

Abstract
In the realm of disease vectors and agricultural pest management, insecticides play a crucial role in preserving global health and ensuring food security. The pervasive use, particularly of organophosphates (OPs), has given rise to a substantial challenge in the form of insecticide resistance. Carboxylesterases emerge as key contributors to OP resistance, owing to their ability to sequester or hydrolyze these chemicals. Consequently, carboxylesterase enzymes become attractive targets for the development of novel insecticides. Inhibiting these enzymes holds the potential to restore the efficacy of OPs against which resistance has developed. This study aimed to screen the FooDB library to identify potent inhibitory compounds targeting carboxylesterase, Ha006a from the agricultural pest Helicoverpa armigera. The ultimate objective is to develop effective interventions for pest control. The compounds with the highest scores underwent evaluation through docking studies and pharmacophore analysis. Among them, four phytochemicals—donepezil, protopine, 3′,4′,5,7-tetramethoxyflavone, and piperine—demonstrated favorable binding affinity. The Ha006a-ligand complexes were subsequently validated through molecular dynamics simulations. Biochemical analysis, encompassing determination of IC50 values, complemented by analysis of thermostability through Differential Scanning Calorimetry and interaction kinetics through Isothermal Titration Calorimetry was conducted. This study comprehensively characterizes Ha006a-ligand complexes through bioinformatics, biochemical, and biophysical methods. This investigation highlights 3′,4′,5,7-tetramethoxyflavone as the most effective inhibitor, suggesting its potential for synergistic testing with OPs. Consequently, these inhibitors offer a promising solution to OP resistance and address environmental concerns associated with excessive insecticide usage, enabling a significant reduction in their overuse.
Keywords: Hydrolase, Insect enzyme, Organophosphate, Insecticides, Insecticide resistance, Phytochemicals
Subject terms: Environmental sciences, Environmental social sciences
Introduction
The necessity of pesticides is paramount in achieving global agricultural productivity, pest management, and ensuring worldwide food security. The significant escalation in the use and dependence on these chemicals over the past few decades has emerged as a major concern. The Nature Geoscience study examined the use of 100 chemicals across 168 countries and found that indiscriminate pesticide application poses a severe worldwide threat1. Besides the significant contributions to crop enhancement and production, unregulated and indiscriminate pesticide application has devastating effects on human health, predator–prey interactions, and biodiversity2,3. Direct or indirect exposure to pesticides has led to increasing health issues such as hormone imbalance, low IQ, immune suppression, neurological defects, behavioral disorders, infertility, and cancer4. Additionally, the extensive use of insecticides, particularly organophosphates (OPs), has resulted in the development of resistance in various insect species5,6. The presence of carboxylesterases (CarEs) nullifies the effect of OP insecticides as this enzyme detoxifies/sequesters the xenobiotic chemicals and builds resistance against those7,8. Some of the disease vectors and insect pests that have reported OP resistance are Lucilia cuprina (blowflies)9, Aedes aegypti (mosquitoes)10, Culex quinquefasciatus, and many others11–13.
Helicoverpa armigera, is a significant agricultural pest, destroying crops like cotton, pulses, tomato, and tobacco globally14. The frequent administration of insecticides in crop fields has resulted in the development of resistance in pests around the world7. Over time, it has rapidly evolved resistance to different classes of insecticides, such as synthetic pyrethroids, organochlorines, organophosphates and carbamates15. The resistance mechanism primarily involves the upregulation of metabolic enzymes, particularly carboxylesterases (CarEs). This enzyme class possesses an active site that facilitates the hydrolysis of carboxyl esters, thus encouraging the catalysis process16,17. In certain other cases, the enzymatic process generates a stable acyl-enzyme complex through the catalytic serine, irreversibly inhibiting the enzyme16. Consequently, the insecticide gets stoichiometrically sequestered by the enzyme as a result of its covalent conjugation and prevents it from reaching its target, acetylcholinesterase (AChE)18,19. Different culicine mosquito species, such as C. quinquefasciatus and all the Culex species, have this CarE-mediated sequestration mechanism20,21.
To surmount insecticide resistance, numerous novel insecticides featuring diverse modes of action have been formulated22,23. Among the various strategies employed, synergists emerge as the most effective approach for mitigating the impact of enzymes, consequently augmenting the efficacy of insecticides with minimal doses. Several such synergists, including piperonyl butoxide, have demonstrated effectiveness by inhibiting cytochrome P450s, thereby intensifying the activity of carbamates and pyrethroids24,25. This methodology aims to counteract enzymes that have evolved to confer metabolic resistance, ultimately reducing the requisite dose of insecticides. The CarEs are therefore the best target to develop inhibitors against it and hence abolish the insecticide resistance.
In this study, we present a comprehensive investigation into the potential inhibitory effects of potent phytochemical compounds on the activity of the CarE, Ha006a. Leveraging a computational approach, we employed homology modeling to predict the structure of Ha006a, which served as the foundation for identifying inhibitory compounds through virtual screening. The screening process yielded four best compounds, namely donepezil, protopine, 3′,4′,5,7-tetramethoxyflavone and piperine. To further elucidate their inhibitory potential, these compounds underwent rigorous examination through molecular dynamics (MD) simulation studies, as well as biochemical and biophysical characterization. These analyses were crucial in assessing the stability of the compounds and their interactions with the CarE, Ha006a. Our findings highlight the structural dynamics and binding affinities of the selected compounds, providing valuable insights into the best compounds that are potential inhibitors of CarE activity. This integrated computational and experimental approach enhances our understanding of the molecular interactions governing the inhibitory effects, showing the way to identify novel inhibitors to fight insecticide resistance.
Methodology
Materials
The reagents were obtained from Sigma and HiMedia. The computational study was conducted on Windows 11 workstation with an Intel Core i7 10700 processor. MD simulation studies were performed on LINUX workstation with Intel Xeon Gold 6226R processor using CHARMM36 force field in GROMACS 2022.2 suite. Several online bioinformatics tools like AlphaFold v2.1.126, RCSB PDB27, NCBI28, Pharmit29, SAVES server30–32, DALI server33, PROSITE34, ChemSketch 1.035 and ExPASy server36 were used for sequence analysis, structure building and its validation. Bioinformatics softwares, such as PyRx 0.837 (https://sourceforge.net/projects/pyrx/files/PyRx/0.8/), Avogadro v1.2.038 (https://sourceforge.net/projects/avogadro/files/avogadro/1.2.0/), PyMOL39 (https://pymol.org/2/), Open Babel40 (https://sourceforge.net/projects/openbabel/) and GROMACS 2022.241 (https://manual.gromacs.org/2022.2/download.html) were used for performing molecular docking and dynamic simulation of the complexes.
Homology modeling
The three-dimensional structure of Ha006a was predicted using AlphaFold v2.1.1. The quality of the model was analyzed using several tools available in the SAVES server v6.0, including VERIFY3D30, PROCHECK and ERRAT31. The stereochemical stability of the modeled protein was evaluated considering the potential energy of the backbone by analyzing angles against the residues of the modeled structure32. The model was further confirmed by ERRAT plot and VERIFY3D tools. The thorough evaluation and visualization of the model was performed in PyMOL39. The best model selected was subjected to structural refinement using CHARMM36 force field by molecular dynamics using GROMACS 2022.2 software.
Choice of ligand library
The ligand structures for virtual screening were retrieved from the FooDB library. Hit screening was carried out using the webserver Pharmit29 based on the drug-likeliness properties and pharmacophore feature hydrophobic type. The best hit compounds obtained were subjected to dock with the modeled Ha006a protein.
Virtual screening and molecular docking
The protein was analyzed using the Conserved Domain Database (CDD), and the substrate binding residues were identified. Further, the multiple sequence alignment was performed and the conserved residues were compared and analyzed with its homologous proteins using Clustal Omega and ESPript 3.042,43 server. The PyRx 0.8 was used as the docking platform and the ligands were pre-processed. The macromolecule was prepared by incorporating polar hydrogen and the Kollman atomic charges and finally converted the compounds to the pdbqt format using AutoDock tools software. The grid dimensions for docking were X: 33.40, Y: 44.67, Z: 40.53, and center X: 47.41, Y: 55.49, Z: 54.10. The screening procedure was executed with rigid docking protocol in the AutoDock Vina by keeping the ligands flexible. For each ligand, 9 conformations were placed in the active site of Ha006a, and the interaction was analyzed in PyMOL to select the best conformation. Among all the ligands, the best pose fitting with the highest binding affinity (kcal/mol) was further selected for molecular dynamics study.
