. PBS only was used as negative control. Treatment and control cohorts included six or eight mice each for all models. Mice were monitored daily for possible toxicities, while tumor growth was measured two times per week with manual caliper. Tumor volume was calculated by the following formula: V ¼ 4/3 × p × (a/2)2 × (b/2), where a is the length and b is the width of the tumor. Mice were sacrificed at the ethical endpoint.
Immunofluorescence and IHC
For in vivo CEA-CAR induction immunofluorescence analysis, CRC0080 xenografts were generated in NOD/SCID mice as described above. When tumors reached a volume of 200 mm3, 5 × 106 NK-92.5F cells pre-labeled with PKH26 (Sigma-Aldrich) were injected directly into the tumor mass. After 3 days from the injection, mice were sacrificed and tumors were explanted and analyzed by immunofluorescence. Briefly, 5-μm sections were cut from optimal cutting temperature-embedded tissues, mounted on slides, fixed with Zinc solution for 10 min, permeabilized with PBS-Triton 0,1% for 10 min, saturated with 1% PBS-BSA at RT for 60 min, and incubated with primary antibody (Alexa Fluor 647 AffiniPure F(ab')2 Fragment Goat Anti-Human IgG, H+L antibody, Jackson Immunoresearch), in a humid chamber overnight 4°C. The following day, after DAPI (D9542, Sigma) staining 5′ at room temperature, sections were mounted on glass slides with mounting solution Mowiol, visualized with a TCS SPE Leica microscope (10×) and analyzed with LAS X Software (Leica). Correlation between the red and the green signal was measured on eight images with Pearson’s correlation by ImageJ software.
CRC xenograft explants after treatment were analyzed by IHC. Samples (5-mm thick) were cut from formalin-fixed, paraffin-embedded tissue sections, mounted on slides, and treated as per standard IHC procedures. Tissue sections were deparaffinized with 100% xylene and rehydrated with decreasing concentrations of ethyl alcohol. Antigen retrieval was performed by boiling the sections in 1 mmol/L EDTA (pH 9.0) for 60 min. Slides were treated with 3% hydrogen peroxide, 1% BSA (Invitrogen), and 5% normal horse serum in TBS [25 mmol/L Tris (pH 7.4) and 150 mmol/L NaCl] containing 0.1% Tween 20 (Sigma-Aldrich). To confirm HER2 expression, slides were incubated in a closed humid chamber overnight at 4°C with the HER2-specific mAb (Ventana anti-HER2 (4B5) Rabbit mAb, Roche). After washing, a secondary anti-mouse IgG xenoantibody was added. Secondary antibodies conjugated to horseradish peroxidase (HRP) were generated and IHC signals were detected with the EnVision1 System-HRP (Dako North America, Inc) and chromogen Diaminobenzidine (DAB) Substrate (DakoCytomation Liquid DABþ Substrate Chromogen System, Dako). Tissue sections were counterstained with Mayer Hematoxylin (Bio-Optica). Tissue sections were mounted on glass slides and visualized with a DM750 Leica Microscope equipped with Leica ICC50W CCD Camera (LAS EZ3.4.0 software).
Statistical analysis
Average, SD, and SEM were calculated using Microsoft Office Excel 2010 software (Microsoft Corporation) and GraphPad Prism 9. Statistical significance was determined using a two-tailed Student’s t test, One-way ANOVA test, two-way ANOVA test, Pearson’s correlation, and log rank test for trend as appropriate. Flow cytometry, immunofluorescence, and IHC figures show one representative experiment.
Data and code availability
All data related to this study are included in this paper and its Supplementary information. Experimental details are available from the corresponding author on reasonable request.
