

Abstract
MafA is a transcription factor that binds to the promoter in the insulin gene and has been postulated to regulate insulin transcription in response to serum glucose levels, but there is no current in vivo evidence to support this hypothesis. To analyze the role of MafA in insulin transcription and glucose homeostasis in vivo, we generated MafA-deficient mice. Here we report that MafA mutant mice display intolerance to glucose and develop diabetes mellitus. Detailed analyses revealed that glucose-, arginine-, or KCl-stimulated insulin secretion from pancreatic β cells is severely impaired, although insulin content per se is not significantly affected. MafA-deficient mice also display age-dependent pancreatic islet abnormalities. Further analysis revealed that insulin 1, insulin 2, Pdx1, Beta2, and Glut-2 transcripts are diminished in MafA-deficient mice. These results show that MafA is a key regulator of glucose-stimulated insulin secretion in vivo.
Insulin is the only polypeptide hormone that is essential for the regulation of blood glucose levels and is synthesized exclusively in β cells of the islets of Langerhans in the pancreas. The molecular mechanisms that control β-cell-specific insulin gene transcription are well characterized. Three conserved cis-regulatory elements within the promoter, E1, A3, and RIPE3b/C1, respectively, appear to be indispensable for proper insulin gene regulation (22, 25). Islet-restricted transcription factors Beta2/NeuroD and Pdx1 bind to the E1 and A3 elements in vitro. Gene disruption experiments in mice have revealed that both Beta2 and Pdx1 play critical roles in insulin gene regulation as well as in islet development and function (1, 8, 21). Furthermore, mutations in both the Beta2 and Pdx1 genes have been identified within populations of patients with type II diabetes (18, 29, 30).
The third regulatory element, RIPE3b/C1, has also been shown to play a critical role in β-cell-specific insulin gene transcription as well as in glucose-regulated expression. Previous studies identified a pancreatic β-cell-restricted factor, called the RIPE3b1 activator, that is enriched in response to glucose in pancreatic β-cell nuclear extracts. Very recently, four groups reported that the RIPE3b1 activator is a member of the Maf family of transcription factors, MafA (10, 12, 20, 26). The large Maf proteins, MafA/L-Maf/SMaf1 (2, 9, 24), MafB (11), c-Maf (23), and Nrl (31), each contain a basic motif followed by a leucine zipper, and all four family members harbor acidic domains that act as transcriptional activation domains. Although a role for MafA in insulin gene regulation was hypothesized, in vivo tests of the hypothesis have not been reported. To elucidate MafA function in insulin gene regulation, we generated MafA-deficient mice.
MATERIALS AND METHODS
Targeted disruption of the mafA gene.
mafA genomic clones were isolated from a 129/SvJ genomic library (Stratagene) using a partial mouse MafA cDNA as a probe. The targeting vector was constructed with the bacterial lacZ gene containing a nuclear localization signal (NLS), and a neomycin resistance (neoR) cassette. NLS-lacZ-neoR genes were inserted into the mafA open reading frame between the Eco47III/EcoRI and EcoRV/Eco47III sites in order to delete both the transactivation and basic motif-leucine zipper domains (Fig. 1A). The diphtheria toxin A gene was inserted at the 3′ end of short arm of the targeting vector for negative selection. Chimeras generated from two correctly targeted embryonic stem (ES) cell clones were bred to ICR mice with resultant germ line transmission.
FIG. 1.

Targeting strategy for mafA mutagenesis and LacZ expression in pancreatic islets in MafA mutant mice. (A) Schematic representation of the wild-type allele, the targeting construct, and the expected product of homologous recombination between them. (B) Southern blot of tail DNA showing NheI-cleaved DNA fragments corresponding to the wild-type (11 kbp) and targeted (16 kbp) mafA alleles. (C) β-Galactosidase expression (blue) in MafA+/− pancreatic β cells. Immunostaining was performed with antiglucagon (green) and anti-insulin (red) antibodies. Nuclear β-galactosidase staining overlaps that of insulin-expressing cells (merged).
Mouse genotyping by PCR/Southern blot analysis.
Genotyping was performed on genomic DNA isolated from ES cells or tails by PCR or Southern blotting. The 5′ and 3′ primers for the mutant allele (∼700 bp amplified) were 5′-ATGCGAAGTGGACCTGGGACCGCGCCGC-3′ and 5′-CTGCGCTGGCGAGGGCTCCCGAGGGAAG-3′ under the following conditions: 30 cycles of 98°C for 10 s, 71°C for 30 s, and 72°C for 30 s, followed by 1 cycle of 72°C for 7 min. The 5′ and 3′ primers for the mafA gene were 5′-GAGGCCTTCCGGGGTCAGAGCTTCGCGG-3′ and 5′-TCTGTTTCAGTCGGATGACCTCCTCCTTGC-3′ under the following conditions: 1 cycle of 94°C for 3 min and 30 cycles of 98°C for 10 s, 71°C for 30 s, and 72°C for 30 s, followed by 1 cycle of 72°C for 7 min, which results in an ∼400-bp-amplified fragment for the wild-type mafA allele. For mafA Southern blot analysis, DNA was digested with NheI and hybridized with probe.