Molecular dynamics (MD) simulation
The MD simulation utilized the top four compounds, each exhibiting optimal poses for their respective ligands as determined by virtual screening adhering to Lipinski’s rule of 544. In this investigation, we have incorporated triphenyl phosphate (TPP), a previously documented inhibitor of CarEs, as the fifth ligand for comprehensive comparative analysis. The GROMACS 2022.2 software was used to understand the MD of the apo Ha006a structure in association with ligands. The CHARMM36 force field and the TIP3P water model were utilized to build the Ha006a topology, and the ligands were prepared by incorporating hydrogen atoms in Avogadro v1.2.0 software. For protein solvation, the SPC water module was used inside a cubic box with padding of 1.0 nm. The ligand structure parameter assignment and the generation of their topology files was performed using the CHARMM General Force Field (CGenFF) server. Each Ha006a-ligand complex was centered inside a cubic box followed by the solvation, neutralization, energy minimization and equilibration of the system. Finally, the system was introduced to a simulation of 100 ns. The trajectory acquired was analyzed employing the trjconv module of the GROMACS incorporating the periodicity in the system. Several GROMACS modules were utilized to understand the structural stability of Ha006a-ligand complexes.
MMPBSA binding free energy
To calculate the binding free energy (∆G) of Ha006a-ligand complexes, a single trajectory protocol in the Molecular Mechanics/Poisson-Boltzmann Surface Area (MMPBSA) method was used by the tools gmx_MMPBSA v1.6.2 and AmberTools2045. The binding energy was calculated considering the contribution of each residue and hence giving insight into the Ha006a-ligand association. The trajectory of Ha006a with the ligand complexes was analyzed to compute free binding energy. The calculation included summation of molecular mechanical energy changes in the gas phase, ∆GGAS (bonded and non-bonded energy components) and solvation energy, ∆GSOLV (polar and non-polar energy components), which gives the enthalpy of binding, ∆H. In this study, the last 1000 frames between 90 and 100 ns were considered to calculate the binding free energy, which was denoted in units of kcal/mol.
Enzyme inhibition assay
Ha006a enzyme was purified to homogeneity using different chromatographic techniques as reported in a previous study46. An inhibition test was conducted in the total volume of 200 µL considering all four ligands independently (donepezil, protopine, 3′,4′,5,7-tetramethoxyflavone, and piperine). The reaction volume consisted of 20 mM sodium phosphate buffer (pH 7.0), 0.2 nM Ha006a enzyme incubated with serial concentration of ligand in the range of 1 µM -103µM and was kept undisturbed for 10 min at 35 °C. In the case of blank, the reaction volume was devoid of enzyme, and all other parameters were kept same. Inhibition assays were terminated by adding a solution containing FBB dye (3 mM) and the substrate, alpha naphthyl acetate, maintained as 10 µM46. The half inhibitory concentrations (IC50) were estimated by comparing the blank subtracted reading obtained with the enzyme activity in the absence of any ligand. Additionally, the positive control values46 of the CarE inhibitor, triphenyl phosphate (TPP), were compared for analyzing the inhibition assay47.
Differential scanning calorimetry
In order to monitor the thermal transitions and stability of protein, DSC experiments were conducted on Microcal VP-DSC Microcalorimeter48. Protein was purified and exchanged against 20 mM sodium phosphate buffer pH 7.0 using PD-10 Sephadex TM G-25 desalting columns. For each scan, protein samples were prepared consisting of 50 μM protein concentration in a total volume of 700 μL. Before the onset of the experiment, the protein sample and the buffer were filtered using a 0.22 μm pore size filter. The solutions were then degassed with MicroCal Thermovac 2 (GE Healthcare, USA) degasser. The thermal unfolding studies was conducted at a rate of 1 °C per minute. Further, binding thermal studies of protein with various ligands were performed allowing an incubation period of 10 min before the scan. The DSC thermograms were evaluated using Origin 7 software (USA) and the data was analyzed as per the non-two-state model fitting.
Isothermal titration calorimetry (ITC)
To unravel the enthralling intricacies inherent in the reaction type, affinity, stoichiometry, and energetics associated with the interaction profile of Ha006a with its ligands, ITC experiment was conducted. This label-free, reliable and robust technique serves to comprehensively understand the thermodynamic kinetics involved during protein–ligand interaction49,50. The experiments were conducted in a MicroCal iTC-200 instrument, maintaining normal jacket temperature. The experiment was based on the mechanism of heat absorbed or released during the interaction, the binding affinity and binding parameters were calculated. Before the commencement of the experiment, the protein sample, titrant solution, and the buffer were filtered using 0.45 µm filter and degassed for 20 min to avoid noise. The protein and the ligand were maintained in the 20 mM sodium phosphate buffer. An average of 20 injections with initial delay of 60 s was set, the first titrant injection was set as 0.4 µL and other subsequent injections were 2.0 µL having a spacing of 200 s. The syringe stirring speed was set at 750 rpm for the homogenous mixture of the two. The binding kinetics and the thermodynamic parameters—enthalpy change (ΔΗ), entropy change(ΔS), association/dissociation constant (ka/kd), and stoichiometry (N) were analyzed using Origin 7.0 software.
Results
Sequence analysis
The protein–protein BLAST of Ha006a against the Protein Data Bank repository revealed homologs having query coverage up to 94%, E-value cut off 10–5 and a maximum identity of 31.39% from Lucilia cuprina, Anopheles gambiae, Culex quinquefasciatus, Manduca sexta and Epiphyas postvittana. To unravel the closest homologs, an analysis against the DALI database with Ha006a protein structure as the query was performed. The DALI search revealed the two closest homologs with a Z-score > 37, having an identity of 25.0–29.0%, and RMSD in the range of 2.4–3.1 Å; both were insect proteins. Employing Clustal Omega, both the identified proteins from Drosophila melanogaster (PDB ID: 5THM51 with 25% identity) and Lucilia cuprina (PDB ID:5IKX52 with 29% identity) were aligned with Ha006a and the signature conserved catalytic triad was highlighted (Fig. 1). The Ha006a protein has a significant abundance of sequence homologs but with diversified functions. The homologs considered in the alignment study were esterase 6 (EST6) of D. melanogaster and alpha esterase -7 carboxylesterase of L. cuprina respectively. The PROSITE analysis further revealed that signature residues of the structure correspond to the classical Carboxylesterases type-B signature53 and hence are hydrolases. The ProtParam tool from ExPASy provided Ha006a information on the molecular weight, the isoelectric point, and the extinction coefficient, which were approximately 60 kDa, 4.5, 74970M-1 cm-1 respectively.
Figure 1.

Sequence alignment of Ha006a with its homologous proteins. The homologous Ha006a proteins from other insects, Drosophila melanogaster (PDB ID: 5THM with 25.0% identity), and Lucilia cuprina (PDB ID:5IKX with 29.0% identity) are aligned, and the catalytic residues are highlighted in the black box. All other conserved residues are highlighted in red. This information in the form of a figure was effectively generated by Clustal Omega and ESPript.