Acknowledgments
The authors thank Simona Corso, Silvia Giordano, and Alberto Bardelli for sharing cell lines; Flavia Martino, Maria Domenica Lupi, and Rossella Pennisi for assistance on immunofluorescence image analysis; Enrico Berrino and Sara Erika Bellomo for help with immunohistochemistry; Emanuela Boccuni for help with gastric cancer cell line culture; Daniela Cantarella, Barbara Martinoglio, Roberta Porporato, Paola Bernabei, Raffaella Albano, and Stefania Giove for technical assistance; and Simona Destefanis for secretarial assistance.
This research was funded by Italian Ministry of Health (CAR T Italia Project RCR-2019-23669115 [E.M.]; “Progetto di Rete ACC 2019” - RCR 2020 [E.M]; “Progetto CAR-T_2 RCR-2023-23684268 [E.M.]; Ricerca Corrente 2024 [E.M.]; GR-2016–02362726 [C.I.]); Fondazione Piemontese per la Ricerca sul Cancro-ONLUS (5x1000 Ministero della Salute 2015 “Immunogenomics” [E.M., L.T.] and 2017 “SEE-HER” [E.M]); Associazione Italiana per la Ricerca sul Cancro (5 per Mille Metastasis - ID 21091 [E.M.,L.T.,A.B.]; under BRIDGE 2022-ID 27321 [S.A.]; IG-2017 ID 20259 [D.S.]).
All procedures in mice adhered to the “Animal Research: Reporting of In Vivo Experiments” (ARRIVE) standards and were approved by the Ethical Commission of the Candiolo Cancer Institute (Candiolo, Torino, Italy), and by the Italian Ministry of Health. All patients provided written informed consent; samples were collected and the study was conducted under the approval of the review boards of the Candiolo Cancer Institute. The study was done in accordance with the principles of the Declaration of Helsinki, the International Conference on Harmonization, and Good Clinical Practice guidelines and General Data Protection Regulation.
Author contributions
M.C.: Design and execution of experiments, analysis, interpretation of data, writing; E.T., A.D., C.P., F.I., and L.F.: execution of experiments; C.D. and V.L.: providing assistance and immunological expertise; S.M.L., V.V., and F.C.: contribution to in vivo experiments; C.I.: transcriptomics profiling; S.A., A.B., L.T., E.V., and D.S.: providing CRC models, expert advice and critical reading of the manuscript; E.M.: Conception and design of the study, supervision, interpretation of data, writing, funding acquisition.
All authors read and approved the final manuscript.
Declaration of interests
The authors declare no potential conflict of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2024.06.023.
Contributor Information
Marco Cortese, Email: marco.cortese@ircc.it.
Enzo Medico, Email: enzo.medico@unito.it.
Supplemental information
References
- 1.Rawla P., Sunkara T., Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019;14:89–103. doi: 10.5114/pg.2018.81072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morgan E., Arnold M., Gini A., Lorenzoni V., Cabasag C.J., Laversanne M., Vignat J., Ferlay J., Murphy N., Bray F. Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN. Gut. 2023;72:338–344. doi: 10.1136/gutjnl-2022-327736. [DOI] [PubMed] [Google Scholar]
- 3.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2020. CA. Cancer J. Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 4.Rumpold H., Niedersüß-Beke D., Heiler C., Falch D., Wundsam H.V., Metz-Gercek S., Piringer G., Thaler J. Prediction of mortality in metastatic colorectal cancer in a real-life population: a multicenter explorative analysis. BMC Cancer. 2020;20:1149. doi: 10.1186/s12885-020-07656-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chau I., Cunningham D. Treatment in Advanced Colorectal Cancer: what, when and How? Preprint. 2009. [DOI] [PMC free article] [PubMed]