Quantitative transcript analysis in pancreatic islets by competitive RT-PCR.
Competitive reverse transcription-PCR (RT-PCR) analysis was performed on total RNA prepared from isolated adult pancreatic islets essentially as previously described (15). Competitor DNA plasmids carrying a small deletion within the respective cDNAs were constructed by appropriate restriction endonuclease digestion, as shown in Table 1. Each PCR product was electrophoresed in a 2% agarose gel and visualized by ethidium bromide staining. The intensity of the amplified fragment was quantified using an NIH image system. To ascertain the efficiency of cDNA preparation from total RNAs, the competitive RT-PCR analysis of hypoxanthine phosphoribosyltransferase (HPRT) transcript was performed in each sample as the internal control. Reactions were plotted on individual standard curves to derive the actual quantity of individual transcripts.
TABLE 1.
Oligonucleotide primers and sizes of PCR products for competitive RT-PCR
| Gene product | Primera | Size of PCR products (bp)
|
Restriction enzyme(s) for construction of competitor DNA | |
|---|---|---|---|---|
| Target | Competitor | |||
| Insulin 1 | 5′-CCAGCTATAATCAGAGACCA-3′ | 377 | 328 | SmaI-BsgI |
| 5′-GGGCCTTAGTTGCAGTAGTT-3′ | ||||
| Insulin 2 | 5′-AGGAAGCCTATCTTCCAGGT-3′ | 401 | 293 | EcoNI-SmaI |
| 5′-ATTCATTGCAGAGGGGTAGG-3′ | ||||
| Glucagon | 5′-CTGGACAATCTTGCCACCAGGGAC-3′ | 413 | 358 | SphI-BstXI |
| 5′-CCACTACGGTTACCAGGTGGTCATGTC-3′ | ||||
| Pdx1 | 5′-TCGCTGGGATCACTGGAGCA-3′ | 365 | 247 | BlpI-MluI |
| 5′-CGGGAGATGTATTTGTTAAATAAGAATTC-3′ | ||||
| Beta2 | 5′-CTCCAGGGTTATGAGATCGTCAC-3′ | 525 | 329 | PpuMI |
| 5′-GATCTCTGACAGAGCCCA-3′ | ||||
| Glut-2 | 5′-TGGGATGAAGAGGAGACTGAA-3′ | 652 | 442 | XcmI-BsrGI |
| 5′-CATCCGTGAAGAGCTGGATCA-3′ | ||||
| Glucokinase | 5′-GATGTATTCCATCCCCGAGGACG-3′ | 410 | 291 | StyI-MscI |
| 5′-GCTCCACATTCTGCATCTCCTCC-3′ | ||||
| HPRT | 5′-GGCTTCCTCCTCAGACCGCTTT-3′ | 702 | 523 | BclI-PpuMI |
| 5′-AGGCTTTGTATTTGGCTTTTCC-3′ | ||||
The sequence of the forward primer is given on the top, and that of the reverse primer is given on the bottom.
Histological analysis.
Immunohistochemistry and light microscopy were performed as follows. Pancreata were dissected, weighed, fixed overnight in 4% paraformaldehyde, and then incubated overnight in 30% sucrose. Frozen sections were mounted on slides. For the quantification of the endocrine mass and determination of the β-cell/α-cell ratio, sections were immunostained with both guinea pig anti-insulin (Linco) and rabbit antiglucagon (DAKO) antibodies. Detection was performed using fluorescein secondary antibodies (Cortex Biochem and ZYMED). Sections were incubated for 5 min with 0.01% Hoechst stain to reveal nuclei. All islets in the sections were photographed, and analyzed with Adobe Photoshop software (Adobe System Inc.). X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) staining was performed as previously described (32).
Glucose tolerance test and insulin release.
Mice were fasted for 12 h and then injected intraperitoneally (i.p.) with glucose (2 g/kg of body weight). Venous blood was obtained from the retro-orbital plexus at 0, 15, 30, 60, and 120 min after the injection. Plasma glucose levels were measured using a Fuji Drichem 3500 (Fuji-Film, Tokyo, Japan). For insulin release, glucose (3 g/kg of body weight) or l-arginine (1 g/kg of body weight) was injected i.p. and venous blood was collected at 0, 2, 5, and 15 min in heparinized tubes. Pancreatic insulin was extracted by the acid-ethanol method as described previously (7). Serum insulin levels and insulin contents of the pancreata were measured with an Ultra-sensitive insulin enzyme-linked immunosorbent assay kit (Morinaga Bioscience, Yokohama, Japan).
Islet isolation and insulin release.
To obtain pancreatic islets, pancreata were removed and islets were isolated by collagenase digestion using the protocol described in reference 16. The islets were individually dissected under a stereomicroscope. Batches of 10 islets of similar size were collected and incubated in RPMI 1640-10% fetal calf serum at 37°C in 5% CO2 for 2 h. These islets were washed and preincubated in 0.5% (wt/vol) bovine serum albumin-Krebs-Ringer HEPES-buffered saline in 2.8 mM glucose at 37°C in 5% CO2 for 30 min and then transferred to 0.5% (wt/vol) bovine serum albumin-Krebs-Ringer HEPES-buffered saline in 2.8 mM glucose, stimulatory 20 mM glucose alone, or 30 mM KCl at 2.8 mM glucose. After incubation at 37°C in 5% CO2 for 30 min, the supernatants were measured for insulin release as described above.