Structure analysis and validation
The Ha006a model was predicted utilizing the AlphaFold v2.1.1, and the modeled structure was visualized in PyMOL and effectively depicted in a schematic illustration in cartoon format (Fig. 2A). To examine the stereochemical stability of the modeled Ha006a, the structure was analyzed in the SAVES v6.0 server (https://saves.mbi.ucla.edu/). The Ramachandran plot analysis of the modeled Ha006a depicted that 99.8% of amino acid residues occupied the allowed region, while 0.2% were situated in the disallowed region (Fig. 2B). Previous investigations have established that a model featuring 90% of residues in the favored region is indicative of stereochemical stability32. Based on both literature findings and the observed values for the allowed region, the predicted 3D model of Ha006a affirms its stereochemical stability. The VERIFY3D analysis revealed 87.62% of amino acids had an average score ≥ 0.1, validating the good quality of the model (Fig. 2C). Additionally, the ERRAT analysis demonstrated an overall quality factor of 95.05% for the modeled structure, confirming the high quality of the model. The generated Ha006a structure was superimposed with the already-known crystal structure of the homologous proteins to compare the software generated Ha006a structure (Fig. S1). The superimposition revealed that the substrate binding residues were oriented in a similar fashion, but prominent displacement was observed. Ha006a exhibited RMSD of 1.964 Å and 2.223 Å when compared with its homologous proteins alpha esterase − 7 carboxylesterase having PDB ID: 5IKX and odorant degrading esterase 6 (EST6) having PDB ID: 5THM, respectively. Thus, Ha006a possesses unique orientation of the residues, which is different from its closest homologous proteins and is validated by protein structure characterization and comparison.
Figure 2.

The model prediction of Ha006a and its structure validation. (A) The model generated by the AlphaFold v2.1.1 platform and the protein structure is illustrated in cartoon format. (B) The Ramachandran plot was generated by the PROCHECK tool of the SAVES server to analyze the predicted Ha006a model. (C) The graph was produced by the VERIFY3D tool of the SAVES server to validate the quality of the Ha006a model.
Virtual Screening
Hit screening based on drug-likeliness properties and pharmacophore feature (hydrophobic) filtered 4500 hit molecules and were retrieved in SDF format. Virtual screening was conducted using AutoDock Vina of PyRx 0.8 platform and the screened compounds along with TPP were considered in docking studies for comparative study. Each compound was meticulously analyzed to assess its docking conformation and its compatibility with the substrate binding site. The binding energy for top compounds was discovered to fall within the range of − 8.0 to − 9.2 kcal/mol, requiring lower binding energy than inhibitor TPP (− 7.6 kcal/mol), as summarized in Table 1. Among nine docking conformations, the optimal pose for the best-screened compound was determined through visualization and rigorous scrutiny using PyMOL. The binding mode of the ligands and the interacting residues of each Ha006a-ligand complex were analyzed and are depicted in Fig. 3.
Table 1.
The most potent phytochemical molecules obtained from the bioinformatic approach are depicted, along with their structures, chemical formulae and molecular weight.
| Phytochemical molecules | AutoDock vina Binding affinity (kcal/mol) |
||
|---|---|---|---|
| Names and chemical formulae | Structures | Molecular weight | |
| Donepezil (C24H29NO3) |  |
379.5 g/mol | − 9.2 |
| Protopine (C20H19NO5) |  |
353.4 g/mol | − 8.0 |
| 3′,4′,5,7-Tetramethoxyflavone (C19H18O6) |  |
342.3 g/mol | − 8.8 |
| Piperine (C17H19NO3) | 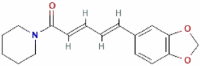 |
285.34 g/mol | − 8.3 |
| Triphenyl phosphate ((C6H5)3PO4) |  |
326.3 g/mol | − 7.6 |
The binding affinity of Ha006a in complex with these molecules is depicted in kcal/mol.
Figure 3.

The interacting profile of Ha006a specific to different ligands. Ha006a in complex with (A) donepezil, (B) protopine, (C) 3′,4′,5,7-tetramethoxyflavone, (D) piperine and (E) triphenyl phosphate. All the interacting residues of Ha006a protein are depicted as sticks (green) and are labeled.
The interacting residues
The interacting amino acids of Ha006a were analyzed using the NCBI CDD platform54. The catalytic triad within the Ha006a protein consists of three residues Ser187, Glu309, and His423, which are highly conserved. Moreover, the substrate binding pocket comprises of the conserved residues such as Gly106, Gly107, Gly108, Glu186, Ser187, Ala188, Ala191, Phe331, Tyr335, Phe336, His372, Ala424 and Leu427. The interacting residues assume a critical role in evaluating the most favorable ligand with strong binding affinity in the in silico study. Thus, the Ha006a-ligand complexes were comprehensively analyzed in PyMOL, and the pose with the best fit was selected for the simulation run. The interacting residues corresponding to each ligand complex are summarized in Table S1.
Molecular dynamics (MD) simulation analysis
To investigate the structural stability of Ha006a apo protein and ligand-bound complexes, an MD simulation run of 100 ns was conducted. The pose of each ligand after a simulation run of 100 ns is depicted in Fig. S2. The investigation of the structural stability of the system involving the examination of substantial deviations from the initial path of the backbone atoms, fluctuations of each residue relative to its initial position, and the assessment of the number of hydrogen bonds between the protein and ligand was performed. Single trajectory protocol of the MMPBSA method was used to study protein–ligand binding interaction and computation of relative binding free energy of ligands.
Root mean square deviation (RMSD)
To discern the dynamic behavior of protein throughout the simulation and unveil the structural changes in context to the Cα-backbone of the protein, an RMSD plot of the complexes against the energy minimized apo protein structure was plotted (Fig. 4A). The RMSD values for Ha006a protein (apo) and Ha006a-ligand complexes viz. Ha006a-donepezil, Ha006a-protopine, Ha006a-3′,4′,5,7-tetramethoxyflavone, Ha006a- piperine and Ha006a-triphenyl phosphate were found to be 0.166 nm, 0.209 nm, 0.169 nm, 0.175 nm, 0.199 nm and 0.163 nm respectively, during the simulation run. The observed RMSD outcome of the protein–ligand complexes signified that the Ha006a generated stable complexes with the screened phytochemicals.
Figure 4.

The structural stability of both the apo Ha006a and its complexes throughout the molecular dynamic simulation run of 100 ns is depicted through graphical representations. The (A) Root Mean Square Deviation (RMSD), (B) Root Mean Square Fluctuation (RMSF), (C) Radius of Gyration (Rg), (D) Solvent Accessible Surface Area (SASA), and (E) Hydrogen Bond analysis of 100 ns trajectories of apo Ha006a and Ha006a-ligand complexes are displayed. The graphs illustrate the behavior of the apo protein (black) and its complexes with donepezil (red), protopine (blue), 3′,4′,5,7-tetramethoxyflavone (green), piperine (purple), and triphenyl phosphate (light brown).
Root mean square fluctuation (RMSF)
To elucidate the dynamic fluctuations in Cα atom coordinates for individual residues in relation to their mean positions throughout the simulation, an RMSF graph was constructed (Fig. 4B). The flexibility of the protein–ligand complexes was analyzed by calculating the RMSF values for the biomolecular system during the simulation. The RMSF values for Ha006a protein (apo) and Ha006a-ligand complexes viz. Ha006a-donepezil, Ha006a-protopine, Ha006a-3′,4′,5,7-tetramethoxyflavone, Ha006a- piperine and Ha006a-triphenyl phosphate were found to be 0.120 nm, 0.129 nm, 0.112 nm, 0.123 nm, 0.111 nm, and 0.113 nm respectively, during the simulation run. The ligands 3′,4′,5,7-tetramethoxyflavone, and donepezil showed more fluctuations when compared to other ligands and apo protein. Thus, these ligands fit well in the binding pocket, as understood from the RMSF plot.
Radius of gyration (Rg)
To quantify the compact packing of protein on the accommodation of ligands to the binding pocket, a Rg graph was constructed (Fig. 4C). The radius of gyration serves as a crucial metric for investigating the protein folding and structural stability throughout the simulation. The Rg values for Ha006a protein (apo) and Ha006a-ligand complexes viz. Ha006a-donepezil, Ha006a-protopine, Ha006a-3′,4′,5,7-tetramethoxyflavone, Ha006a- piperine and Ha006a-triphenyl phosphate were found to be 2.37 nm, 1.98 nm, 1.89 nm, 1.90 nm, 1.91 nm and 1.90 nm respectively, during the simulation run. The complexes depicted lower Rg values as compared to the apo structure, implying that the ligands were compactly fitted and hence formed stable complexes during the run.