- 6.Shin A.E., Giancotti F.G., Rustgi A.K. Metastatic colorectal cancer: mechanisms and emerging therapeutics. Trends Pharmacol. Sci. 2023;44:222–236. doi: 10.1016/j.tips.2023.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roy-Chowdhuri S., Davies K.D., Ritterhouse L.L., Snow A.N. ERBB2 (HER2) Alterations in Colorectal Cancer. J. Mol. Diagn. 2022;24:1064–1066. doi: 10.1016/j.jmoldx.2022.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Oh D.Y., Bang Y.J. HER2-targeted therapies — a role beyond breast cancer. Preprint Nat. Res. 2020 doi: 10.1038/s41571-019-0268-3. [DOI] [PubMed] [Google Scholar]
- 9.Siena S., Sartore-Bianchi A., Marsoni S., Hurwitz H.I., McCall S.J., Penault-Llorca F., Srock S., Bardelli A., Trusolino L. Preprint at Oxford University Press; 2018. Targeting the Human Epidermal Growth Factor Receptor 2 (HER2) Oncogene in Colorectal Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laurent-Puig P., Balogoun R., Cayre A., Le Malicot K., Tabernero J., Mini E., Folprecht G., van Laethem J.-L., Thaler J., Petersen L.N., et al. ERBB2 alterations a new prognostic biomarker in stage III colon cancer from a FOLFOX based adjuvant trial (PETACC8) Ann. Oncol. 2016;27 doi: 10.1093/annonc/mdw370.08. [DOI] [Google Scholar]
- 11.Yonesaka K., Zejnullahu K., Okamoto I., Satoh T., Cappuzzo F., Souglakos J., Ercan D., Rogers A., Roncalli M., Takeda M., et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci. Transl. Med. 2011;3 doi: 10.1126/scitranslmed.3002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sartore-Bianchi A., Amatu A., Porcu L., Ghezzi S., Lonardi S., Leone F., Bergamo F., Fenocchio E., Martinelli E., Borelli B., et al. HER2 Positivity Predicts Unresponsiveness to EGFR-Targeted Treatment in Metastatic Colorectal Cancer. Oncologist. 2019;24:1395–1402. doi: 10.1634/theoncologist.2018-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertotti A., Migliardi G., Galimi F., Sassi F., Torti D., Isella C., Corà D., Di Nicolantonio F., Buscarino M., Petti C., et al. A Molecularly Annotated Platform of Patient-Derived Xenografts (“Xenopatients”) Identifies HER2 as an Effective Therapeutic Target in Cetuximab-Resistant Colorectal Cancer. Cancer Discov. 2011;1:508–523. doi: 10.1158/2159-8290.CD-11-0109. [DOI] [PubMed] [Google Scholar]
- 14.Kavuri S.M., Jain N., Galimi F., Cottino F., Leto S.M., Migliardi G., Searleman A.C., Shen W., Monsey J., Trusolino L., et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5:832–841. doi: 10.1158/2159-8290.CD-14-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lazzari L., Corti G., Picco G., Isella C., Montone M., Arcella P., Durinikova E., Zanella E.R., Novara L., Barbosa F., et al. Patient-derived xenografts and matched cell lines identify pharmacogenomic vulnerabilities in colorectal cancer. Clin. Cancer Res. 2019;25:6243–6259. doi: 10.1158/1078-0432.CCR-18-3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sartore-Bianchi A., Trusolino L., Martino C., Bencardino K., Lonardi S., Bergamo F., Zagonel V., Leone F., Depetris I., Martinelli E., et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17:738–746. doi: 10.1016/S1470-2045(16)00150-9. [DOI] [PubMed] [Google Scholar]
- 17.Sartore-Bianchi A., Lonardi S., Martino C., Fenocchio E., Tosi F., Ghezzi S., Leone F., Bergamo F., Zagonel V., Ciardiello F., et al. Pertuzumab and trastuzumab emtansine in patients with HER2-amplified metastatic colorectal cancer: The phase II HERACLES-B trial. ESMO Open. 2020;5 doi: 10.1136/esmoopen-2020-000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siena S., Di Bartolomeo M., Raghav K., Masuishi T., Loupakis F., Kawakami H., Yamaguchi K., Nishina T., Fakih M., Elez E., et al. Trastuzumab deruxtecan (DS-8201) in patients with HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2021;22:779–789. doi: 10.1016/S1470-2045(21)00086-3. [DOI] [PubMed] [Google Scholar]