RESULTS
MafA is expressed in pancreatic β cells.
A positive or negative targeting vector was constructed in which the bacterial β-galactosidase gene containing a nuclear localization signal and a phosphoglycerate kinase promoter-neomycin resistance (PGK-neoR) cassette was used to replace MafA coding sequences (Fig. 1A and B). Chimeras obtained from two correctly targeted ES cell clones were bred to ICR mice and resulted in germ line transmission. β-Galactosidase expression in these heterozygous mutant mice was detected in the lens, somites, and olfactory bulb (data not shown). In addition, strong X-Gal staining was observed in pancreatic islets. β-Galactosidase expression overlapped anti-insulin immunostaining, but not antiglucagon staining, indicating that MafA is expressed exclusively in pancreatic β cells (Fig. 1C).
We intercrossed MafA+/− mice to determine the viability of homozygous MafA mutant mice. Analysis of offspring from the first line of heterozygous intercrosses revealed that MafA−/− mice were recovered, but the frequency was low (data not shown). To confirm the results, we analyzed the offspring from a second independent line of MafA+/− mutant intercrosses. Analysis of 223 offspring from 26 litters demonstrated that 59 mice (26.5%) were homozygous mutant. Since the phenotypes of MafA-deficient mice from the two lines were indistinguishable, we concluded that MafA deficiency does not confer embryonic lethality. MafA−/− mice survived until adulthood.
MafA-deficient mice develop diabetes mellitus.
Since it was suggested that MafA is a primary candidate regulator for insulin gene transcription, we screened the MafA−/− mutant mice for blood glucose levels. Fasting blood glucose levels of MafA−/− female mice were significantly higher than those of wild-type female littermates at 4 weeks of age (wild-type glucose level was 121 ± 10.9 mg/dl, MafA+/− glucose level was 150 ± 5.1 mg/dl, and MafA−/− glucose level was 152 ± 8.0 mg/dl, respectively) (Fig. 2A), and at 8 weeks of age, both male and female MafA −/− mice had high blood glucose levels (P < 0.05). The differences in blood glucose became more significant at 12 weeks postnatally. Although there was no significant difference in the mean body weight of MafA−/− and wild-type male mice, the mutant female animals displayed significant growth retardation postnatally from 8 weeks onward (Fig. 2B).
FIG. 2.

Development of diabetes mellitus in MafA−/− mice. (A) Fasting blood glucose levels of offspring derived from intercrosses of MafA+/− mice were determined using a semiautomated analyzer. Results represent the mean ± standard error of the mean. (B) Mean body weight ± standard error of the mean of offspring derived from intercrosses of MafA +/− mice at the indicated ages (weeks). Both sets of data are from 7 to 22 animals of each genotype. * indicates P < 0.05, while ** represents P < 0.01.
We followed MafA−/− mice to determine whether or not they developed overt diabetes mellitus. Under random feeding conditions, the blood glucose levels of 5/11 male and 1/9 female MafA−/− mice were over 300 mg/dl at 12 weeks of age. Five of 19 adult MafA−/− male mice became urinary glucose positive by 20 weeks of age, and the mean blood glucose levels of these mice reached ca. 600 mg/dl. By 50 weeks, 1/3 male and 1/5 female MafA−/− mice had over 500 mg/dl blood glucose. Our preliminary results suggest that there was no significant difference in the rate of survival between MafA−/− and wild-type mice until 50 weeks. These data show that the MafA-deficient mouse is a new model for overt diabetes mellitus.
Glucose-stimulated insulin secretion (GSIS) is impaired in MafA-deficient mice.
To elucidate developmental mechanisms that might contribute to diabetes mellitus, we tested the glucose tolerance of 8-week-old MafA−/− mice by an i.p. glucose tolerance test (ipGTT). Blood glucose levels were rapidly induced in MafA−/− mutants after i.p. glucose injection and were sustained at high levels for at least 2 h following glucose administration. Plasma glucose levels of MafA-deficient mice were 521 ± 31.7 mg/dl, while the level in wild-type mice was 243 ± 16.9 mg/dl 2 h after glucose injection (P < 0.01). Furthermore, MafA heterozygous mutant mice also had significantly higher blood glucose levels (417 ± 29.8 mg/dl) than wild-type littermates, demonstrating a MafA haploinsufficiency in pancreatic function (Fig. 3A).
FIG. 3.

Glucose tolerance and arginine tolerance tests and their effects on insulin production. (A) Glucose tolerance tests (ipGTT) after intraperitoneal loading with 2 g d-glucose/kg were performed on 8-week-old male animals of the indicated genotypes following a 12-h fast. Each symbol represents the following: *, P < 0.05, MafA−/− versus MafA+/+; **, P < 0.01, MafA−/− versus MafA+/+; ***, P < 0.01, MafA−/− versus MafA+/−; #, P < 0.05, MafA+/− versus MafA+/+; ##, P < 0.01, MafA+/− versus MafA+/+. (B) Level of plasma insulin of each MafA genotype during ipGTT. **, P < 0.01, MafA −/− versus MafA+/+; ***, P < 0.05, MafA−/− versus MafA+/−; #, P < 0.05, MafA+/− versus MafA+/+. (C) Level of plasma insulin after intraperitoneal arginine administration of each MafA genotype. *, P < 0.05, MafA−/− versus MafA+/+. (D) Insulin content of wild-type, MafA+/−, and MafA−/− mice. All data represent the mean values ± standard error for at least five male mice (8 to 14 weeks of age) of each genotype.