Solvent accessible surface area (SASA)
To determine the surface area of the protein that is accessible to the solvent, SASA graph was plotted (Fig. 4D). The dynamic nature of the protein in apo and in the complex state was evaluated by monitoring the solvent interacting with the protein surface thereby, providing insights into the stability. The SASA values for Ha006a protein (apo) and Ha006a-ligand complexes viz. Ha006a-donepezil, Ha006a-protopine, Ha006a-3′,4′,5,7-tetramethoxyflavone, Ha006a-piperine and Ha006a-triphenyl phosphate were found to be 240.55 nm2, 241.46 nm2, 237.69 nm2, 243.20 nm2, 239.82 nm2 and 239.77 nm2 respectively, during the 100 ns MD simulation run. The ligands 3′,4′,5,7-tetramethoxyflavone and donepezil showed higher SASA values, implying that the introduction of these ligands to the binding pocket made more residues exposed to solvent and made favorable contact.
Hydrogen bond analysis
The stability of the protein–ligand system is augmented through intricate interactions characterized by hydrogen bonds. In order to comprehensively understand the stability, dynamics and structure conformational alterations of the system, the hydrogen bond was assessed throughout the simulation run. The number of hydrogen bonds formed between Ha006a and its ligands ranged from 0 to 3. The highest intermolecular hydrogen bond was observed with 3′,4′,5,7-tetramethoxyflavone ligand, denoting a robust association. The optimal hydrogen bonds between Ha006a and its ligands is graphically represented in Fig. 4E, thereby providing a comprehensive insight into the intricate intermolecular forces that contribute to the stability and conformational dynamics of the protein–ligand complex.
MMPBSA calculations
The MMPBSA analysis utilizes a single trajectory protocol to monitor the relative binding free energy of the ligands from the MD trajectories on dismissal of the term conformation entropy (ΔS), providing information about the enthalpy of binding45. The MD trajectories from the last 1000 frames of all Ha006a-ligand complexes were used to calculate the enthalpy of binding (∆H), which is the sum of gas phase molecular mechanical energy, ∆GGAS and solvation energy, ∆GSOLV. ∆GGAS contributes to the bonded (internal energy), and ∆GSOLV contributes to polar and non-polar components. In single trajectory protocol, internal energy change (bond, angle, dihedral energies) is canceled, and the non-bonded terms contributed to van der Waal energy, ∆EvdW, and electrostatic energy, ∆Eele. Polar solvation energy, ∆GPB, is calculated by the Poisson-Boltzmann equation, and non-polar contribution includes the solvation energy in the cavity formation ∆Gcavity, which is correlated to SASA. The enthalpy of binding values obtained for Ha006a-ligand complexes viz. Ha006a-donepezil, Ha006a-protopine, Ha006a-3′,4′,5,7-tetramethoxyflavone, Ha006a- piperine and Ha006a-triphenyl phosphate were − 16.76 kcal/mol, − 11.21 kcal/mol, − 3.58 kcal/mol, − 15.09 kcal/mol and − 16.45 kcal/mol respectively (Table 2). The van der Waal energy forces showed 3′,4′,5,7-tetramethoxyflavone > piperine > donepezil of − 35.48 kcal/mol, − 35.19 kcal/mol, − 34.50 kcal/mol respectively, which is comparable to the natural inhibitor, triphenyl phosphate with − 36.46 kcal/mol. The ligand donepezil showed the highest electrostatic energy of − 7.77 kcal/mol when compared to 3′,4′,5,7-tetramethoxyflavone, protopine, and piperine, i.e., − 6.92 kcal/mol, − 2.86 kcal/mol, and − 2.57 kcal/mol, respectively. The ligands donepezil and 3′,4′,5,7-tetramethoxyflavone required the highest non-bonded interaction energy of − 42.27 kcal/mol and − 42.40 kcal/mol, respectively.
Table 2.
MMPBSA energy summary of Ha006a complexed with ligands.
| Complexes | Non-bonded interaction energy, ∆Enon-bonded (kcal/mol) | Solvation energy, ∆GSOLV (kcal/mol) |
Enthalpy of binding, ∆H (kcal/mol) | ||
|---|---|---|---|---|---|
| van der Waal energy, EvdW | Electrostatic energy, Eele | Polar component, Gpolar/GPB |
Non-polar component, Gnonpolar/Gcavity |
||
| Donepezil | − 34.50 | − 7.77 | 29.39 | − 3.88 | − 16.76 |
| Protopine | − 17.77 | − 2.86 | 11.84 | − 2.42 | − 11.21 |
| 3′,4′,5,7-Tetramethoxyflavone | − 35.48 | − 6.92 | 42.74 | − 3.92 | − 3.58 |
| Piperine | − 35.19 | − 2.57 | 26.57 | − 3.90 | − 15.09 |
| Triphenyl phosphate | − 36.46 | − 13.87 | 37.31 | − 3.44 | − 16.45 |
Inhibition assay and IC50 calculation
A representative substrate, α-NA (α-naphthyl acetate), was employed to evaluate the impact of screened ligands on the enzymatic activity of Ha006a. In a previous study, the IC50 value (the concentration at which the inhibitor reduces the enzyme activity by half) of the positive inhibitor TPP towards Ha006a was reported as 425.67 ± 5.65 μM46. The ligands under investigation, namely donepezil, 3′,4′,5,7-tetramethoxyflavone, protopine and piperine, demonstrated significantly higher inhibitory effects on Ha006a activity compared to TPP. The respective IC50 values for these ligands were found to be 81.26 ± 8.05 μM, 31.26 ± 1.22 μM, 210.53 ± 40.58 μM and 355.06 ± 33.05 μM, respectively (Fig. 5).
Figure 5.

Inhibition assay to calculate the IC50 values of Ha006a with various phytochemical molecules. The inhibition linear plots illustrating the effects of (A) Donepezil, (B) 3′,4′,5,7-Tetramethoxyflavone, (C) Protopine and (D) Piperine on Ha006a are presented. The measurements were taken in triplicate, and the averaged values, obtained by subtracting the blank, are depicted in the plot. The horizontal bar on the graph indicates error bars, representing the standard deviation.
The results revealed a significant difference in the inhibitory profiles of all four ligands against protein, with 3′,4′,5,7-tetramethoxyflavone being the most potent inhibitor, followed by donepezil. The observation of heightened sensitivity of Ha006a from H. armigera to the screened ligands, as compared to TPP, suggests a potential for these ligands to be more effective inhibitors of Ha006a enzymatic activity. This information could be valuable for understanding the enzyme behavior and developing strategies for targeted inhibition of Ha006a or other similar insect proteins.
Differential Scanning Calorimetry (DSC)
DSC experiment was conducted to monitor the thermal profile of Ha006a protein in the apo state and in the ligand-bound state. To understand the stability and thermal variability of Ha006a protein (apo) and Ha006a-ligand complexes viz. Ha006a-donepezil, Ha006a-3′,4′,5,7-tetramethoxyflavone, Ha006a-protopine, Ha006a- piperine and Ha006a-triphenyl phosphate, DSC studies were carried out. The DSC scan from 20 to 120 ℃ was monitored as a function of temperature against molar heat capacity. The evaluation of the melting curve for apo Ha006a showed a multistage unfolding curve with three thermal transitions with melting temperatures (Tm) of 48.13 ± 0.49, 73.13 ± 0.18 and 90.06 ± 0.17 °C, analyzed by a non-two state model fitting (Fig. 6A). The curve showed that the second and third transition were wider as compared to the first transition among the three Tm. This difference in the peaks of the three transitions is also supported by the ΔH values signifying molar enthalpy and ΔHV values demonstrating van’t Hoff enthalpy changes. The result demonstrated that Ha006a possesses a stable conformation with a Tm at 48.13 °C. Changes in heat capacity (Cp) are attributed to disruptions in forces maintaining native protein structure, including hydrogen bonds, van der Waals, electrostatic interactions, hydration of exposed residues, and hydrophobic interactions55. Thermodynamic parameters from DSC experiments are highly sensitive to biomolecular structure, and any conformational changes upon protein–ligand interaction are effectively monitored in DSC thermograms48,56. In the case of Ha006a-ligand complexes, an increase in the melting temperature was observed when Ha006a was bound to the ligands, thus validating the conformational changes due to the binding of two entities (Fig. 6B–F). Among all the ligands, Ha006a when bound to 3′,4′,5,7-tetramethoxyflavone revealed significant increase in all the melting temperatures (Table 3). Furthermore, the calculated ratio of ΔHV/ΔH for each transition was consistently < 1, indicating a multistage unfolding of Ha006a leading to denaturation and the formation of stable intermediates57. The DSC analysis demonstrated distinct conformational changes upon ligand binding, affirming the interaction and stability of the complexes.