- 19.Tosi F., Sartore-Bianchi A., Lonardi S., Amatu A., Leone F., Ghezzi S., Martino C., Bencardino K., Bonazzina E., Bergamo F., et al. Long-term Clinical Outcome of Trastuzumab and Lapatinib for HER2-positive Metastatic Colorectal Cancer. Clin. Colorectal Cancer. 2020;19:256–262.e2. doi: 10.1016/j.clcc.2020.06.009. [DOI] [PubMed] [Google Scholar]
- 20.Casak S.J., Marcus L., Fashoyin-Aje L., Mushti S.L., Cheng J., Shen Y.-L., Pierce W.F., Her L., Goldberg K.B., Theoret M.R., et al. FDA Approval Summary: Pembrolizumab for the First-line Treatment of Patients with MSI-H/dMMR Advanced Unresectable or Metastatic Colorectal Carcinoma. Clin. Cancer Res. 2021;27:4680–4684. doi: 10.1158/1078-0432.CCR-21-0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borelli B., Antoniotti C., Carullo M., Germani M.M., Conca V., Masi G. Immune-Checkpoint Inhibitors (ICIs) in Metastatic Colorectal Cancer (mCRC) Patients beyond Microsatellite Instability. Cancers (Basel) 2022;14:4974. doi: 10.3390/cancers14204974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiu M.-Z., He C.-Y., Yang X.-H., Yang L.-Q., Lin J.-Z., Zhou D.-L., Long Y.-K., Guan W.-L., Jin Y., Li Y.-H., et al. Relationship of HER2 Alteration and Microsatellite Instability Status in Colorectal Adenocarcinoma. Oncologist. 2021;26:e1161–e1170. doi: 10.1002/onco.13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gross G., Waks T., Eshhar Z. 1989. Expression of Immunoglobulin-T-Cell Receptor Chimeric Molecules as Functional Receptors with Antibody-type Specificity. (chimeric genes/antibody variable region) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenberg S.A., Restifo N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X., Zhu L., Zhang H., Chen S., Xiao Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.927153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marofi F., Motavalli R., Safonov V.A., Thangavelu L., Yumashev A.V., Alexander M., Shomali N., Chartrand M.S., Pathak Y., Jarahian M., et al. Preprint at BioMed Central Ltd; 2021. CAR T Cells in Solid Tumors: Challenges and Opportunities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang R.F., Wang H.Y. Preprint at Nature Publishing Group; 2017. Immune Targets and Neoantigens for Cancer Immunotherapy and Precision Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chruściel E., Urban-Wójciuk Z., Arcimowicz Ł., Kurkowiak M., Kowalski J., Gliwiński M., Marjański T., Rzyman W., Biernat W., Dziadziuszko R., et al. Adoptive Cell Therapy—Harnessing Antigen-Specific T Cells to Target Solid Tumours. Cancers (Basel) 2020;12:683. doi: 10.3390/cancers12030683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of t cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morsut L., Roybal K.T., Xiong X., Gordley R.M., Coyle S.M., Thomson M., Lim W.A. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell. 2016;164:780–791. doi: 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roybal K.T., Rupp L.J., Morsut L., Walker W.J., McNally K.A., Park J.S., Lim W.A. Precision Tumor Recognition by T Cells with Combinatorial Antigen-Sensing Circuits. Cell. 2016;164:770–779. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roybal K.T., Williams J.Z., Morsut L., Rupp L.J., Kolinko I., Choe J.H., Walker W.J., McNally K.A., Lim W.A. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell. 2016;167:419–432.e16. doi: 10.1016/j.cell.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kopan R., Ilagan M.X.G. Preprint at Elsevier B.V.; 2009. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hyrenius-Wittsten A., Su Y., Park M., Garcia J.M., Alavi J., Perry N., Montgomery G., Liu B., Roybal K.T. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.abd8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choe J.H., Watchmaker P.B., Simic M.S., Gilbert R.D., Li A.W., Krasnow N.A., Downey K.M., Yu W., Carrera D.A., Celli A., et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.abe7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moghimi B., Muthugounder S., Jambon S., Tibbetts R., Hung L., Bassiri H., Hogarty M.D., Barrett D.M., Shimada H., Asgharzadeh S. Preclinical assessment of the efficacy and specificity of GD2-B7H3 SynNotch CAR-T in metastatic neuroblastoma. Nat. Commun. 2021;12 doi: 10.1038/s41467-020-20785-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tseng H.C., Xiong W., Badeti S., Yang Y., Ma M., Liu T., Ramos C.A., Dotti G., Fritzky L., Jiang J.G., et al. Efficacy of anti-CD147 chimeric antigen receptors targeting hepatocellular carcinoma. Nat. Commun. 2020;11 doi: 10.1038/s41467-020-18444-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen P.-C., Yeh Y.-M., Chu C.-T., Su P.-F., Chiu P.-H., Lin B.-W., Chen S.-H., Lin P.-C., Lee C.-T., Chen H.H.W., Chen C.C. HER2 amplification in colorectal cancer with brain metastasis: A propensity score matching study. Eur. J. Cancer. 2023;181:62–69. doi: 10.1016/j.ejca.2022.12.019. [DOI] [PubMed] [Google Scholar]
- 39.Hou Y., Nitta H., Li Z. HER2 Intratumoral Heterogeneity in Breast Cancer, an Evolving Concept. Cancers (Basel) 2023;15:2664. doi: 10.3390/cancers15102664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carter P., Presta L., Gormant C.M., Ridgwayt J.B.B., Hennert D., Lee W., Wongt T., Rowlandf A.M., Kotts C., Carvert M.E., et al. 1992. Humanization of an Anti-p185HER2 Antibody for Human Cancer Therapy. (antibody engineering/site-directed mutagenesis/c-erbB-2/neu) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gong J.H., Maki G., Klingemann H.G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652–658. [PubMed] [Google Scholar]
- 42.Suck G., Odendahl M., Nowakowska P., Seidl C., Wels W.S., Klingemann H.G., Tonn T. NK-92: an ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2016;65:485–492. doi: 10.1007/s00262-015-1761-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tonn T., Schwabe D., Klingemann H.G., Becker S., Esser R., Koehl U., Suttorp M., Seifried E., Ottmann O.G., Bug G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15:1563–1570. doi: 10.1016/j.jcyt.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 44.Klingemann H., Boissel L., Toneguzzo F. Preprint at Frontiers Media S.A; 2016. Natural Killer Cells for Immunotherapy - Advantages of the NK-92 Cell Line over Blood NK Cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chacim S., Monjardino T., Cunha J.L., Medeiros P., Redondo P., Bento M.J., Mariz J.M. Costs, effectiveness, and safety associated with Chimeric Antigen Receptor (CAR) T-cell therapy: Results from a comprehensive cancer center. PLoS One. 2022;17 doi: 10.1371/journal.pone.0278950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arai S., Meagher R., Swearingen M., Myint H., Rich E., Martinson J., Klingemann H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy. 2008;10:625–632. doi: 10.1080/14653240802301872. [DOI] [PubMed] [Google Scholar]