To elucidate the mechanism of this impaired glucose tolerance, we analyzed plasma insulin levels in the ipGTT mice. Plasma insulin levels in the MafA−/− mutants were significantly lower than in wild-type littermates. Very interestingly, there was almost no acute insulin secretion in response to the elevated blood glucose levels in MafA−/− mice (Fig. 3B). Plasma insulin levels of wild-type, MafA+/−, and MafA−/− at 2 min after glucose injection were 1.51 ± 0.25 ng/ml, 0.84 ± 0.12 ng/ml, 0.51 ± 0.08 ng/ml, respectively. We also measured serum insulin levels after arginine stimulation to ascertain whether or not insulin secretion from β cells was affected. Serum insulin levels in the MafA−/− mutants were significantly lower than in the wild-type mice, and the animals displayed almost no response to arginine administration (Fig. 3C). These results indicated two possibilities: either dysfunction of the insulin secretory machinery or decreased insulin content in β cells. To clarify the reason for the impaired response, we measured the amounts of insulin in the pancreata of these animals. The absolute insulin content in MafA−/− mutant pancreata was comparable to that of wild-type mice (insulin contents in wild-type, MafA+/−, and MafA−/− mice were 148 ± 23.5 μg/g, 140 ± 7.8 μg/g, and 128 ± 13.0 μg/g, respectively; Fig. 3D). These results summarily demonstrate that MafA is a vital regulator of glucose-stimulated insulin secretion but not of insulin production.
Isolated MafA-deficient islets display abnormalities of GSIS.
To analyze the underlying defect in glucose-stimulated insulin secretion in the MafA mutants, pancreatic islets from each mouse were isolated and measured for insulin secretion in response to glucose or KCl stimulation in vitro. Islets from wild-type mice secreted insulin in response to glucose administration (2.66 ± 0.46 ng/islet/h), but islets from the MafA-deficient mice did not (1.12 ± 0.19 ng/islet/h; P < 0.05). Islets recovered from MafA heterozygous mice also displayed a impaired response to glucose (1.40 ± 0.68 ng/islet/h; Fig. 4), although it was not significant. In addition, KCl stimulation had very little effect on insulin secretion in MafA-deficient islets (P < 0.01 versus wild type; Fig. 4). The results clearly indicate that MafA-deficient islets have a β-cell autonomous defect in GSIS.
FIG. 4.

Insulin secretion from isolated pancreatic islets in vitro. Insulin secretion in response to the indicated secretagogues. Values are expressed in nanograms of insulin islet−1 h−1, as the mean ± standard error of the mean of at least three male mice (8 to 12 weeks of age) per genotype. * indicates P < 0.05, while ** represents P < 0.01.
Abnormal architecture of pancreatic islets in adult MafA-deficient mice.
Since MafA−/− mutant mice displayed impaired GSIS and developed diabetes mellitus, and since targeted disruption of other β-cell-affiliated transcription factors such as Beta2, Pdx1, and Pax6 (28) cause disruption of islet cell development, we next analyzed histologically the pancreatic islets of MafA mutant mice. Using anti-insulin and antiglucagon antibodies, the number and size of islets were measured in wild-type, MafA+/−, and MafA−/− littermates at P1 and at 12 weeks of age. There was no significant difference in the architecture of pancreatic islets observed in equivalent cross-sections of the islets at P1 (Fig. 5A). The ratio of β to α cells, as determined by histologically assessing simultaneous expression of insulin and glucagon, in the MafA−/− mouse pancreas was comparable to that of wild-type mice at P1 (Fig. 5B). In contrast, a significant difference was observed in the structure of islets and also in the ratio of β to α cells in the MafA mutant and wild-type mice at 12 weeks of age (Fig. 5A and C), although there was no obvious difference in the diameters of islets among these three genotyped mice (Fig. 5D). These results indicate that MafA is dispensable for embryonic pancreatic development but is indispensable for the maintenance of adult pancreatic architecture and function.
FIG. 5.

Histological analysis of pancreatic islets. (A) Insulin (red) and glucagon (green) immunoreactivity in wild-type (+/+) and MafA homozygous mutant (−/−) mice at P1 or 12 weeks of age. Scale bar, 20 μm. (B and C) β-Cell/α-cell ratio of the pancreatic islets from mice of each genotype at P1 (B) and 12 weeks of age (C) (male mice). Pancreatic sections were double stained with anti-insulin and antiglucagon antibodies. Data are the mean β-cell/α-cell ratios ± standard error of the mean for at least three mice of each genotype. * indicates P < 0.05, while ** represents P < 0.01. (D) Morphometric analysis of islet diameter in pancreata from wild-type (+/+), heterozygous (+/−), and MafA homozygous mutant (−/−) 12-week-old male mice. Data represent the mean ratios ± standard error of at least three male mice of each genotype.