Figure 6.

The DSC thermograms emphasizing on the thermodynamic characterization of both apo Ha006a and Ha006a-ligand complexes. The graphs of (A) Apo Ha006a, the protein in complex with (B) Donepezil, (C) Protopine, (D) 3′,4′,5,7-tetramethoxyflavone, (E) Piperine and (F) Triphenyl phosphate are displayed.
Table 3.
The DSC thermograms were used to derive the thermodynamic parameters for apo Ha006a and Ha006a-ligand complexes.
| Tm (°C) ± SE | ΔH (kcal/mol) | ΔHv (kcal/mol) | ΔHv/ΔH | |
|---|---|---|---|---|
| Apo Ha006a | Tm1 48.13 ± 0.49 | 2.49E+05 | 5.29E+04 | 0.21 |
| Tm2 73.13 ± 0.18 | 3.41E+06 | 4.13E+04 | 0.01 | |
| Tm3 90.06 ± 0.17 | 2.21E+06 | 5.33E+04 | 0.02 | |
| Donepezil | Tm1 50.12 ± 0.45 | 1.17E+05 | 4.71E+04 | 0.40 |
| Tm2 63.42 ± 0.62 | 2.08E+05 | 3.51E+04 | 0.17 | |
| Tm3 82.46 ± 0.79 | 1.24E+05 | 3.52E+04 | 0.28 | |
| Protopine | Tm1 49.93 ± 0.55 | 6.30E+05 | 4.32E+04 | 0.07 |
| Tm2 66.76 ± 0.33 | 1.87E+06 | 3.15E+04 | 0.02 | |
| Tm3 86.29 ± 0.67 | 7.83E+05 | 4.01E+04 | 0.05 | |
| 3′,4′,5,7-Tetramethoxyflavone | Tm1 51.52 ± 0.25 | 5.84E+05 | 3.40E+04 | 0.06 |
| Tm2 79.01 ± 0.98 | 9.57E+05 | 3.53E+04 | 0.04 | |
| Tm3 91.76 ± 0.43 | 3.58E+05 | 5.88E+04 | 0.16 | |
| Piperine | Tm1 51.30 ± 0.32 | 2.18E+05 | 4.33E+04 | 0.20 |
| Tm2 65.47 ± 0.33 | 2.09E+05 | 4.08E+04 | 0.20 | |
| Tm3 80.80 ± 0.36 | 4.46E+04 | 4.09E+04 | 0.92 | |
| Triphenyl phosphate | Tm1 51.32 ± 1.70 | 4.57E+05 | 3.59E+04 | 0.08 |
| Tm2 67.66 ± 0.26 | 1.31E+06 | 6.68E+04 | 0.05 | |
| Tm3 78.21 ± 0.44 | 4.72E+05 | 8.95E+04 | 0.19 |
The displayed values include van’t Hoff enthalpy ΔHv (kcal/mol), molar heat enthalpy ΔH (kcal/mol) and the ΔHv/ΔH ratio. Additionally, the table presents the thermal transition temperature (Tm) in the form of molar heat capacity.
Isothermal titration calorimetry (ITC)
The ITC experiment was conducted to validate and quantify the thermodynamic parameters governing the protein–ligand interaction. The ITC isotherm revealed that the Ha006a-ligand complexes viz. Ha006a-donepezil, Ha006a-protopine, Ha006a-3′,4′,5,7-tetramethoxyflavone, Ha006a- piperine and Ha006a-triphenyl phosphate were enthalpy-driven and exothermic in nature (Fig. 7).
Figure 7.

The Isothermal Titration Calorimetry (ITC) graphs for analyzing the interaction mechanism of Ha006a with various ligands. The top plot depicts the differential power versus time, while the bottom plot illustrates the injection heat versus molar ratio within the cell. The graphs depict the binding profile with (A) Donepezil, (B) Protopine, (C) 3′,4′,5,7-Tetramethoxyflavone, (D) Piperine, and (E) Triphenyl phosphate. To avoid any mismatch, the ligands and the Ha006a were consistently kept in identical buffers in every experiment.
The analysis further yielded thermodynamic parameters stating dissociation constant (kd), stoichiometry (N), enthalpy change (ΔH) and entropy change (ΔS) with each ligand (Table 4). Overall, this experiment provided invaluable insights into the protein–ligand interaction, contributing to understanding the binding mechanism involved. In the context of biomolecular studies, the potency of binding energies underscores the robustness and specificity of the binding affinity between the two entities58,59. The isotherm analysis revealed that the protein–ligand binding involved a multitude of interactions, including hydrogen bond, van der Waal forces and hydrophobic interactions, thus signifying higher binding affinity. The dissociation constant (kd) values obtained for the ligands were in the range of 19–83 µM, whereas TPP showed kd value of 90 µM. It is established that the lower kd value signifies higher affinity and it was observed that all the screened ligands showed higher affinity as compared to TPP. Moreover, the enthalpy change associated with the binding process implied that the reactions are exothermic, signifying the presence of favorable interactions. Furthermore, the entropy change was negative for each ligand interacting with Ha006a, depicting an ordered and stable system upon binding with minimal conformational changes. Additionally, it was observed that the stoichiometry of each binding reaction was consistently unity. Considering the results, it can be inferred that the binding interaction was specific, favorable, and robust between Ha006a and the ligands under investigation, and the binding efficiency of 3′,4′,5,7-tetramethoxyflavone > donepezil > protopine > piperine > TPP.
Table 4.
The thermodynamic values derived from the isothermogram obtained through the ITC experiment are summarized.
| Ligands | N | ΔH (cal/mol) | ΔS (cal/mol/deg) | kd (μM) |
|---|---|---|---|---|
| Donepezil | 1 | − 3.290E5 | − 1.08E3 | 43 |
| Protopine | 1 | − 1.432E5 | − 461 | 56 |
| 3′,4′,5,7-Tetramethoxyflavone | 1 | − 2.286E5 | − 745 | 19 |
| Piperine | 1 | − 4.516E6 | − 1.51E4 | 83 |
| Triphenyl phosphate | 1 | − 1.412E6 | − 4.71E3 | 90 |
The table includes values for stoichiometry (N), enthalpy change (ΔH), entropy change (ΔS), and dissociation constant (kd) observed during the binding studies.
Discussion
The widespread and injudicious use of insecticides has sparked global concern due to the emergence of insect resistance to various chemicals15. Organophosphates and pyrethroids are distinctly reported classes of insecticides mostly used for pest control worldwide. The presence of insect enzymes such as carboxylesterases (CarEs) renders insecticides ineffective, as this enzyme fosters resistance5,7. Over the past 50 years, more than 50 instances of pesticide resistance have been associated to CarEs5. Some of the disease vectors and insect pests that have reported CarEs mediated insecticide resistance are Anopheles gambiae60, Musca domestica61, Plutella xylostella62, Helicoverpa armigera63,64 and Spodoptera litura65. Helicoverpa armigera (Noctuidae: Lepidoptera) is a highly destructive pest that poses a significant threat to economically important crops worldwide48. This insect has been reported to cause severe damage to over 180 plant species from 47 different families50,66. To revitalize the effectiveness of OP insecticides, developing new pesticides and using synergists to combat resistance phenomena is a viable alternative strategy and an urgent necessity. Phytochemicals, or bioactive compounds, are crucial in developing environmentally friendly insecticides either in their natural state or as templates for synthetic modifications67,68. Due to their lower toxicity compared to conventional insecticides, phytochemicals serve as promising alternatives, particularly in synergistic approaches that may require lower doses for effective pest control69.