- 47.Williams B.A., Law A.D., Routy B., denHollander N., Gupta V., Wang X.-H., Chaboureau A., Viswanathan S., Keating A. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget. 2017;8:89256–89268. doi: 10.18632/oncotarget.19204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang X., Yang L., Li Z., Nalin A.P., Dai H., Xu T., Yin J., You F., Zhu M., Shen W., et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018;8:1083–1089. [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang C., Wang Z., Yang Z., Wang M., Li S., Li Y., Zhang R., Xiong Z., Wei Z., Shen J., et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA+ Metastatic Colorectal Cancers. Mol. Ther. 2017;25:1248–1258. doi: 10.1016/j.ymthe.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Q., Zhang H., Ding J., Liu H., Li H., Li H., Lu M., Miao Y., Li L., Zheng J. Combination therapy with EpCAM-CAR-NK-92 cells and regorafenib against human colorectal cancer models. J. Immunol. Res. 2018;2018 doi: 10.1155/2018/4263520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y., Chen M., Wu Z., Tong C., Dai H., Guo Y., Liu Y., Huang J., Lv H., Luo C., et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology. 2018;7 doi: 10.1080/2162402X.2018.1440169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Posey A.D., Clausen H., June C.H. Preprint at Cell Press; 2016. Distinguishing Truncated and Normal MUC1 Glycoform Targeting from Tn-MUC1-specific CAR T Cells: Specificity Is the Key to Safety. [DOI] [PubMed] [Google Scholar]
- 53.EGFR CART. Cells for Patients With Metastatic Colorectal Cancer. https://classic.clinicaltrials.gov/ct2/show/NCT03152435
- 54.Fagerberg L., Hallström B.M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K., et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics. 2014;13:397–406. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., et al. Tissue-based map of the human proteome. Science. 2015;1979 doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 56.Karlsson M., Zhang C., Méar L., Zhong W., Digre A., Katona B., Sjöstedt E., Butler L., Odeberg J., Dusart P., et al. A single–cell type transcriptomics map of human tissues. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abh2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nap M., Mollgard K., Burtin P., Fleuren G.J. Immunohistochemistry of Carcino-Embryonic Antigen in the Embryo, Fetus and Adult. Tumour Biol. 1988;9:145–153. doi: 10.1159/000217555. [DOI] [PubMed] [Google Scholar]
- 58.Gold P., Freedman S.O. SPECIFIC CARCINOEMBRYONIC ANTIGENS OF THE HUMAN DIGESTIVE SYSTEM. J. Exp. Med. 1965;122:467–481. doi: 10.1084/jem.122.3.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ilantzis C., Jothy S., Alpert L.C., Draber P., Stanners C.P. Cell-surface levels of human carcinoembryonic antigen are inversely correlated with colonocyte differentiation in colon carcinogenesis. Lab. Invest. 1997;76:703–716. [PubMed] [Google Scholar]
- 60.Hoadley K.A., Yau C., Hinoue T., Wolf D.M., Lazar A.J., Drill E., Shen R., Taylor A.M., Cherniack A.D., Thorsson V., et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell. 2018;173:291–304.e6. doi: 10.1016/j.cell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cascianelli S., Barbera C., Ulla A.A., Grassi E., Lupo B., Pasini D., Bertotti A., Trusolino L., Medico E., Isella C., Masseroli M. Multi-label transcriptional classification of colorectal cancer reflects tumor cell population heterogeneity. Genome Med. 2023;15:37. doi: 10.1186/s13073-023-01176-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brockhoff G., Heiss P., Schlegel J., Hofstaedter F., Knuechel R. Epidermal growth factor receptor, c-erbB2 and c-erbB3 receptor interaction, and related cell cycle kinetics of SK-BR-3 and BT474 breast carcinoma cells. Cytometry. 2001;44:338–348. doi: 10.1002/1097-0320(20010801)44. [DOI] [PubMed] [Google Scholar]