Gene expression in MafA-deficient islets.
In order to identify the underlying molecular mechanisms for impaired GSIS and the abnormal architecture of adult pancreatic islets, we next analyzed the levels of several other well-characterized β-cell effectors in the MafA−/− mutants (Fig. 6). The quantitative RT-PCR data using RNA recovered from isolated pancreatic islets of each genotype show that MafA mRNA is not detected in MafA−/− mutant islets, and about half the amount of MafA mRNA was detected in MafA+/− mutant islets, as expected (data not shown). The amounts of insulin 1, insulin 2, or Glut-2 mRNAs were reduced in the MafA−/− islets in comparison to wild-type islets, while no difference was observed in glucagon or glucokinase mRNA levels. Moreover, there was a tendency toward reduced expression of Pdx1 or Beta2 mRNAs in the MafA−/− islets as compared to wild type.
FIG. 6.
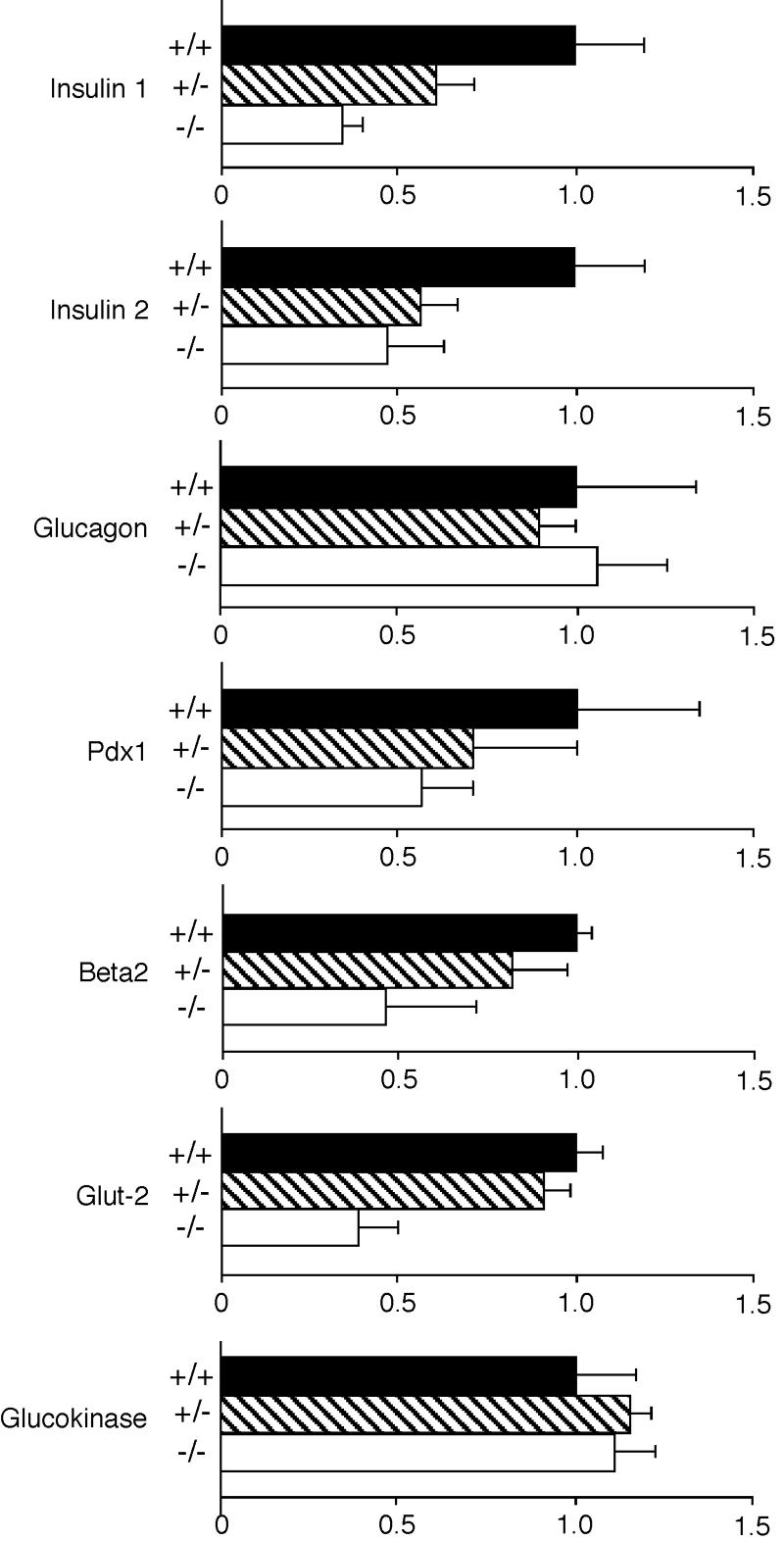
Comparison of gene expression in pancreatic islets. For quantitative analysis using competitive RT-PCR, pancreatic islets from 8-week-old male mice were used. The amount of each transcript was normalized to the amount of HPRT transcript. Data represent the mean ratios ± standard error of three mice of each genotype.