The current study mainly focuses on structure-based ligand design and pharmacophore modeling for screening potential hit compounds. The three-dimensional model for the Ha006a was predicted using AlphaFold v2.1.1 and validated using various bioinformatics tools to analyze the stereochemical stability and quality of the model. FooDB library (70,926 compounds) was used for the hit screening utilizing pharmacophore-based modeling to obtain 4500 filtered compounds, followed by virtual screening to find the best possible hits. The virtual screening workflow identified the top four phytochemicals as potential Ha006a inhibitors. We have selected the compounds showing the best binding pose, score, and intermolecular interaction with Ha006a for in-depth investigation, aiming to elucidate structural stability and protein–ligand interaction. As a reference compound, the natural inhibitor TPP was included in the study. The four investigated compounds are donepezil, 3′,4′,5,7-tetramethoxyflavone, protopine and piperine. Donepezil hydrochloride, a piperidine derivative, is a reversible acetylcholinesterase (AChE) inhibitor and is approved to be used for the treatment of Alzheimer's disease70. The second compound, 3′,4′,5,7-tetramethoxyflavone, is a flavonoid, it is also an AChE inhibitor and associated with Alzheimer's disease71. The third compound, protopine is an alkaloid found in papaveraceae plants and has been reported to possess AChE inhibitory, hepatoprotective, and anticancer activity72. Lastly, piperine, a plant-produced alkaloid found in Piper longum (Piperaceae), has been reported to possess anti-amoebic, anti-cancer, anti-asthmatic, anti-ulcer, anti-oxidant, anti-inflammatory, and anti-mutagenic properties73. The physicochemical characteristics and pharmacophore parameters indicate that all these compounds meet the criteria for drug-likeness. These compounds have previously demonstrated the ability to impede AChE activity. However, this study marks the first instance of a thorough examination of their interaction and specificity with the Ha006a enzyme. The stability of the protein is attributed to the presence of hydrogen bonds and hydrophobic interactions forming Ha006a-ligand complexes.
MD simulation analysis showed that ligand structures formed a stable complex with Ha006a protein throughout the production run of 100 ns. The protein–ligand complexes viz. Ha006a-donepezil and Ha006a-3′,4′,5,7-tetramethoxyflavone showed higher RMSF and SASA values throughout the simulation run, inferring effective binding and conformational stability on fitting to the binding pocket. Additionally, ligand 3′,4′,5,7-tetramethoxyflavone showed the highest polar contacts throughout a timescale of 100 ns. Further, MMPBSA analysis reveals that ligand donepezil requires the lowest energy of − 16.76 kcal/mol for the binding, considering both gas-phase and solvation energy. On assessing each energy term calculated: 3′,4′,5,7-tetramethoxyflavone, donepezil, and triphenyl phosphate showed the highest non-bonded interaction energy; 3′,4′,5,7-tetramethoxyflavone showed highest solvation energy of 38.82 kcal/mol i.e., favorable non-polar solvation energy and polar solvation energy of − 3.92 kcal/mol and 42.74 kcal/mol respectively.
Additionally, the ITC results validated the spontaneous and exothermic nature of the binding interactions between Ha006a and various compounds, exhibiting diverse dissociation constant values. Notably, 3′,4′,5,7-tetramethoxyflavone demonstrated the lowest dissociation constant, signifying its higher affinity for Ha006a. Subsequent compounds exhibiting noteworthy affinity included donepezil (kd value of 43 µM), protopine (kd 56 µM), piperine (kd 83 µM), and TPP (kd 90 µM). This affinity trend was consistent with the insecticide inhibition assay results, providing additional support for the reliability of the findings. Furthermore, the DSC isothermogram clearly indicated a distinctive increase in the transition temperature upon binding of these phytochemicals to Ha006a compared to the apo protein. It is crucial to highlight that, so far, this study stands as a pioneering effort in furnishing thermodynamic binding interaction study, shedding light on the inhibitory characteristics of phytochemicals against Ha006a from H. armigera.
Ensuring environmental safety is a fundamental necessity for the practical applicability of synergists. The Ha006a protein was meticulously investigated for proteins sharing similarities in structure using several bioinformatics tools. Analysis of both sequence and structure indicated a notably low similarity (> 30%) with the homologous proteins. This diminished structural resemblance substantiates the assertion that these phytochemicals will specifically interact with the Ha006a protein and may not contribute to toxicity caused by off-target effects. Furthermore, historical validation exists for the safe use of other such compounds as a synergist for pyrethroids, even when the compounds demonstrated the ability to inhibit non-target human metabolic enzymes. For instance, cytochrome P450s was inhibited by piperonyl butoxide as a synergist for pyrethroids and has been applied for decades without causing detrimental effects25. Thus, similar approaches can be implemented to safely utilize phytochemicals as synergists with organophosphates (OP), aiming to mitigate the indiscriminate application of OP insecticides for pest control worldwide. This, in turn, will promote both human well-being and environmental safety.
Conclusion
The findings of this study represent a significant stride forward in the application of virtual screening for the identification of inhibitors targeting insecticide resistance. Through this research, we identified phytochemical inhibitors of a key resistance enzyme, Ha006a. Our comprehension of the protein–ligand interaction was enhanced through the bioinformatics approach, corroborated by biochemical assays and biophysical techniques. The compounds presented in this study are strategically selected to enhance the effectiveness of OP insecticides against H. armigera, underscoring the viability of targeting CarE-based resistance mechanisms. In the current study, we have investigated the inhibitory potential of four phytochemical compounds, i.e., donepezil, 3′,4′,5,7-tetramethoxyflavone, protopine and piperine against Ha006a protein. The amalgamation of data notably reinforces the assertion that 3′,4′,5,7-tetramethoxyflavone and donepezil serve as potent inhibitors for Ha006a and can be used as synergists with OP. An in-depth research is needed to elucidate the functionality of these inhibitors in in vivo conditions, such as conducting insect bioassay becomes imperative. Consequently, the integration of structural analysis and in vivo studies will contribute to discerning the physiological role of these inhibitors.
Supplementary Information
Acknowledgements
Harry Kaur thanks the Department of Science and Technology (DST-INSPIRE), Government of India, for providing fellowship. The authors thank the Institute Instrumentation Centre (IIC) at the Indian Institute of Technology, Roorkee for the ITC experimental facility. We thank the Bioinformatics Center (BIC), supported by the Department of Biotechnology (DBT), the Government of India, for providing computational facility (reference number BT/PR40141/BTIS/ 137/16/2021). Dr. Simranjeet Singh thanks RCB Haryana for RA- fellowship letter No DBT-RA/2022/July/N/2044 & MoE-STARS/STARS-2/2023-0714, dated September 26, 2022.
Author contributions
Conceptualization, Methodology, Visualization, Data curation, Formal analysis, Writing – Original draft: Harry Kaur, Simranjeet Singh; Methodology, Writing & Editing: Sandra Kathott Prakash, Surabhi Rode, Sapna Lonare, Rakesh Kumar, Joginder Singh, Nadeem A Khan; Software, Writing & Editing: Pravindra Kumar, Praveen C Ramamurthy; Conceptualization, Visualization, Investigation, Software, Validation & Supervision: Ashwani Kumar Sharma. All the authors discussed the results and approved the final manuscript.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Data availability
The data that support the findings of this study are available from [Harry Kaur]. Still, restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. However, data are available from the authors upon reasonable request and with permission of [Harry Kaur].
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-69497-y.
References
- 1.Raj, A., Dubey, A., Malla, M. A. & Kumar, A. Pesticide pestilence: Global scenario and recent advances in detection and degradation methods. J. Environ. Manage.338, 117680 (2023). 10.1016/j.jenvman.2023.117680 [DOI] [PubMed] [Google Scholar]
- 2.Chen, H. et al. Adsorption and biodegradation of carbaryl on montmorillonite, kaolinite and goethite. Appl. Clay Sci.46, 102–108 (2009). 10.1016/j.clay.2009.07.006 [DOI] [Google Scholar]
- 3.Chevillard, A., Angellier-Coussy, H., Guillard, V., Gontard, N. & Gastaldi, E. Investigating the biodegradation pattern of an ecofriendly pesticide delivery system based on wheat gluten and organically modified montmorillonites. Polym. Degrad. Stab.97, 2060–2068 (2012). 10.1016/j.polymdegradstab.2012.02.017 [DOI] [Google Scholar]
- 4.Sharma, A. et al. Worldwide pesticide usage and its impacts on ecosystem. SN Appl. Sci.1, 1446 (2019). 10.1007/s42452-019-1485-1 [DOI] [Google Scholar]
- 5.Oakeshott, J. G., Claudianos, C., Campbell, P. M., Newcomb, R. D. & Russell, R. J. Biochemical genetics and genomics of insect esterases. In Comprehensive Molecular Insect Science 309–381 (Elsevier, 2005). 10.1016/B0-44-451924-6/00073-9.