- 63.Leto S.M., Sassi F., Catalano I., Torri V., Migliardi G., Zanella E.R., Throsby M., Bertotti A., Trusolino L. Sustained Inhibition of HER3 and EGFR Is Necessary to Induce Regression of HER2-Amplified Gastrointestinal Carcinomas. Clin. Cancer Res. 2015;21:5519–5531. doi: 10.1158/1078-0432.CCR-14-3066. [DOI] [PubMed] [Google Scholar]
- 64.Cibrián D., Sánchez-Madrid F. CD69: from activation marker to metabolic gatekeeper. Eur. J. Immunol. 2017;47:946–953. doi: 10.1002/eji.201646837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li C., Liu F., Sun L., Liu Z., Zeng Y. Natural killer cell-related gene signature predicts malignancy of glioma and the survival of patients. BMC Cancer. 2022;22:230. doi: 10.1186/s12885-022-09230-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alter G., Malenfant J.M., Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 67.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E., et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ughetto S., Migliore C., Pietrantonio F., Apicella M., Petrelli A., D’Errico L., Durando S., Moya-Rull D., Bellomo S.E., Rizzolio S., et al. Personalized therapeutic strategies in HER2-driven gastric cancer. Gastric Cancer. 2021;24:897–912. doi: 10.1007/s10120-021-01165-w. [DOI] [PubMed] [Google Scholar]
- 69.Krishnamurti U., Silverman J.F. HER2 in breast cancer: a review and update. Adv. Anat. Pathol. 2014;21:100–107. doi: 10.1097/PAP.0000000000000015. [DOI] [PubMed] [Google Scholar]
- 70.Hammarström S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol. 1999;9:67–81. doi: 10.1006/scbi.1998.0119. [DOI] [PubMed] [Google Scholar]
- 71.Ozawa T., Matsuda K., Ishihara S., Fukushima Y., Shimada R., Hayama T., Nozawa K., Hashiguchi Y. The robust performance of carcinoembryonic antigen levels after adjuvant chemotherapy for the recurrence risk stratification in patients with colorectal cancer. J. Surg. Oncol. 2021;124:97–105. doi: 10.1002/jso.26497. [DOI] [PubMed] [Google Scholar]
- 72.Konishi T., Shimada Y., Hsu M., Tufts L., Jimenez-Rodriguez R., Cercek A., Yaeger R., Saltz L., Smith J.J., Nash G.M., et al. Association of Preoperative and Postoperative Serum Carcinoembryonic Antigen and Colon Cancer Outcome. JAMA Oncol. 2018;4:309–315. doi: 10.1001/jamaoncol.2017.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Arnaud J.P., Koehl C., Adloff M. Carcinoembryonic antigen (CEA) in diagnosis and prognosis of colorectal carcinoma. Dis. Colon Rectum. 1980;23:141–144. doi: 10.1007/BF02587615. [DOI] [PubMed] [Google Scholar]
- 74.Parkhurst M.R., Yang J.C., Langan R.C., Dudley M.E., Nathan D.-A.N., Feldman S.A., Davis J.L., Morgan R.A., Merino M.J., Sherry R.M., et al. T Cells Targeting Carcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer but Induce Severe Transient Colitis. Mol. Ther. 2011;19:620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thistlethwaite F.C., Gilham D.E., Guest R.D., Rothwell D.G., Pillai M., Burt D.J., Byatte A.J., Kirillova N., Valle J.W., Sharma S.K., et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017;66:1425–1436. doi: 10.1007/s00262-017-2034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang L., Ma N., Okamoto S., Amaishi Y., Sato E., Seo N., Mineno J., Takesako K., Kato T., Shiku H. Efficient tumor regression by adoptively transferred CEA-specific CAR-T cells associated with symptoms of mild cytokine release syndrome. Oncoimmunology. 2016;5 doi: 10.1080/2162402X.2016.1211218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hernandez-Lopez R.A., Yu W., Cabral K.A., Creasey O.A., Lopez Pazmino M.D.P., Tonai Y., de Guzman A., Mäkelä A., Saksela K., Gartner Z.J., Lim W.A. T cell circuits that sense antigen density with an ultrasensitive threshold. Science. 