DISCUSSION
Here we report the generation and initial characterization of MafA−/− mutant mice and show that they display abnormal GSIS and adult islet structure, thus leading to pathological development of diabetes mellitus. The data demonstrate that transcription factor MafA is a crucial regulator of insulin secretion and of islet structural maintenance but is not required for β-cell development per se.
Three major observations on pancreatic function are reported here. First, MafA-deficient mice display almost normal insulin content in pancreata, although transcription of insulin 1 and insulin 2 is markedly reduced in MafA-deficient mice. Since MafA has been identified as a transcription factor that binds to a promoter element of the insulin gene and is thought to regulate insulin transcription in response to serum glucose levels (10, 12, 13, 19, 20, 26), we expected that insulin transcription and insulin content would be diminished in MafA-deficient mice. As expected, insulin 1 and insulin 2 transcription is markedly reduced in MafA-deficient mice. These results indicate that MafA is an important regulator of insulin transcription in vivo, as well as in vitro. The reduction of Pdx1 and Beta2 may also have a synergistic effect on diminished insulin transcription. Alternatively, the insulin content of a MafA−/− pancreas is not significantly diminished in comparison to that in the wild-type sibling pancreas. This paradoxical observation could be explained by either of two hypotheses. An abnormality of GSIS may be one possibility. Since GSIS of MafA-deficient mice is impaired as we demonstrated here, the secretion of insulin might be diminished, and thus the steady-state insulin content in the MafA−/− pancreas is not significantly affected. Another possibility is posttranscriptional regulation of insulin synthesis. Leroux et al. reported that the amount of insulin 2 protein in insulin 1-deficient mice is augmented as compared with that of wild-type mice, even though the amount of insulin 2 transcript is unchanged (17). These results indicate the existence of posttranscriptional regulation of insulin synthesis and suggest that the insulin content in MafA mutant mice may be regulated by posttranscriptional mechanism.
The second major observation is that MafA deficiency had no effect on embryonic development of pancreatic islets. This is in striking contrast to the consequences of Pdx1 or Beta2 mutation, which are also known to be important for insulin gene expression in vitro, and loss of these factors led to severe islet development abnormalities in vivo. Pdx1 deficiency leads to pancreatic agenesis, while Beta2-deficient mice display developmental arrest between embryonic day 14.5 (E14.5) and E17.5, a period characterized by a major expansion of the β-cell population (8, 21). Matsuoka et al. recently reported that MafA is expressed initially in insulin-expressing cells at E13.5 but is not detected in Nkx6.1-null mutant pancreata (19). These results may indicate that MafA is hypostatic to Nkx6.1 during pancreatic islet development.
In MafA mutant adult mice, the α cells are located inside of the pancreatic islets and the islet structure becomes abnormal. While we have not identified the specific molecular mechanism leading to this aberrant structural anomaly, diminished Pdx1 expression is implicated, since Pdx1+/− mice display similar structural defects in adult islets but not in newborn mice (1). As abnormal islet structure is often accompanied by impaired glucose-stimulated insulin secretion, as seen in Glut-2-null or glucokinase-null mutant mice (5, 33), aberrant islet architecture itself may cause moderate impairment of insulin secretion.
The third important observation reported here is that MafA is a key regulator of GSIS in vivo. The data demonstrate that MafA-deficient mice and islets are unable to respond to glucose, arginine, or KCl administration. GSIS consists of two stimulatory pathways, ionic and nonionic. Whereas the glucose-induced ionic pathway (i.e., closure of K+ATP channels, membrane depolarization, activation of L-type voltage-dependent Ca2+ channels, Ca2+ influx, elevation of cytosol-free Ca2+) is the major signaling pathway in β-cell insulin secretion, the nonionic glucose activity (termed K+ATP channel-independent action of glucose) has significant physiological relevance. The activation of a cyclic AMP-protein kinase A pathway in β cells by GLP-1 augments Ca2+-stimulated insulin release but also appears to enhance insulin secretion of a distal event, beyond the elevation of Ca2+ influx (14). Arginine or KCl, like GLP-1, potentiates insulin secretion in the presence, but not in the absence, of glucose. Arginine or KCl directly depolarizes the β-cell membrane and thereby elicits Ca2+-dependent electrical activity, Ca2+ entry, and insulin secretion. Thus, the unresponsiveness of MafA-deficient mice or isolated islets to glucose, arginine, or KCl stimulation indicates that both the ionic and nonionic pathways are affected by MafA deficiency. Since Pdx1 mRNA is diminished in MafA-deficient mice, Pdx1 could be the one of the causes of the observed unresponsiveness to glucose, since Pdx1+/− islets display abnormal response to glucose and KCl accompanied with decreased protein levels of Glut-2 and glucokinase (3). Samaras et al. also reported that Pdx1 expression is regulated by MafA in β cells (27). Accordingly, we hypothesize that abnormal GSIS observed in MafA-deficient mice may be partially explained by this down-regulation of Pdx1. Further studies, especially the comparison of gene expression profiles using a DNA microarray, must performed to elucidate the target genes of MafA in the β cells of MafA-deficient mice.