- 6.Whalon, M. E., Mota-Sanchez, D., & Hollingworth, R. M. Global Pesticide Resistance in Arthropods. (CABI, 2008).
- 7.Farnsworth, C. A. et al. Esterase-based metabolic resistance to insecticides in heliothine and spodopteran pests. J. Pest. Sci.35, 275–289 (2010). 10.1584/jpestics.R10-13 [DOI] [Google Scholar]
- 8.Gunning, R. V., Moores, G. D. & Devonshire, A. L. Esterases and fenvalerate resistance in a field population of Helicoverpa punctigera (Lepidoptera: Noctuidae) in Australia. Pest. Biochem. Physiol.58, 155–162 (1997). 10.1006/pest.1997.2295 [DOI] [Google Scholar]
- 9.Parker, A. G., Russell, R. J., Delves, A. C. & Oakeshott, J. G. Biochemistry and physiology of esterases in organophosphate-susceptible and -resistant strains of the Australian sheep blowfly, Lucilia cuprina. Pest. Biochem. Physiol.41, 305–318 (1991). 10.1016/0048-3575(91)90085-Z [DOI] [Google Scholar]
- 10.Marcombe, S. et al. Insecticide resistance in the dengue vector Aedes aegypti from martinique: Distribution, mechanisms and relations with environmental factors. PLoS ONE7, e30989 (2012). 10.1371/journal.pone.0030989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun, L., Zhou, X., Zhang, J. & Gao, X. Polymorphisms in a carboxylesterase gene between organophosphate-resistant and -susceptible Aphis gossypii (Homoptera: Aphididae). J. Econ. Entom.98, 1325–1332 (2005). 10.1603/0022-0493-98.4.1325 [DOI] [PubMed] [Google Scholar]
- 12.Ketterman, A. J., Jayawardena, K. G. & Hemingway, J. Purification and characterization of a carboxylesterase involved in insecticide resistance from the mosquito Culex quinquefasciatus. Biochem. J.287, 355–360 (1992). 10.1042/bj2870355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu, Y., Zhang, H., Qiao, C., Lu, X. & Cui, F. Correlation between carboxylesterase alleles and insecticide resistance in Culex pipiens complex from China. Parasit. Vectors4, 236 (2011). 10.1186/1756-3305-4-236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tay, W. T. et al. A brave new world for an old world pest: Helicoverpa armigera (Lepidoptera: Noctuidae) in Brazil. PLoS ONE8, e80134 (2013). 10.1371/journal.pone.0080134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang, Y., Li, Y. & Wu, Y. Current status of insecticide resistance in Helicoverpa armigera after 15 years of Bt cotton planting in China. J. Econ. Entom.106, 375–381 (2013). 10.1603/EC12286 [DOI] [PubMed] [Google Scholar]
- 16.Aldridge, W. N. Some properties of specific cholinesterase with particular reference to the mechanism of inhibition by diethyl p -nitrophenyl thiophosphate (E 605) and analogues. Biochem. J.46, 451–460 (1950). 10.1042/bj0460451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aranda, J. et al. The catalytic mechanism of carboxylesterases: A computational study. Biochemistry53, 5820–5829 (2014). 10.1021/bi500934j [DOI] [PubMed] [Google Scholar]
- 18.Gordon, J. R. & Ottea, J. Association of esterases with insecticide resistance in Culex quinquefasciatus (Diptera: Culicidae). J. Econ. Entom.105, 971–978 (2012). 10.1603/EC11224 [DOI] [PubMed] [Google Scholar]
- 19.Karunaratne, S. H. P. P., Jayawardena, K. G., Hemingway, J. & Ketterman, A. J. Characterization of a B-type esterase involved in insecticide resistance from the mosquito Culex quinquefasciatus. Biochem. J.294, 575–579 (1993). 10.1042/bj2940575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devonshire, A. L. et al. The evolution of insecticide resistance in the peach–potato aphid, Myzus persicae. Phil. Trans. R. Soc. Lond. B353, 1677–1684 (1998). 10.1098/rstb.1998.0318 [DOI] [Google Scholar]
- 21.Mouchès, C. et al. Amplification of an esterase gene is responsible for insecticide resistance in a california Culex mosquito. Science233, 778–780 (1986). 10.1126/science.3755546 [DOI] [PubMed] [Google Scholar]
- 22.Mao, Y.-B. et al. Silencing a cotton bollworm P450 monooxygenase gene by plant-mediated RNAi impairs larval tolerance of gossypol. Nat. Biotechnol.25, 1307–1313 (2007). 10.1038/nbt1352 [DOI] [PubMed] [Google Scholar]
- 23.Swale, D. R. et al. An insecticide resistance-breaking mosquitocide targeting inward rectifier potassium channels in vectors of Zika virus and malaria. Sci. Rep.6, 36954 (2016). 10.1038/srep36954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matthews, H. B. & Casida, J. E. Properties of housefly microsomal cytochromes in relation to sex, strain, substrate specificity, and apparent inhibition and induction by synergist and insecticide chemicals. Life Sci.9, 989–1001 (1970). 10.1016/0024-3205(70)90122-0 [DOI] [PubMed] [Google Scholar]
- 25.Casida, J. E. Mixed-function oxidase involvement in the biochemistry of insecticide synergists. J. Agric. Food Chem.18, 753–772 (1970). 10.1021/jf60171a013 [DOI] [PubMed] [Google Scholar]
- 26.Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature596, 583–589 (2021). 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.wwPDB consortium et al. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res.47, D520–D528 (2019). [DOI] [PMC free article] [PubMed]
- 28.Database resources of the National Center for Biotechnology Information. Nucleic Acids Res44, D7–D19 (2016). [DOI] [PMC free article] [PubMed]
- 29.Sunseri, J. & Koes, D. R. Pharmit: Interactive exploration of chemical space. Nucleic Acids Res.44, W442–W448 (2016). 10.1093/nar/gkw287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eisenberg, D., Lüthy, R. & Bowie, J. U. VERIFY3D: Assessment of protein models with three-dimensional profiles. In Methods in Enzymology, vol. 277 396–404 (Elsevier, 1997). [DOI] [PubMed]
- 31.Colovos, C. & Yeates, T. O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci.2, 1511–1519 (1993). 10.1002/pro.5560020916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr.26, 283–291 (1993). 10.1107/S0021889892009944 [DOI] [Google Scholar]
- 33.Holm, L. & Sander, C. Dali/FSSP classification of three-dimensional protein folds. Nucleic Acids Res.25, 231–234 (1997). 10.1093/nar/25.1.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Castro, E. et al. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res.34, W362–W365 (2006). 10.1093/nar/gkl124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hunter, A. D. ACD/ChemSketch 1.0 (freeware); ACD/ChemSketch 2.0 and its Tautomers, Dictionary, and 3D Plug-ins; ACD/HNMR 2.0; ACD/CNMR 2.0. J. Chem. Educ.74, 905 (1997).
- 36.Gasteiger, E. et al. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res.31, 3784–3788 (2003). 10.1093/nar/gkg563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dallakyan, S. & Olson, A. J. Small-molecule library screening by docking with PyRx. In Chemical Biology, vol. 1263 (eds. Hempel, J. E. et al.) 243–250 (Springer, 2015). [DOI] [PubMed]
- 38.Hanwell, M. D. et al. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform.4, 17 (2012). 10.1186/1758-2946-4-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.8. Schrodinger LLC (2015).