2021;371:1166–1171. doi: 10.1126/science.abc1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Labanieh L., Majzner R.G., Klysz D., Sotillo E., Fisher C.J., Vilches-Moure J.G., Pacheco K.Z.B., Malipatlolla M., Xu P., Hui J.H., et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell. 2022;185:1745–1763.e22. doi: 10.1016/j.cell.2022.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.De Palma M., Montini E., Santoni de Sio F.R., Benedicenti F., Gentile A., Medico E., Naldini L. Promoter trapping reveals significant differences in integration site selection between MLV and HIV vectors in primary hematopoietic cells. Blood. 2005;105:2307–2315. doi: 10.1182/blood-2004-03-0798. [DOI] [PubMed] [Google Scholar]
- 80.Srivastava S., Salter A.I., Liggitt D., Yechan-Gunja S., Sarvothama M., Cooper K., Smythe K.S., Dudakov J.A., Pierce R.H., Rader C., Riddell S.R. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell. 2019;35:489–503.e8. doi: 10.1016/j.ccell.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frigault M.J., Lee J., Basil M.C., Carpenito C., Motohashi S., Scholler J., Kawalekar O.U., Guedan S., McGettigan S.E., Posey A.D., et al. Identification of Chimeric Antigen Receptors That Mediate Constitutive or Inducible Proliferation of T Cells. Cancer Immunol. Res. 2015;3:356–367. doi: 10.1158/2326-6066.CIR-14-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zuo P., Li Y., He C., Wang T., Zheng X., Liu H., Wu Z., Zhang J., Liao X., Zhang L. Anti-tumor efficacy of anti-GD2 CAR NK-92 cells in diffuse intrinsic pontine gliomas. Front. Immunol. 2023;14 doi: 10.3389/fimmu.2023.1145706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cao B., Liu M., Huang J., Zhou J., Li J., Lian H., Huang W., Guo Y., Yang S., Lin L., et al. Development of mesothelin-specific CAR NK-92 cells for the treatment of gastric cancer. Int. J. Biol. Sci. 2021;17:3850–3861. doi: 10.7150/ijbs.64630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.The Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Isella C., Terrasi A., Bellomo S.E., Petti C., Galatola G., Muratore A., Mellano A., Senetta R., Cassenti A., Sonetto C., et al. Stromal contribution to the colorectal cancer transcriptome. Nat. Genet. 2015;47:312–319. doi: 10.1038/ng.3224. [DOI] [PubMed] [Google Scholar]
- 86.Ma Q., DeMarte L., Wang Y., Stanners C.P., Junghans R.P. Carcinoembryonic antigen-immunoglobulin Fc fusion protein (CEA-Fc) for identification and activation of anti-CEA immunoglobulin-T-cell receptor-modified T cells, representative of a new class of Ig fusion proteins. Cancer Gene Ther. 2004;11:297–306. doi: 10.1038/sj.cgt.7700685. [DOI] [PubMed] [Google Scholar]
- 87.Hombach A., Hombach A.A., Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010;17:1206–1213. doi: 10.1038/gt.2010.91. [DOI] [PubMed] [Google Scholar]
- 88.Vigna E., Cavalieri S., Ailles L., Geuna M., Loew R., Bujard H., Naldini L. Robust and Efficient Regulation of Transgene Expression in Vivo by Improved Tetracycline-Dependent Lentiviral Vectors. Mol. Ther. 2002;5:252–261. doi: 10.1006/mthe.2002.0542. [DOI] [PubMed] [Google Scholar]
- 89.Montagner I.M., Penna A., Fracasso G., Carpanese D., Dalla Pietà A., Barbieri V., Zuccolotto G., Rosato A. Anti-PSMA CAR-Engineered NK-92 Cells: An Off-the-Shelf Cell Therapy for Prostate Cancer. Cells. 2020;9:1382. doi: 10.3390/cells9061382. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data related to this study are included in this paper and its Supplementary information. Experimental details are available from the corresponding author on reasonable request.