Since other regulators of insulin gene expression (Pdx1 and Beta2) are associated in some populations of patients with type 2 diabetes and mature onset diabetes of the young (4, 6, 18, 29), it will be of significant interest to determine whether mutations in the human MafA gene are associated with disease susceptibility. Finally, we suspect that these MafA−/− mutant mice will serve as a very useful new model to develop novel therapies for treating human diabetes mellitus.
Acknowledgments
We would like to thank N. Minegishi, T. Yokomizo, M. Ema, H. Shimano, M Ishikawa, and H. Sone (Tsukuba University) for helpful discussions. We are grateful to A. Godo for excellent assistance.
This work was supported in part by the NIH (R01 CA80088), a Grant-in-Aid from the Ministry of Education, Science, Sports and Culture, and the Environmental Response Project of JST-ERATO.
REFERENCES
- 1.Ahlgren, U., J. Jonsson, L. Jonsson, K. Simu, and H. Edlund. 1998. β-Cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the β-cell phenotype and maturity onset diabetes. Genes Dev. 12:1763-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benkhelifa, S., S. Provot, O. Lecoq, C. Pouponnot, G. Calothy, and M. P. Felder-Schmittbuhl. 1998. mafA, a novel member of the maf proto-oncogene family, displays developmental regulation and mitogenic capacity in avian neuroretina cells. Oncogene 17:247-254. [DOI] [PubMed] [Google Scholar]
- 3.Brissova, M., M. Shiota, W. E. Nicholson, M. Gannon, S. M. Knobel, D. W. Piston, C. V. Wright, and A. C. Powers. 2002. Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J. Biol. Chem. 277:11225-11232. [DOI] [PubMed] [Google Scholar]
- 4.Furuta, H., Y. Horikawa, N. Iwasaki, M. Hara, L. Sussel, M. M. Le Beau, E. M. Davis, M. Ogata, Y. Iwamoto, M. S. German, and G. I. Bell. 1998. β-Cell transcription factors and diabetes: mutations in the coding region of the BETA2/NeuroD1 (NEUROD1) and Nkx2.2 (NKX2B) genes are not associated with maturity-onset diabetes of the young in Japanese. Diabetes 47:1356-1358. [DOI] [PubMed] [Google Scholar]
- 5.Guillam, M. T., E. Hummler, E. Schaerer, J. I. Yeh, M. J. Birnbaum, F. Beermann, A. Schmidt, N. Deriaz, and B. Thorens. 1997. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat. Genet. 17:327-330. [DOI] [PubMed] [Google Scholar]
- 6.Hansen, L., J. N. Jensen, S. Urioste, H. V. Petersen, F. Pociot, H. Eiberg, O. P. Kristiansen, T. Hansen, P. Serup, J. Nerup, O. Pedersen et al. 2000. NeuroD/BETA2 gene variability and diabetes: no associations to late-onset type 2 diabetes but an A45 allele may represent a susceptibility marker for type 1 diabetes among Danes. Diabetes 49:876-878. [DOI] [PubMed] [Google Scholar]
- 7.im Walde, S. S., C. Dohle, P. Schott-Ohly, and H. Gleichmann. 2002. Molecular target structures in alloxan-induced diabetes in mice. Life Sci. 71:1681-1694. [DOI] [PubMed] [Google Scholar]
- 8.Jonsson, J., L. Carlsson, T. Edlund, and H. Edlund. 1994. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 371:606-609. [DOI] [PubMed] [Google Scholar]
- 9.Kajihara, M., S. Kawauchi, M. Kobayashi, H. Ogino, S. Takahashi, and K. Yasuda. 2001. Isolation, characterization, and expression analysis of zebrafish large Mafs. J. Biochem. (Tokyo) 129:139-146. [DOI] [PubMed] [Google Scholar]
- 10.Kajihara, M., H. Sone, M. Amemiya, Y. Katoh, M. Isogai, H. Shimano, N. Yamada, and S. Takahashi. 2003. Mouse MafA, homologue of zebrafish somite Maf 1, contributes to the specific transcriptional activity through the insulin promoter. Biochem. Biophys. Res. Commun. 312:831-842. [DOI] [PubMed] [Google Scholar]
- 11.Kataoka, K., K. T. Fujiwara, M. Noda, and M. Nishizawa. 1994. MafB, a new Maf family transcription activator that can associate with Maf and Fos but not with Jun. Mol. Cell. Biol. 14:7581-7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kataoka, K., S. I. Han, S. Shioda, M. Hirai, M. Nishizawa, and H. Handa. 2002. MafA is a glucose-regulated and pancreatic β-cell-specific transcriptional activator for the insulin gene. J. Biol. Chem. 277:49903-49910. [DOI] [PubMed] [Google Scholar]
- 13.Kataoka, K., S. Shioda, K. Ando, K. Sakagami, H. Handa, and K. Yasuda. 2004. Differentially expressed Maf family transcription factors, c-Maf and MafA, activate glucagon and insulin gene expression in pancreatic islet alpha- and β-cells. J. Mol. Endocrinol. 32:9-20. [DOI] [PubMed] [Google Scholar]