- 40.O’Boyle, N. M. et al. Open babel: An open chemical toolbox. J. Cheminform.3, 33 (2011). 10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Der Spoel, D. et al. GROMACS: Fast, flexible, and free. J. Comput. Chem.26, 1701–1718 (2005). 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]
- 42.Gouet, P., Courcelle, E., Stuart, D. I. & Motoz, F. ESPript: Analysis of multiple sequence alignments in PostScript. Bioinformatics15, 305–308 (1999). 10.1093/bioinformatics/15.4.305 [DOI] [PubMed] [Google Scholar]
- 43.Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol7, 539 (2011). 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.64, 4–17 (2012). 10.1016/j.addr.2012.09.019 [DOI] [PubMed] [Google Scholar]
- 45.Valdés-Tresanco, M. S., Valdés-Tresanco, M. E., Valiente, P. A. & Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput.17, 6281–6291 (2021). 10.1021/acs.jctc.1c00645 [DOI] [PubMed] [Google Scholar]
- 46.Kaur, H. et al. Heterologous expression, biochemical characterization and prospects for insecticide biosensing potential of carboxylesterase Ha006a from Helicoverpa armigera. Pesticide Biochem. Physiol.2024, 105844. 10.1016/j.pestbp.2024.105844 (2024). 10.1016/j.pestbp.2024.105844 [DOI] [PubMed] [Google Scholar]
- 47.Glenn, D. C., Hoffmann, A. A. & McDonald, G. Resistance to pyrethroids in Helicoverpa armigera (Lepidoptera: Noctuidae) from corn: Adult resistance, larval resistance, and fitness effects. J. Econ. Entomol.87, 1165–1171 (1994). 10.1093/jee/87.5.1165 [DOI] [Google Scholar]
- 48.Kumar, P. et al. Mutation studies and structure-based identification of potential inhibitor molecules against periplasmic amino acid binding protein of Candidatus Liberibacter asiaticus (CLasTcyA). Int. J. Biol. Macromol.147, 1228–1238 (2020). 10.1016/j.ijbiomac.2019.09.250 [DOI] [PubMed] [Google Scholar]
- 49.Rajarathnam, K. & Rösgen, J. Isothermal titration calorimetry of membrane proteins—Progress and challenges. Biochim. Biophys. Acta BBA Biomembranes1838, 69–77 (2014). 10.1016/j.bbamem.2013.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar, R. et al. Farnesol dehydrogenase from Helicoverpa armigera (Hübner) as a promising target for pest management: Molecular docking, in vitro and insect bioassay studies using geranylgeraniol as potential inhibitor. Biotech13, 175 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Younus, F. et al. Molecular basis for the behavioral effects of the odorant degrading enzyme Esterase 6 in Drosophila. Sci. Rep.7, 46188 (2017). 10.1038/srep46188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fraser, N. J. et al. Evolution of protein quaternary structure in response to selective pressure for increased thermostability. J. Mol. Biol.428, 2359–2371 (2016). 10.1016/j.jmb.2016.03.014 [DOI] [PubMed] [Google Scholar]
- 53.Myers, M., Richmond, R. C. & Oakeshott, J. G. On the origins of esterases. Mol. Biol. Evol.5, 113–119 (1988). [DOI] [PubMed] [Google Scholar]
- 54.Lu, S. et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res.48, D265–D268 (2020). 10.1093/nar/gkz991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gill, P., Moghadam, T. T. & Ranjbar, B. Differential scanning calorimetry techniques: Applications in biology and nanoscience. J. Biomol. Tech.21, 167–193 (2010). [PMC free article] [PubMed] [Google Scholar]
- 56.Verma, P., Kar, B., Varshney, R., Roy, P. & Sharma, A. K. Characterization of AICAR transformylase/ IMP cyclohydrolase (ATIC ) from Staphylococcus lugdunensis. FEBS J.284, 4233–4261 (2017). 10.1111/febs.14303 [DOI] [PubMed] [Google Scholar]
- 57.Durowoju, I. B., Bhandal, K. S., Hu, J., Carpick, B. & Kirkitadze, M. Differential scanning calorimetry—a method for assessing the thermal stability and conformation of protein antigen. JoVE2017, 55262. 10.3791/55262 (2017). 10.3791/55262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bissantz, C., Kuhn, B. & Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem.53, 5061–5084 (2010). 10.1021/jm100112j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hedstrom, L. Enzyme specificity and selectivity. In Encyclopedia of Life Sciences (Wiley, 2010). 10.1002/9780470015902.a0000716.pub2.
- 60.Mutunga, J. M. et al. Carbamate and pyrethroid resistance in the akron strain of Anopheles gambiae. Pest. Biochem. Physiol.121, 116–121 (2015). 10.1016/j.pestbp.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Feng, X. & Liu, N. Functional analyses of house fly carboxylesterases involved in insecticide resistance. Front. Physiol.11, 595009 (2020). 10.3389/fphys.2020.595009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang, M. et al. Two carboxylesterase genes in Plutella xylostella associated with sex pheromones and plant volatiles degradation. Pest Manage. Sci.77, 2737–2746 (2021). 10.1002/ps.6302 [DOI] [PubMed] [Google Scholar]
- 63.Bai, L. et al. Identification and biochemical characterization of carboxylesterase 001G associated with insecticide detoxification in Helicoverpa armigera. Pesticide Biochem. Physiol.157, 69–79 (2019). 10.1016/j.pestbp.2019.03.009 [DOI] [PubMed] [Google Scholar]
- 64.Li, Y.-Q. et al. Functional characterization of two carboxylesterase genes involved in pyrethroid detoxification in Helicoverpa armigera. J. Agric. Food Chem.68, 3390–3402 (2020). 10.1021/acs.jafc.9b06690 [DOI] [PubMed] [Google Scholar]
- 65.Shi, Y., Li, W., Zhou, Y., Liao, X. & Shi, L. Contribution of multiple overexpressed carboxylesterase genes to indoxacarb resistance in Spodoptera litura. Pest Manage. Sci.78, 1903–1914 (2022). 10.1002/ps.6808 [DOI] [PubMed] [Google Scholar]
- 66.Alam, M. S. et al. In silico identification of potential phytochemical inhibitors targeting farnesyl diphosphate synthase of cotton bollworm (Helicoverpa armigera). J. Biomol. Struct. Dyn.41, 1978–1987 (2023). 10.1080/07391102.2022.2025904 [DOI] [PubMed] [Google Scholar]
- 67.Zhang, T., Jayachandran, M., Ganesan, K. & Xu, B. Black truffle aqueous extract attenuates oxidative stress and inflammation in STZ-induced hyperglycemic rats via Nrf2 and NF-κB pathways. Front. Pharmacol.9, 1257 (2018). 10.3389/fphar.2018.01257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jayachandran, M. et al. Isoquercetin upregulates antioxidant genes, suppresses inflammatory cytokines and regulates AMPK pathway in streptozotocin-induced diabetic rats. Chemico-Biol. Interact.303, 62–69 (2019). 10.1016/j.cbi.2019.02.017 [DOI] [PubMed] [Google Scholar]
- 69.Amoabeng, B. W., Gurr, G. M., Gitau, C. W. & Stevenson, P. C. Cost:benefit analysis of botanical insecticide use in cabbage: Implications for smallholder farmers in developing countries. Crop Protect.57, 71–76 (2014). 10.1016/j.cropro.2013.11.019 [DOI] [Google Scholar]
- 70.Seltzer, B. Donepezil: A review. Expert Opin. Drug Metabol. Toxicol.1, 527–536 (2005). 10.1517/17425255.1.3.527 [DOI] [PubMed] [Google Scholar]
- 71.Natsume, N. et al. Effect of methoxyflavones contained in Kaempferia parviflora on CRE-mediated transcription in PC12D cells. Bioorg. Med. Chem. Lett.30, 127606 (2020). 10.1016/j.bmcl.2020.127606 [DOI] [PubMed] [Google Scholar]
- 72.Mathew, B. et al. Protopine. In Naturally Occurring Chemicals Against Alzheimer’s Disease 167–174 (Elsevier, 2021). 10.1016/B978-0-12-819212-2.00014-1.
- 73.Meghwal, M., Devu, S., Singh, H. & Goswami, T. K. Piperine and curcumin. In A Centum of Valuable Plant Bioactives 589–612 (Elsevier, 2021). 10.1016/B978-0-12-822923-1.00013-3.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from [Harry Kaur]. Still, restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. However, data are available from the authors upon reasonable request and with permission of [Harry Kaur].