- 14.Komatsu, M., T. Schermerhorn, T. Aizawa, and G. W. Sharp. 1995. Glucose stimulation of insulin release in the absence of extracellular Ca2+ and in the absence of any increase in intracellular Ca2+ in rat pancreatic islets. Proc. Natl. Acad. Sci. USA 92:10728-10732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kudo, T., Y. Ikehara, A. Togayachi, K. Morozumi, M. Watanabe, M. Nakamura, S. Nishihara, and H. Narimatsu. 1998. Up-regulation of a set of glycosyltransferase genes in human colorectal cancer. Lab. Investig. 78:797-811. [PubMed] [Google Scholar]
- 16.Lacy, P. E., and M. Kostianovsky. 1967. Method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes 16:35-39. [DOI] [PubMed] [Google Scholar]
- 17.Leroux, L., P. Desbois, L. Lamotte, B. Duvillie, N. Cordonnier, M. Jackerott, J. Jami, D. Bucchini, and R. L. Joshi. 2001. Compensatory responses in mice carrying a null mutation for Ins1 or Ins2. Diabetes 50(Suppl. 1):S150-S153. [DOI] [PubMed] [Google Scholar]
- 18.Malecki, M. T., U. S. Jhala, A. Antonellis, L. Fields, A. Doria, T. Orban, M. Saad, J. H. Warram, M. Montminy, and A. S. Krolewski. 1999. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat. Genet. 23:323-328. [DOI] [PubMed] [Google Scholar]
- 19.Matsuoka, T. A., I. Artner, E. Henderson, A. Means, M. Sander, and R. Stein. 2004. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc. Natl. Acad. Sci. USA 101:2930-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuoka, T.-A., L. Zhao, I. Artner, H. W. Jarrett, D. Friedman, A. Means, and R. Stein. 2003. Members of the large Maf transcription family regulate insulin gene transcription in islet β cells. Mol. Cell. Biol. 23:6049-6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naya, F. J., H. P. Huang, Y. Qiu, H. Mutoh, F. J. DeMayo, A. B. Leiter, and M. J. Tsai. 1997. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 11:2323-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naya, F. J., C. M. Stellrecht, and M. J. Tsai. 1995. Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes Dev. 9:1009-1019. [DOI] [PubMed] [Google Scholar]
- 23.Nishizawa, M., K. Kataoka, N. Goto, K. T. Fujiwara, and S. Kawai. 1989. v-maf, a viral oncogene that encodes a “leucine zipper” motif. Proc. Natl. Acad. Sci. USA 86:7711-7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogino, H., and K. Yasuda. 1998. Induction of lens differentiation by activation of a bZIP transcription factor, L-Maf. Science 280:115-118. [DOI] [PubMed] [Google Scholar]
- 25.Ohlsson, H., K. Karlsson, and T. Edlund. 1993. IPF1, a homeodomain-containing transactivator of the insulin gene. EMBO J. 12:4251-4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olbrot, M., J. Rud, L. G. Moss, and A. Sharma. 2002. Identification of β-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc. Natl. Acad. Sci. USA 99:6737-6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Samaras, S. E., L. Zhao, A. Means, E. Henderson, T. A. Matsuoka, and R. Stein. 2003. The islet β cell-enriched RIPE3b1/Maf transcription factor regulates pdx-1 expression. J. Biol. Chem. 278:12263-12270. [DOI] [PubMed] [Google Scholar]
- 28.Sander, M., and M. S. German. 1997. The β cell transcription factors and development of the pancreas. J. Mol. Med. 75:327-340. [DOI] [PubMed] [Google Scholar]
- 29.Stoffers, D. A., J. Ferrer, W. L. Clarke, and J. F. Habener. 1997. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat. Genet. 17:138-139. [DOI] [PubMed] [Google Scholar]
- 30.Stoffers, D. A., N. T. Zinkin, V. Stanojevic, W. L. Clarke, and J. F. Habener. 1997. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat. Genet. 15:106-110. [DOI] [PubMed] [Google Scholar]
- 31.Swaroop, A., J. Z. Xu, H. Pawar, A. Jackson, C. Skolnick, and N. Agarwal. 1992. A conserved retina-specific gene encodes a basic motif/leucine zipper domain. Proc. Natl. Acad. Sci. USA 89:266-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takahashi, S., K. Onodera, H. Motohashi, N. Suwabe, N. Hayashi, N. Yanai, Y. Nabesima, and M. Yamamoto. 1997. Arrest in primitive erythroid cell development caused by promoter-specific disruption of the GATA-1 gene. J. Biol. Chem. 272:12611-12615. [DOI] [PubMed] [Google Scholar]
- 33.Terauchi, Y., H. Sakura, K. Yasuda, K. Iwamoto, N. Takahashi, K. Ito, H. Kasai, H. Suzuki, O. Ueda, N. Kamada, K. Jishage, K. Komeda, M. Noda, Y. Kanazawa, S. Taniguchi, I. Miwa, Y. Akanuma, T. Kodama, Y. Yazaki, T. Kadowaki. 1995. Pancreatic β-cell-specific targeted disruption of glucokinase gene. Diabetes mellitus due to defective insulin secretion to glucose. J. Biol. Chem. 270:30253-30256. [DOI] [PubMed] [Google Scholar]